
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBPTI folding revisited: switching a disulfide into methylene thioacetal reveals a previously hidden path†
Reem
Mousa
,
Shifra
Lansky
,
Gil
Shoham
and
Norman
Metanis
 *
*
Institute of Chemistry, The Hebrew University of Jerusalem, Jerusalem, 91904, Israel. E-mail: metanis@mail.huji.ac.il
First published on 2nd May 2018
Abstract
Bovine pancreatic trypsin inhibitor (BPTI) is a 58-residue protein that is stabilized by three disulfide bonds at positions 5–55, 14–38 and 30–51. Widely studied for about 50 years, BPTI represents a folding model for many disulfide-rich proteins. In the study described below, we replaced the solvent exposed 14–38 disulfide bond with a methylene thioacetal bridge in an attempt to arrest the folding pathway of the protein at its two well-known intermediates, N′ and N*. The modified protein was expected to be unable to undergo the rate-determining step in the widely accepted BPTI folding mechanism: the opening of the 14–38 disulfide bond followed by rearrangements that leads to the native state, N. Surprisingly, instead of halting BPTI folding at N′ and N*, we uncovered a hidden pathway involving a direct reaction between the N* intermediate and the oxidizing reagent glutathione (GSSG) to form the disulfide-mixed intermediate N*–SG, which spontaneously folds into N. On the other hand, N′ was unable to fold into N. In addition, we found that the methylene thioacetal bridge enhances BPTI stability while fully maintaining its structure and biological function. These findings suggest a general strategy for enhancing protein stability without compromising on function or structure, suggesting potential applications for future therapeutic protein production.
Introduction
Protein folding still represents one of the most mysterious puzzles in science, and mistakes in this process (i.e. protein misfolding) are linked to many pathological conditions.1 Despite significant advances in recent years, the study of protein folding is still considered rather challenging. It is less complicated to study in vitro oxidative protein folding, in which the cysteine residues in the reduced, unfolded protein are oxidized to form disulfide bonds and a three-dimensional native state.2–4 Through alkylation or acid quenching, intermediates may be isolated and characterized, providing step-by-step snapshots of the entire oxidative folding mechanism. Beginning with RNase A,5 this method has been successfully applied to study a wide range of proteins and has provided most of the current knowledge related to oxidative protein folding.4A protein that has been studied intensively for about 50 years is bovine pancreatic trypsin inhibitor (BPTI), which represents a folding model for many disulfide-rich proteins.2,3 BPTI is a small protein, consisting of 58 residues and containing three disulfide bonds, positioned between the 5–55, 14–38 and 30–51 cysteine pairs (Fig. 1a). The oxidative folding of BPTI, mapped as a bifurcated pathway (Fig. 1b), is widely believed to proceed predominantly through the formation of native intermediates and native disulfide bonds, that is, via intermediates that closely resemble the native structure, containing disulfide bonds that are present in the native state, N.6–8 According to the classical mechanism, two major intermediates are formed during the folding process, each containing two disulfide bonds, N′ [14–38; 30–51] and N* [5–55; 14–38].6–10 While N* has been referred to as a dead-end9 or non-productive11 intermediate, N′ was proposed to rearrange into a third two-disulfide containing intermediate NSHSH [5–55; 30–51] (Fig. 1b) through substantial unfolding and intramolecular thiol–disulfide exchange reactions. This rate-determining rearrangement step is believed to be independent of an external oxidant.6,9 Notably, the formation of NSHSH, which is oxidized readily to form the N state, requires breaking the solvent exposed 14–38 disulfide bond in N′ and N*. Based on these assumptions, we speculated that locking the 14–38 disulfide bond with a stable methylene thioacetal bridge (MT, also known as methylenedithioether12) (Fig. 1c) should halt the folding pathway at the two corresponding trapped intermediates, N′ and N*, preventing the formation of N (Fig. 1d).
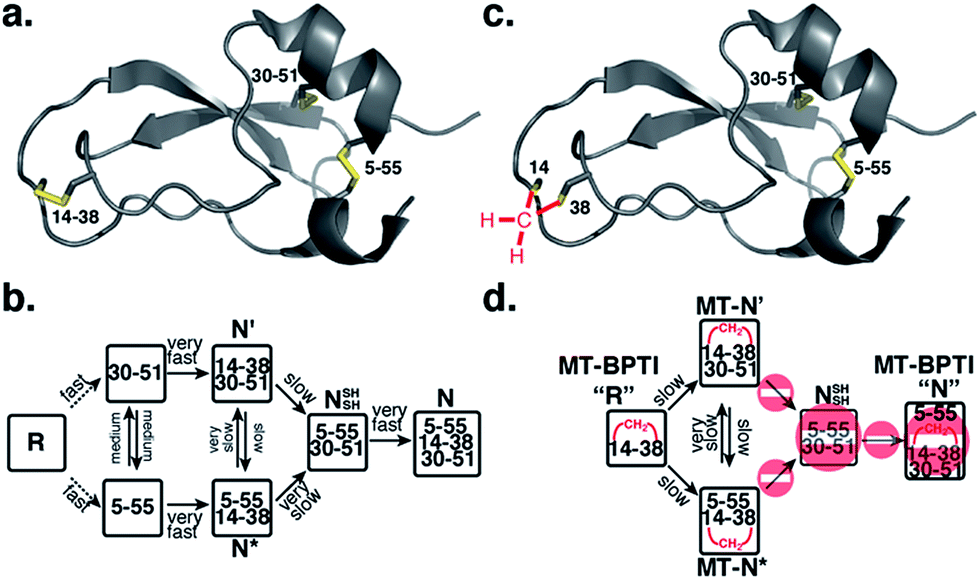 | ||
| Fig. 1 Structure and folding of WT-BPTI and MT-BPTI. (a) A secondary structure representation of the reported crystal structure of WT-BPTI (PDB:1BPI); (b) the proposed folding mechanism of reduced WT-BPTI, as suggested by Weissman and Kim;7 (c) the proposed 3D model of folded MT-BPTI; the methylene thioacetal (MT) moiety is shown in red; and (d) the predicted folding mechanism of reduced MT-BPTI. | ||
In BPTI, 14–38 is the only solvent exposed disulfide bond. It is the first disulfide to form during oxidative folding but rapidly rearranges into the other disulfide bonds 30–51 and 5–55,10,13 and the first to be reduced in the reductive unfolding.14–17 Crieghton has shown that when Cys14 and Cys38 were alkylated with iodoacetic acid, the modified protein was more stable to unfolding, but after complete unfolding, the protein could not readily regain the native state.18 Goldenberg and coworkers have tested the global conformation of BPTI and the Tyr35Gly mutant in which the 14–38 disulfide was reduced and alkylated with methyl iodide to give the doubly methylated protein at Cys14 and Cys38.19 The overall conformation of the two proteins was retained, and this modification was reported to have relatively little influence on backbone motion.19 Additionally, we have shown previously20 that the substitution of the 14–38 disulfide bond with a diselenide led to accelerated folding. However, the final yield of the formed N state was significantly lower, which was attributed to precipitation, a result of aggregation from the solvent-exposed and rather reactive 14–38 diselenide bond.
In the present work we demonstrate that replacing the solvent exposed 14–38 disulfide bond in BPTI with a stable MT moiety reveals a hidden pathway for the formation of the N state. Subsequent studies on MT-BPTI suggest that the MT substitution is an excellent disulfide bond mimic, which has great potential to enhance the stability of therapeutic disulfide-rich proteins, while retaining their overall structure and full biological activity.
Results
Preparation of reduced WT-BPTI and reduced MT-BPTI
The oxidative folding of BPTI has been extensively studied over the years and is generally accepted to follow a bifurcated pathway, as described in Fig. 1b.7 Of particular interest is the conversion of the two trapped intermediates, N′ and N*, into NSHSH, as this is the rate-determining step and requires substantial unfolding, coupled with opening of the solvent exposed 14–38 disulfide bond (Fig. 1b).In order to examine this proposed mechanism of folding, and in an attempt to confirm it unequivocally, we undertook a new synthetic approach, converting the 14–38 disulfide bond into a stable methylene thioacetal (MT) moiety. Such a conversion, in principle, should halt the folding pathway at the N′ and N* intermediates, preventing the final folding into the native state, N (Fig. 1d).
The objective of this synthetic effort was to produce the reduced form of MT-BPTI, in which the MT moiety is located between Cys14 and Cys38, while the other Cys residues are in the reduced form (Fig. S1†). Toward this end, we dissolved folded WT-BPTI in a phosphate buffer at pH 8 and treated it with 1.5 equiv. of a reductant (tris(2-carboxyethyl)phosphine, TCEP) (Fig. S1 and S2†). The progress of the reaction was monitored by HPLC and ESI-MS, which indicated that within 3 h, the 14–38 disulfide was selectively reduced, giving NSHSH (Fig. S2b†).19 Upon completion of the reduction step, a large excess of diiodomethane (CH2I2) was added and the reaction was stirred at 35 °C under argon for 22 h.12 The resulting MT-BPTI protein was isolated by HPLC (Fig. S2c†) in 70% yield, which co-eluted with minor amounts of folded WT-BPTI (roughly 8%). This mixture was then reduced in a buffer containing 6 M GdmCl and a large excess of DTT. We found that this reduction step also required heating to 55 °C, due to the significantly higher stability of the MT-BPTI protein (vide infra). The reduced MT-BPTI (Fig. S2d†), possessing a single MT moiety at position 14–38 and four free cysteines (Cys5, Cys30, Cys51 and Cys55), was isolated as the major product. All details concerning the preparation procedures of reduced WT-BPTI and reduced MT-BPTI are provided in the ESI.†
Oxidative folding of reduced WT-BPTI and reduced MT-BPTI
We first tested the oxidative folding of reduced WT-BPTI and reduced MT-BPTI under standard conditions, as described previously.7,20,21 In brief, the two reduced proteins were folded in parallel in phosphate buffer at pH 7.3 under anaerobic conditions and in the presence of GSSG as an oxidant (5 equiv. relative to the protein). For reduced WT-BPTI (Fig. S3a†), formation of the two one-disulfide intermediates [30–51] and [5–55] is followed by the formation of the two-disulfide intermediates N′ and N*, which are stable due to their quasi-native structures.22,23 At neutral pH, N* is significantly more populated due to a slow intramolecular thiol–disulfide rearrangement, while N′ rearranges more quickly into NSHSH, which is rapidly oxidized to form N.7,10 As a result, N* becomes a long-lived species (referred also as a “dead-end” species),9 which remains stable for over 22 h. The final yield of native WT-BPTI under these conditions is below 40% (Fig. S3a†).On the other hand, we expected the oxidative folding of reduced MT-BPTI to proceed via a lower population of species, leading to blocked intermediates (Fig. 1d). As the MT moiety is located at position 14–38, the only native disulfides that can form are 30–51, giving MT-N′, and 5–55, giving MT-N*. The formation of MT-N′ and MT-N* from a reduced MT-BPTI is slower than the formation of N′ and N* from [30–51] and [5–55] in WT-BPTI (Fig. S3b†), which is in a good agreement with earlier studies.7,9,10 In the case of the [30–51] and [5–55] intermediates in WT-BPTI, which exhibit native-like structures,24,25 the oxidation of the solvent exposed Cys14 and Cys38, to form N′ and N*, is relatively rapid. In contrast, although the reduced MT-BPTI is expected to have a native-like structure, as in the case of the [14–38] intermediate of WT-BPTI,26 the free Cys residues at positions 5, 30, 51, and 55 are considerably more buried, causing the direct formation of either the 30–51 or 5–55 disulfides to be much slower.
Furthermore, in WT-BPTI, the N′ conversion into N* and NSHSH proceeds at comparable rates.7,9 Yet, MT-N′ cannot be converted into NSHSH, and can, in principle, give only MT-N*, which should be for all intents and purposes a dead-end. This seems to be the case at first glance (Fig. S3b†). Surprisingly, however, roughly 4% of the reduced MT-BPTI reached the N state after 22 h. This result was unexpected since the pathway to the N state normally proceeds via reduction, followed by rearrangement through an intramolecular thiol–disulfide exchange reaction, and re-oxidation of the 14–38 disulfide bond, which is not possible in the case of reduced MT-BPTI. This unexpected observation suggested that there is an alternative, previously overlooked, path to the N state, and that the N* (or MT-N*) state is not a dead-end intermediate under these conditions.
Encouraged by these results, we decided to explore further the course of oxidative folding in the case of reduced MT-BPTI. Under basic conditions, proteins containing disulfide bonds fold considerably faster, as the rate of thiol–disulfide exchange reactions is significantly enhanced.4 Indeed, at pH 8.7, reduced WT-BPTI folds much faster along the same pathway observed at pH 7.3.7,20,21 At this pH, ∼90% of the reduced WT-BPTI reached the N state after 22 h, and only less than 10% of the N* intermediate was present (Fig. 2a).
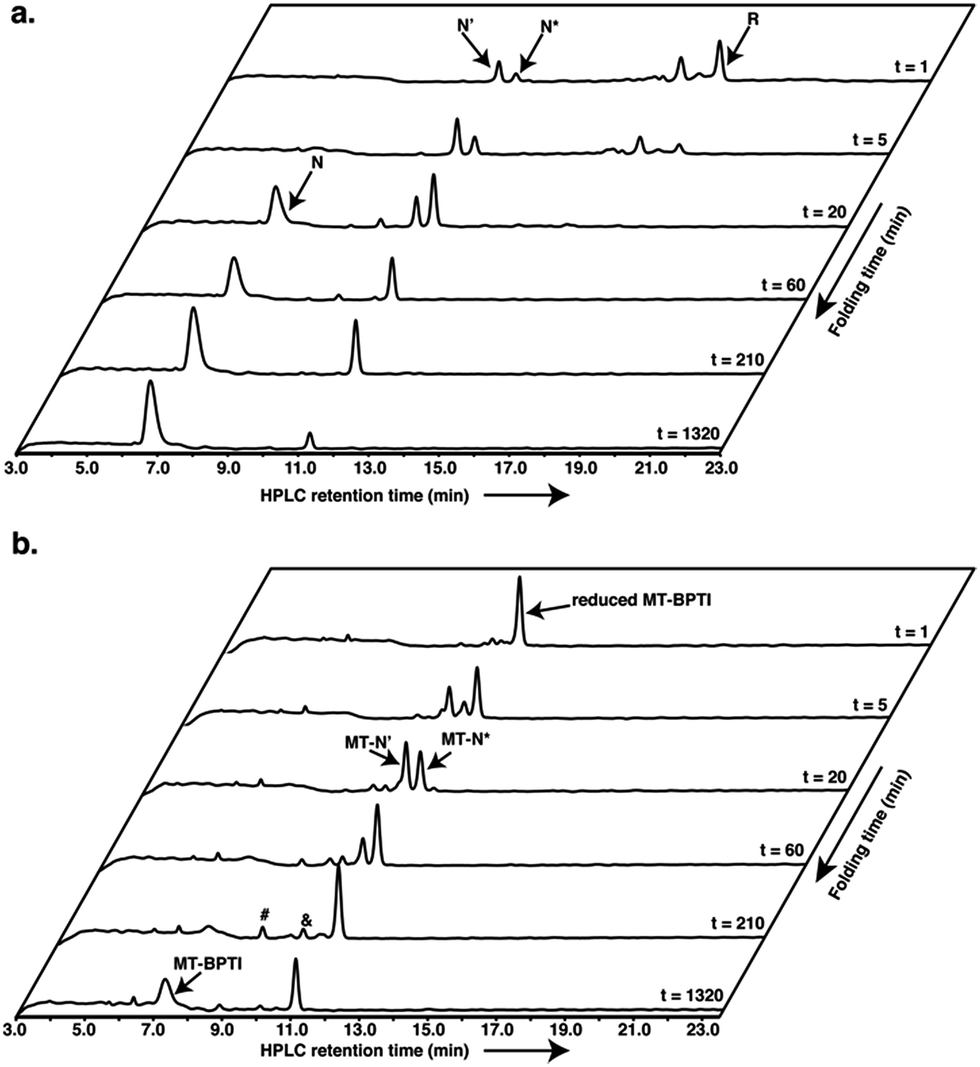 | ||
| Fig. 2 Anaerobic oxidative folding at pH 8.7 of (a) 30 μM reduced WT-BPTI with 150 μM GSSG, and (b) 20 μM reduced MT-BPTI with 100 μM GSSG. The & and # signs indicate the MT-N*–SG and MT-N*–(SG)2 species, respectively. | ||
The folding of reduced MT-BPTI at pH 8.7 seemed to be much clearer. Both of the MT-N′ and MT-N* intermediates formed faster at a higher pH, such that after 3.5 h, MT-N* was the major species observed (Fig. 2b). As the reaction proceeded, the HPLC chromatogram showed two species, at ∼9.9 min and at ∼8.8 min. ESI-MS analysis indicated that these two peaks contained the singly and doubly glutathionylated (SG) two-disulfide intermediates, respectively (Fig. S4†). At 22 h, over 50% of the reduced MT-BPTI had folded into the N state, while MT-N* was still evident.
Since the final N state of MT-BPTI started to emerge mostly after the complete rearrangement of MT-N′ to MT-N*, it is quite conceivable that MT-N* refolds into the N state through direct reaction with oxidized glutathione (GSSG) to form the MT-N*–SG species (appearing at ∼9.9 min in the HPLC chromatogram). This result also agrees with our previous findings in the selenoglutathione (GSeSeG)-mediated oxidative folding of WT-BPTI,27,28 in which the trapped intermediate N* was found to be rescued, as it forms N*–SeG, and then folds into the N state.28 In contrast, GSeSeG was found to have a negligible effect on the conversion of N′ into N,28 in agreement with previous conclusions by Weissman and Kim that the direct reaction between N′ and GSSG to give the N state is inhibited by the native structure of N′.10 The difference observed in the reactivity of the two intermediates, N′ and N*, can be attributed, at least in part, to the slight solvent exposure of the free thiol of Cys30 in N* and in MT-N* (Fig. S5†).28,29 This residue is suggested to react with the oxidant (GSSG or GSeSeG).28,30 Once N*–SG or MT-N*–SG is formed, the remaining free thiol of Cys51 attacks the mixed disulfide (or selenosulfide in N*–SeG) bond and replaces the SG (or SeG) group, forming the final disulfide bond 30–51 and giving the N state. The doubly glutathionylated species observed by HPLC at ∼8.8 min is likely to be the MT-N*–(SG)2 intermediate (analogous to the previously observed N*–(SeG)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28), which is a nonproductive intermediate that must be reduced before converting into the N state.
28), which is a nonproductive intermediate that must be reduced before converting into the N state.
These results indicate that for both WT-BPTI and MT-BPTI the conversion of the N* (or MT-N*) intermediate into the final N state does not exclusively require intramolecular thiol–disulfide exchange reactions that are coupled with substantial unfolding, as was previously suggested.6,9 Alternatively, a direct reaction with an oxidant can potentially lead to final folding via an intermolecular reaction.30 If such an alternative route was operative, an increase in the concentration of the oxidant should enhance folding. Indeed, folding was accelerated when we increased the GSSG concentration from 5 to 25 equivalents for the WT-BPTI and MT-BPTI folding at both pH 7.3 (Fig. S6 and S8†) and pH 8.7 (Fig. S7 and S9†). Our results (Fig. S6–S9†) clearly indicate that with an increased concentration of GSSG the yield of the N state increases in both cases.
It is worth noting that in the case of WT-BPTI a significant increase in the formation of N occurs in the second stage, where N* reacts directly with GSSG, after the complete conversion of N′ to N, the latter conversion being independent of the oxidant (Fig. S9†). On the other hand, a higher concentration of GSSG led to a more significant increase in all stages of MT-BPTI folding (Fig. S9†). Additionally, the chromatogram peaks corresponding to N*–SG and N*–(SG)2, as well as to those of MT-N*–SG and MT-N*–(SG)2, are more populated, as would be expected from a direct reaction with GSSG. As a result, the reduced MT-BPTI is converted into the N state (about 90%) after 22 h under these conditions, while for WT-BPTI it took only 8 h (Fig. S7†). These findings are supported by previous studies done by Kibria and Lees, in which efficient folding was achieved under balanced conditions of GSSG and GSH concentrations, which minimized the non-productive folding pathways, and enhanced the intermolecular conversion of N* and N′ into N.30
Rearrangement of MT-N′ and MT-N*
Next we investigated the rearrangement behavior of the intermediates MT-N′ and MT-N* in buffer solution in comparison with earlier work reported by Weissman and Kim.7,9 First, we isolated these acid-quenched intermediates by HPLC and allowed them to rearrange in buffer solution at pH 8.7 under anaerobic conditions and in the absence of oxidizing reagents. For WT-BPTI, previous folding studies reported the low-level formation of two non-native intermediates [5–38, 30–51] and [5–14, 30–51], which were suggested, in some of the previous studies, to be crucial for the conversion of N′ and N* into NSHSH.9,18,31 In the case of MT-BPTI, such conversions are prohibited.In good agreement with the literature, our results (Fig. S10†) show that MT-N′ rearranges into only the MT-N* intermediate, a process that takes about 1 h (Fig. S10a†). This observation demonstrates that such a conversion is independent of oxidizing agents, and also that it occurs without the formation of the two non-native intermediates described above. Our results also confirm that MT-N* is a dead-end intermediate in the absence of an oxidizing agent (Fig. S10b†), further supporting the mechanism where the conversion to the N state is obtained via direct reaction between MT-N* and GSSG.
Stability and activity of MT-BPTI
Next, we studied the effect of MT substitution on the protein stability. The three disulfide bonds of WT-BPTI and its compact structure make this protein highly stable. WT-BPTI is resistant to proteolysis and denaturation by chaotropic agents, including guanidine hydrochloride and urea, is stable at extreme pH conditions, and has a melting temperature of approximately 100 °C.32–34 In the past, the stabilization effect of substituting a disulfide bond with an MT moiety had only been tested on short peptides, such as oxytocin and other small natural products,12,35,36 but not on a full protein such as BPTI.To compare the reductive unfolding behavior of folded WT-BPTI and MT-BPTI, we incubated the two proteins in a phosphate buffer with 2 M GdmCl and 100 equiv. of DTT at room temperature. Under these conditions, folded WT-BPTI is reduced first to NSHSH and then to the fully reduced, unfolded state (R). After 30 h, only 10% of the starting WT-BPTI protein remained in its original N state (Fig. 3a and S11a†). In contrast, the MT-BPTI protein was completely stable under these conditions, with no traces of reduction or unfolding observed even after 30 h (Fig. 3a and S11b†). This experiment demonstrates that the MT substitution contributes significantly to the enhanced structural stability of the resulting MT-BPTI derivative, making such a derivative an attractive replacement strategy for a wide range of applications.
Furthermore, from a functional perspective, WT-BPTI acts as an excellent inhibitor for many proteases, displaying inhibition constants in the nanomolar to femtomolar concentration range.37–39 The inhibition activity of WT-BPTI and MT-BPTI against the representative protease chymotrypsin was therefore compared. Previous studies showed that the BPTI–chymotrypsin complex is thermodynamically stable, with an inhibition constant Ki of approximately 11 nM.37 Our current studies demonstrated that WT-BPTI and MT-BPTI show very similar protease inhibition characteristics, with practically identical Ki values toward chymotrypsin of 12.7 ± 0.8 nM and 12.6 ± 0.7 nM, respectively (Fig. 3b).
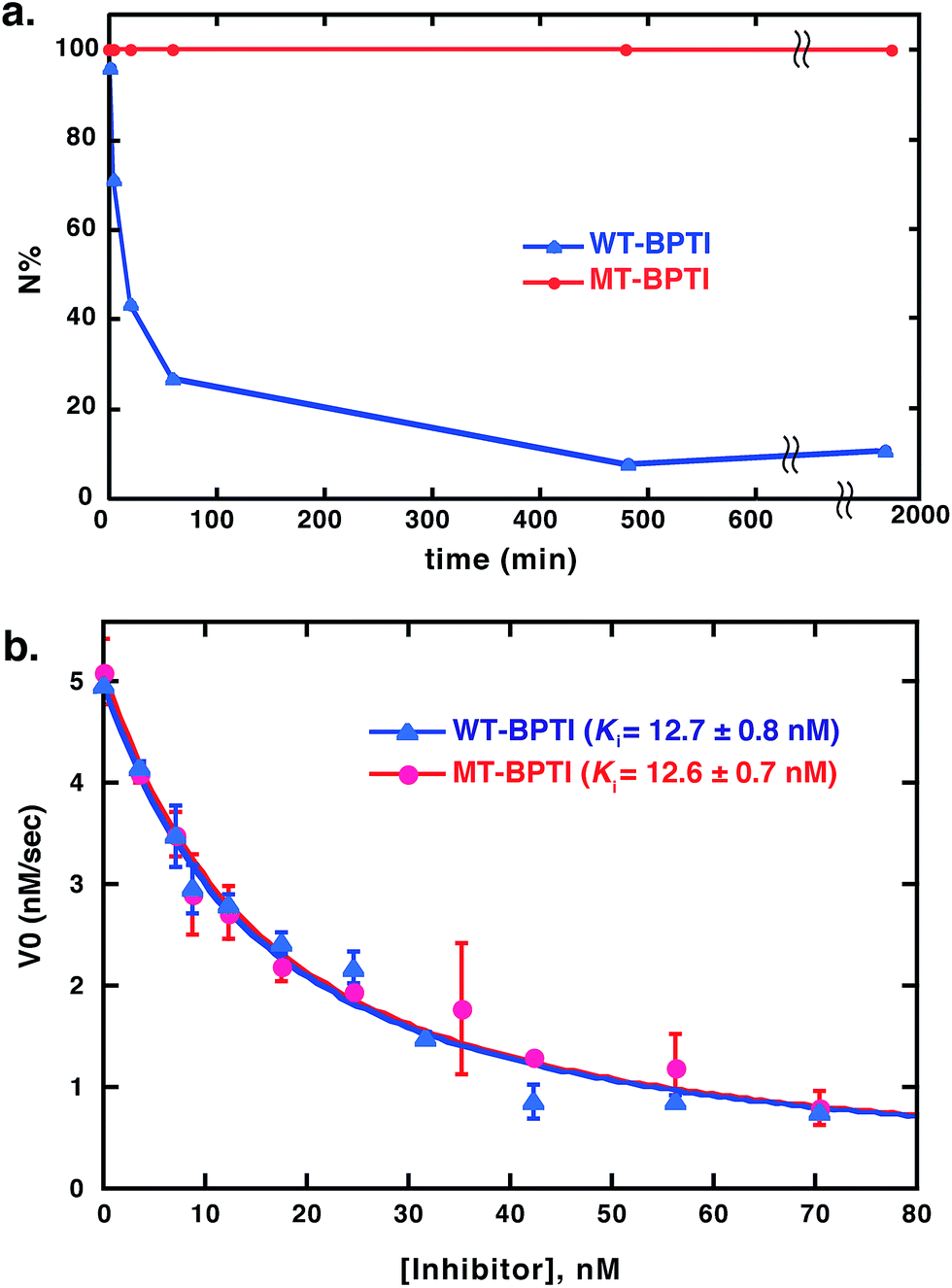 | ||
| Fig. 3 Stability and activity comparison between WT-BPTI and MT-BPTI. (a) Kinetic traces of the reductive unfolding reactions of 30 μM folded BPTI proteins at pH 7, in the presence of 2 M GdmCl and 100 equiv. of DTT. Compared here is WT-BPTI (blue) with MT-BPTI (red). The lines connecting the data points are shown only for clarity and do not represent a data fit. (b) Inhibition studies of bovine α-chymotrypsin by WT-BPTI (blue) and MT-BPTI (red). The s.d. values were calculated from experiments done in triplicate, and were fitted as described previously to give the calculated Ki values indicated.37 | ||
Structural studies of MT-BPTI
Encouraged by these results, we found it critical to study the effect of the MT moiety on the overall 3D structure of BPTI. For this purpose we used both circular dichroism (CD) and X-ray crystallography. The far-UV CD spectra obtained for WT-BPTI and MT-BPTI were almost completely overlapped, with very minor changes observed, indicating that the introduction of the MT moiety into BPTI caused only negligible conformational changes (Fig. S12†).This result was unequivocally confirmed by the high-resolution crystal structure determination of MT-BPTI (Fig. 4). MT-BPTI was crystallized under conditions similar to those for native WT-BPTI,40 allowing detailed 3D structural analysis at 1.72 Å resolution (further described in the ESI†).
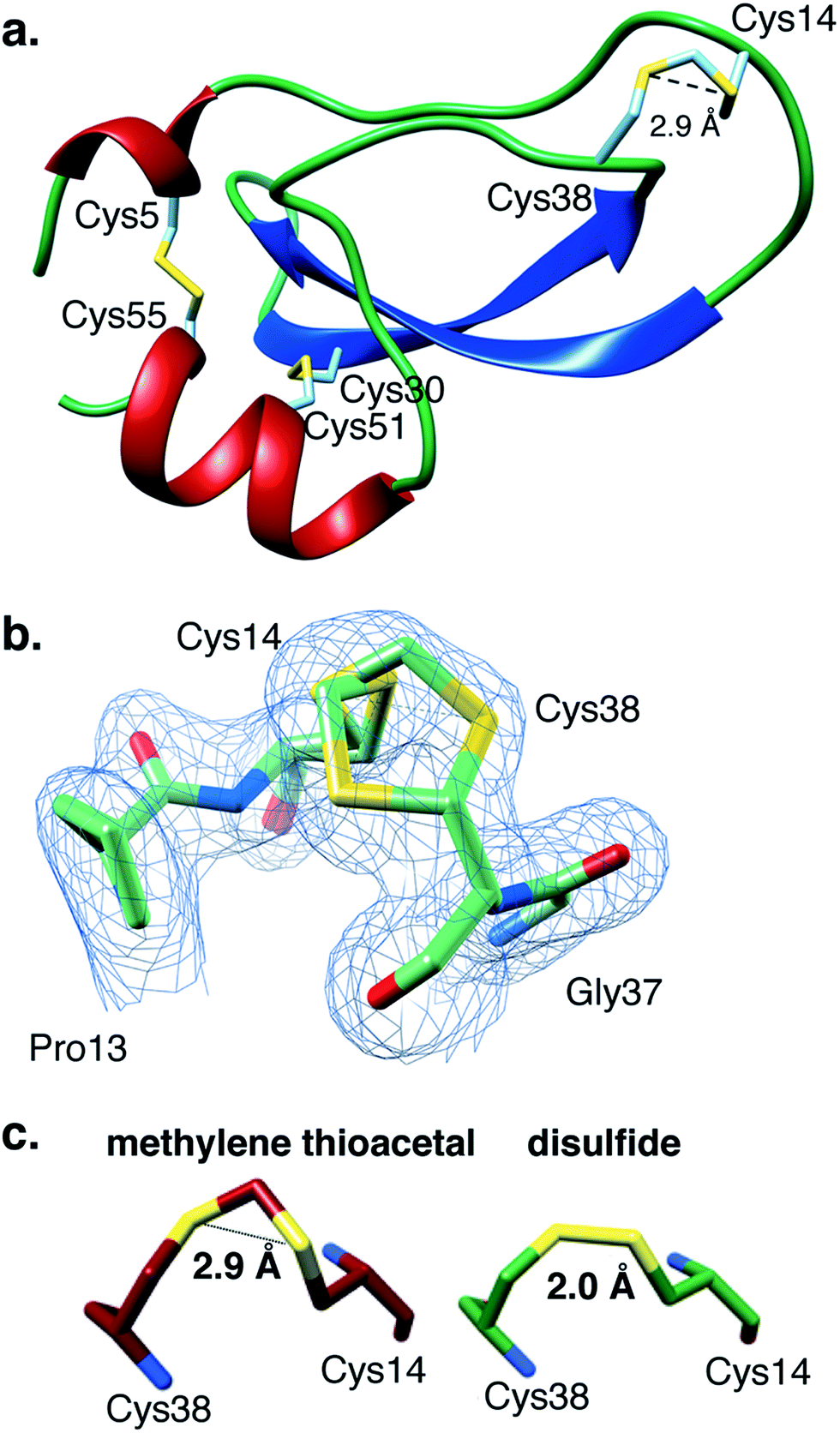 | ||
| Fig. 4 The present crystal structure of MT-BPTI (PDB: 6F1F). (a) A scheme of the crystal structure of MT-BPTI as determined here at 1.72 Å resolution. α-Helices are presented in red, β-sheets in blue and sulfur atoms in yellow. The cysteine residues involved in the disulfide bonds and in the MT bridge are indicated. Only one conformation is shown for the 14–38 MT bridge, for clarity. (b) A zoom-in of the region around the 14–38 MT bridge, showing the two alternate conformations observed for the C–S–C–S–C segment. The corresponding crystallographic electron density is shown in light blue, demonstrating the clear fitting of the model to the experimental map. (c) The average distances between the two sulfur atoms in the MT vs. the disulfide bridge 14–38 observed in the respective MT-BPTI and WT-BPTI structures; WT-BPTI (PDB code 2HEX, green) and MT-BPTI (6F1F, dark red). | ||
Similar to WT-BPTI (PDB code 2HEX40), MT-BPTI crystallized as a decameric assembly, containing five independent monomers in the crystallographic asymmetric unit. The unique MT moiety could be clearly identified in the resulting crystallographic electron density maps (Fig. 4b). Interestingly, however, in all five MT-BPTI monomers, the 14–38 MT bridge had to be modelled in two alternate conformations. In retrospect, this observation is not surprising, as similar alternate conformations have also been observed for the 14–38 disulfide bond in the corresponding WT-BPTI structure.41
A comparison between the 3D structures of WT-BPTI and MT-BPTI demonstrates an almost complete overlap (Fig. S13a and b†), with root mean square deviations (RMSD) of only 0.12–0.29 Å between the two structures (monomer vs. monomer).
Almost no differences are observed in the loop around the MT moiety, except for a slight difference in the positions of the Arg39 and Lys15 side chains (Fig. S13b†). The average distance between the two sulfur atoms of the MT moiety in MT-BPTI is 2.89 ± 0.03 Å, compared with 2.03 ± 0.03 Å for the 14–38 disulfide bond in WT-BPTI (Fig. 4c). These values reflect a significant difference in the relative positions of these key sulfur atoms, but surprisingly this has only an insignificant effect on the overall structure of the protein. To the best of our knowledge, this is the first crystallographic structure of a full protein that contains a methylene thioacetal bridge.
Discussion
The indistinguishable biological activity of the two proteins is not surprising, based on their highly similar structures, as demonstrated above by CD and X-ray crystallography. Thus, the structural and activity studies outlined above confirm that the methylene thioacetal substitution did not cause any major conformational or functional changes in the protein.All observations outlined above strongly suggest that the canonical mechanism proposed for BPTI folding cannot explain some of our results and therefore requires an important modification. Our current proposal for a modified folding mechanism (Fig. 5) combines the large wealth of previous studies7–10,18,28 and the previously proposed mechanism together with our new results. In this modified mechanism, each step that is indicated as “major” implies that the path is relatively fast or most-favored, while “minor” implies that the path is relatively slow or less-favored.
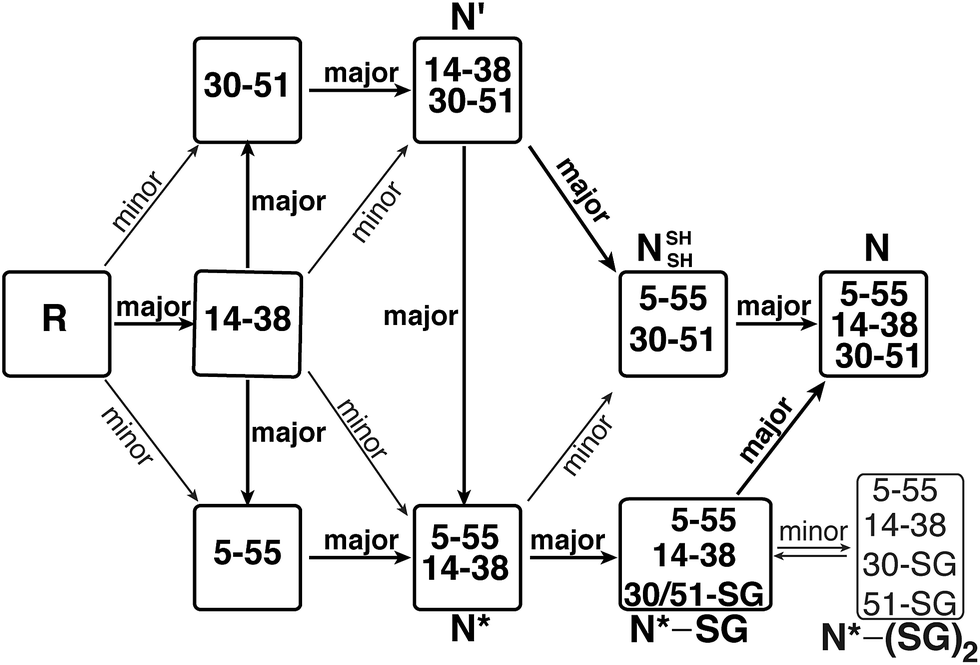 | ||
| Fig. 5 The modified oxidative folding mechanism of BPTI, reflecting the alternative path proposed here for the conversion of N* via the N*–SG intermediate into the final N state. “major” indicates a preferred (or fast) path, while “minor” indicates a less preferred (or slow) path. | ||
The reduced, unfolded protein, R, is first oxidized to form intermediates with a single disulfide bond. Formation of the [14–38] intermediate is favored,13 which is a species that assumes a native-like structure,26 but it is short-lived and rapidly rearranges to form the [30–51] or the [5–55] intermediate.13 Next, these single-disulfide intermediates assume a 14–38 disulfide bond to form N′ and N*, respectively. We found that a reduced MT-BPTI, in which the MT moiety “locks in” the 14–38 bridge, could also form the corresponding intermediates MT-N′ and MT-N*. Nonetheless, such folding occurs at a slower rate compared with the formation of N′ and N* in WT-BPTI, which could explain the order of disulfide bond formation. Although the [14–38] intermediate of WT-BPTI contains four free Cys residues at positions 5, 30, 51, and 55, all these SH groups are largely buried in the protein interior. As a result, direct oxidation of these SH groups to form the 30–51 disulfide (resulting in the N′ intermediate) or the 5–55 disulfide (resulting in the N* intermediate) is not preferred and therefore occurs slowly in the case of MT-BPTI. In WT-BPTI, the [30–51] and [5–55] intermediates contain native-like structures;24,25 hence, formation of the solvent exposed 14–38 disulfide bond occurs rapidly to give N′ and N*, respectively.
In the next step, N′ rearranges into N* and NSHSH (at similar rates) by intramolecular thiol–disulfide exchange reactions, which is independent of the oxidant.7,9 This step also appears to be independent of the formation of the low-populated non-native intermediates [5–14, 30–51] and [5–38, 30–51]. At this point, the NSHSH intermediate is readily oxidized to the final N state. On the other hand, the conversion of the N* intermediate (which is very stable) into the NSHSH intermediate is not preferred. Rather, the free SH group of Cys30, which is slightly solvent exposed (Fig. S5†) in the N* intermediate, reacts with the oxidant GSSG to form N*–SG mixed disulfide. In this intermediate, Cys51 replaces the glutathione to form the last 30–51 disulfide bond, resulting in the final native state, N, thereby completing the oxidative folding of the protein.
Conclusions
To summarize, our main goal in this study was to gain a deeper insight into the oxidative folding mechanism of BPTI, specifically with regard to the role of the 14–38 disulfide bond in the folding process of the major intermediates N′ and N*. In addition to the important mechanistic findings outlined above, our results demonstrate that the methylene thioacetal moiety has virtually no effect on the overall structure and function of the modified protein, serving as an excellent mimic to disulfide bonds. Moreover, this substitution appears to stabilize proteins against reductive unfolding, especially when replacing a solvent exposed disulfide bond. These findings suggest a useful potential strategy for future production of therapeutic proteins with enhanced stability and prolonged physiological efficacy.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank Prof. Donald Hilvert for helpful discussions, Dr Danna Reichmann for help in recording the HR-MS of the proteins, and Daniel Hervitz for help in protein purifications. N. M. acknowledges the financial support of the Israel Science Foundation (1072/14) and G. S. acknowledges the support of both the Israel Science Foundation (1905/15) and The Israeli Ministry of Science (3-12484/15). R. M. acknowledges the support of the Dalia and Dan Maydan Fellowship and S. L. is grateful to the Azrieli Foundation for the award of an Azrieli Fellowship.References
- C. M. Dobson, Nature, 2003, 426, 884–890 CrossRef CAS PubMed.
- J. Y. Chang, Biochemistry, 2011, 50, 3414–3431 CrossRef CAS PubMed.
- J. L. Arolas, F. X. Aviles, J. Y. Chang and S. Ventura, Trends Biochem. Sci., 2006, 31, 292–301 CrossRef CAS PubMed.
- B. S. Mainathambika and J. C. Bardwell, Annu. Rev. Cell Dev. Biol., 2008, 24, 211–235 CrossRef PubMed.
- C. B. Anfinsen, Science, 1973, 181, 223–230 CAS.
- T. E. Creighton, Prog. Biophys. Mol. Biol., 1978, 33, 231–297 CrossRef CAS PubMed.
- J. S. Weissman and P. S. Kim, Science, 1991, 253, 1386–1393 CrossRef CAS PubMed.
- T. E. Creighton and D. P. Goldenberg, J. Mol. Biol., 1984, 179, 497–526 CrossRef CAS PubMed.
- J. S. Weissman and P. S. Kim, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 9900–9904 CrossRef CAS.
- J. S. Weissman and P. S. Kim, Nat. Struct. Biol., 1995, 2, 1123–1130 CrossRef CAS PubMed.
- T. E. Creighton, N. J. Darby and J. Kemmink, FASEB J., 1996, 10, 110–118 CrossRef CAS PubMed.
- C. M. B. K. Kourra and N. Cramer, Chem. Sci., 2016, 7, 7007–7012 RSC.
- M. Dadlez and P. S. Kim, Nat. Struct. Biol., 1995, 2, 674–679 CrossRef CAS PubMed.
- J. A. Mendoza, M. B. Jarstfer and D. P. Goldenberg, Biochemistry, 1994, 33, 1143–1148 CrossRef CAS PubMed.
- L. C. Ma and S. Anderson, Biochemistry, 1997, 36, 3728–3736 CrossRef CAS PubMed.
- D. P. Goldenberg, Trends Biochem. Sci., 1992, 17, 257–261 CrossRef CAS PubMed.
- D. P. Goldenberg, L. S. Bekeart, D. A. Laheru and J. D. Zhou, Biochemistry, 1993, 32, 2835–2844 CrossRef CAS PubMed.
- T. E. Creighton, J. Mol. Biol., 1977, 113, 275–293 CrossRef CAS PubMed.
- S. A. Beeser, T. G. Oas and D. P. Goldenberg, J. Mol. Biol., 1998, 284, 1581–1596 CrossRef CAS PubMed.
- N. Metanis and D. Hilvert, Chem. Sci., 2015, 6, 322–325 RSC.
- N. Metanis and D. Hilvert, Angew. Chem., Int. Ed., 2012, 51, 5585–5588 CrossRef CAS PubMed.
- D. J. States, C. M. Dobson, M. Karplus and T. E. Creighton, J. Mol. Biol., 1984, 174, 411–418 CrossRef CAS PubMed.
- C. P. M. van Mierlo, N. J. Darby, D. Neuhaus and T. E. Creighton, J. Mol. Biol., 1991, 222, 353–371 CrossRef CAS PubMed.
- J. P. Staley and P. S. Kim, Protein Sci., 1994, 3, 1822–1832 CrossRef CAS PubMed.
- N. J. Darby, C. P. M. van Mierlo and T. E. Creighton, FEBS Lett., 1991, 279, 61–64 CrossRef CAS PubMed.
- E. Barbar, G. Barany and C. Woodward, Biochemistry, 1995, 34, 11423–11434 CrossRef CAS PubMed.
- J. Beld, K. J. Woycechowsky and D. Hilvert, Biochemistry, 2007, 46, 5382–5390 CrossRef CAS PubMed.
- N. Metanis, C. Foletti, J. Beld and D. Hilvert, Isr. J. Chem., 2011, 51, 953–959 CrossRef CAS.
- S. Parkin, B. Rupp and H. Hope, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1996, 52, 18–29 CrossRef CAS PubMed.
- F. M. Kibria and W. J. Lees, J. Am. Chem. Soc., 2008, 130, 796–797 CrossRef CAS PubMed.
- M. Qin, W. Wang and D. Thirumalai, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11241–11246 CrossRef CAS PubMed.
- G. I. Makhatadze, K. S. Kim, C. Woodward and P. L. Privalov, Protein Sci., 1993, 2, 2028–2036 CrossRef CAS PubMed.
- J. P. Vincent, R. Chicheportiche and M. Lazdunski, Eur. J. Biochem., 1971, 23, 401–411 CrossRef CAS PubMed.
- E. Moses and H. J. Hinz, J. Mol. Biol., 1983, 170, 765–776 CrossRef CAS PubMed.
- Y. B. Kim, Y. H. Kim, J. Y. Park and S. K. Kim, Bioorg. Med. Chem. Lett., 2004, 14, 541–544 CrossRef CAS PubMed.
- S. Lindman, G. Lindeberg, A. Gogoll, F. Nyberg, A. Karlen and A. Hallberg, Bioorg. Med. Chem., 2001, 9, 763–772 CrossRef CAS PubMed.
- M. J. M. Castro and S. Anderson, Biochemistry, 1996, 35, 11435–11446 CrossRef CAS PubMed.
- H. Fritz and G. Wunderer, Arzneim. Forsch., 1983, 33(1), 479–494 CAS.
- G. Hewlett, Biotechnology, 1990, 8, 565–566 CAS.
- J. Lubkowski and A. Wlodawer, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 335–337 CrossRef CAS PubMed.
- H. Czapinska, J. Otlewski, S. Krzywda, G. M. Sheldrick and M. Jaskolski, J. Mol. Biol., 2000, 295, 1237–1249 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01110a |
| This journal is © The Royal Society of Chemistry 2018 |