
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA three body problem: a genuine heterotrimetallic molecule vs. a mixture of two parent heterobimetallic molecules†
Haixiang
Han
a,
Zheng
Wei
a,
Matthew C.
Barry
a,
Jesse C.
Carozza
a,
Melisa
Alkan
b,
Andrey Yu
Rogachev
b,
Alexander S.
Filatov
c,
Artem M.
Abakumov
d and
Evgeny V.
Dikarev
 *a
*a
aDepartment of Chemistry, University at Albany, SUNY, Albany, NY 12222, USA. E-mail: edikarev@albany.edu
bDepartment of Chemistry, Illinois Institute of Technology, Chicago, IL 60616, USA
cDepartment of Chemistry, The University of Chicago, IL 60637, USA
dCenter for Electrochemical Energy Storage, Skolkovo Institute of Science and Technology, Moscow 143026, Russia
First published on 8th May 2018
Abstract
This work raises a fundamental question about the “real” structure of molecular compounds containing three different metals: whether they consist of genuine heterotrimetallic species or of a mixture of parent heterobimetallic species. Heterotrimetallic complex Li2CoNi(tbaoac)6 (1, tbaoac = tert-butyl acetoacetate) has been designed based on the model tetranuclear structure featuring two transition metal sites in order to be utilized as a molecular precursor for the low-temperature preparation of the LiCo0.5Ni0.5O2 battery cathode material. An investigation of the structure of 1 appeared to be very challenging, since the Co and Ni atoms have very similar atomic numbers, monoisotopic masses, and radii as well as the same oxidation state and coordination number/environment. Using a statistical analysis of heavily overlaid isotope distribution patterns of the [Li2MM′L5]+ (M/M′ = Co2, Ni2, and CoNi) ions in DART mass spectra, it was concluded that the reaction product 1 contains both heterotrimetallic and bimetallic species. A structural analogue approach has been applied to obtain Li2MMg(tbaoac)6 (M = Co (2) and Ni (3)) complexes that contain lighter, diamagnetic magnesium in the place of one of the 3d transition metals. X-ray crystallography, mass spectrometry, and NMR spectroscopy unambiguously confirmed the presence of three types of molecules in the reaction mixture that reaches an equilibrium, Li2M2L6 + Li2Mg2L6 ↔ 2Li2MMgL6, upon prolonged reflux in solution. The equilibrium mixture was shown to have a nearly statistical distribution of the three molecules, and this is fully supported by the results of theoretical calculations revealing that the stabilization energies of heterotrimetallic assemblies fall exactly in between those for the parent heterobimetallic species. The LiCo0.5Ni0.5O2 quaternary oxide has been obtained in its phase-pure form by thermal decomposition of heterometallic precursor 1 at temperatures as low as 450 °C. Its chemical composition, structure, morphology, and transition metal distribution have been studied by X-ray and electron diffraction techniques and compositional energy-dispersive X-ray mapping with nanometer resolution. The work clearly illustrates the advantages of heterometallic single-source precursors over the corresponding multi-source precursors.
Introduction
The rapid development of energy-related materials has brought about a great need for new design strategies to improve their performance.1–4 It is well known that the properties of materials, such as their electrochemical, catalytic, or magnetic properties, are closely related to the status of 3d transition metals in the host structure.5–11 The advance of compounds that incorporate two or more different transition metals has been primarily achieved on the basis of the well-established single-transition-metal-containing archetypes12–14 and has significantly expanded the research field of both new materials and modified known phases.15–18 Since the transition metal–oxygen framework typically holds the key to the overall structure type, each single transition metal is capable of endowing compounds with unique properties.19–21 At the same time, the combination of different 3d transition metals affords the materials with overall more balanced properties, while compensating for the drawbacks brought about by individual metals as well.22,23 Moreover, variables such as transition metal types and ratios are extra parameters that allow one to subtly tune the properties of target materials based on specific needs.24–27 Among the numerous cases showing the benefits of combining two different 3d transition metals, there is an interesting LiCo0.5Ni0.5O2 oxide that is considered as a cathode material for lithium-ion batteries.1,6,28,29 By substituting a half portion of Co with Ni in LiCoO2 (LCO), this layered phase has been found to be superior to both corresponding ternary oxides by demonstrating higher capacity and reversibility over cycling.22,29–32One of the biggest challenges in the preparation of materials containing two or more 3d transition metals is that synthetic methods are typically limited to conventional solid state reactions.7,17,33–35 Preparation of stoichiometric trimetallic oxides like LiCo0.5Ni0.5O2 usually requires harsh synthetic conditions, e.g. high calcination temperatures and long annealing times.22,28,31,33 Such synthetic routes impose difficulties for manipulating nanostructured particles with a narrow size distribution and often cause the loss of Li resulting in the appearance of nonstoichiometric impurities like the Ni-rich LixNi2−xO2 (0 < x < 1).20,29,30 In addition, an irregular morphology and inhomogeneous chemical composition/transition metal distribution are commonly encountered problems,36,37 especially when certain thermodynamically favoured binary oxides are formed during the synthetic process.37–39
One of the ways to avoid the above problems is the application of single-source precursors (SSPs) – molecules containing all the necessary metals in an appropriate ratio and decomposable in a controllable manner under mild conditions to yield phase-pure target materials.40–43 Single-source precursors having an intimate mixing of elements at the molecular level undergo a clean low-temperature thermolysis to produce nanomaterials with a homogeneous metal distribution throughout the product, as a result of rapid interactions between constituent elements, and reduce the possibility of generating unwanted intermediates.41,44–46
The design of a heterotrimetallic molecular precursor for the LiCo0.5Ni0.5O2 oxide material is not a trivial task. It requires, at least, a tetranuclear molecule containing Li, Co, and Ni in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio. We proposed to utilize heterobimetallic analogue Li2M2L6, which is a tetranuclear cyclic molecule (Scheme 1) with two transition metal sites. Molecules of that type were shown to be readily prepared on a large scale and to be capable of accommodating a number of divalent metals that prefer an octahedral environment, including even main group metals like Mg.41,47 Construction of such cyclic molecules requires the utilization of unsymmetric ligands (diketonates and beta-ketoesters) that feature different (small/bulky) substituents at the two ligand ends. The ligand oxygen located under small (CH3, CF3) substituents acts as a chelating–bridging, while the other one, under bulky (OtBu, tBu) groups, is purely chelating. The bulky ligand tails face outward of the heterometallic assembly and prevent further oligomerization of the metal core, ensuring an impressive stability of Li2M2L6 molecules in the solid state, gas phase, and solution. Importantly, the above heterobimetallic precursors proved to be efficient in producing phase-pure LiMO2 layered oxides, with L = tbaoac (tert-butyl acetoacetate; R = Me, R′ = OtBu) leading the way as the most efficient ligand for clean, low-temperature decomposition.
:1:1 ratio. We proposed to utilize heterobimetallic analogue Li2M2L6, which is a tetranuclear cyclic molecule (Scheme 1) with two transition metal sites. Molecules of that type were shown to be readily prepared on a large scale and to be capable of accommodating a number of divalent metals that prefer an octahedral environment, including even main group metals like Mg.41,47 Construction of such cyclic molecules requires the utilization of unsymmetric ligands (diketonates and beta-ketoesters) that feature different (small/bulky) substituents at the two ligand ends. The ligand oxygen located under small (CH3, CF3) substituents acts as a chelating–bridging, while the other one, under bulky (OtBu, tBu) groups, is purely chelating. The bulky ligand tails face outward of the heterometallic assembly and prevent further oligomerization of the metal core, ensuring an impressive stability of Li2M2L6 molecules in the solid state, gas phase, and solution. Importantly, the above heterobimetallic precursors proved to be efficient in producing phase-pure LiMO2 layered oxides, with L = tbaoac (tert-butyl acetoacetate; R = Me, R′ = OtBu) leading the way as the most efficient ligand for clean, low-temperature decomposition.
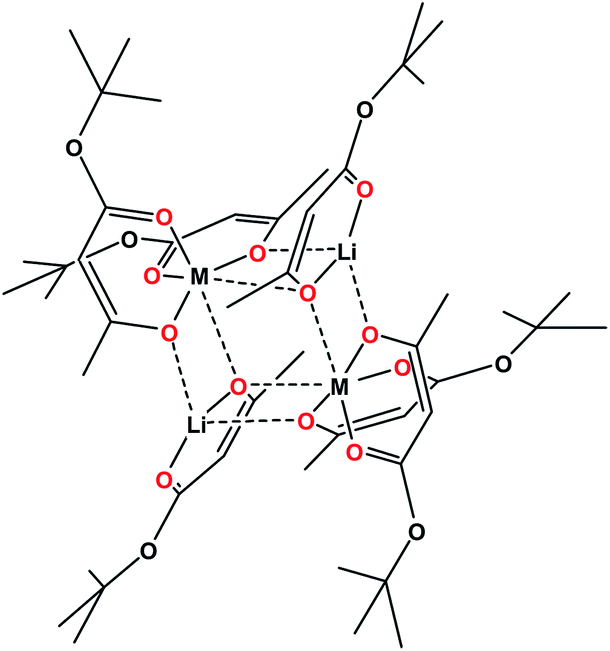 | ||
| Scheme 1 | ||
 | ||
| Scheme 2 | ||
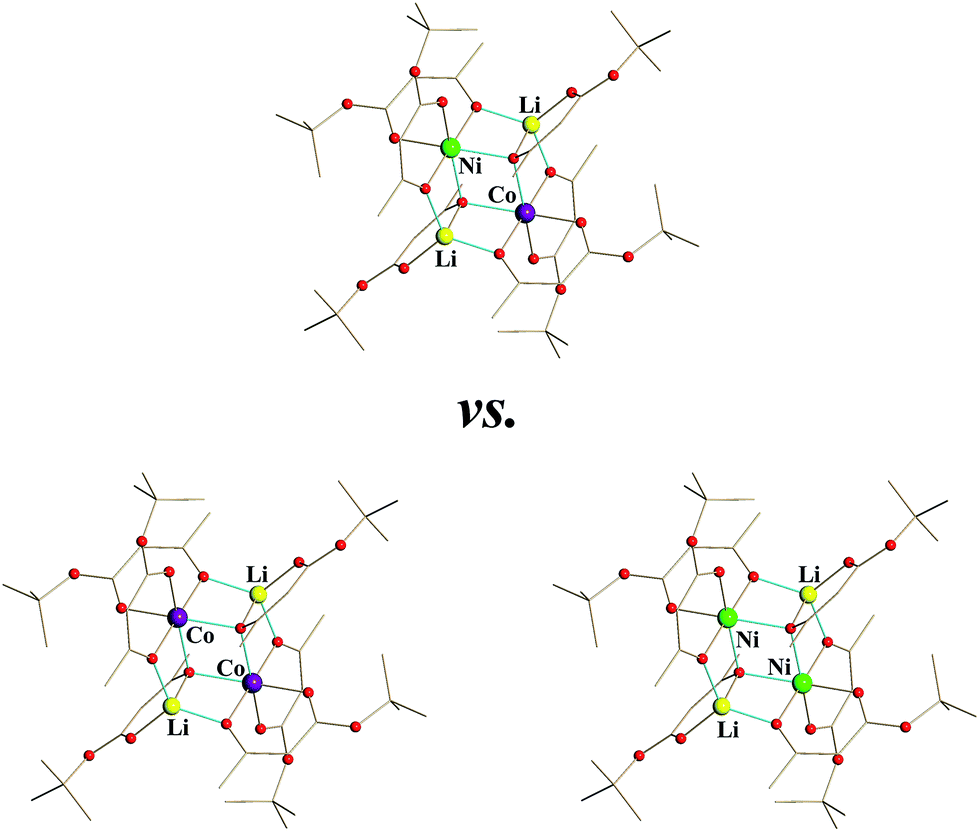
In this work, we have successfully employed the model tetranuclear structure (Scheme 1) to obtain heterotrimetallic precursor Li2CoNi(tbaoac)6 (1), which was successfully utilized to produce the phase-pure target oxide LiCo0.5Ni0.5O2 upon low-temperature thermolysis. Despite this accomplishment, our research has raised a fundamental issue regarding the structure of heterotrimetallic precursor 1: whether it consists of genuine heterotrimetallic molecules or contains a statistical mixture of two heterobimetallic units (Scheme 2). Herein we describe our approaches to decode the structure of such heterotrimetallic molecules in what can perhaps be regarded as one of the most complex cases.
Heterotrimetallic precursor Li2CoNi(tbaoac)6 (1)
Synthesis and properties
Heterotrimetallic precursor Li2CoNi(tbaoac)6 (1) was prepared by routes represented by eqn (1) and (2). Both reactions can be run in the solid state or in solution and employ commercially/readily available starting reagents. The solution approach affords the product on a large scale, in nearly quantitative yield (see the ESI† for full synthetic details). The first preparative technique (eqn (1)) is a one-step reaction of anhydrous transition metal(II) chlorides with an excess of Li(tbaoac). The heterometallic product is readily separable from LiCl based on their different solubilities in dichloromethane. The second technique represents a stoichiometric addition of homometallic lithium and divalent transition metal tbaoac salts and is performed in non-coordinating solvents such as 1,2-dichloroethane (DCE). While this reaction affords the heterometallic precursor as the sole product, it requires the preparation of M(tbaoac)2 complexes as an additional step. Synthetic conditions such as solvent, temperature, and time were shown to have a significant influence on the composition of the final product (vide infra).| 6Li(tbaoac) + CoCl2 + NiCl2 → Li2CoNi(tbaoac)6 + 4LiCl | (1) |
| 2Li(tbaoac) + Co(tbaoac)2 + Ni(tbaoac)2 → Li2CoNi(tbaoac)6 | (2) |
Heterotrimetallic precursor 1 was isolated in the form of fine crystalline powder. It can be further purified by sublimation in dynamic vacuum (cold finger) at 140–160 °C. The absence of crystalline impurities in the bulk sample of 1 was confirmed by comparison of its experimental X-ray powder pattern with the one calculated from the single crystal data (ESI, Fig. S2 and Table S5†). ICP-MS analysis of the reaction product provided the ratio of Co:Ni as 0.51(1):0.49(1). The product displays a homogeneous brown colour distribution that is clearly different from both pure heterobimetallic Li2Co2(tbaoac)6 (purple) and Li2Ni2(tbaoac)6 (green). Compound 1 cannot be sublimed at the static vacuum conditions (sealed, evacuated glass ampule), which is strikingly different from the behaviour of Li2Co2(tbaoac)6, which is volatile at 140 °C under the same conditions. The other properties of 1 are very similar to those of the parent heterobimetallic complexes: it can be quantitatively resublimed under dynamic vacuum without any visible indications of differently coloured components, it is highly soluble in all common solvents, and stable in open air for a reasonable period of time. In addition, the crystals of 1, regardless of growing conditions, always appear as allotwins48 consisting of triclinic and monoclinic polytypes that differ by mutual packing of the Li2M2L6 molecules (ESI, Fig. S2†).
Crystal structure
Single crystal X-ray investigation of heterotrimetallic precursor 1 revealed that its tetranuclear structure (Fig. 1) is isomorphous to both heterobimetallic Li2Co2(tbaoac)6 and Li2Ni2(tbaoac)6. It conforms to the centrosymmetric triclinic space group with an inversion centre located in the middle of the heterometallic assembly. Thus, only a single transition metal position is crystallographically independent. Unsurprisingly, the refinement of this position as 50:50 Co/Ni as well as pure Co or Ni did not result in an alteration of the R-value, standard deviations, or thermal parameters beyond statistically significant values (ESI, Table S12†). Careful inspection of the M–O bond distances in the structure of 1 indicates that the lengths of all three different types of interactions (chelating, chelating–bridging, and bridging) fall in between the corresponding characteristics established for the Co–O and Ni–O bonds in heterobimetallic analogues (Table 1).
 | ||
| Fig. 1 Solid state structure of Li2CoNi(tbaoac)6 (1). The lithium–oxygen and transition metal–oxygen bonds to the tbaoac ligands involved in the bridging interactions are shown in blue. Hydrogen atoms are omitted for clarity. The full view of the structure drawn with thermal ellipsoids can be found in ESI, Fig. S6.† | ||
An analysis of the structural features in 1 brings up an important question: does the structure of the trimetallic product consist only of heterotrimetallic Li2CoNi(tbaoac)6 molecules in their alternative orientations or does it contain a statistical mixture of heterobimetallic Li2Co2(tbaoac)6 and Li2Ni2(tbaoac)6 assemblies (Scheme 3). In terms of the application aspect, the query can be formulated as follows: does the heterometallic compound represent a single-source or a multi-source precursor?
 | ||
| Scheme 3 | ||
This is a common problem that should be accounted for in any heteromultimetallic molecule, especially when two of its metal atoms have the same oxidation state and coordination number/environment as well as very similar radii and atomic numbers/masses. The situation becomes even more complicated in the case when there is an extra symmetry within the molecule, and the metal atoms in question are not crystallographically independent. Apparently, other diffraction methods such as neutron and resonant diffraction that we have successfully applied before49 for analysis of site occupancy by 3d transition metal atoms cannot resolve the above problem, other than to confirm mixed-occupancy and to provide “elemental analysis” data for a particular single crystal. Thus we turned to mass spectrometry in order to analyse the composition of heterometallic molecules based on the rich isotope distribution patterns of accessible mother ions.
Mass spectrometry investigation
DART (Direct Analysis in Real Time) mass spectrometry has already been established as a powerful tool for investigating the gas phase behaviour of volatile heterometallic compounds42,43,47 as well as for confirmation of transition metal oxidation states.49 In this work, we extended the application scope of the technique for analysis of heterotrimetallic compounds primarily based on their isotope distribution patterns. In accord with their heterocyclic tetranuclear structure, the title molecules exhibit high volatility along with a sufficiently large temperature window between sublimation and decomposition, as well as sound structural stability upon retaining the heterometallic assembly in the gas phase.In the positive mode mass spectra of heterotrimetallic precursor 1, the [M–L]+ (M = Li2M′2L6; M′2 = CoNi, Co2, and Ni2; L = tbaoac) ion appears as a dominant peak. Analysis of the [M–L]+ ion isotope distribution pattern can provide a basis for recognition of molecular Li2Co2(tbaoac)6, Li2Ni2(tbaoac)6, and Li2CoNi(tbaoac)6 species that might be present in the bulk heterotrimetallic product. Unfortunately, the severe overlap in the isotope distribution patterns (Table 2) of the three possible [M–L]+ ions makes the quantitative analysis quite problematic.
| m/z | [Li2Co2L5]+ | [Li2Ni2L5]+ | [Li2CoNiL5]+ |
|---|---|---|---|
| 915 | 100.0% | 15.5% | |
| 916 | 15.6% | 55.2% | 100% |
| 917 | 55.2% | 89.6% | 43.3% |
| 918 | 100% | 49.4% | 51.0% |
| 919 | 12.3% | 40.0% | 21.8% |
Despite the observed overlap, a careful examination of the isotope distribution pattern “shape” in the mass spectrum of precursor 1 obtained by the above-described reaction (eqn (2)) at room temperature enabled us to make an important conclusion: it represents a mixture of heterotrimetallic and both heterobimetallic ions. Indeed, the experimental isotope distribution pattern (Fig. 2) matches neither the calculated profile for pure trimetallic [Li2CoNiL5]+ ions, nor the one for a mixture of bimetallic [Li2Co2L5]+/[Li2Ni2L5]+ ions taken in any ratio.
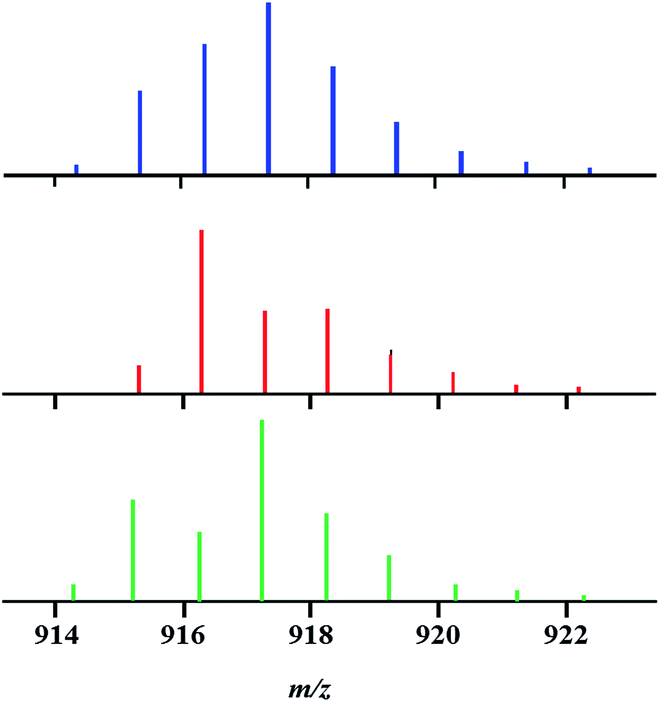 | ||
| Fig. 2 Isotope distribution patterns for the [M–L]+ ions in the mass spectra of bulk product 1 (experimental, top); [Li2CoNiL5]+ (calculated, middle); and an equimolar mixture of Li2Co2L6 and Li2Ni2L6 complexes (experimental, bottom). | ||
Having this important conclusion established on a qualitative level, we attempted to analyse mass spectra for quantification of three [M–L]+ ions in four major samples (Fig. 3): (a) an equimolar physical mixture of heterobimetallic Li2Co2(tbaoac)6 and Li2Ni2(tbaoac)6 complexes dissolved in 1,2-dichloroethane (DCE) at room temperature; (b) product 1 obtained from the stoichiometric reaction of Li, Co, and Ni tbaoac complexes (eqn (2)) in DCE at room temperature for 24 h; (c) the same reaction performed in DCE at reflux conditions for 24 h; and (d) reflux of the reaction mixture in DCE for 2 weeks. Five high-intensity peaks with m/z = 915, 916, 917, 918, and 919 were utilized to calculate the individual ion ([Li2Co2L5]+, [Li2Ni2L5]+, and [Li2CoNiL5]+) percentage ratios (Table 3).
 | ||
| Fig. 3 Isotope distribution patterns for the [M–L]+ (L = tbaoac) ions in positive ion DART mass spectra: (a) an equimolar mixture of heterobimetallic Li2M2(tbaoc)6 (M2 = Co2 and Ni2) complexes; (b) initial reaction (eqn (2)) in DCE at room temperature for 24 h; (c) reflux in DCE for 24 h; and (d) reflux in DCE for 2 weeks. The black lines are simulated isotope distribution patterns that were calculated based on the ratios of [Li2Co2L5]+, [Li2Ni2L5]+, and [Li2CoNiL5]+ species shown in Table 3. | ||
| [Li2Ni2L5]+ | [Li2CoNiL5]+ | [Li2Co2L5]+ | |
|---|---|---|---|
| (a) | 61% | 0 | 39% |
| (b) | 38% | 38% | 24% |
| (c) | 34% | 44% | 22% |
| (d) | 17% | 72% | 11% |
The calculations revealed a clear trend in the relative intensities of the three ions upon changing the reaction conditions. Thus, an equimolar mixture of parent Li2Co2(tbaoac)6 and Li2Ni2(tbaoac)6 complexes in DCE (Fig. 3a) yields an isotope distribution pattern that can be described by the presence of only [Li2Ni2L5]+ and [Li2Co2L5]+ ions in a ratio of 61:39, indicating no interaction between heterobimetallic molecules at such conditions. It also rules out any “recombination reactions” taking place in the mass spectrometer, and so all other spectra were recorded at exactly the same conditions. As we have already mentioned, the mass spectrum of 1 obtained by the reaction (eqn (2)) in DCE at room temperature (Fig. 3b) contains both heterotrimetallic [Li2CoNiL5]+ and heterobimetallic [Li2Ni2L5]+ and [Li2Co2L5]+ species, whose abundances were calculated to be 38:38:24. Running the same reaction at reflux conditions increases the content of heterotrimetallic ions to 44% (Fig. 3c). Apparently, the scrambling takes place and the content of the target Li2CoNi(tbaoac)6 molecules keeps growing upon extending the reaction time. After a two week reflux of the reaction mixture in DCE, the percentage of the heterotrimetallic [Li2CoNiL5]+ ion in the mass spectrum gradually reaches its maximum at 72% (Fig. 3d) and does not grow any further despite elevating the temperature by using higher boiling solvent.
It should be underlined that the relative percentage ratio of ions extracted from the DART-MS data does not directly represent the actual molar ratio of the corresponding parent molecules in the solid sample, since their appearance is highly influenced by many factors, the most important of them being volatility, ionization, and the gas phase thermal stability. Notably, in all four spectra, regardless of the sample preparation conditions, the intensity ratio of [Li2Ni2L5]+/[Li2Co2L5]+ ions is consistently about 3:2, while any of the solid samples under investigation should contain equimolar amounts of Li2Ni2(tbaoac)6 and Li2Co2(tbaoac)6 molecules.
The structural analogue approach
Design of structural analogues
Analysing the structure and composition of heterotrimetallic products like Li2CoNi(tbaoac)6 (1) proved to be a very complicated task as (i) Co and Ni have similar atomic numbers and radii, and therefore, they cannot be distinguished by X-ray diffraction, especially in the case of mixed-occupancy; (ii) very similar monoisotopic masses of cobalt and nickel (58.9332 and 57.9353, respectively) result in a severe overlap of the isotope distribution patterns for the [M–L]+ ions; and (iii) both divalent metal ions are high-spin, thus preventing the use of NMR spectroscopy for characterization of tetranuclear molecules in solution.Considering all these issues, a structural analogue strategy was adopted in order to rationalize the trends in the formation of heterotrimetallic assemblies. We proposed to replace one of the 3d transition metals with another element that (i) in its +2 oxidation state exhibits the same coordination behaviour as Co and Ni to ensure no changes in the connectivity pattern within the tetranuclear Li2M2(tbaoac)6 assembly; (ii) has an atomic number that is different enough from Co and Ni to be unambiguously distinguished by X-ray diffraction analysis; (iii) has a monoisotopic mass sufficiently different from those of Co and Ni in order to eliminate the possible overlap of heterobi- and heterotrimetallic ions in the mass spectra; (iv) is diamagnetic, so that the presence of its heterobimetallic Li2M2L6 molecules in solution can be detected by multinuclear NMR spectroscopy.
After an extensive search for the most appropriate metal, we settled on magnesium as the best (though not ideal) candidate fulfilling the above requirements in order to allow an investigation into the formation and structure of heterotrimetallic compounds Li2MMg(tbaoac)6 (M = Co or Ni). Magnesium was found to support the tetranuclear type of structure Li2Mg2L6 with a number of unsymmetric ligands, all isomorphous to those of 3d transition metals.47 Divalent Mg can be neither reduced nor oxidized in the presence of divalent Ni and Co. Its atomic number and monoisotopic mass (23.9850) are far from those of the 3d transition metals of interest. Finally, the Li2Mg2(tbaoac)6 molecule has been thoroughly characterized47 in solutions of non-coordinating solvents by multinuclear NMR spectroscopy.
Synthesis and structure of the Mg-containing analogues
Heterotrimetallic compounds Li2CoMg(tbaoac)6 (2) and Li2NiMg(tbaoac)6 (3) have been prepared with nearly quantitative yields by the reactions described above for the synthesis of Li2CoNi(tbaoac)6 (1) (see the ESI† for more details). Compounds 2 and 3 appear very similar to the parent heterobimetallic complexes in terms of crystal shape, solubility, and thermal stability. They have almost the same colour as the corresponding Co and Ni complexes, but not as the Mg one, which is colourless. The only obvious difference is the volatility of compound 2, which cannot be sublimed in a sealed evacuated ampule as its dicobalt counterpart.Single crystal X-ray diffraction investigation confirmed that both Li2MMg(tbaoac)6 compounds (M = Co (2) and Ni (3)) are indeed the structural analogues of Li2CoNi(tbaoac)6 (1) as well as of the parent heterobimetallic complexes. That was expected, since the Mg–O distances in the structure of Li2Mg2(tbaoac)6 are similar to those of Co–O and Ni–O. Perhaps, this is one of the major reasons why magnesium is able to support the title tetranuclear structure. As of today, we were unable to find the divalent metal that readily forms the Li2M2L6 structure while exhibiting M–O distances that are significantly different from those of 3d transition metals (Mn–Zn). The metal–oxygen distances in 2 and 3 predictably fall within the range established for the parent Li2M2(tbaoac)6 (M2 = Co2, Ni2, and Mg2) molecules (ESI, Table S15†). However, in the case of Mg-containing compounds 2 and 3 (Fig. 4), a mixed occupancy in the sole crystallographically independent metal position has been successfully refined resulting in M:Mg ratios of 53:47 (M = Co) and 47:53 (M = Ni). Several single crystals taken from the same batch typically gave M:Mg ratios within 3% of 50:50. The largest deviations from parity (up to Ni:Mg = 41:59) are characteristic of the crystals grown from the bulk product that was obtained by the reactions at room temperature, while those derived from the compounds prepared at long-time reflux conditions show M:Mg ratios closer to 1:1. The departure from an ideal composition is reflective of the fact that compounds 2 and 3 do not consist of heterotrimetallic Li2MMg(tbaoac)6 molecules alone. The deviations result from the differences (even small) in solubilities of heterobimetallic molecules Li2Mg2(tbaoac)6 and Li2M2(tbaoac)6.
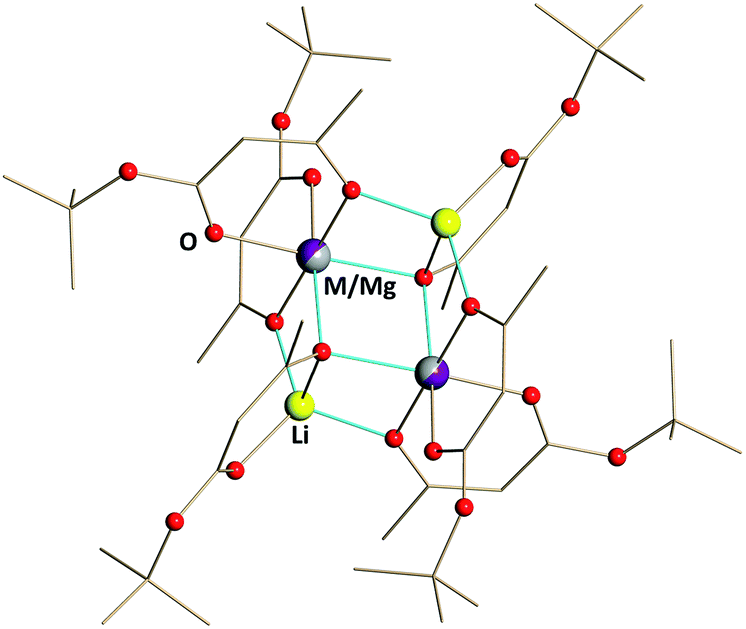 | ||
| Fig. 4 Solid state structure of Li2MMg(tbaoac)6 (M = Co (2) and Ni (3)) products. The metal–oxygen bonds involved in the bridging interactions are shown in blue. Hydrogen atoms are omitted for clarity. The full views of the structures drawn with thermal ellipsoids as well as the bond distances can be found in ESI, Fig. S7 and S8, and Tables S13 and S14.† | ||
Mass spectrometry investigation of Mg-containing analogues
DART mass spectrometry has provided unambiguous evidence for the concomitant presence of heterotrimetallic Li2MMg(tbaoac)6 (M = Co or Ni) and heterobimetallic parent compounds Li2M2(tbaoac)6 and Li2Mg2(tbaoac)6 in the structural analogue products 2 and 3. As can be seen from Fig. 5, physical mixtures of heterobimetallic complexes Li2M2(tbaoac)6 and Li2Mg2(tbaoac)6 give only bimetallic [Li2M2L5]+ peaks with no trimetallic [Li2MMg(tbaoac)5]+ counterparts present. The latter clearly appear in the spectra of products obtained in the reaction described by eqn (2) in DCE at room temperature. Now, three ions [Li2Mg2L5]+, [Li2MMgL5]+, and [Li2M2L5]+ (M = Co or Ni) show up in the spectra without any overlap. In both cases, the relative intensity of heterotrimetallic [LiMMgL5]+ peaks increases obviously upon increasing the reaction temperature and time. Similar to the case of Li2CoNi(tbaoac)6 (1), the intensities of the [LiMMgL5]+ peaks reach their maximum at 51% and 50% for the Co- and Ni-containing compounds, respectively, after two weeks of reflux in DCE and exhibit no further meaningful progression after that point, thus indicating that the equilibrium Li2M2(tbaoac)6 + Li2Mg2(tbaoac)6 ↔ 2Li2MMg(tbaoac)6 has been established. As we have mentioned earlier in the case of compound 1, the intensity ratios of [Li2Mg2L5]+/[Li2M2L5]+ ions in all four spectra are almost the same at ca. 1.3 and 1.6 for M = Co and Ni, respectively. An estimation of the molar content of heterotrimetallic species in the reaction mixture at equilibrium gives 51% and 50% for products 2 and 3, respectively.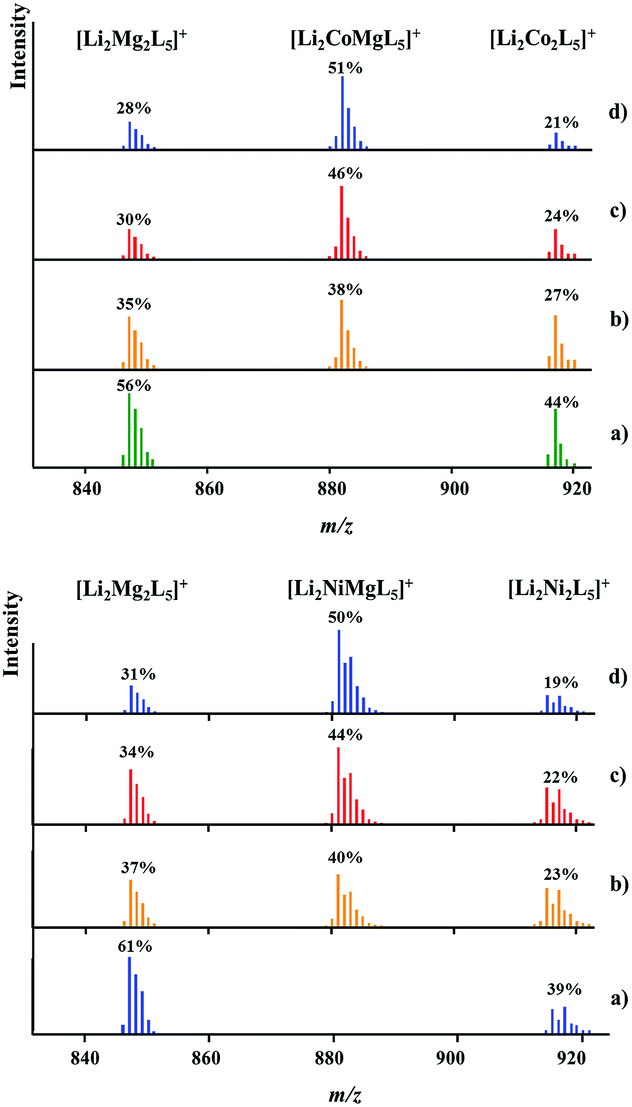 | ||
| Fig. 5 Isotope distribution patterns for the [Li2MMgL5]+ ions in the positive ion DART mass spectra of Li2CoMg(tbaoac)6 (2, top) and Li2NiMg(tbaoac)6 (3, bottom) products: (a) an equimolar mixture of heterobimetallic Li2M2(tbaoc)6 (M = Co or Ni) and Li2Mg2(tbaoac)6 complexes; (b) initial reaction (eqn (2)) in DCE at room temperature for 24 h; (c) reflux in DCE for 24 h; and (d) reflux in DCE for 2 weeks. The intensity percentages were calculated based on the monoisotopic masses of the corresponding ions. | ||
1H NMR investigation of structural analogue Li2NiMg(tbaoac)6 (3)
One of the reasons behind selecting magnesium to design the structural analogues of the Li2CoNi(tbaoac)6 (1) complex was the prospect of using NMR spectroscopy in order to unambiguously confirm/disprove the presence of diamagnetic Li2Mg2(tbaoac)6 molecules in the products and to estimate the molar ratio (as well as its change based on synthetic conditions) of heterobimetallic and heterotrimetallic molecules in the reaction mixture.While the Li2CoMg(tbaoac)6 (2) analogue was found to be nearly impossible to analyse, the Ni-counterpart (3) revealed meaningful features in its proton NMR spectra. The 1H NMR spectrum of pure Li2Mg2(tbaoac)6 in CDCl3 clearly shows two sets of singlets for CH, CH3 and OC(CH3)3 protons (ca. 1:3:9), which correspond to the 1:2 distribution of tbaoac ligands chelating lithium and magnesium, respectively (Fig. 6a). Importantly, this spectrum provides a basis for understanding the spectral features of the other two participants, Li2Ni2(tbaoac)6 and Li2NiMg(tbaoac)6 (3).
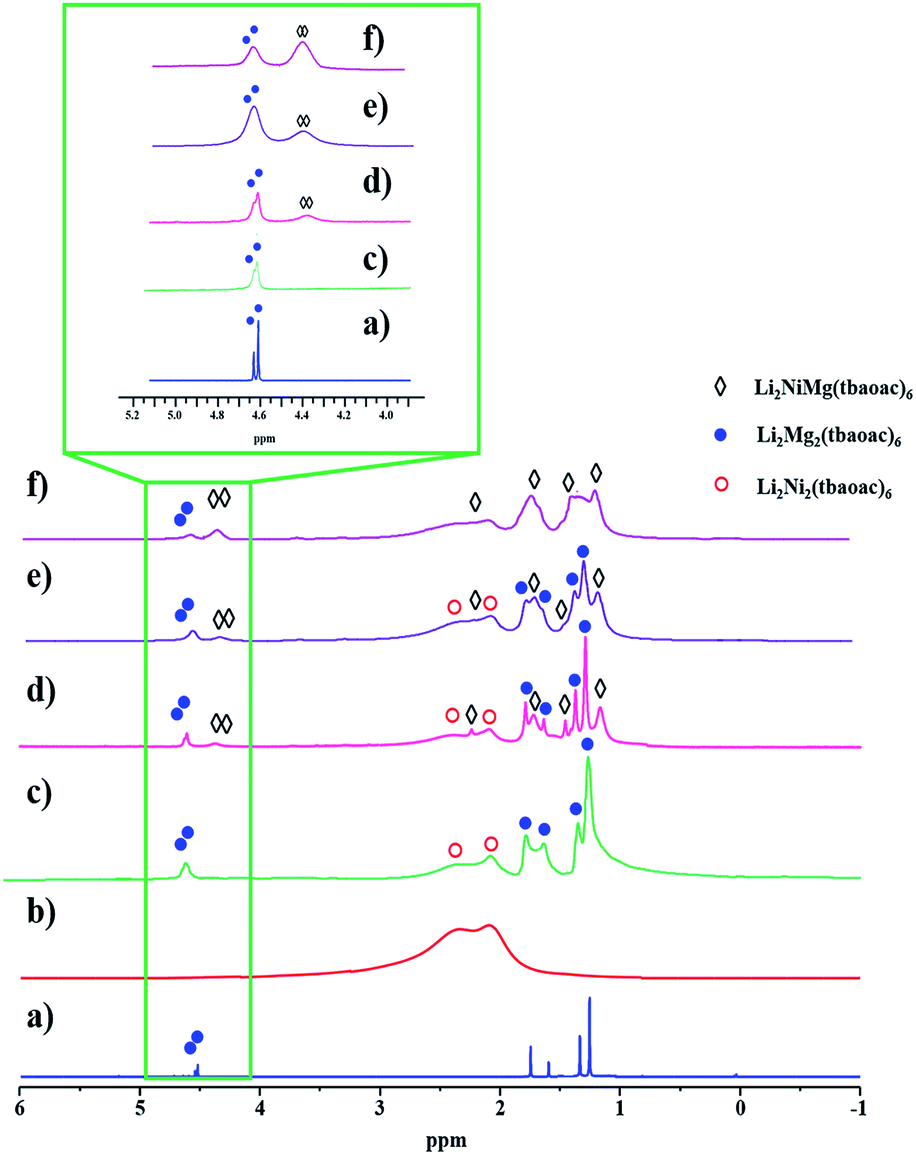 | ||
| Fig. 6 1H NMR spectra of heterometallic tetranuclear complexes in CDCl3 recorded at room temperature: (a) Li2Mg2(tbaoac)6; (b) Li2Ni2(tbaoac)6; (c) an equimolar mixture of Li2Ni2(tbaoac)6 and Li2Mg2(tbaoac)6; (d) the product isolated from the initial reaction (eqn (2)) in DCE at room temperature for 24 h; (e) reflux in DCE for 24 h; and (f) reflux in DCE for 2 weeks. The aromatic proton region is shown in the inset. | ||
While the spectrum of Li2Ni2(tbaoac)6 displays just an overlaid “bump” due to the presence of two high-spin divalent nickel centres (Fig. 6b), in the spectrum of an equimolar mixture of heterobimetallic Li2Ni2(tbaoac)6 and Li2Mg2(tbaoac)6 compounds, some of the visibly broadened proton peaks can be reasonably assigned (Fig. 6c). Compared to the latter spectrum, the one recorded for the bulk product of Li2NiMg(tbaoac)6 (3) synthesized at room temperature clearly shows the extra peaks (Fig. 6d) that can be assigned to the heterotrimetallic species. Upon increasing the reaction temperature (Fig. 6e) and time (Fig. 6f), the latter peaks keep growing, while the corresponding signals of Li2Mg2(tbaoac)6 become weaker. This trend can be seen at its best for the aromatic CH protons (Fig. 6, inset) of Li2NiMg(tbaoac)6 (3) and Li2Mg2(tbaoac)6, whose integration ratio changes from ca. 0.4 to 2.3 on going from spectrum (d) to (f). Further increase of the reaction temperature and/or time beyond this point does not visibly affect the NMR spectrum, pointing out that the equilibrium Li2Ni2(tbaoac)6 + Li2Mg2(tbaoac)6 ↔ 2Li2NiMg(tbaoac)6 has been reached. The rough estimation gives an almost statistical distribution with the molar content of heterotrimetallic species at equilibrium at about 53.5%. Therefore, the NMR study decidedly supports the results of mass spectrometry investigation of structural analogues 2 and 3.
Theoretical calculations of the Li2MM′(tbaoac)6 molecules
Theoretical evaluation of thermodynamic stability of tetranuclear molecules was performed using the B2PLYP-D/TZVP/ZORA approach (see the ESI† for more details) by deconstruction of the Li2MM′(tbaoac)6 assemblies into monomeric units, namely, Li(tbaoac), M(tbaoac)2, and M′(tbaoac)2. As can be seen from the results listed in Table 4, the stabilization energy for the heterotrimetallic Li2CoNi(tbaoac)6 (1) molecule falls exactly in between those for its heterobimetallic counterparts, Li2Co2(tbaoac)6 and Li2Ni2(tbaoac)6. A similar situation is found for the structural analogue Li2NiMg(tbaoac)6 (3), whose stabilization energy is approximately equal to the average value for two parent molecules. Thus, the calculated thermodynamic stabilization energy gives no preference for heterotrimetallic systems over the mixture of heterobimetallic ones. These results support the observation that all three types of species are present in the equilibrium mixture Li2M2(tbaoac)6 + Li2M′2(tbaoac)6 ↔ 2Li2MM′(tbaoac)6 at a roughly statistical 25:25:50% distribution of components.
| Li2MM′(tbaoac)6 | E (bonding, kcal mol−1) |
|---|---|
| Li2Co2(tbaoac)6 | −134.11 |
| Li2Ni2(tbaoac)6 | −149.31 |
| Li2CoNi(tbaoac)6 (1) | −141.75 |
| Li2Mg2(tbaoac)6 | −147.31 |
| Li2NiMg(tbaoac)6 (3) | −148.35 |
Thermal decomposition of the heterometallic precursor Li2CoNi(tbaoac)6 (1)
According to X-ray powder diffraction analysis, thermal decomposition of the bulk Li2CoNi(tbaoac)6 (1) product obtained from reflux in DCE for 2 weeks yields a phase-pure layered LiCo0.5Ni0.5O2 oxide at temperatures as low as 450 °C (Fig. 7a). The crystallinity of the residue is greatly improved upon elevating the thermolysis temperature up to 750 °C (Fig. 7b). The unit cell parameters of decomposition residues derived from the Le Bail fit correspond well with the literature data31 for LiCo0.5Ni0.5O2, while being clearly different from those for the heterobimetallic oxides LiMO2 (M = Co and Ni) (Table 5). Traces of the target oxide material start to appear in the X-ray powder pattern at as low as 350 °C, while the Ni-rich LixNi2−xO2 (0 < x < 1) phase becomes visible in the samples obtained above 750 °C. Under the same decomposition conditions, the equimolar mixture of heterobimetallic Li2Co2(tbaoac)6 and Li2Ni2(tbaoac)6 complexes prepared by evaporation of DCE solution at room temperature produces a very complex multi-phase residue (Fig. 7c) that contains nickel-rich Li0.4Ni1.6O2, LiCoO2, and some unidentified compounds (ESI, Fig. S19†). The latter represents an important observation that clearly demonstrates the advantage of single-source precursors over multiple-source precursors.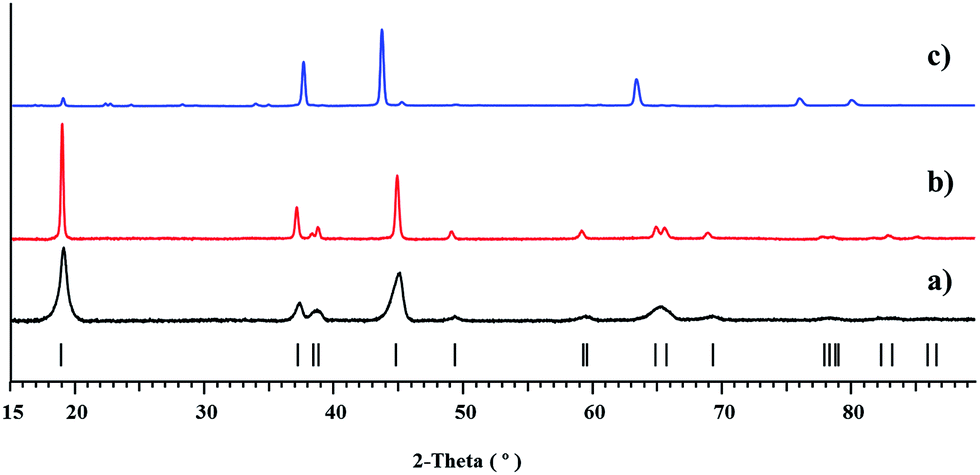 | ||
| Fig. 7 X-ray powder diffraction patterns of residues obtained by thermal decomposition of (a) the bulk product Li2CoNi(tbaoac)6 (1) at 450 °C and (b) at 750 °C; and (c) an equimolar mixture of Li2Co2(tbaoac)6 and Li2Ni2(tbaoac)6 at 750 °C. The theoretical peak positions of LiCo0.5Ni0.5O2 oxide are shown at the bottom as black bars. | ||
To the best of our knowledge, 450 °C is the lowest temperature utilized so far to prepare a LiCo0.5Ni0.5O2 material that is free of impurities. The typical solid-state preparative methods for LiCo0.5Ni0.5O2 cathode materials that employ oxides, carbonates, or acetates as starting reagents require high annealing temperatures of ca. 700 °C in order to obtain phase-pure materials.22,28,30,31,50 Several “soft chemistry” routes such as sol–gel51 as well as other multi-source precursor (assisted precipitation) approaches29 have also been introduced. While in some of those techniques calcination temperatures as low as 500 °C have been attempted, it was found that the phase-pure LiCo0.5Ni0.5O2 can only be obtained at 600 °C.29,51
The X-ray powder diffraction identification of the LiCo0.5Ni0.5O2 oxide phase by its unit cell parameters and the absence of visible crystalline impurities are not enough for thorough characterization of the target material appearance. The morphology of the particles, their chemical composition, and the homogeneous distribution of Co and Ni represent even more important characteristics for practical applications. Therefore, the decomposition residues have been analysed by recording electron diffraction (ED) patterns, high angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images, and energy dispersive X-ray (EDX) spectra. The samples were found to consist of agglomerated crystals with a size of 50–300 nm. The electron diffraction patterns of the oxide phase (Fig. 8) can be indexed in the space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m with unit cell parameters a ≈ 2.8 and c ≈ 14.1 Å, typical of the layered LiCoO2-type structure.52 EDX compositional maps reveal a predominantly homogeneous distribution of transition metals (Fig. 9) with an averaged Co:Ni atomic ratio of 49.0(16):51.0(16). An extensive analysis of electron diffraction images showed some inhomogeneity in the Co/Ni distribution, which was observed only very locally, in the form of small 5–10 nm Ni-enriched (less than 5%) clusters and occasional Co-rich and Co-depleted crystallites (ESI, Fig. S20–S22†). Such inhomogeneity can be attributed to the presence of heterobimetallic molecules Li2Co2(tbaoac)6 and Li2Ni2(tbaoac)6 in the bulk precursor. Again, this observation clearly underscores the advantages of a single-source precursor over a multi-source precursor.
m with unit cell parameters a ≈ 2.8 and c ≈ 14.1 Å, typical of the layered LiCoO2-type structure.52 EDX compositional maps reveal a predominantly homogeneous distribution of transition metals (Fig. 9) with an averaged Co:Ni atomic ratio of 49.0(16):51.0(16). An extensive analysis of electron diffraction images showed some inhomogeneity in the Co/Ni distribution, which was observed only very locally, in the form of small 5–10 nm Ni-enriched (less than 5%) clusters and occasional Co-rich and Co-depleted crystallites (ESI, Fig. S20–S22†). Such inhomogeneity can be attributed to the presence of heterobimetallic molecules Li2Co2(tbaoac)6 and Li2Ni2(tbaoac)6 in the bulk precursor. Again, this observation clearly underscores the advantages of a single-source precursor over a multi-source precursor.
 | ||
| Fig. 8 Electron diffraction patterns of the LiCo0.5Ni0.5O2 residue indexed in the Rm space group with unit cell parameters a ≈ 2.8 and c ≈ 14.1 Å. | ||
 | ||
| Fig. 9 HAADF-STEM image, the EDX elemental maps for Co, Ni, and O, and the mixed compositional maps showing a homogeneous distribution of Co and Ni in the decomposition residue. | ||
Conclusions
A heterotrimetallic tetranuclear molecular precursor containing two different 3d transition metals, Li2CoNi(tbaoac)6 (1), has been synthesized and successfully applied for the low-temperature preparation of a layered LiCo0.5Ni0.5O2 oxide which is an important cathode material for lithium ion batteries. The work raises a fundamental question regarding the “real” structure of heterotrimetallic compounds: do they consist of genuine heterotrimetallic species, contain a statistical mixture of two heterobimetallic molecules, or feature all of the above, similar to what was found in this study. We believe that any multimetallic molecule (containing three or more different metals) should be thoroughly analysed taking this point into account.This study clearly demonstrates the need for the single-source precursor consisting of only heterotrimetallic molecules for the preparation of target materials with a perfect homogeneous distribution of metals. The real challenge is how to design such a heterotrimetallic molecule that contains at least two different metal atoms with very similar characteristics? We envision two major ways to attain the structural distinctions for such atoms: different oxidation states and/or different coordination environments. In the particular case of the title precursor Li2CoNiL6, the former approach translates into making the cationic assembly [Li2CoIIINiIIL6]+. While this seems to be the most viable synthetic option, two practical points should be taken into account. First, this route will not yield a volatile precursor; instead, it will yield the one that can be applied for bulk or solution decomposition only. Second, a careful consideration of the counteranion chemical composition is required. It should not contain the elements that may affect the purity of decomposition residues, so that several convenient options for anion such as X−, BF4− and PF6− are impractical. The second strategy, which involves changing the coordination environment of transition metal centres in the title precursor, implies the application of a mixed-ligand approach. It relies upon using two different ligands with one of them having electron-withdrawing properties to preferentially coordinate the more electron-rich Ni centre, and the other one with electron-donating (or less electron-withdrawing) characteristics to be attached to the Co ion. However, from a practical perspective this approach would likely require as many as three different ligands, since the most electron-donating one will be exclusively located on the Li ion, leaving two transition metals to compete for the rest.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from the National Science Foundation (CHE-1152441) (ED) and from the Illinois Institute of Technology (AR) is gratefully acknowledged. The authors thank CRDF Global (FSCX-16-62133-0, AA) and (OISE-16-62134-0, ED) for funding this collaborative project. ChemMatCARS Sector 15 is principally supported by the Divisions of Chemistry (CHE) and Materials Research (DMR), National Science Foundation, under grant number NSF/CHE-1346572. Use of the PILATUS3 X CdTe 1M detector was supported by the National Science Foundation under grant number NSF/DMR-1531283. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357.References
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef PubMed.
- J. B. Goodenough, J. Power Sources, 2007, 174, 996–1000 CrossRef.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef PubMed.
- A. S. Arico, P. Bruce, B. Scrosati, J.-M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef PubMed.
- B. L. Ellis, K. T. Lee and L. F. Nazar, Chem. Mater., 2010, 22, 691–714 CrossRef.
- L. Croguennec and M. R. Palacin, J. Am. Chem. Soc., 2015, 137, 3140–3156 CrossRef PubMed.
- C. Yada, A. Ohmori, K. Ide, H. Yamasaki, T. Kato, T. Saito, F. Sagane and Y. Iriyama, Adv. Energy Mater., 2014, 4, 1301416 CrossRef.
- J. Hessels, R. J. Detz, M. T. M. Koper and J. N. H. Reek, Chem.–Eur. J., 2017, 23, 16413–16418 CrossRef PubMed.
- C. Zheng and S.-L. You, Chem, 2016, 1, 830–857 Search PubMed.
- D. Banham, S. Ye, K. Pei, J.-i. Ozaki, T. Kishimoto and Y. Imashiro, J. Power Sources, 2015, 285, 334–348 CrossRef.
- S. M. Aldoshin, D. V. Korchagin, A. V. Palii and B. S. Tsukerblat, Pure Appl. Chem., 2017, 89, 1119–1143 CrossRef.
- J. C. Knight, S. Therese and A. Manthiram, J. Electrochem. Soc., 2015, 162, A426–A431 CrossRef.
- S. Levasseur, M. Menetrier and C. Delmas, J. Power Sources, 2002, 112, 419–427 CrossRef.
- S. Levasseur, M. Menetrier and C. Delmas, Chem. Mater., 2002, 14, 3584–3590 CrossRef.
- P. K. Nayak, E. M. Erickson, F. Schipper, T. R. Penki, N. Munichandraiah, P. Adelhelm, H. Sclar, F. Amalraj, B. Markovsky and D. Aurbach, Adv. Energy Mater., 2017, 1702397 Search PubMed.
- M. Sathiya, K. Hemalatha, K. Ramesha, J. M. Tarascon and A. S. Prakash, Chem. Mater., 2012, 24, 1846–1853 CrossRef.
- Y. K. Yoon, C. W. Park, H. Y. Ahn, D. H. Kim, Y. S. Lee and J. Kim, J. Phys. Chem. Solids, 2007, 68, 780–784 CrossRef.
- K. Saravanan, A. Jarry, R. Kostecki and G. Chen, Sci. Rep., 2015, 5, 8027 CrossRef PubMed.
- H. Xia, Y. Wan, W. Assenmacher, W. Mader, G. Yuan and L. Lu, NPG Asia Mater., 2014, 6, e126 CrossRef.
- W. Liu, P. Oh, X. Liu, M.-J. Lee, W. Cho, S. Chae, Y. Kim and J. Cho, Angew. Chem., Int. Ed., 2015, 54, 4440–4457 CrossRef PubMed.
- M. M. Thackeray, Prog. Solid State Chem., 1997, 25, 1–71 CrossRef.
- M. Y. Song, H. Rim and H. R. Park, Ceram. Int., 2013, 39, 6937–6943 CrossRef.
- H.-J. Kweon, G. B. Kim, H. S. Lim, S. S. Nam and D. G. Park, J. Power Sources, 1999, 83, 84–92 CrossRef.
- X. Wang, M. Tamaru, M. Okubo and A. Yamada, J. Phys. Chem. C, 2013, 117, 15545–15551 Search PubMed.
- B. Mortemard de Boisse, D. Carlier, M. Guignard and C. Delmas, J. Electrochem. Soc., 2013, 160, A569–A574 CrossRef.
- B. Mortemard de Boisse, PhD thesis, Université de Bordeaux, 2014.
- F. Zhou, M. Cococcioni, K. Kang and G. Ceder, Electrochem. Commun., 2004, 6, 1144–1148 CrossRef.
- S.-J. Lee, J.-K. Lee, D.-W. Kim, H.-K. Baik and S.-M. Lee, J. Electrochem. Soc., 1996, 143, L268–L270 CrossRef.
- C. Julien, C. Letranchant, S. Rangan, M. Lemal, S. Ziolkiewicz, S. Castro-Garcia, L. El-Farh and M. Benkaddour, Mater. Sci. Eng., B, 2000, 76, 145–155 CrossRef.
- I. Belharouak, H. Tsukamoto and K. Amine, J. Power Sources, 2003, 119–121, 175–177 CrossRef.
- M. V. Reddy, G. V. S. Rao and B. V. R. Chowdari, J. Phys. Chem. C, 2007, 111, 11712–11720 Search PubMed.
- R. Stoyanova, E. Zhecheva, R. Alcántara, J. L. Tirado, G. Bromiley, F. Bromiley and T. Boffa Ballaran, Solid State Ionics, 2003, 161, 197–204 CrossRef.
- G. X. Wang, J. Horvat, D. H. Bradhurst, H. K. Liu and S. X. Dou, J. Power Sources, 2000, 85, 279–283 CrossRef.
- J. Cho and B. Park, J. Power Sources, 2001, 92, 35–39 CrossRef.
- J. M. Wang, J. P. Hu, C. Y. Ouyang, S. Q. Shi and M. S. Lei, Solid State Commun., 2011, 151, 234–237 CrossRef.
- P. T. Barton, Y. D. Premchand, P. A. Chater, R. Seshadri and M. J. Rosseinsky, Chem.–Eur. J., 2013, 19, 14521–14531 CrossRef PubMed.
- E. Antolini and M. Ferretti, Mater. Lett., 1997, 30, 59–63 CrossRef.
- E. Antolini, J. Mater. Chem., 1998, 8, 2783–2786 RSC.
- S. Yang, H. Yue, Y. Yin, J. Yang and W. Yang, Electrochim. Acta, 2006, 51, 4971–4976 CrossRef.
- A. Navulla, L. Huynh, Z. Wei, A. S. Filatov and E. V. Dikarev, J. Am. Chem. Soc., 2012, 134, 5762–5765 CrossRef PubMed.
- Z. Wei, H. Han, A. S. Filatov and E. V. Dikarev, Chem. Sci., 2014, 5, 813–818 RSC.
- M. C. Barry, Z. Wei, T. He, A. S. Filatov and E. V. Dikarev, J. Am. Chem. Soc., 2016, 138, 8883–8887 CrossRef PubMed.
- Z. Wei, A. S. Filatov and E. V. Dikarev, J. Am. Chem. Soc., 2013, 135, 12216–12219 CrossRef PubMed.
- T. J. Boyle, M. L. Neville, C. A. Apblett, S. M. Hoppe and M. Gembicky, Polyhedron, 2013, 65, 89–97 CrossRef.
- M. Tiitta and L. Niinistou, Chem. Vap. Deposition, 1997, 3, 167–182 CrossRef.
- V. G. Kessler, Chem. Commun., 2003, 1213–1222 RSC.
- H. Han, Z. Wei, M. C. Barry, A. S. Filatov and E. V. Dikarev, Dalton Trans., 2017, 46, 5644–5649 RSC.
- M. Nespolo, G. Ferraris and H. Takeda, Acta Crystallogr., Sect. A: Found. Adv., 2000, 56, 132–148 CrossRef.
- C. M. Lieberman, M. C. Barry, Z. Wei, A. Y. Rogachev, X. Wang, J.-L. Liu, R. Clérac, Y.-S. Chen, A. S. Filatov and E. V. Dikarev, Inorg. Chem., 2017, 56, 9574–9584 CrossRef PubMed.
- V. Subramanian, K. Karki and B. Rambabu, Solid State Ionics, 2004, 175, 315–318 CrossRef.
- Y.-K. Sun, I.-H. Oh and K. Y. Kim, J. Mater. Chem., 1997, 7, 1481–1485 RSC.
- J. Akimoto, Y. Gotoh and Y. Oosawa, J. Solid State Chem., 1998, 141, 298–302 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, synthetic details, X-ray powder diffraction patterns, IR and mass spectra, phase analysis of thermal decomposition traces of the heterometallic precursor. CCDC 1823874–1823877. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc00917a |
| This journal is © The Royal Society of Chemistry 2018 |