
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Taming a monomeric [Cu(η6-C6H6)]+ complex with silylene†‡
Nasrina
Parvin
a,
Shiv
Pal
a,
Jorge
Echeverría
 *b,
Santiago
Alvarez
*b and
Shabana
Khan
*a
*b,
Santiago
Alvarez
*b and
Shabana
Khan
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research Pune, Dr Homi Bhaba Road, Pashan, Pune-411008, India. E-mail: shabana@iiserpune.ac.in
bInorganic Chemistry Department, Facultat de Química, Universitat de Barcelona, Diagonal, 645, 08028 Barcelona, Spain. E-mail: jorge.echeverria@qi.ub.es; santiago.alvarez@qi.ub.es
First published on 13th April 2018
Abstract
Previous theoretical and experimental endeavors suggested that [Cu(C6H6)]+ prefers the η1/η2 mode over the η6 mode due to the augmented repulsion between the benzene ring and metal d-electrons. Nevertheless, the use of silylene as a neutral ligand has led to the isolation of the first monomeric copper cation, [{PhC(NtBu)2SiN(SiMe3)2}Cu(η6-C6H6)]+[SbF6]− (3), where a copper atom is bound to the benzene ring in an unsupported η6 fashion. However, the use of IPr (1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) in place of silylene results in the formation of [IPr·Cu(η2-C6H6)]+[SbF6]− (6), where the copper atom is bound to the benzene ring in the η2 mode. The discrepancy in hapticities is also reflected when hexamethylbenzene is employed as the arene ring. The silylene supported copper cation continues to bind in the η6 mode in 2 while the NHC copper cation displays an η3 bonding mode in 5. DFT calculations are carried out to understand how the use of silylene led to the η6 binding mode and why IPr afforded the η2 binding mode.
Introduction
Synthetic chemists often find fascination in isolating a compound that has been theoretically predicted as well as observed in the gas phase but never realized under laboratory conditions. However, access to such compounds often poses a formidable synthetic challenge. One such moiety is [Cu(η6-C6H6)]+. It is well evident from the literature that group 11 metal–arene complexes strongly prefer the η2 binding mode.1,2 Armentrout and coworkers reasoned that the preference of the η2 bonding mode over η6 is due to the increase of repulsion between the metal d-electrons and the benzene ligand in the latter.3 Cu–arene complexes with the η6 bonding mode have also been reported albeit in small numbers, when using tethered arene rings in order to create a cavity between the two arene rings by diminishing the repulsion.4 These experimental results were further computationally supported by Guo and co-workers, who found that in the gas phase free Cu+ may form η6 type of complexation with benzene but in the condensed phase the propensity of Cu+ to form η2 complexes with benzene drastically increases in the presence of a counter-anion.5A major breakthrough in this research was recently achieved by Hayton and coworkers, who isolated two half sandwich complexes [(η6-C6Me6)Cu(PR3)][PF6] (R = Ph, OPh) where the C6Me6 ring is bound to the Cu ion in the η6 coordination mode.6 However, they have also theoretically shown that when benzene is employed instead of hexamethylbenzene as an arene, the η2 mode is preferred, and hence surmised that the preference for the η6 mode over the η2 mode is exclusively due to steric repulsion between Me groups and PR3 units. Additionally, they calculated the relative energies for [Cu(C6H6)]+ in gas as well as condensed phases and found the preference for the η2 mode in both phases but more in the condensed phase, as previously predicted by Guo et al. These studies consequently lead to the question: is it even possible to isolate [Cu(η6-C6H6)]+ in the condensed phase?
It is apparent now that one of the main factors responsible for the success or failure of the synthesis of [Cu(η6-arene)]+ is the ligand with an appropriate substituent. Being better σ-donors than phosphines, silylenes have recently been found to attract widespread interest as ligands for transition metals.7 For this challenging work, we turned our attention towards a [PhC(NtBu)2SiN(SiMe3)2]8 supported copper bromide complex, [{PhC(NtBu)2}Si{N(SiMe3)2}]2Cu2Br2 (1).9 An appealing facet of [PhC(NtBu)2SiN(SiMe3)2] is that it accepts electron density from the metal as evidenced in its coinage metal complexes.9,10 We postulate that such back-donation can diminish the electrostatic repulsion between metal d-electrons and arene rings, which may facilitate the formation of the η6 mode. This potential has been duly realized through the isolation and characterization of an unprecedented [Cu(η6-C6H6)]+ complex. For a direct systematic comparison, we carried out the same reactions with N-heterocyclic carbene in place of 1. Our results are reported herein.
Results and discussion
A simple synthetic protocol was designed to generate the desired copper cations. To check the credentials of 1 as a ligand, we commenced our investigation by probing the reaction of 1 with AgSbF6 in the presence of hexamethylbenzene with the assumption that it would furnish [(η6-C6Me6)Cu]+ analogous to Hayton's results. Gratifyingly, the abstraction of bromide ions from a dichloromethane solution of 1 with AgSbF6 in the presence of hexamethylbenzene results in the formation of [{PhC(NtBu)2SiN(SiMe3)2}Cu(η6-C6Me6)]+[SbF6]− (2) (see S1 in the ESI for experimental details†) (Scheme 1).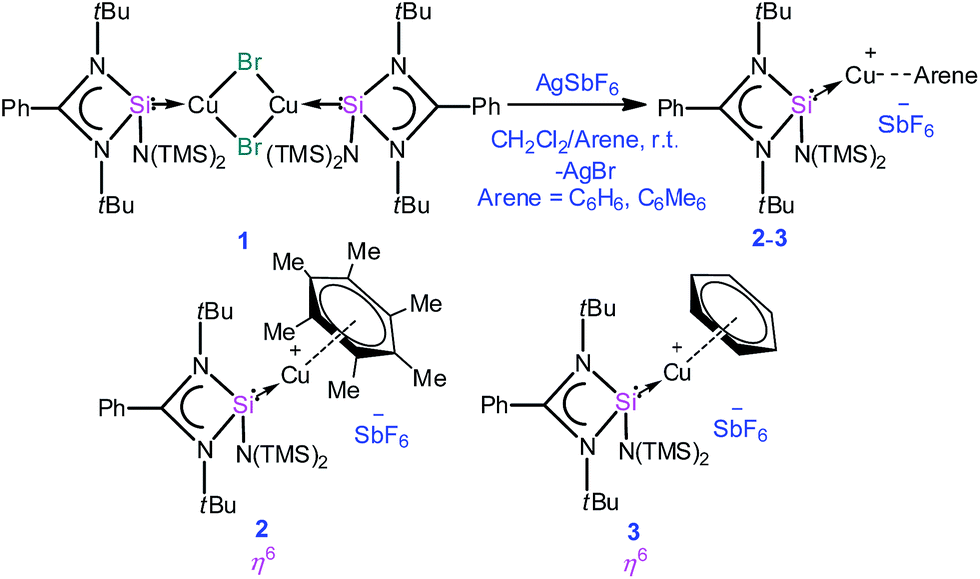 | ||
| Scheme 1 Synthesis of complexes 2 and 3. | ||
The molecular structure of 2 is shown in Fig. 1, which revealed the η6 mode of the arene ring. The Si atom adopts a distorted tetrahedral geometry with a Si→Cu bond length of 2.219(1) Å, which is in good accordance with that in 1 [2.222(2) Å].10 The Cu–C(arene) bond lengths varies from 2.310(4) to 2.449(5) Å, reflecting a slightly unsymmetrical binding of the Cu with respect to the ring. The C–C bond lengths in the arene ring are more or less the same ranging from 1.408(6) to 1.420(6) Å. The distance between the Cu atom and the centroid of the arene ring is 1.920 Å.
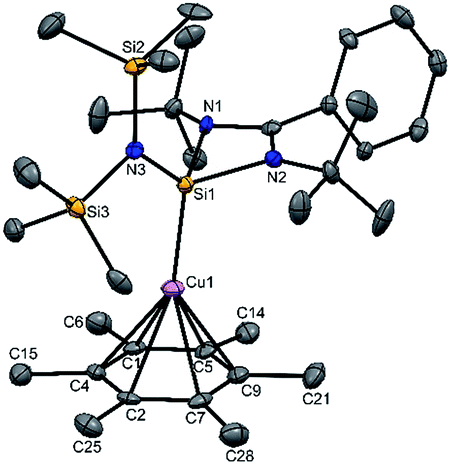 | ||
| Fig. 1 The molecular structure of 2 (ellipsoids are shown at the probability level of 50%). Counter anion SbF6− and hydrogen atoms are omitted for clarity. Selected bond lengths (Å): Si1–Cu1 2.219(1), Si1–N1 1.837(3), Si1–N2 1.834(3), Cu1–C1 2.449(5), Cu1–C2 2.372(4), Cu1–C4 2.443(5), Cu1–C5 2.407(4), Cu1–C7 2.318(4), Cu1–C9 2.310(4), C1–C4 1.408(6), C4–C2 1.414(5), C2–C7 1.420(6), C7–C9 1.410(6), C9–C5 1.409(6), and C5–C1 1.414(6). | ||
Next, we turned our endeavours towards our primary objective of isolating [Cu(η6-C6H6)]+. A similar synthetic protocol was adopted to access [{PhC(NtBu)2SiN(SiMe3)2}Cu(η6-C6H6)]+[SbF6]− (3). Complex 3 crystallizes in the monoclinic space group P21/n. The molecular structure of 3 (Fig. 2) reveals the η6 coordination mode of benzene to the Cu center. The Cu–Cbenzene bond distances range from 2.342(9) to 2.477(8) Å, with an average of 2.404 Å, which is longer than those reported for [Cu(η6-C6Me6)]+.6 Similarly, the distance between the Cu atom and the centroid of the benzene ring (Cu–Ccentroid 1.960 Å) in 3 is slightly longer than those in Hayton's [Cu(η6-C6Me6)]+ complexes (1.800(3) and 1.775(6) Å),6 but significantly shorter than those reported for the tethered Cu(arene) complexes such as Cu(I)-cyclophanes or 9,10-anthracene derived endo-cyclic Cu(I) complexes (∼2.5–3.0 Å).4 The [SbF6] anion in the asymmetric unit shows no significant bonding interaction with the Cu+ atom and the closest approach between the F atom and the Cu center (Cu⋯F) is 4.96(1) Å, which rules out any possibility of interaction between them. The average C–C bond length of the C6H6 ligand in 3 is 1.39 Å (range 1.38(1)–1.40(1) Å) (C–CC6H6(non-bound): 1.40 Å; C–CMe6C6(non-bound): 1.41 Å). The Si(II) atom assumes a distorted tetrahedral geometry with a Si(II)→Cu bond length of 2.231(2) Å, which is similar to that in 1 and 2.
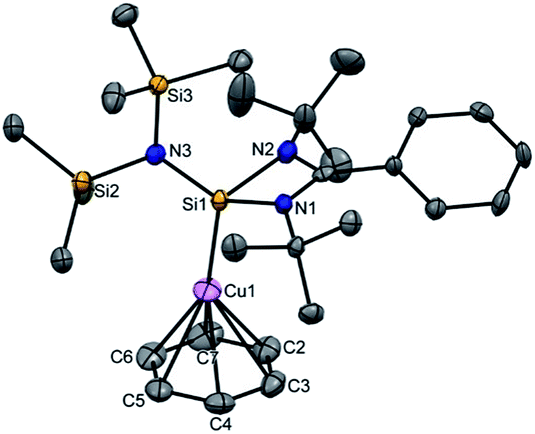 | ||
| Fig. 2 The molecular structure of 3 (ellipsoids are shown at the probability level of 50%). Counter anion SbF6− and hydrogen atoms are omitted for clarity. Selected bond lengths (Å): N2–Si1 1.844(4), N1–Si1 1.847(5), N3–Si1 1.737(5), Cu1–Si1 2.231(2), Cu1–C2 2.454(7), Cu1–C3 2.477(8), Cu1–C4 2.413(9), Cu1–C5 2.359(9), Cu1–C6 2.342(9), Cu1–C7 2.379(8), C2–C3 1.38(1), C3–C4 1.39(1), C4–C5 1.39(1), C5–C6 1.39(1), C6–C7 1.40(1), C7–C2 1.40(1), and Cu1–centroid of the benzene ring 1.960. | ||
All analytical and spectroscopic data of 2 and 3 are consistent with the proposed structures. The binding of benzene to the Cu atom in 3 resulted in a slight downfield shift of the C6H6 protons (δ 7.46 ppm). The appearance of two signals for the trimethylsilyl groups in 1H (δ 0.24 and 0.39 ppm) as well as 29Si NMR (δ 7.21 and 7.65 ppm) of 3 indicates that they are not equivalent and the diastereotopicity arises from the bulky substituents around the Si(II) atom. The Si(II) center resonates at δ 4.41 ppm, which is marginally upfield relative to that in 1 (δ 5.72 ppm) in the 29Si NMR spectrum (Scheme 2).10
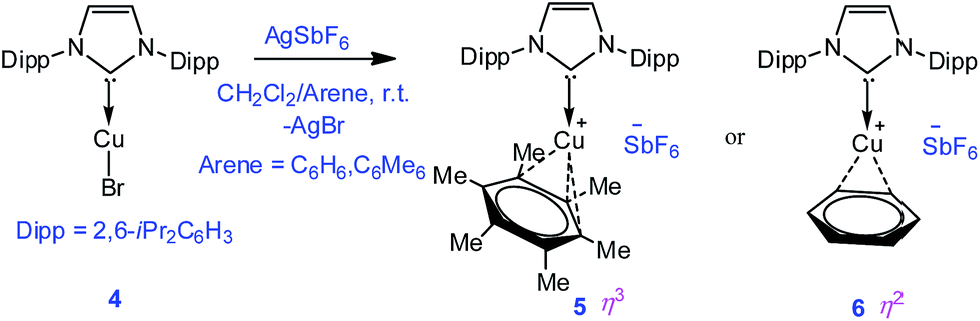 | ||
| Scheme 2 Synthesis of complexes 5 and 6. | ||
To extend the analogous chemistry with N-heterocyclic carbenes we reacted the previously reported IPr·CuBr (4)11 with AgSbF6 in the presence of hexamethylbenzene and benzene, which afforded [IPr·Cu(η3-C6Me6)]+[SbF6]− (5) and [IPr·Cu(η2-C6H6)]+[SbF6]− (6), respectively. Single crystal X-ray studies on 5 and 6 indicated η3 and η2 coordination12 of the Cu atom with the arene rings, respectively (Fig. 3) (please see S3 in the ESI† for the deduction of hapticities in 5 and 6). The CIPr–Cu bond lengths in 5 and 6 are 1.890(3) and 1.886(5) Å, respectively. The Cu–Carene bond lengths in 5 range from 2.114(4) to 2.319(4) Å, and from 2.129(6) to 2.217(5) Å for 6. The methyl protons of the C6Me6 ring appear at δ 1.8 ppm with an integration of 18 protons.
 | ||
| Fig. 3 Molecular structure of 5 and 6 (ellipsoids are shown at the probability level of 50%). Counter anion SbF6− and hydrogen atoms are omitted for clarity. Selected bond lengths (Å) 5: N1–C3 1.353(4), N2–C3 1.356(4), Cu1–C3 1.890(3), Cu1–C18 2.114(4), Cu1–C22 2.289(4), Cu1–C26 2.319(4), Cu1–C21 2.678(4), Cu1–C20 2.715(4), and Cu1–C19 2.894(4). 6: C1–N1 1.356(7), C1–N2 1.349(6), C1–Cu1 1.886(5), Cu1–C29 2.129(6), Cu1–C30 2.217(5), Cu1–C28 2.456(6), Cu1–C31 2.621(5), Cu1–C33 2.813(6), and Cu1–C32 2.892(6). | ||
In order to understand the different hapticities observed experimentally, the geometries of 2, 3, 5, and 6 have been optimized at the DFT level (see the ESI† for a detailed description of the computational procedure and for the atomic coordinates of the optimized structures).
The hapticities and the most relevant bond distances, calculated at the B3LYP-D3 level, are shown in Table 1, together with the experimental values. It can be seen that the experimental Cu–L (L = Si, C) distances are reproduced within 0.02 Å and the Cu–arene ones within 0.1 Å. The η6 coordination in the silylene complexes is well reproduced by our calculations. The hapticities for other cases for which η1, η2 and η3 coordinations can be hard to distinguish are deduced from the values of the distance ratios of the three shortest Cu–Carene distances (d1 < d2 < d3), ρ1 (d2/d1) and ρ2 (d3/d1).12 For the carbene complexes, the calculated hapticity is η3 for 5 and η2 for 6, as indicated by the corresponding values of ρ1 and ρ2. It is worth mentioning here that carbene complexes evolve to η3/η2 when starting the optimization from an η3 geometry, whereas silylene complexes behave conversely.
| Molecule | Hapticity (ρ1, ρ2) | Cu–L (Å) | Shortest Cu–Carene (Å) |
|---|---|---|---|
| 2 | Exp. η6 | 2.219 | 2.310 |
| Calcd. η6 | 2.206 | 2.298 | |
| 3 | Exp. η6 | 2.231 | 2.342 |
| Calcd. η6 | 2.209 | 2.272 | |
| 5 | Exp. η3(1.05, 1.09) | 1.890 | 2.114 |
| Calcd. η3(1.08, 1.13) | 1.886 | 2.078 | |
| 6 | Exp. η2 (1.04, 1.15) | 1.886 | 2.129 |
| Calcd. η2 (1.01, 1.33) | 1.887 | 2.105 |
We have performed an NBO analysis of the benzene complexes 3 and 6 to try to rationalize their different behavior in terms of arene coordination. Second order perturbation analysis revealed that bonding between Cu and the carbene ligand is a donor–acceptor interaction from the carbene lone pair to an empty Cu orbital (nC→nCu*, E = 109.1 kcal mol−1). Cu–silylene donor–acceptor interactions in 3, however, are not clearly determined because the complex could not be decomposed into the same fragments as in 6. On the other hand, the coordination of the benzene ring to the metal atom is associated with donor–acceptor interactions involving a mixture of s and p benzene orbitals (98 and 84 kcal mol−1 in 3 and 6, respectively). Moreover, for the carbene complex there is π-back donation towards the benzene ring (nCu→πC–C*, E = 19.3 kcal mol−1). Another relevant result is that the atomic charge on the donor atoms is −0.06 for CIPr in 6, but +1.26 for the Si atom in 3. These values are consistent with the zero-valent nature of the carbenoid carbon atom and the formal positive charge of the Si atom in the zwitterionic Lewis structure of the ligand (Scheme 3), calculated to be +1.18 for the free ligand. The calculated charge on Si in 3 is thus the result of a formal positive charge increased by σ donation, partially compensated by π back-donation from Cu.
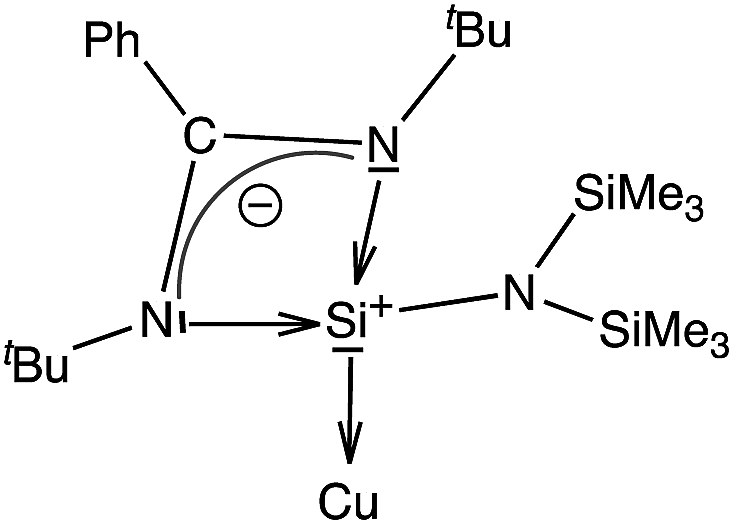 | ||
| Scheme 3 Zwitterionic form of the silylene ligand. | ||
The presence of H⋯H attractive interactions,13 involving the arene's hydrogen atoms on one side, and those of the iPr and SiMe3 groups of the carbene and silylene ligands on the other side, might also have some effect on the different stabilities of the η6 coordination in the two cases. The optimized complexes present numerous dihydrogen contacts between the arene and the silylene ligand at distances in the range of 2.3–2.5 Å (consistent with C⋯C distances of 3.5–3.9 Å in the crystal structures). Such intramolecular interactions have been shown to stabilize otherwise unstable systems, as for example in the case of molecules with very long C–C bonds14 or the cis form of a substituted azobenzene.15
We have performed NCI (non-covalent interaction) calculations16 (see the NCI maps of 2, 3, 5 and 6 in ESI, S4†), observing regions of attractive non-covalent interactions between the hydrogen atoms of the benzene and the methyl groups of the silylene in 3 (also in the hexamethylbenzene complex 2) and between the benzene hydrogens and the carbene iPr groups in 6. Indeed, AIM analysis of 3 discloses a bond path between the H atoms of the coordinated C2H2 moiety of benzene and those of the iPr groups of the carbene, with an electron density at a bond critical point of 0.003 au, similar to previously reported dihydrogen interactions.17
To further test the relative influence of steric repulsions and non-covalent interactions on the hapticity of the coordinated arenes we have carried out energy decomposition analysis (EDA) for 3 and 6 (Table 2), as well as for the hypothetical complex 6′ in which the benzene is forced to be coordinated in an η6 mode. In this constrained model complex, which is not a minimum of the potential energy surface, the Cu–Carene distances were set to those of 3. In general, the interaction energy and its decomposition was evaluated between two molecular fragments: the C6H6 ring and the Cu(IPr) and Cu(silylene) fragments, respectively. It can be seen that the results for 6′ indicate a much stronger Pauli repulsion between the two ligands than in 3, which is relieved by its slippage to an η2 coordination. Even if slippage also reduces the stabilizing components, the balance yields a more favorable interaction energy due to the dramatic decrease of the Pauli repulsion term. EDA analysis of the interaction between benzene and the complementary ligands in the absence of the Cu centre for the three compounds in Table 2 allows us to estimate a Pauli term that calibrates the part of the steric repulsion that comes from benzene–ligand interactions, and we can also roughly estimate the benzene–Cu repulsion as the difference between the total and the benzene–ligand Pauli terms. The important Pauli repulsion between the η6-C6H6 molecule and the Cu ion can be attributed to the interaction between the occupied π(e1g) orbitals of benzene and the dxz and dyz orbitals of Cu. Hence, the smaller repulsion in η6-3 compared to η6-6′ must also be attributed to the longer Cu–Si/Ccarbene distance in the former case (2.21 Å in 3vs. 1.89 Å in 6′). Clearly, slippage of the benzene ring from the η6 coordination in 6′ to the η2 mode in 6 results in a significant decrease in both the benzene–carbene and benzene–copper Pauli repulsions, and explains the preference for the η2 mode in the latter, in contrast with the preference for the η6 coordination in 3.
| Cpd. | ΔEint | ΔEelect | ΔEdisp | ΔEpol | ΔECT | ΔEPauli | ||
|---|---|---|---|---|---|---|---|---|
| Total | C6H6·L | C6H6·Cu | ||||||
| 3 (η6) | −23.3 | −51.7 | −15.4 | −25.0 | −27.5 | 96.4 | 10.6 | 85.8 |
| 6′ (η6) | −17.4 | −70.6 | −22.0 | −27.0 | −32.8 | 134.9 | 37.5 | 97.4 |
| 6 (η2) | −38.9 | −36.4 | −15.0 | −19.7 | −23.0 | 55.4 | 7.2 | 48.2 |
The dispersion interaction contributes significantly to the bonding between the C6H6 ring and the CuL fragments (L = {PhC(NtBu)2SiN(SiMe3)2} and IPr for 3 and 6, respectively), and outweighs in both cases the benzene–ligand steric repulsions, contributing some 5–8 kcal mol−1 to the bonding between the benzene and the CuL fragment. It must be noted that the dispersion contribution is similar in the two cases and therefore has a negligible effect on the hapticity.
Conclusions
This study was undertaken to synthesize the first copper cation bound to the benzene ring in an unsupported η6 mode. The silylene supported copper cation was found to be bound with both benzene and hexamethylbenzene in an η6 mode. DFT calculations revealed that the positive charge on silylene favors back-donation from the Cu atom, thus relieving the repulsions between the benzene π-system and the Cu d-electrons. Furthermore, the long Cu–Si bond distance places the tBu substituents of the silylene at a longer distance from the arene hydrogens, thereby significantly reducing the steric repulsion that prevents the η6 coordination in the case of the NHC complexes. We conclude that, to favor an η6 coordination mode, the complementary ligands must have a π-acceptor character, with a third row donor atom to minimize steric repulsions, and with a relatively small cone angle. Based on these principles, we have carried out test calculations on the so far unprepared [(C6H6)Cu(CN)] and [(C6H6)Cu(CO)]+ complexes that disclose an η6 coordination in both cases and similar bonding parameters to those in compound 3 reported herein.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SK thanks SERB (India) and DST-FIST for support. NP and SP thank UGC for providing fellowships. SA and JE thank the Spanish MINECO for funds (CTQ2015-64579-C3-1-P) and for a Juan de la Cierva fellowship (IJC-2014-20097), and CSUC for computational resources. We thank the reviewers for their critical inputs to improve the quality of the manuscript.Notes and references
- (a) E. Herrero-Gómez, C. Nieto-Oberhuber, S. López, J. Benet-Buchholz and A. M. Echavarren, Angew. Chem., 2006, 118, 5581–5585 ( Angew. Chem., Int. Ed. , 2006 , 45 , 5455–5459 ) CrossRef; (b) V. Lavallo, G. D. Frey, S. Kousar, B. Donnadieu and G. Bertrand, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13569–13573 CrossRef CAS PubMed; (c) K. Shelly, D. C. Finster, Y. J. Lee, W. R. Scheidt and C. A. Reed, J. Am. Chem. Soc., 1985, 107, 5955–5959 CrossRef CAS.
- (a) M. B. Dines and P. H. Bird, J. Chem. Soc., Chem. Commun., 1973, 12 RSC; (b) R. R. Conry, Chem. Commun., 1998, 555–556 RSC; (c) R. R. Conry, W. S. Striejewske and A. A. Tipton, Inorg. Chem., 1999, 38, 2833–2843 CrossRef CAS PubMed; (d) A. M. Lee, S. J. Na, H. Y. Kwon, B. Y. Lee and S. O. Kang, Organometallics, 2004, 23, 5382–5385 CrossRef; (e) R. W. Turnerla and E. L. Amma, J. Am. Chem. Soc., 1966, 88, 1877–1882 CrossRef.
- F. Meyer, F. A. Khan and P. B. Armentrout, J. Am. Chem. Soc., 1995, 117, 9740–9748 CrossRef CAS.
- (a) M. Mascal, J.-L. Kerdelhu, A. J. Blake and P. A. Cooke, Angew. Chem., 1999, 111, 2094–2096 ( Angew. Chem., Int. Ed. , 1999 , 38 , 1968–1971 ) CrossRef; (b) F.-B. Xu, Q.-S. Li, L.-Z. Wu, X.-B. Leng, Z.-C. Li, X.-S. Zeng, Y. L. Chow and Z.-Z. Zhang, Organometallics, 2003, 22, 633–640 CrossRef CAS.
- S.-L. Zhang, L. Liu, Y. Fu and Q.-X. Guo, J. Mol. Struct., 2005, 757, 37–46 CrossRef CAS.
- A. M. Wright, B. J. Irving, G. A. Wu, J. H. M. Meijer and T. W. Hayton, Angew. Chem., 2015, 127, 3131–3134 ( Angew. Chem., Int. Ed. , 2015 , 54 , 3088–3091 ) CrossRef.
- (a) C. I. Someya, M. Haberberger, W. Wang, S. Enthaler and S. Inoue, Chem. Lett., 2013, 42, 286–288 CrossRef CAS; (b) A. Brück, D. Gallego, W. Wang, E. Irran, M. Driess and J. F. Hartwig, Angew. Chem, 2012, 124, 11645–11649 ( Angew. Chem., Int. Ed. , 2012 , 51 , 11478–11482 ) CrossRef; (c) L. Alvarez-Rodriguez, J. A. Cabeza, P. Garcia-Alvarez and P. Polo, Coord. Chem. Rev., 2015, 300, 1–28 CrossRef CAS; (d) B. Blom, M. Stoelzel and M. Driess, Chem.–Eur. J., 2013, 19, 40–62 CrossRef CAS PubMed; (e) S. Khan, S. K. Ahirwar, S. Pal, N. Parvin and N. Kathewad, Organometallics, 2015, 34, 5401–5406 CrossRef CAS; (f) S. Khan, S. Pal, N. Kathewad, I. Purushothaman, S. De and P. Parameswaran, Chem. Commun., 2016, 52, 3880–3882 RSC; (g) W. Wang, S. Inoue, S. Enthaler and M. Driess, Angew. Chem., 2012, 124, 6271–6275 ( Angew. Chem., Int. Ed. , 2012 , 51 , 6167–6171 ) CrossRef; (h) W. Wang, S. Inoue, E. Irran and M. Driess, Angew. Chem., 2012, 124, 3751–3754 ( Angew. Chem., Int. Ed. , 2012 , 51 , 3691–3694 ) CrossRef; (i) W. Wang, S. Inoue, S. Yao and M. Driess, J. Am. Chem. Soc., 2010, 132, 15890–15892 CrossRef CAS PubMed; (j) D. Gallego, A. Brück, E. Irran, F. Meier, M. Kaupp, M. Driess and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 15617–15626 CrossRef CAS PubMed.
- S. S. Sen, J. Hey, R. Herbst-Irmer, H. W. Roesky and D. Stalke, J. Am. Chem. Soc., 2011, 133, 12311–12316 CrossRef CAS PubMed.
- N. Parvin, R. Dasgupta, S. Pal, S. S. Sen and S. Khan, Dalton Trans., 2017, 46, 6528–6532 RSC.
- N. Parvin, S. Pal, S. Khan, S. Das, S. K. Pati and H. W. Roesky, Inorg. Chem., 2017, 56, 1706–1712 CrossRef CAS PubMed.
- S. Díez-González, E. C. Escudero-Adán, J. Benet-Buchholz, E. D. Stevens, A. M. Z. Slawin and S. P. Nolan, Dalton Trans., 2010, 39, 7595–7606 RSC.
- A. Falceto, E. Carmona and S. Alvarez, Organometallics, 2014, 33, 6660–6668 CrossRef CAS.
- J. Echeverría, G. Aullón, D. Danovich, S. Shaik and S. Alvarez, Nat. Chem., 2011, 3, 323–330 CrossRef PubMed.
- P. R. Schreiner, L. V. Chernish, P. A. Gunchenko, E. Y. Tikhonchuk, H. Hausmann, M. Serafin, S. Schlecht, J. E. P. Dahl, R. M. K. Carlson and A. A. Fokin, Nature, 2011, 477, 308–311 CrossRef CAS PubMed.
- L. Schweighauser, M. A. Strauss, S. Bellotto and H. A. Wegner, Angew. Chem., 2015, 127, 13636–13639 ( Angew. Chem., Int. Ed. , 2015 , 54 , 13436–13439 ) CrossRef.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- J. Echeverría, G. Aullón and S. Alvarez, Int. J. Quantum Chem., 2017, 117, e25432 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details of 2, 3, 5 and 6, their single crystal X-ray data, and details of theoretical calculations. CCDC 1540046, 1540047, 1547791, and 1547792. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc00459e |
| ‡ Dedicated to Prof. Krishna N. Ganesh on the occasion of his 65th birthday. |
| This journal is © The Royal Society of Chemistry 2018 |