DOI:
10.1039/C8SC01197D
(Edge Article)
Chem. Sci., 2018,
9, 5949-5956
A high performance catalyst of shape-specific ruthenium nanoparticles for production of primary amines by reductive amination of carbonyl compounds†
Received
14th March 2018
, Accepted 14th June 2018
First published on 18th June 2018
Abstract
The creation of metal catalysts with highly active surfaces is pivotal to meeting the strong economic demand of the chemical industry. Specific flat-shaped pristine fcc ruthenium nanoparticles having a large fraction of atomically active {111} facets exposed on their flat surfaces have been developed that act as a highly selective and reusable heterogeneous catalyst for the production of various primary amines at exceedingly high reaction rates by the low temperature reductive amination of carbonyl compounds. The high performance of the catalyst is attributed to the large fraction of metallic Ru serving as active sites with weak electron donating ability that prevail on the surface exposed {111} facets of flat-shaped fcc Ru nanoparticles. This catalyst exhibits a highest turnover frequency (TOF) of ca. 1850 h−1 for a model reductive amination of biomass derived furfural to furfurylamine and provides a reaction rate approximately six times higher than that of an efficient and selective support catalyst of Ru-deposited Nb2O5 (TOF: ca. 310 h−1).
Introduction
Heterogeneous catalysis based on metal nanoparticles (NPs) constitutes a key technology due to the innumerable applications for the production of indispensable chemical resources.1–5 Metal NPs deposited on substrates with high surface area, i.e. support catalysts, have commonly been used as metal catalysts;6–11 however, they have limited catalytic performance due to the existence of most stable atomic structures (atomically smooth, less active) on the surfaces of these conventional spherical NPs, as predicted by surface science.12–14 The development of structurally controlled metal NPs with catalytically highly active domains (atomically rough) is one of the most crucial issues in the field of heterogeneous catalysis.15–17 Significant attention has recently been devoted towards the size, shape and electronic properties of ruthenium (Ru)-based metal catalysts,18–23 which exhibit significant potential for various applications including ammonia synthesis/decomposition,22,23 Fischer–Tropsch synthesis,24,25 CO oxidation,20,21 and CO/CO2 methanation.19,26,27 In these cases, an activity–structure relationship has been evidenced through the combination of experiments and theory. We recently observed that Ru NPs can be self-organized on calcium amide (Ca(NH2)2) into a specific structure comprising anchored flat Ru particles by strong interaction between Ru and N.22 The Ru/Ca(NH2)2 catalyst exhibited activity for ammonia synthesis that was an order of magnitude higher than that of conventional Ru catalysts.22 While catalysis over Ru NPs with such specific morphology has not been investigated to date, this morphology is also expected to exhibit high catalytic performance over a broad platform for the synthesis of valuable chemicals/intermediates.
Primary amines are key intermediates in the chemical industry with extensive applications in the manufacture of polymers, pharmaceuticals, dyes and detergents.28–32 The catalytic reductive amination of carbonyl compounds with ammonia (NH3) and hydrogen (H2) as the nitrogen source and reductant, respectively, is the most practical and valuable approach for the synthesis of primary amines.33–50 To date, metal catalysts have been reported as the prime candidates for the reductive amination of carbonyl compounds with NH3 and H2, mainly based on several support catalysts of Ni,39,40 Cu,41 Pd,42 Ru,43–47 Rh,44,48 and Pt,49 which only provide moderate reaction rates, even under high H2 pressure (≥4 MPa). Moreover, the selective reductive amination of carbonyl compounds (except simple aryl and alkyl aldehydes) to primary amines is typically hindered by the formation of secondary and tertiary amines and/or undesired further-hydrogenated products. We focus on the specific morphology and atomic structure of a metal catalyst to realize high intrinsic activity and selective response for the quantitative production of primary amines at markedly high reaction rates compared with conventional support catalysts, which is a difficult and challenging goal. Herein, we first report a new specific flat-shaped pristine fcc Ru-NP based catalyst having active sites with weak electron-donating power, which enables highly efficient and selective reductive amination of various carbonyl compounds. The Ru-NP catalyst provides higher reaction rates that are approximately six times that of an efficient support catalyst of Ru-deposited Nb2O5.47
Results and discussion
Characterization of the structure of Ru NPs
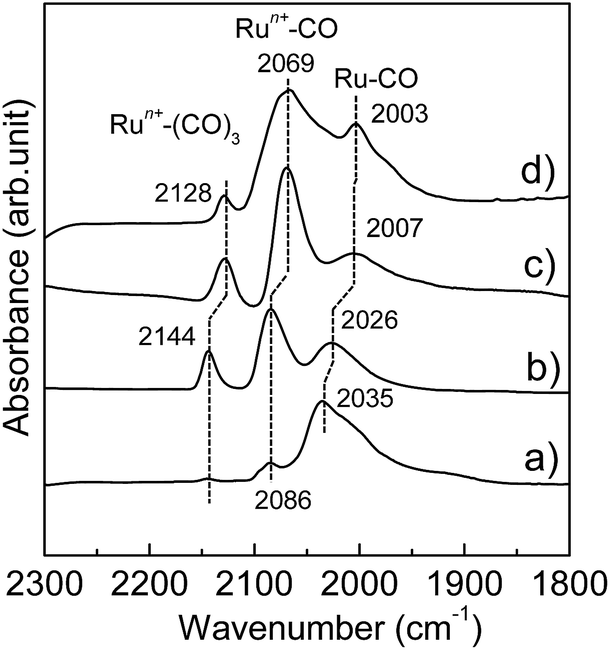
The Ru-NP catalysts were prepared from Ru-deposited Ca(NH2)2. The Ru/Ca(NH2)2 catalyst cannot be used under ambient conditions, because of the extreme instability of Ca(NH2)2. The removal of Ca(NH2)2 was confirmed by the lack of a detectable X-ray photoelectron spectroscopy (XPS) peak in the Ca 2p region for the as-obtained Ru-NPs (see ESI, Fig. S1†). High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) analysis was conducted to investigate the microstructure of the Ru NPs. Fig. 1a shows small and flat Ru patches over a large domain. No conventional round shaped grains of Ru were observed. However, after the removal of Ca(NH2)2, the Ru particles tended to aggregate. The average Ru particle size in Ru/Ca(NH2)2 was estimated to be ca. 2 nm with a narrow particle size distribution.22 In contrast, larger particles were observed after the removal of Ca(NH2)2, as shown in Fig. 1a. This was also confirmed by particle size estimation using CO adsorption on the Ru NP surfaces, as shown in Table 1, where the particle sizes of the Ru-NPs were estimated to exceed 10 nm. The lattice spacing in the Ru NPs (Fig. 1b) was observed to be 2.20 Å, which is close to that (d111 = 2.21 Å) of the face-centered-cubic (fcc) (111) plane of Ru,21 and differs from that for the conventional hexagonal-close-packed (hcp) structure (d002 = 2.142 Å) of Ru particles.18,19 This was also confirmed by powder X-ray diffraction (XRD) analysis of the Ru NPs (Fig. 2a), where broad diffraction peaks due to fcc Ru appear. The morphology of the Ru NPs was not changed significantly compared to that before isolation from Ca(NH2)2 (Fig. 1c). There was also no noteworthy difference in the lattice spacing (fcc, d111 = 2.23 Å) of the Ru NPs pre-formed on the Ca(NH2)2 support22 in Ru/Ca(NH2)2 (Fig. 1d), and the NP structure was successfully preserved in pristine Ru-NPs, without stabilization by the interaction between Ru-sites and N after Ca(NH2)2 removal. The possibility of any trace of N element remaining bound in the pristine Ru-NPs has been ruled out by the lack of detectable XPS peaks in the N 1s region (Fig. S2†). Hence, the Ru-sites bound to Ca(NH2)2 in Ru/Ca(NH2)2 are expected to be available in the pristine Ru-NP catalyst. It has been proposed that the presence of abundant {111} facets is essential in fcc Ru nanocrystals for high activity.21,24 These flat-shaped fcc Ru NPs are composed of a few atomic layers and could be able to generate a large fraction of open {111} facets exposed on the flat NP surfaces, as suggested by the microstructural analysis. Flat fcc Ru NPs (fcc Ru is a metastable phase compared with hcp Ru21) are considered to be stabilized by the strong adhesion between Ru and Ca(NH2)2 during formation, which prevents the growth of large particles (Fig. S3†). This is also supported by the major growth of gradually larger hcp crystallites (alongside anchored fcc Ru particles) for Ru-NPs prepared from 15 and 20 wt% Ru/Ca(NH2)2, as evident from XRD measurements (Fig. 2), presumably due to an increase in the numbers of non-interacting Ru NPs over Ca(NH2)2. The HRTEM image (Fig. S4†) revealed that flat-shaped Ru NPs are intermingled with large particles of hcp Ru grown alongside flat-shaped Ru NPs in Ru-NP (20), Ru-NPs prepared from 20 wt% Ru/Ca(NH2)2.51 Strong adhesion energy between the metal and the support has also been reported as a reason for dominant 2D growth (flat-shaped particles) over 3D growth (spherical particles) of other supported metal NPs.52,53 The Ru NP sizes obtained from CO chemisorption (Table 1) did not vary significantly for different Ru-NPs and Ru-NPs were larger than those analysed from the HAADF-STEM images, due to aggregation of the NPs after the removal of Ca(NH2)2. XPS analysis (Fig. S1†) in the Ru 3d region shows two intense narrow peaks at binding energies of 279.9 and 284.0 eV for Ru 3d5/2 and 3d3/2, respectively. These peaks can be assigned to metallic Ru according to the literature,54–56 which indicates that the composition of the NPs is mainly Ru0. In diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy with CO as a probe molecule, a weak band due to linearly adsorbed CO on partially oxidized Ru [Run+–CO; 2069–2086 cm−1] was observed on CO-adsorbed Ru-NPs; a small fraction of the Ru surface has been oxidized (Fig. 4, see below).
 |
| | Fig. 1 (a) HAADF-STEM and (b) high magnification HAADF-STEM images of Ru-NPs prepared from 10 wt% Ru-deposited Ca(NH2)2. (c) HAADF-STEM and (d) high magnification HAADF-STEM images of 10 wt% Ru/Ca(NH2)2. | |
Table 1 Physicochemical properties and catalytic performance of various Ru catalysts
| Entry |
Catalysta |
S
BET
(m2 g−1) |
S
Ru
(m2 g−1) |
D
(%) |
d
(nm) |
2a yieldf (%) |
TOF (h−1) |
|
Ru-NPs recovered from different weight percentages of Ru loaded on Ca(NH2)2, as shown in parentheses.
Specific surface area (SBET) obtained from N2 sorption analysis.
Ru surface area (SRu).
Ru dispersion (D) determined by CO chemisorption and the stoichiometry of CO/Ru = 0.6 was assumed.23
Ru particle sizes estimated from dispersion.
Reaction conditions: catalyst (0.2 mg for Ru-NPs and Ru-HCP; 20 mg for 1 wt% Ru/Nb2O5 and Ru/SiO2), 1a (0.5 mmol), MeOH (5 mL), NH3 (8 mmol), and H2 (2 MPa), 363 K, 2 h.
TOF calculated from 2a yield at 30 min.
|
| 1g |
Ru-NP (10) |
108 |
46 |
11 |
11.6 |
99 |
1850 |
| 2g |
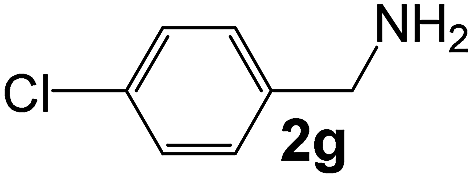
Ru-NP (15) |
101 |
44 |
11 |
11.9 |
95 |
1670 |
| 3g |
Ru-NP (20) |
98 |
49 |
12 |
11.0 |
93 |
1400 |
| 4 |
Ru-HCP |
86 |
62 |
17 |
7.9 |
39 |
290 |
| 5 |
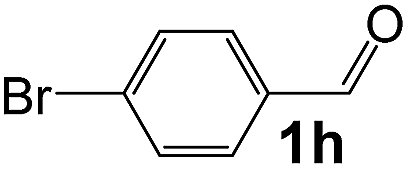
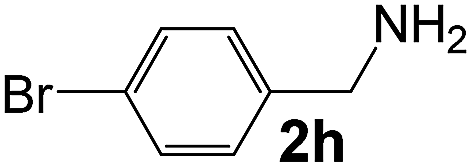
Ru/Nb2O5 |
112 |
88 |
24 |
5.5 |
59 |
310 |
| 6 |
Ru/SiO2 |
260 |
151 |
41 |
3.3 |
62 |
190 |
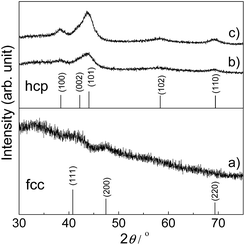 |
| | Fig. 2 XRD patterns of Ru-NP samples prepared from (a) 10 wt%, (b) 15 wt% and (c) 20 wt% Ru-deposited Ca(NH2)2. JCPDF card numbers are (hcp) 01-070-0274 and (fcc) 01-088-2333. | |
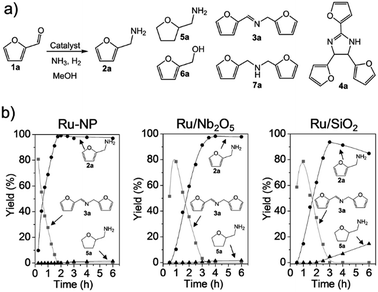 |
| | Fig. 3 (a) Catalytic reductive amination of furfural (1a) to furfurylamine (2a) and related byproducts.46–48N-furfurylidenefurfurylamine (3a), 2,4,5-tris(2-furyl)imidazoline (4a), tetrahydrofurfurylamine (5a), furfuryl alcohol (6a) and difurfurylamine (7a) are the main byproducts. (b) Time courses for the reductive amination of 1a over Ru-NP, Ru/Nb2O5 and Ru/SiO2 catalysts (Ru: 1 wt% for Ru/Nb2O5 and Ru/SiO2). Reaction conditions: catalyst (0.2 mg for Ru-NP; 20 mg for Ru/Nb2O5 and Ru/SiO2), 1a (0.5 mmol), MeOH (5 mL), NH3 (8 mmol), and H2 (2 MPa), 363 K. | |
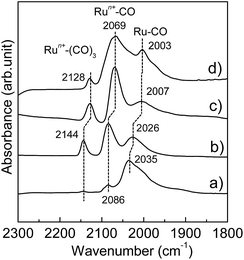 |
| | Fig. 4 Difference DRIFT spectra for adsorption of CO onto (a) Ru-NP, (b) Ru/Nb2O5, (c) Ru/SiO2 and (d) Ru-HCP samples at 103 K. | |
Catalyst screening for model reductive amination of furfural
The conversion of biomass-derived57,58 furfural (1a) to furfurylamine (2a) was investigated as a model reaction for reductive amination (Fig. 3a). 1a cannot be completely converted into 2a over most heterogeneous catalysts (except Ru-deposited Nb2O5, which gives a 2a yield of 99%47), because the formation of 2a is thermodynamically unfavourable compared to other byproducts (see the ESI†). Ru-NPs prepared from Ru-deposited Ca(NH2)2 samples with different loading amounts of Ru (10, 15 and 20 wt%) were tested for initial screening of the catalyst (Tables 1 and S1†). Ru-NPs prepared from 10 wt% Ru-loaded Ca(NH2)2 provided an almost quantitative yield (99%) of 2a in 2 h without significant formation of other byproducts (Table S1,† entry 1). Ru-NPs prepared from 15 and 20 wt% Ru-loaded Ca(NH2)2 also gave 2a in high yield (95 and 93%, respectively), although a ring hydrogenated product (tetrahydrofurfurylamine; 5a) was formed to a significant extent with a total yield of 3–5% along with a small amount of secondary amine (7a) through hydrogenation of the dimeric imine 3a (Table S1,† entries 2 and 3). Turnover frequencies (TOFs) for the formation of 2a were estimated using the dispersion of surface Ru atoms obtained from CO chemisorption (Table 1).23 The less-selective reductive amination and gradually decreasing TOFs with Ru-NPs obtained from higher Ru loading are perhaps due to the effect of a larger amount of hcp Ru particles with fcc Ru, as suggested by XRD analysis. In fact, a pristine hcp Ru NP (Ru-HCP, see ESI Fig. S5†) catalyst24 gives a 2a yield of only 39% at 2 h with a much lower TOF (Table 1) and large amount of byproducts (Table S1,† entries 4 and 5). In the case of Ru/Nb2O5 and Ru/SiO2, the 2a yield did not reach 70% at 2 h. Ru-NPs prepared from 10 wt% Ru-loaded Ca(NH2)2 were employed in the following experiments.
Performance of Ru-NPs beyond conventional efficient catalysts
The time course for the reductive amination of 1a over Ru-NPs was compared with those for efficient supported metal catalysts of Ru/Nb2O5 and Ru/SiO2 (Fig. 3b).47,59 In the case of Ru-NPs, 1a was rapidly converted, and only after approximately 15 min the yield of dimeric imine 3a reached a maximum. The yield of 2a gradually increased to 99% at approximately 105 min with a decrease in the yield of 3a. Hydrogenation products such as 5a, 6a and 7a were not formed to any substantial extent, even after the catalytic test was conducted for 6 h. Ru/Nb2O5 also gave a 2a yield of 98% at 4 h without significant progress of other side-reactions, although Ru/Nb2O5 had a much lower rate of 2a formation than Ru-NPs. This is also supported by the production rate of dimeric imine 3a. 3a was observed even after 3 h over Ru/Nb2O5. On the other hand, it disappeared completely before 2 h in the case of Ru-NPs. Intermediate 3a produced by the reversible reaction of 1a and 2a is expected to be selectively converted into 2a under the reaction conditions.47 The Ru/SiO2 catalyst exhibited a similar reaction rate to that of Ru/Nb2O5; however, in a sharp contrast, subsequent hydrogenation of 2a into 5a was observed. It should be noted that an approximately 40% yield of 2a was achieved within 30 min over Ru-NPs, which is an approximately three times higher rate than that for a similar yield (at 90 min) of 2a obtained over either Ru/Nb2O5 or Ru/SiO2. 4a is formed when the hydrogenation rate of the intermediate furfurylimine (8a) to 2a is not sufficiently high (see the ESI†).47 The formation of 4a was not observed at all over Ru-NPs, whereas the yield of 4a over Ru/SiO2 exceeded 6%. The TOF for 2a over Ru-NPs (1850 h−1) was much higher than that over Ru/Nb2O5 (310 h−1) and Ru/SiO2 (190 h−1) irrespective of the lower surface area of Ru in the Ru-NP catalyst (Table 1), which signifies the presence of highly active Ru-sites on the surface of the flat-shaped Ru-NPs. TOF estimation was also carried out on the basis of active site-poisoning by using 1,10-phenanthroline (see ESI, Fig. S6†),61,62 and the TOFs for Ru-NP (flat-shaped fcc Ru catalyst), Ru/Nb2O5 (efficient supported metal catalyst of hcp Ru) and Ru-HCP (conventional unsupported hcp Ru nanoparticles) were estimated to be 1580 h−1, 370 h−1 and 270 h−1 respectively. These results are consistent with those estimated by CO adsorption (Table 1). To the best of our knowledge, the Ru-NP catalyst exhibited the highest TOF among the metal catalysts reported to date for the reductive amination of 1a to produce 2a (Table 2).45,47,48 Reuse experiments with the Ru-NP catalyst for the reductive amination of 1a (Fig. S7†) showed no noticeable decrease in activity, even after four reuses, which indicates the robustness of the catalyst.
Table 2 Reductive amination of furfural (1a) to furfurylamine (2a) over various metal catalysts
| Entry |
Catalyst |
Temp. (°C) |
pH2 (MPa) |
Time (h) |
2a yield (%) |
TOF (h−1) |
Ref. |
| 1 |
Ru-NP |
90 |
2 |
2 |
99 |
1850 |
This work |
| 2 |
Ru-HCP |
90 |
2 |
5 |
71 |
290 |
This work |
| 3 |
Ru/Nb2O5 |
90 |
2 |
4 |
98 |
310 |
This work |
| 4 |
Ru/SiO2 |
90 |
2 |
4 |
91 |
190 |
This work |
| 5 |
Ru/Nb2O5 |
90 |
4 |
2 |
99 |
520 |
47
|
| 6 |
Ru/TiO2 |
90 |
4 |
4 |
97 |
110 |
47
|
| 7 |
Rh/Al2O3 |
80 |
2 |
2 |
92 |
990 |
48
|
| 8 |
Ru/γ-Al2O3 |
80 |
3 |
2 |
75 |
100 |
45
|
| 9 |
RANEY® Ni |
100 |
6 |
3 |
56 |
1 |
60
|
| 10 |
CoReMo |
75 |
9 |
3 |
88 |
— |
60
|
| 11 |
Ru/HAP |
100 |
0.3 |
2 |
60 |
6 |
46
|
Origin of the high performance catalysis over Ru-NPs
The Ru-NP catalyst is largely superior to Ru/Nb2O5 in terms of the rate of 2a formation. However, there is no significant difference in selectivity between the catalysts. A recent report revealed that the yield of 2a from 1a is dependent on the electron-donating power of Ru on different supports.47 Selective reductive amination over Ru/Nb2O5 arises from the weak electron-donating capability of Ru particles on the Nb2O5 surface.47 Apart from this, only an increase of electron donation to Ru from the support materials was demonstrated.63–65 However, high performance catalysis over Ru-NPs cannot be explained by such a support-to-metal electronic contribution. To understand the phenomenon, the electronic state of Ru in the Ru-NPs was examined and compared with that in Ru-HCP, Ru/Nb2O5 and Ru/SiO2 using DRIFT spectroscopy with CO as a probe molecule. As shown in Fig. 4, bands were assignable to linearly adsorbed CO on Ru0 [Ru0–CO; 2003–2035 cm−1], linearly adsorbed CO on partially oxidized Ru [Run+–CO; 2069–2086 cm−1] and tricarbonyl species on partially oxidized Ru [Run+–(CO)3; 2128–2144 cm−1].66,67 The bands due to CO bonded to Ru/Nb2O5 (2026, 2086, and 2144 cm−1) were blue-shifted more than those of Ru-HCP (2003, 2069, and 2128 cm−1) and Ru/SiO2 (2007, 2069, and 2128 cm−1),47 due to a decrease of the electron density of Ru by Nb2O5 as an acidic (weak electron donating) material.68 In the case of Ru-NPs, the band for Ru0–CO (2035 cm−1) was slightly more blue-shifted than that for Ru/Nb2O5 (2026 cm−1), although there was no significant difference in the position of the bands for Run+–CO (2086 cm−1) and Run+–(CO)3 (2144 cm−1), which indicates that both the catalysts have comparably weak electron-donating power and would promote selective reductive amination. However, in sharp contrast, the Ru0–CO band was observed with much higher intensity than Run+–CO and Run+–(CO)3 for the Ru-NPs, relative to those observed for Ru-HCP, Ru/Nb2O5 and Ru/SiO2. This implies a noticeably large fraction of metallic Ru (rather than oxidized Ru species) as the active sites on the Ru-NPs, which is also in agreement with the XPS analysis. Metallic Ru-species are considered to play an important role for efficient reductive amination. The reductive amination of 1a did not proceed over a catalyst of RuO2-deposited Nb2O5. However, the high TOF obtained for the Ru-NPs cannot be explained by the large fraction of metallic Ru and the electronic state of Ru-NP. One possible explanation for the catalysis over the Ru-NPs is the atomic structure of the flat-shaped Ru NPs. It has been demonstrated by computational studies that 2D (flat) Ru particles are catalytically more active than hemispherical Ru particles.23 From computational studies and experiments, it was recently established that the CO dissociation barrier for Fischer–Tropsch synthesis is sufficiently low on the abundant {111} facets of fcc Ru nanocrystals, which results in high mass-specific catalytic activity.24 The CO dissociation barrier on fcc {111} is 1.13 eV, whereas it varies from 0.94 to 2.37 eV for different facets of hcp Ru.24 The hcp {0001} facet has a lower barrier for CO dissociation, but its site density is rather low.19,24 On the other hand, fcc Ru has abundant {111} facets with a slightly higher barrier.24 Recently, higher activity of fcc Ru compared to hcp Ru was also reported in a hydrogenation reaction.69 This is also clearly demonstrated in the TOF obtained with the Ru-NP catalyst (Table 1). The XRD patterns in Fig. 2 reveal that hcp Ru in the obtained Ru-NPs is increased with the amount of Ru-loading on Ca(NH2)2; excess Ru-loading forms hcp Ru on flat-shaped fcc Ru and decreases the surface exposed {111} facets of fcc Ru. An increase in hcp Ru on Ru-NPs decreases the TOF of Ru-NPs for the reductive amination of furfural, as shown in Table 1. For flat-shaped pristine Ru-NPs, the large fraction of surface exposed {111} facets (after Ca(NH2)2 removal, Ru-sites become completely free) and electronic state of the Ru-sites possibly affect the adsorption of the substrates and/or the activity of adsorbed hydrogen atoms, which could result in high intrinsic activity for highly efficient reductive amination.
Substrate scope for high performance reductive amination
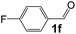
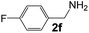
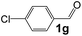
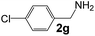
Ru-NPs are also effective for the reductive amination of several carbonyl compounds. For example, an important biomass-derived aldehyde, 5-hydroxymethylfurfural (1b; a platform chemical for synthesis of several derivatives having potential industrial demand),57,58,70 is efficiently converted into 5-hydroxymethylfurfurylamine (2b) in 95% yield (Fig. 5). The reaction proceeds with a much higher rate under 4 MPa H2 to give a 92% yield of 2b in only 1.5 h, which is approximately 2.5 times higher than that recently reported for an efficient supported metal catalyst (Ru/Nb2O5) employed under identical conditions.47 A broad range of aldehydes and ketones that contain different reduction sensitive functional groups (Table 3), such as alkyl/aryl substituents, halogens and heterocycles, were also successfully tolerated, and the corresponding primary amines were readily produced in noticeably high yields, mostly irrespective of the electronic effects from the functional groups. In comparison with the efficient supported metal catalyst Ru/Nb2O5 (Table 3), Ru-NP showed prominently higher TOFs and provided a reaction rate approximately 5–7 times higher for production of various primary amines by reductive amination of these carbonyl compounds.
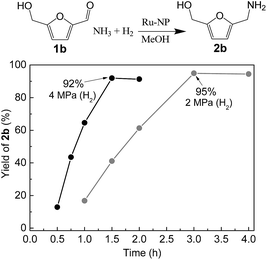 |
| | Fig. 5 Time courses of reductive amination of 5-hydroxymethylfurfural (1b) over Ru-NP. Reaction conditions: catalyst (0.2 mg), 1b (0.5 mmol), MeOH (5 mL), NH3 (8 mmol), and H2 (2 and 4 MPa), 363 K. | |
Table 3 Comparison of primary amine production from various aldehydes and ketones over the Ru-NP and Ru/Nb2O5 catalystsa
| Entry |
Substrate |
Time (h) |
Product |
Yieldb (%) |
TOF (h−1) |
R
Ru-NP
|
| Ru-NP |
Ru/Nb2O5 |
Ru-NP |
Ru/Nb2O5 |
|
Reaction conditions: catalyst (0.2 mg for Ru-NPs; 20 mg for 1 wt% Ru/Nb2O5), substrate (0.5 mmol), MeOH (5 mL), NH3 (8 mmol), H2 (2 MPa), 363 K.
GC yield.
R
Ru-NP defines the ratio of TOFs for Ru-NP and Ru/Nb2O5.
|
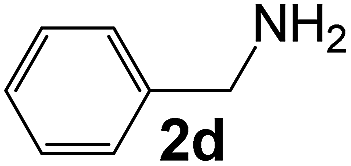
| 1 |
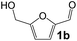
|
3 |
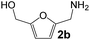
|
95 |
39 |
730 |
140 |
5.2 |
| 2 |
|
2 |
|
68 |
29 |
780 |
150 |
5.2 |
| 3 |
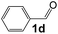
|
4 |
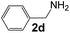
|
95 |
34 |
550 |
90 |
6.1 |
| 4 |
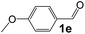
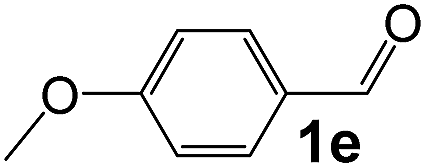
|
4 |
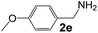
|
92 |
35 |
530 |
90 |
5.9 |
| 5 |
|
2 |
|
94 |
30 |
1080 |
160 |
6.7 |
| 6 |
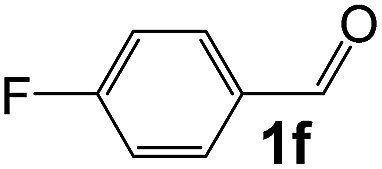
|
2 |
|
90 |
28 |
1030 |
150 |
6.9 |
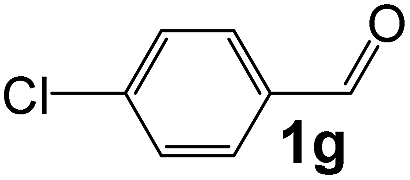
| 7 |
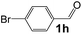
|
2 |
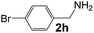
|
93 |
33 |
1070 |
170 |
6.3 |
| 8 |
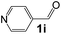
|
2 |
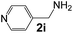
|
88 |
31 |
1010 |
160 |
6.3 |
| 9 |
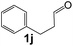
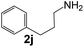
|
2 |
|
97 |
35 |
1110 |
180 |
6.2 |
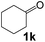
| 10 |
|
1 |
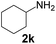
|
97 |
43 |
2230 |
450 |
5.0 |
| 11 |
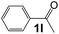
|
3 |
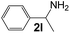
|
98 |
38 |
750 |
130 |
5.8 |
Conclusions
In summary, flat-shaped pristine fcc Ru NPs, consisting of a large fraction of atomically active {111} facets on their flat surfaces, were constructed as a high performance catalyst. The specific flat-shaped morphology and surface electronic structure of Ru NPs clearly contribute to their outstanding catalytic performance, as reflected by the exceptionally high TOFs observed for the reductive amination of several carbonyl compounds, compared to conventional efficient supported metal catalysts. The Ru-NP catalyst also exhibits excellent durability and selectivity during prolonged recycling. Shape-specific Ru NPs are available as a benchmark catalyst for the efficient production of various primary amines, particularly the important biomass-based amine intermediates. The present Ru-NP catalyst is also expected to be widely applicable for the metal-catalyzed production of several indispensable chemical resources, which is still a challenge for the chemical industry with conventional supported metal catalysts.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the ALCA Grant (JPMJAL1205) of the Japan Science and Technology Agency (JST), and Grants-in-Aid for Japan Society for the Promotion of Science (JSPS) Fellows and for Scientific Research from the Ministry of Education, Culture, Science, Sports, and Technology (MEXT) of Japan (18H05251).
Notes and references
- L. He, F. Weniger, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 12582–12594 CrossRef PubMed.
- B. C. Gates, Chem. Rev., 1995, 95, 511–522 CrossRef.
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef PubMed.
- P. Munnik, P. E. de Jongh and K. P. de Jong, Chem. Rev., 2015, 115, 6687–6718 CrossRef PubMed.
- M. Hara, M. Kitano and H. Hosono, ACS Catal., 2017, 7, 2313–2324 CrossRef.
- S. Crossley, J. Faria, M. Shen and D. E. Resasco, Science, 2010, 327, 68–72 CrossRef PubMed.
- N. Zheng and G. D. Stucky, J. Am. Chem. Soc., 2006, 128, 14278–14280 CrossRef PubMed.
- C. Xiao, T.-W. Goh, Z. Qi, S. Goes, K. Brashler, C. Perez and W. Huang, ACS Catal., 2016, 6, 593–599 CrossRef.
- R. Fang, H. Liu, R. Luque and Y. Li, Green Chem., 2015, 17, 4183–4188 RSC.
- P. Pachfule, X. Yang, Q.-L. Zhu, N. Tsumori, T. Uchida and Q. Xu, J. Mater. Chem. A, 2017, 5, 4835–4841 RSC.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S.-W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef PubMed.
- C. T. Campbell, Acc. Chem. Res., 2013, 46, 1712–1719 CrossRef PubMed.
- C. T. Campbell and J. R. V. Sellers, Chem. Rev., 2013, 113, 4106–4135 CrossRef PubMed.
- R. Schlögl, Angew. Chem., Int. Ed., 2015, 54, 3465–3520 CrossRef PubMed.
- K. An and G. A. Somorjai, ChemCatChem, 2012, 4, 1512–1524 CrossRef.
- A. T. Bell, Science, 2003, 299, 1688–1691 CrossRef PubMed.
- G. Ertl, Angew. Chem., Int. Ed., 2008, 47, 3524–3535 CrossRef PubMed.
- J. Ohyama, T. Sato, Y. Yamamoto, S. Arai and A. Satsuma, J. Am. Chem. Soc., 2013, 135, 8016–8021 CrossRef PubMed.
- A.-X. Yin, W.-C. Liu, J. Ke, W. Zhu, J. Gu, Y.-W. Zhang and C.-H. Yan, J. Am. Chem. Soc., 2012, 134, 20479–20489 CrossRef PubMed.
- S. H. Joo, J. Y. Park, J. R. Renzas, D. R. Butcher, W. Huang and G. A. Somorjai, Nano Lett., 2010, 10, 2709–2713 CrossRef PubMed.
- K. Kusada, H. Kobayashi, T. Yamamoto, S. Matsumura, N. Sumi, K. Sato, K. Nagaoka, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2013, 135, 5493–5496 CrossRef PubMed.
- Y. Inoue, M. Kitano, K. Kishida, H. Abe, Y. Niwa, M. Sasase, Y. Fujita, H. Ishikawa, T. Yokoyama, M. Hara and H. Hosono, ACS Catal., 2016, 6, 7577–7584 CrossRef.
- A. M. Karim, V. Prasad, G. Mpourmpakis, W. W. Lonergan, A. I. Frenkel, J. G. Chen and D. G. Vlachos, J. Am. Chem. Soc., 2009, 131, 12230–12239 CrossRef PubMed.
- W.-Z. Li, J.-X. Liu, J. Gu, W. Zhou, S.-Y. Yao, R. Si, Y. Guo, H.-Y. Su, C.-H. Yan, W.-X. Li, Y.-W. Zhang and D. Ma, J. Am. Chem. Soc., 2017, 139, 2267–2276 CrossRef PubMed.
- J. Kang, S. Zhang, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2009, 48, 2565–2568 CrossRef PubMed.
- A. Chen, T. Miyao, K. Higashiyama, H. Yamashita and M. Watanabe, Angew. Chem., Int. Ed., 2010, 49, 9895–9898 CrossRef PubMed.
- T. Abe, M. Tanizawa, K. Watanabe and A. Taguchi, Energy Environ. Sci., 2009, 2, 315–321 RSC.
- K. S. Hayes, Appl. Catal., A, 2001, 221, 187–195 CrossRef.
- S. Imm, S. Bähn, L. Neubert, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 8126–8129 CrossRef PubMed.
- B. Chen, U. Dingerdissen, J. G. E. Krauter, H. G. J. Lansink Rotgerink, K. Möbus, D. J. Ostgard, P. Panster, T. H. Riermeier, S. Seebald, T. Tacke and H. Trauthwein, Appl. Catal., A, 2005, 280, 17–46 CrossRef.
- M. Pera-Titus and F. Shi, ChemSusChem, 2014, 7, 720–722 CrossRef PubMed.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef PubMed.
-
P. Roose, K. Eller, E. Henkes, R. Rossbacher and H. Höke, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2015, pp. 1–55 Search PubMed.
- J. L. Klinkenberg and J. F. Hartwig, Angew. Chem., Int. Ed., 2011, 50, 86–95 CrossRef PubMed.
- J. Kim, H. J. Kim and S. Chang, Eur. J. Org. Chem., 2013, 3201–3213 CrossRef.
- A. K. Holzer, K. Hiebler, F. G. Mutti, R. C. Simon, L. Lauterbach, O. Lenz and W. Kroutil, Org. Lett., 2015, 17, 2431–2433 CrossRef PubMed.
- T. Gross, A. M. Seayad, M. Ahmad and M. Beller, Org. Lett., 2002, 4, 2055–2058 CrossRef PubMed.
- J. Gallardo-Donaire, M. Ernst, O. Trapp and T. Schaub, Adv. Synth. Catal., 2016, 358, 358–363 CrossRef.
- C. F. Winans, J. Am. Chem. Soc., 1939, 61, 3564–3565 CrossRef.
- C. Schäfer, B. Nişanci, M. P. Bere, A. Daştan and B. Török, Synthesis, 2016, 48, 3127–3133 CrossRef.
- K. V. R. Chary, K. K. Seela, D. Naresh and P. Ramakanth, Catal. Commun., 2008, 9, 75–81 CrossRef.
- A. W. Heinen, J. A. Peters and H. v. Bekkum, Eur. J. Org. Chem., 2000, 2501–2506 CrossRef.
- S. Gomez, J. A. Peters, J. C. van der Waal, W. Zhou and T. Maschmeyer, Catal. Lett., 2002, 84, 1–5 CrossRef.
- J. Bódis, L. Lefferts, T. E. Müller, R. Pestman and J. A. Lercher, Catal. Lett., 2005, 104, 23–28 CrossRef.
- B. Dong, X. Guo, B. Zhang, X. Chen, J. Guan, Y. Qi, S. Han and X. Mu, Catalysts, 2015, 5, 2258 CrossRef.
- S. Nishimura, K. Mizuhori and K. Ebitani, Res. Chem. Intermed., 2016, 42, 19–30 CrossRef.
- T. Komanoya, T. Kinemura, Y. Kita, K. Kamata and M. Hara, J. Am. Chem. Soc., 2017, 139, 11493–11499 CrossRef PubMed.
- M. Chatterjee, T. Ishizaka and H. Kawanami, Green Chem., 2016, 18, 487–496 RSC.
- Y. Nakamura, K. Kon, A. S. Touchy, K.-i. Shimizu and W. Ueda, ChemCatChem, 2015, 7, 921–924 CrossRef.
- S. R. Kirumakki, M. Papadaki, K. V. R. Chary and N. Nagaraju, J. Mol. Catal. A: Chem., 2010, 321, 15–21 CrossRef.
- When CO was adsorbed on Ru-NP (20), a band was observed at 2027 cm−1 with a shoulder (ca. 2035 cm−1) assignable to CO adsorbed on flat-shaped fcc Ru NPs (Fig. S8†). These results suggest that flat-shaped Ru NPs are mixed with large hcp Ru NPs in Ru-NP (20); Ru-NP (20) has a higher TOF than Ru-HCP (Table 1) although the diffraction peaks due to hcp Ru are mainly observed in the XRD pattern of Ru-NP (20)..
- X. Shao, S. Prada, L. Giordano, G. Pacchioni, N. Nilius and H.-J. Freund, Angew. Chem., Int. Ed., 2011, 50, 11525–11527 CrossRef PubMed.
- J. Liu, ChemCatChem, 2011, 3, 934–948 CrossRef.
- E. A. Carbonio, M. J. Prieto, A. d. Siervo and R. Landers, J. Phys. Chem. C, 2014, 118, 28679–28688 CrossRef.
- K. Qadir, S. H. Joo, B. S. Mun, D. R. Butcher, J. R. Renzas, F. Aksoy, Z. Liu, G. A. Somorjai and J. Y. Park, Nano Lett., 2012, 12, 5761–5768 CrossRef PubMed.
- K. Qadir, S. M. Kim, H. Seo, B. S. Mun, F. A. Akgul, Z. Liu and J. Y. Park, J. Phys. Chem. C, 2013, 117, 13108–13113 CrossRef.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef PubMed.
- M. Hara, K. Nakajima and K. Kamata, Sci. Technol. Adv. Mater., 2015, 16, 034903 CrossRef PubMed.
- The kinetic parameters were estimated by fitting the time course of the concentration of 3a and 2a on the assumption that the reaction is sequential. Attempts to reproduce the reaction profile were unsuccessful likely because the present reductive amination cannot be simply explained by the sequential reaction and the reaction mechanism is complicated as mentioned in our previous paper. The detailed reaction mechanism and kinetic parameters including the reaction order and activation energy of each step are currently under investigation..
-
T. Ayusawa, S. Mori, T. Aoki and R. Hamana, US Pat. 4598159, 1986.
- E. Bayram, J. C. Linehan, J. L. Fulton, J. A. S. Roberts, N. K. Szymczak, T. D. Smurthwaite, S. Özkar, M. Balasubramanian and R. G. Finke, J. Am. Chem. Soc., 2011, 133, 18889–18902 CrossRef PubMed.
- E. Bayram and R. G. Finke, ACS Catal., 2012, 2, 1967–1975 CrossRef.
- S. Kanbara, M. Kitano, Y. Inoue, T. Yokoyama, M. Hara and H. Hosono, J. Am. Chem. Soc., 2015, 137, 14517–14524 CrossRef PubMed.
- T.-N. Ye, J. Li, M. Kitano, M. Sasase and H. Hosono, Chem. Sci., 2016, 7, 5969–5975 RSC.
- M. Kitano, S. Kanbara, Y. Inoue, N. Kuganathan, P. V. Sushko, T. Yokoyama, M. Hara and H. Hosono, Nat. Commun., 2015, 6, 6731 CrossRef PubMed.
- S. Y. Chin, C. T. Williams and M. D. Amiridis, J. Phys. Chem. B, 2006, 110, 871–882 CrossRef PubMed.
- E. Guglielminotti, F. Boccuzzi, M. Manzoli, F. Pinna and M. Scarpa, J. Catal., 2000, 192, 149–157 CrossRef.
- K. Nakajima, R. Noma, M. Kitano and M. Hara, J. Phys. Chem. C, 2013, 117, 16028–16033 CrossRef.
- Y. Yancai, H. D. Sheng, L. Yue, F. Xiaoqian, W. Xin, Y. Peiqun, H. Xun, Z. Gang, W. Yuen and L. Yadong, Angew. Chem., 2016, 128, 5591–5595 CrossRef.
- B. Liu and Z. Zhang, ChemSusChem, 2016, 9, 2015–2036 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, one table, XPS spectrum, TEM image, reuse experiment data, and computational free energy diagram. See DOI: 10.1039/c8sc01197d |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Yasunori
Inoue
bc,
Masato
Sasase
cd,
Masaaki
Kitano
*a,
Yasunori
Inoue
bc,
Masato
Sasase
cd,
Masaaki
Kitano