
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn iridium(III/IV/V) redox series featuring a terminal imido complex with triplet ground state†
Markus
Kinauer
 a,
Martin
Diefenbach
b,
Heiko
Bamberger
c,
Serhiy
Demeshko
a,
Edward J.
Reijerse
d,
Christian
Volkmann
a,
Christian
Würtele
a,
Joris
van Slageren
c,
Bas
de Bruin
*e,
Max C.
Holthausen
*b and
Sven
Schneider
*a
a,
Martin
Diefenbach
b,
Heiko
Bamberger
c,
Serhiy
Demeshko
a,
Edward J.
Reijerse
d,
Christian
Volkmann
a,
Christian
Würtele
a,
Joris
van Slageren
c,
Bas
de Bruin
*e,
Max C.
Holthausen
*b and
Sven
Schneider
*a
aUniversität Göttingen, Institut für Anorganische Chemie, Tammannstr. 4, 37077 Göttingen, Germany. E-mail: sven.schneider@chemie.uni-goettingen.de
bInstitut für Anorganische und Analytische Chemie, Goethe-Universität, Max-von-Laue-Str. 7, 60438 Frankfurt am Main, Germany
cInstitut für Physikalische Chemie, Universität Stuttgart, Pfaffenwaldring 55, D-70569 Stuttgart, Germany
dMax-Planck-Institut für Chemische Energiekonversion, Stiftstr. 34-36, 45470 Mülheim an der Ruhr, Germany
evan 't Hoff Institute for Molecular Sciences (HIMS), University of Amsterdam, The Netherlands. E-mail: b.debruin@uva.nl
First published on 17th April 2018
Abstract
The iridium(III/IV/V) imido redox series [Ir(NtBu){N(CHCHPtBu2)2}]0/+/2+ was synthesized and examined spectroscopically, magnetically, crystallographically and computationally. The monocationic iridium(IV) imide exhibits an electronic doublet ground state with considerable ‘imidyl’ character as a result of covalent Ir–NtBu bonding. Reduction gives the neutral imide [Ir(NtBu){N(CHCHPtBu2)2}] as the first example of an iridium complex with a triplet ground state. Its reactivity with respect to nitrene transfer to selected electrophiles (CO2) and nucleophiles (PMe3), respectively, is reported.
Introduction
Transient metal oxo (LnM=O) and imido (LnM=NR) species are pivotal intermediates in challenging catalytic oxidation reactions, such as oxo-/nitrene-insertion into unactivated C–H bonds or epoxidation/aziridination of olefins.1 Two mechanisms are generally associated with these reactions, i.e. concerted electrophilic O/NR-group transfer and stepwise radical mechanisms, respectively. In any case, the thermochemical basis for such species to be involved in group transfer reactivity are comparatively weak M–O/NR bonds within the oxo/imido intermediates. The reactivity of such species is therefore often associated with the occupation of M–O/N σ- and/or π-antibonding molecular orbitals (MOs), i.e. expressed by the prevalence of electron rich late transition metal catalysts. These simple considerations are illustrated by the ‘oxo wall’, which was defined by Ballhausen and Gray (B + G) as the border for stable oxo complexes with tetragonal symmetry.2 The B + G MO-model (Scheme 1) indicates that valence electron configurations beyond d2 grow increasingly unstable. Accordingly, well-characterized or even isolable low-valent group 8 oxo/nitride/imido complexes are rare,3 and group 9 metals are frequently utilized in oxo/nitrene transfer catalysis.4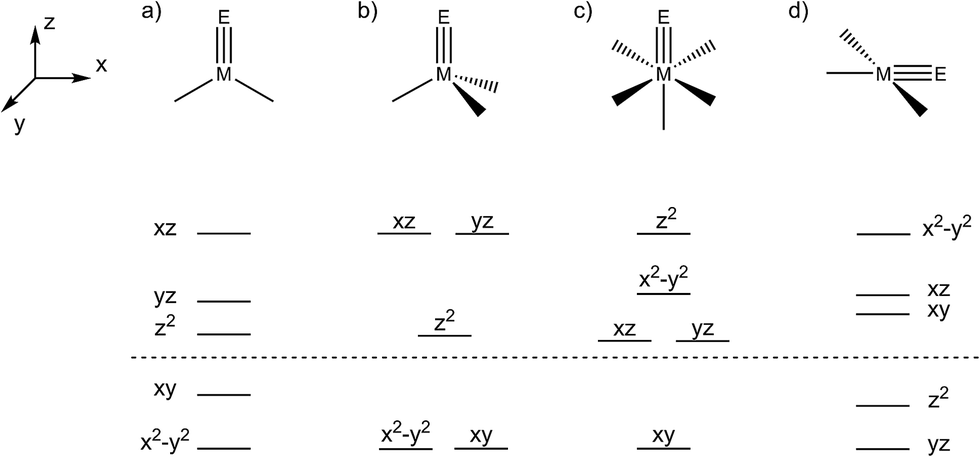 | ||
| Scheme 1 Qualitative d-orbital splitting of complexes with twofold π-donating ligands (E = O, NR, N) in trigonal-planar (C2v) (a), pseudo-tetrahedral (C3v) (b), tetragonal (C4v) (c) and square-planar (C2v) (d) ligand fields. Orbitals below the dashed line are non-bonding, and above anti-bonding in character. | ||
Despite great efforts, characterization of transient oxo/imido intermediates in catalysis remains challenging and mechanistic rationalization heavily relies on computational support.5 Furthermore, well-characterized examples of terminal oxo/imido/nitride complexes beyond group 8 that could be utilized for synthetic modeling and computational benchmarking are scarce.6 Stabilization of such species can arise from O/N-interaction with Lewis or Brønsted acids.7 Otherwise, synthetic strategies generally rely on low metal coordination numbers (Scheme 1) to host both strongly π-donating ligands and high metal valence electron counts. These considerations are exemplified by pseudo-tetrahedral [IrV(O)(Mes)3],8 distorted square-planar [PtIV(O)(LPCN)]+ (LPCN = C6H3[CH2P(tBu)2](CH2)2N(CH3)2),9 or linearly coordinated [NiII(NR)(IPr*)] (IPr* = 1,3-bis(2,6-bis(diphenylmethyl)-4-methylphenyl)imidazol-2-ylidene),10 respectively. Isolable group 9 imido complexes are dominated by pseudo-tetrahedral examples.11 In contrast to the larger body of work for cobalt, Wilkinson's [Ir(O)(Mes)3] and the imides reported by Bergman and Tejel (Fig. 1), respectively, are the only known oxo/imido complexes for the higher homologues Rh and Ir.
 | ||
| Fig. 1 Group 9 imido complexes reported in the literature. | ||
All isolable group 9 imides except Deng's cobalt imide (Fig. 1), exhibit electronic d6 low-spin ground states but nitrene insertion reactivity via radical pathways is generally associated with accessible open shell states.12 For example, Theopold's and Betley's cobalt imides (Fig. 1), which undergo intramolecular nitrene C–H insertion, exhibit thermally accessible open shell excited states.11c,e Furthermore, some cobalt imido catalyst species with open-shell ground states and considerable spin-delocalization onto the NR ligand could be spectroscopically detected.4b,13 Heavier oxo/imido species relevant to catalysis with open-shell ground or low lying excited states, such as [Cp*IrV(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E)X(L)]+ (E = O, NR), were also proposed computationally.14 However, spectroscopically characterized or even isolable examples remain elusive.
E)X(L)]+ (E = O, NR), were also proposed computationally.14 However, spectroscopically characterized or even isolable examples remain elusive.
We recently reported the characterization of the terminal nitride complexes [IrV(N)(PNP)]+ (1), [IrIV(N)(PNP)] (2), [RhIV(N)(PNP)] and [(PNN)RhII(μ-N)RhIII(PNN)] (PNP = N(CHCHPtBu2)2; PNN = NC5H3-2-CHPtBu2-5-py), respectively.15 Attempts to prepare the parent imide [IrIII(NH)(PNP)] (3) gave mixtures of amide [IrII(NH2)(PNP)] (4) and dinitrogen bridged [N2{IrI(PNP)}2] (5), as a result of rapid imide disproportionation and nitride coupling (Scheme 2).4c DFT modeling of 3 indicated several almost degenerate electronic states preventing a reliable description of the ground state. In continuation of this work, we sought to stabilize an iridium imide by N-substitution. Here, the isolation and characterization of the iridium(III/IV/V) imido redox series [Ir(NtBu)(PNP)]n+ (n = 0–2) is reported.
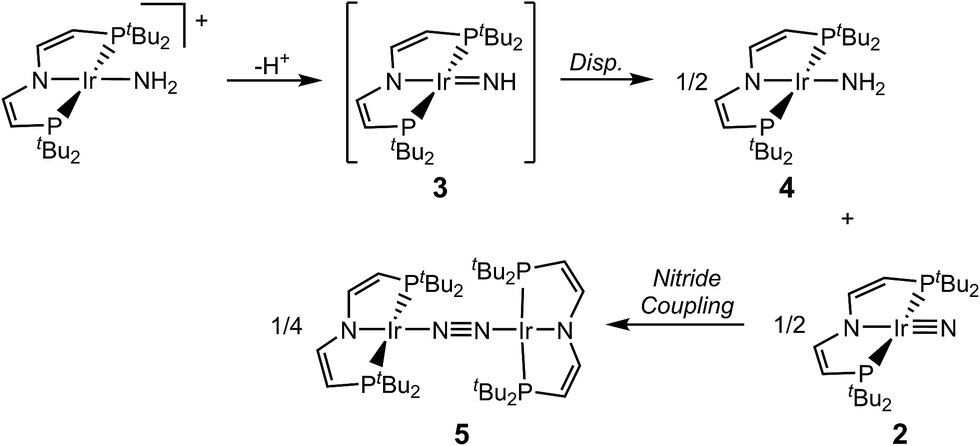 | ||
| Scheme 2 Disproportionation of proposed parent imido complex 3. | ||
Results and discussion
Syntheses of the imido redox series [IrIII/IV/V(NtBu)(PNP)]n+ (n = 0, 1, 2)
Salt metathesis of [IrIICl(PNP)] (6)16 with an excess of lithium tert-butylamide in H2NtBu as solvent gives the green amide complex [IrII(NHtBu)(PNP)] (7; Scheme 3).‡1H NMR spectroscopy indicates near quantitative yields, but the isolation in analytical purity was hampered by the high solubility of 7 and some minor side products. However, in situ oxidation of 7 (ETHF1/2 = −0.8 V vs. Fc/Fc+) with AgPF6 affords the blue, diamagnetic, square-planar iridium(III) amide complex [IrIII(NHtBu)(PNP)]PF6 (8) in analytical purity (Scheme 3).‡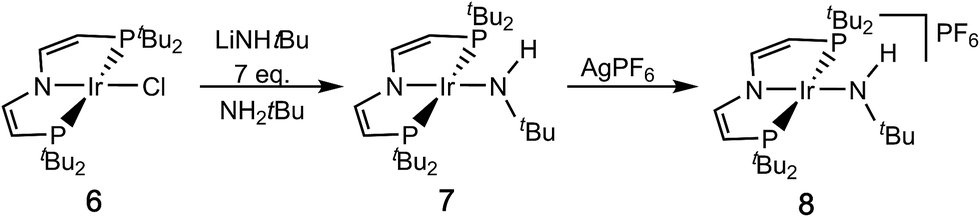 | ||
| Scheme 3 Syntheses of the amido complexes 7 and 8. | ||
All attempts to obtain an iridium(III) imide by deprotonation of 8 resulted in intractable mixtures of several products. Therefore, an iridium(IV) imide was targeted.§ In fact, the violet imide complex [Ir(NtBu)(PNP)]CF3CO2 (9) can be isolated directly from 6 in a one-pot synthesis that couples amide deprotonation with oxidation (Scheme 4). In situ generation of 7 with LiNHtBu (2.7 eq.) and subsequent addition of excess AgO2CCF3 (4.2 eq.) gave the best isolated yields in 9 (57%). Interestingly, the characterization of 9 by cyclic voltammetry (CV) in THF shows both reversible oxidation (E1/2 = +0.4 V vs. Fc+/Fc) and reduction (E1/2 = −0.9 V vs. Fc+/Fc) at remarkably mild potentials, respectively.‡,¶ This observation is in line with the necessity for an excess in oxidant to obtain acceptable yields in 9. However, the mechanism of the 2e−/1H+ oxidation reaction was not examined in detail.
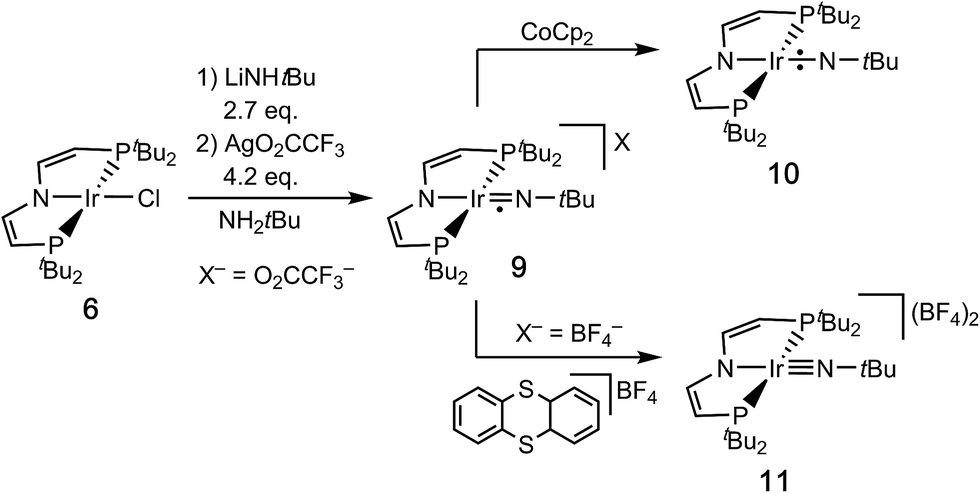 | ||
| Scheme 4 Syntheses of the imido complexes 9–11. | ||
The neutral imido complex [Ir(NtBu)(PNP)] (10) was isolated in yields over 60% upon reduction of 9 with cobaltocene (Scheme 4). Complex 10 is sufficiently stable to be handled at room temperature up to 40–50 °C. Even after repeated crystallization some impurities of iridium(II) amide 7 with almost identical solubility could not be fully separated (see below).
The oxidation of 9 with thianthrenium tetrafluoroborate in MeCN at −30 °C gives the green dicationic iridium(V) imide [Ir(NtBu)(PNP)](BF4)2 (11) in 70% isolated yield (Scheme 4). In contrast to 9 and 10, 11 is only soluble in very polar solvents, like acetonitrile or nitrobenzene and decomposes in solution at temperatures above −10 °C.
Characterization of [IrIII/IV/V(NtBu)(PNP)]n+ (9–11)
The two imide complexes 9 and 10 were characterized crystallographically (Fig. 2). In both cases, the metal ions exhibit approximately square-planar coordination. While the imido moiety in 9 is close to linearity (Ir–N–C: 171.3(3)°), it is more strongly bent in 10 (Ir–N–C: 157.2(2)°). This observation suggests weaker π-bonding for 10 resulting in a shallower imide bending potential. Accordingly, the Ir–NtBu distance of 10 (1.868(2) Å) is longer than in parent 9 (1.805(2) Å). The longer Ir–NtBu bond together with slightly shortened Ir–P bonds (by about 0.04 Å) in 10 indicates considerable covalent effects rather than merely changes in metal ionic radii. Both Ir–NtBu bonds are significantly shorter than in the amides [IrII/III(NH2)(PNP)]n+ (IrII: 1.9521(17) Å; IrIII: 1.900(2) Å) and longer than in nitride 1 (1.678(4) Å),13a Burger's nitride (1.646(9) Å)17 and Bergman's imide [Cp*Ir(NtBu)] (1.712(7) Å),9 which all exhibit Ir![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bonding character. These comparisons support π bonding character intermediate between a single and a triple bond for the Ir–NtBu moiety in 9 and 10, in line with a (yz)2(z2)2(π*1/π*2)x electronic configuration (x = 1 (9), 2 (10); Scheme 1).
N triple bonding character. These comparisons support π bonding character intermediate between a single and a triple bond for the Ir–NtBu moiety in 9 and 10, in line with a (yz)2(z2)2(π*1/π*2)x electronic configuration (x = 1 (9), 2 (10); Scheme 1).
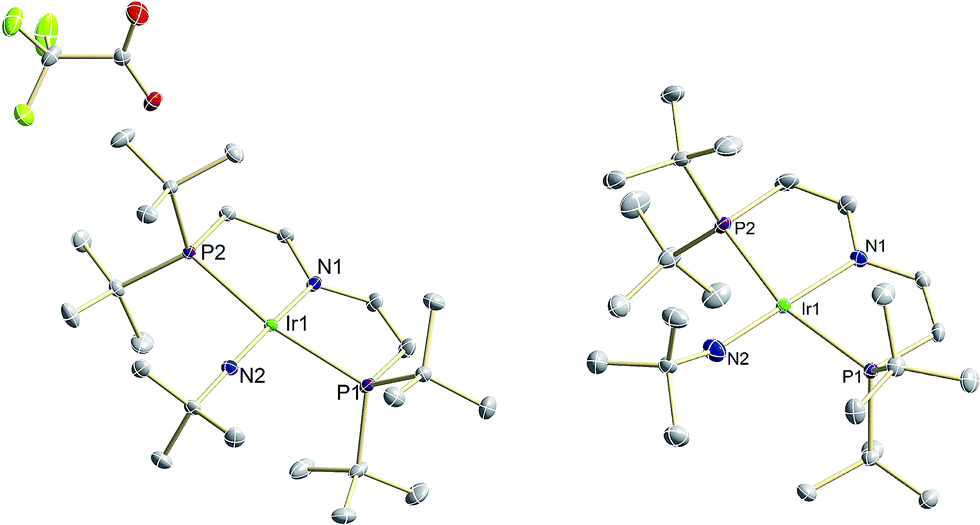 | ||
| Fig. 2 Molecular structures of 9, (left) and 10 (right) derived by single X-ray diffraction. ORTEP plots with anisotropic displacement parameters set at 50% probability. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°] of 9: Ir1–N1 1.979(2), Ir1–N2 1.805(2), N2–C21 1.444(2); Ir1–N2–C21 171.3(2) N1–Ir1–N2 175.55(5). 10: Ir1–N1 2.059(2), Ir1–N2 1.868(2), N2–C21 1.438(3); Ir1–N2–C21 157.2(2), N1–Ir1–N2 178.61(9). | ||
Further information about the electronic structures of 9–11 was obtained from spectroscopic characterization. The NMR spectra of dicationic imide 11 at room temperature support a C2v-symmetric structure with a closed-shell, (yz)2(z2)2 ground state like the isoelectronic iridium(V) nitride 1.15a The sharp 11B and 19F NMR signals of the BF4-anions in solution give no indication for fluoride coordination to the metal. Despite the electronic unsaturation, low metal Lewis acidity at the vacant coordination sites presumably results from the filled dz2 orbital.
The paramagnetic iridium(IV) imide 9 exhibits a magnetic moment in solution derived by Evans' method (μeff = 1.6 ± 0.2 μB) close to the spin-only value for S = 1/2. The doublet ground state is confirmed by X-band EPR spectroscopy (see ESI†). A broad, rhombic EPR signal is observed with g values well below ge (g = [1.709, 1.625, 1.332]), resembling that of iridium(IV) nitride 2 (g = [1.862, 1.582, 1.321]).13a In that case, the unusual g-anisotropy was rationalized in terms of heavy SOMO/LUMO-mixing due to near degeneracy of the perpendicular M–N π*-MOs and the large spin–orbit coupling (SOC) constant of iridium. The high IrN bond covalency of 2 results in considerable spin delocalization to the nitride (‘non-innocent’ behavior) as substantiated by the rhombic 14N hyperfine interaction (HFI) derived from Davies-ENDOR (electron nuclear double resonance) spectroscopy (A(14N) = [−19, +49, −60] MHz). In the present case, ENDOR spectroscopy was unsuccessful, presumably due to anisotropic signal broadening. However, the imide nitrogen HFI tensor of 9 could be obtained from electron–electron double resonance detected NMR (ELDOR-NMR) spectra at W-band (Fig. 3) with the 15N-imide isotopologue of 9 (15N–9). The features below 60 MHz in the spectrum at 5.0 T are dominated by contributions from 191Ir and 193Ir (I = 3/2), while the peaks up to 140 MHz can be assigned to the 31P HFI as supported by DFT computations (see ESI†). Signals attributable to a high frequency transition of a strongly coupled imido 15N nucleus could be simulated with the HFI tensor A(14N) = [+30, +102, −111] MHz. The experimental g- and A-tensors are sufficiently well reproduced by a computational model (see below). In analogy to iridium(IV) nitride 2,15a the rhombicity of the A-tensor and the vanishing isotropic HFI support the picture of an Ir–NtBu π-radical with cylindrical spin density delocalization around the Ir–N π-bond.
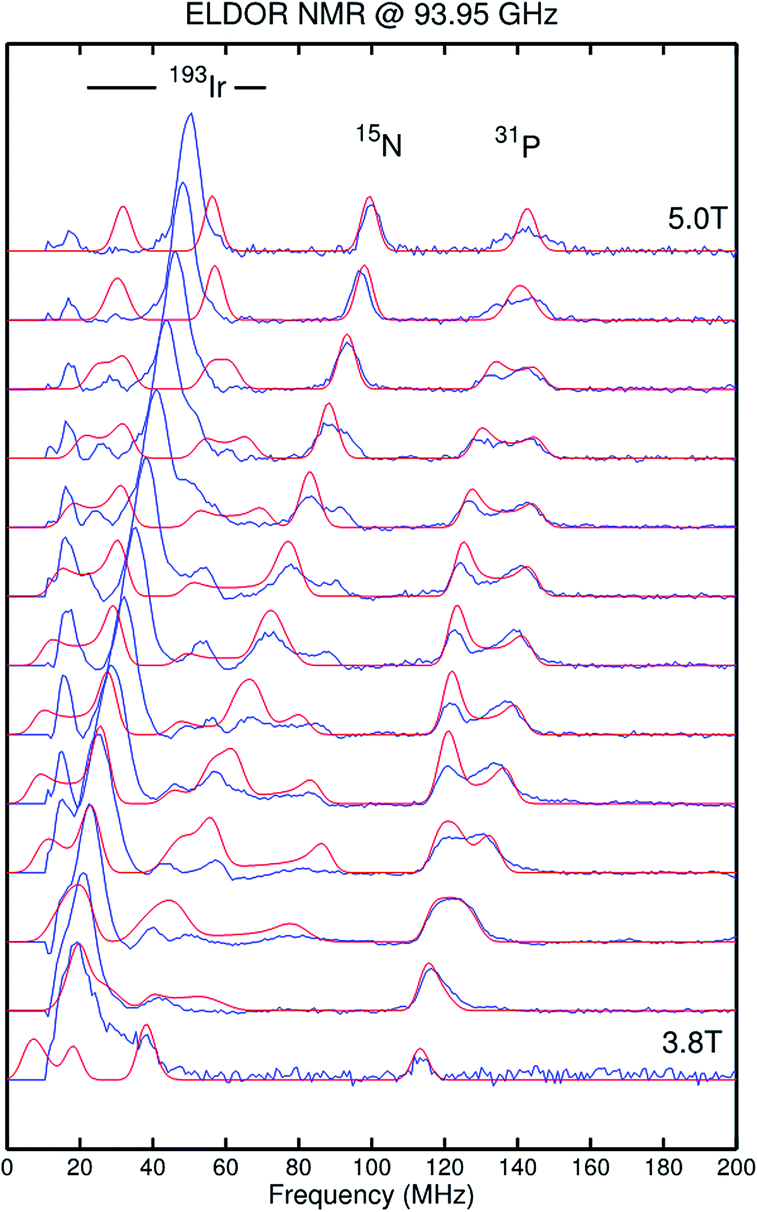 | ||
| Fig. 3 Electron–electron double resonance detected NMR (ELDOR-NMR) spectra of [Ir(15NtBu)(PNP)]+ (15N–9) recorded at W-band (blue line) and simulation (red line). Simulation parameters are provided in the ESI.† | ||
In contrast to the diamagnetic iridium(V) imide 11, no 31P signal was found for the fully reduced iridium(III) imide 10 and the four 1H NMR signals exhibit strongly paramagnetically shifted (+20 to −60 ppm at r.t.), yet relatively sharp (Δν1/2(CH3) = 29 Hz) signals as compared with 9 (Δν1/2(CH3) = 65 Hz). Small amounts of 7 (around 4%) are detected by 1H NMR spectroscopy despite repeated recrystallization. However, quantification from NMR might be misleading due to the much broader lines of iridium(II). The distinct temperature dependence of the 1H NMR chemical shifts of 10 (Δδ up to 26 ppm@173–293 K; Fig. 4) rules out purely temperature independent paramagnetism (TIP) from excited state admixture into a closed-shell ground state, as in case of square-planar osmium(II) complex [OsCl(PNP)].18 Furthermore, the non-linear Curie-plot of the 1H NMR temperature dependence (Fig. 4) indicate thermal population of more than one accessible electronic state within this temperature range.
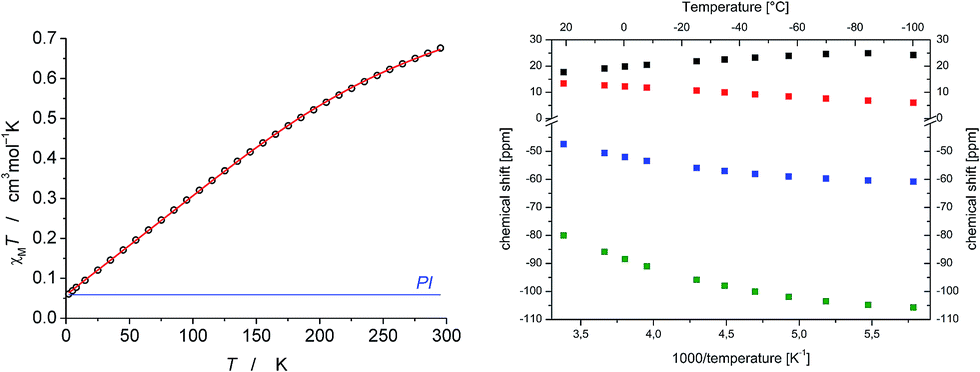 | ||
| Fig. 4 Left: χMT vs. T plot of 10 (dots) measured by SQUID magnetometry, fitting curve (red line) for g = 1.98, D = +466 cm−1 and PI = 15.6% (S = 1/2); right: 1H NMR chemical shifts of 10 in d8-THF as a function of inverse temperature. | ||
The magnetic moment of 10 in solution at r.t. (μeff = 2.3 μB, Evans' method) is considerably lower than the expected spin-only value for a spin triplet (2.83 μB) indicating strong orbital contributions. SQUID magnetometry of a powdered sample (0.68 cm3 mol−1 K or 2.33 μB at 295 K, Fig. 4) confirms the magnetic moment derived by the Evans method, thus ruling out strong solvent effects. On lowering the temperature, the χMT product decreases and below ca. 150 K the χMT curve becomes linear. This low temperature regime of the χMT curve is reminiscent of the magnetic data found for [OsClPNP],18 which exhibits large temperature independent paramagnetism (TIP = 1.03 10−4 cm3 mol−1) in the range of 2–295 K. Supported by multireference NEVPT2/QDPT computations, this data was rationalized with a triplet electronic ground state that is strongly split due to large spin–orbit coupling, which results in a thermally well separated ms = 0 ground level without magnetic moment. However, in case of 10 the intercept of the χMT curve has a finite value of the magnetic moment. To further elucidate these results, magnetic circular dichroism (MCD) measurements of frozen solution samples in 2-MeTHF were carried out (Fig. 5). The MCD spectral band positions in the visible range resemble the UV/vis spectrum of 10. However, complexes 10 and 7 show strong spectral overlap in this range, while only compound 7 exhibits a broad NIR band at λmax = 1190 nm (Fig. S12†). In fact, nesting behavior of the MCD intensity, pointing to contributions of a system with appreciable zero-field splitting of the ground state (complex 10), is only observed for the bands in the visible range (inset Fig. 5). In contrast, the NIR MCD band, i.e. characteristic of complex 7, does not show nesting behavior as expected for an isolated Kramers doublet. Hence, the MCD data point to an S = 1 electronic ground state of complex 10 and NMR and MCD data both support small levels of impurities of 7. Accordingly, good fits are obtained for the SQUID data of 10 using a Zero-Field-Splitting (ZFS) Spin-Hamiltonian (SH) for a spin triplet (S = 1) with g values of ca. 2.0 when considering a paramagnetic impurity (PI) such as 7 (S = 1/2) of up to 15% (Fig. 4). Importantly, D-values around +470 cm−1 were consistently obtained, which places the ms = 0 state well below (ΔE = D) ms = ±1. This axial ZFS splitting parameter is unusually high, yet in a similar range for the square-planar ruthenium(II) complex [RuCl(PNP)] (D = 209 cm−1) and related compounds.19,20 However, the simple SH-formalism treats the spin–orbit interaction as a perturbation of the spin eigenstates |S, Ms〉 and should thus be used with caution if ZFS in that range indicates strong mixing of the ground state with excited states through SOC.21 In such a case, multi-reference calculations are required to rationalize the electronic structure, which are presented in the next section.
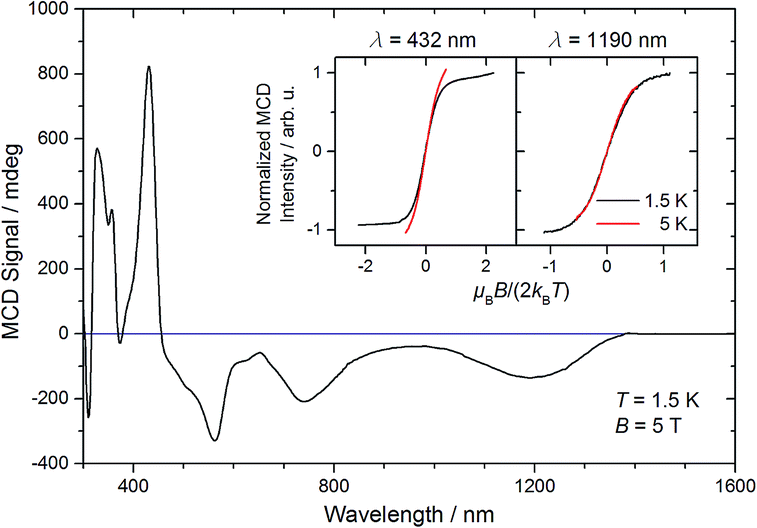 | ||
| Fig. 5 MCD spectrum of 10 recorded in a 2-MeTHF glass at 1.5 K and 5 T. The inset depicts the MCD intensities at 432 nm and 1190 nm divided by the intensity at 1.5 K and maximum field, respectively, as a function of reduced field at different temperatures as indicated. | ||
Computational analysis of [Ir(NtBu)(PNP)]n+ (9–11)
The molecular structure of 9 obtained by DFT computations is in excellent agreement with the experimentally derived structure.‡ The Ir–NtBu multiple bonding character is expressed by the Ir–N Wiberg/Mayer bond indices (WBI/MBI = 1.54/1.67). With reference to nitride 2 (WBI/MBI = 2.07/2.25) the lower values are attributed to weaker donation of the NR+vs. N ligand. The experimental g- and A-tensors of 9 (see above) are sufficiently well reproduced by unrestricted (Zeeman corrected) relativistic (ZORA) calculations (gDFT = (1.881, 1.708, 1.357); A(14N)DFT = [+10, +108, −51] MHz). A qualitative representation of the π-type MO-interactions (Fig. 6a) with (π1)2(π2)2(yz)2(z2)2(π*1)1 occupation resembles the isoelectronic ‘nitridyl’ radical complex 2.13a The computed SOMO (Fig. 6c) exhibits predominantly Ir–NtBu π-antibonding character. Complex 9 has strong nitrogen centered radical character, with a calculated spin density (Fig. 6d) of +80% at the ‘imidyl’ nitrogen atom (2: +51%). This picture suggests a stronger contribution from the {IrIII–NtBu} than from the {IrIV = NtBu} Lewis representation, which is in agreement with the smaller Ir–N WBI/MBI and the Ir–N bond elongation compared with 2. Complex 9 is probably best described as an iridium(III) ‘nitrene radical’ complex, containing a one-electron reduced Fischer-type nitrene ligand (RN˙−).6b Near linearity of the NtBu ligand implies a small energetic spacing of the two π*-MOs, resulting in strong SOMO/LUMO mixing due to the large SOC for this metal, which is expressed in the distinct rhombicities of the g- and A-tensors.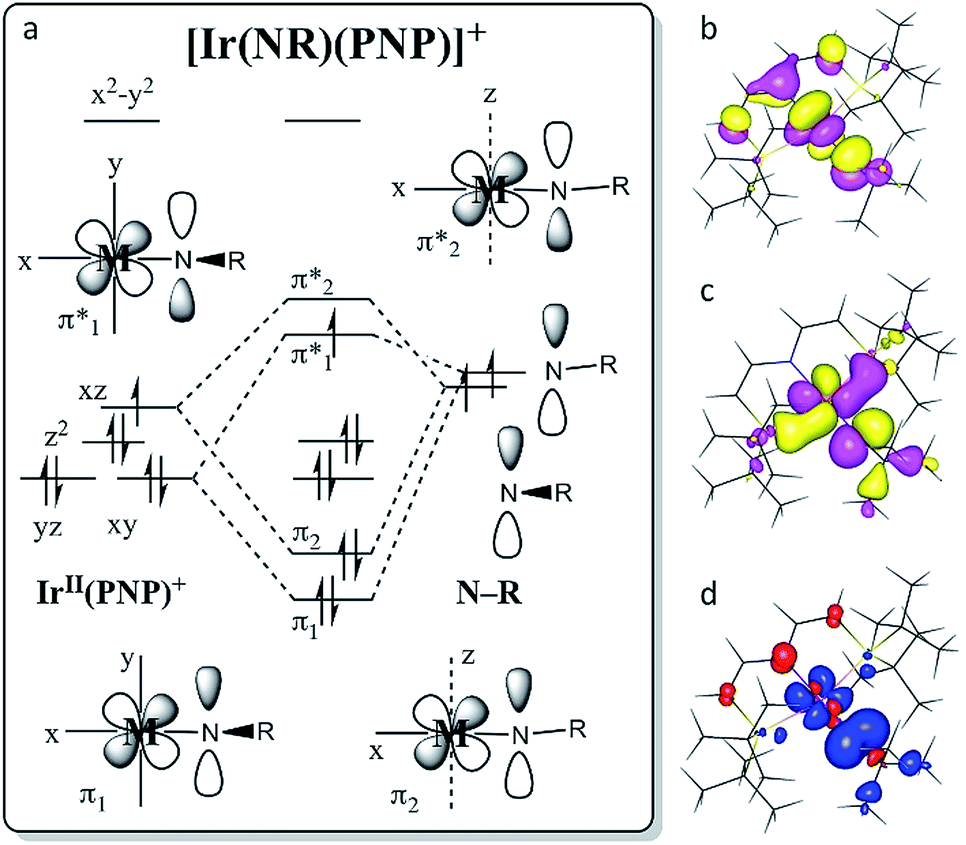 | ||
| Fig. 6 (a) Schematic MO interaction diagram for the Ir(PNP)+ and nitrene fragments of 9. (b) DFT calculated π-LUMO. (c) π-SOMO. (d) Spin density plot with Mulliken spin density distribution: Ir: +15%, NtBu: +80%, NPNP: −5%, CPNP: −5%. | ||
The closed shell ground-state of dicationic imide 11 (and of cationic nitride 1) is in agreement with the picture that evolves from Fig. 6a upon one-electron oxidation. The more intriguing question arises for the nature of reduction product 10. We previously used DFT to compute the electronic structure of the elusive, parent imide 3. Energies of the triplet (T; (yz)2(z2)2(π*1)1(π*2)1) and closed-shell singlet (S; (yz)2(z2)2(π*1)2(π*2)0) states and for an open-shell singlet (OSS) with antiferromagnetically coupled electrons in the π*1/π*2 MOs were all within error precluding a definitive assignment of the ground state multiplicity.6c The DFT model of 10 favors the electronic T over the S state by ΔE = −7.2 kcal mol−1, but correcting for electron correlation with benchmark coupled-cluster ONIOM computations reduces the S/T gap to below 2 kcal mol−1.‡ However, the different electronic states of 10 (and of 3) feature distinctly different Ir–N–tBu imide bending angles (S10: 136°, T10: 160°). In fact, the T model closely resembles the experimental structure (157.2(2)°), while the singlet state computed at this geometry is about ΔE = 12 kcal mol−1 higher in energy. Within the simple MO picture (Fig. 6a), stronger bending of this angle reduces the Ir–NtBu π-overlap in one direction thereby stabilizing a (π1)2(π2)2(yz)2(z2)2(π*1)2 singlet state.
This picture was further refined with multi-reference computations.‡ Based on the DFT-optimized triplet-state geometry of 10, the energies of the non-relativistic singlet and triplet eigenstates were calculated with NEVPT2 (Fig. 7) and corrected for spin–orbit effects employing quasi-degenerate perturbation theory (QDPT). According to these calculations the triplet lies about 4200 cm−1 below the singlet state (about 12 kcal mol−1, fully consistent with the coupled-cluster results). The triplet root is further split by SOC into an isolated ground-state that is placed about 450 cm−1 below two quasi-degenerate states. This picture supports the SH parametrization model for the experimental susceptibility data, which implies a triplet state that is split by strong axial ZFS (D = +466 cm−1) into the |1, 0〉 ground and |1, ±1〉 excited states. Notably, complex 10 is the first isolable iridium compound with a non-low-spin (S > 1/2) ground state.
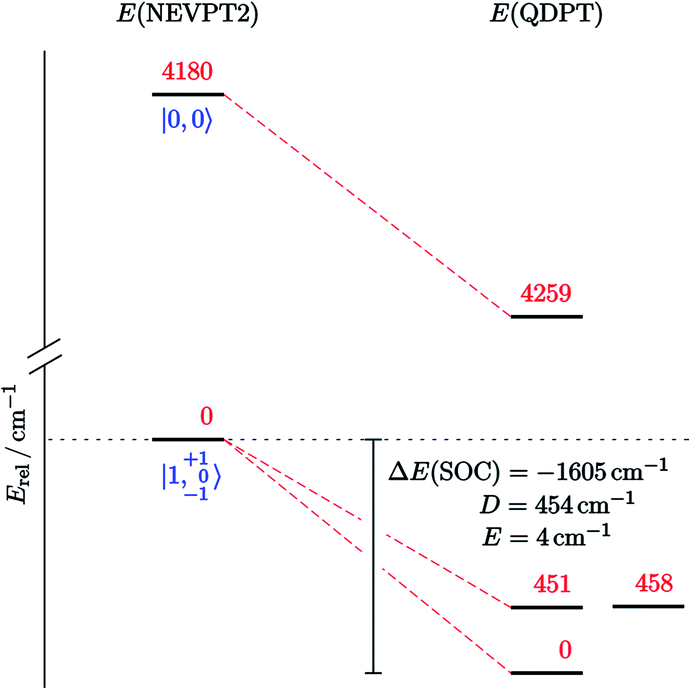 | ||
| Fig. 7 Computed state-energy diagram of 10. Relative energies of the lowest non-relativistic (left) and spin–orbit states (right) in red (cm−1) and corresponding |S, MS〉 labels in blue. | ||
Reactivity of [Ir(NtBu)(PNP)]n+ (9–11)
Linear imido ligands generally exhibit electrophilic behavior as a result of the M–NR triple bonding character.22 In contrast, Bergman's closed-shell iridium imide [(Cp*)Ir(NtBu)] shows distinct N-centered nucleophilic reactivity, e.g. with MeI or CO2.11a Furthermore, the transient, photochemically generated imide complex [WII(NPh)(CO)5], which is formally isolobal with square-planar 10, undergoes nitrene transfer with both nucleophiles and electrophiles.23 This ambiphilic reactivity was rationalized with a small π*1/π*2 HOMO/LUMO gap.Reactivity studies of dicationic imide 11 were hampered by its low thermal stability and poor solubility. Clean and solvent independent decay to the cationic nitride 1 and equimolar amounts of isobutene proceeds within a few minutes at r.t. The fate of the eliminated proton could not be clarified. This decomposition route prevented the examination of nitrene group transfer reactivity at elevated temperatures. In contrast, imide 9 is remarkably stable despite considerable N-radical character (see above) and does not react with selected nucleophiles (PMe3), electrophiles (CO2, olefins) or benzylic C–H bonds (toluene), respectively. Reaction with MeMgCl mainly results in reduction to 10.
In comparison with 9, the nitrene moiety of reduced 10 is more reactive as expected from the additional population of an Ir–N π* MO. The enhanced nucleophilicity of 10 is expressed by the selective reactivity under 1 bar of CO2 at −10 °C to the green, diamagnetic complex 12 (Scheme 5). NMR spectra of 12 indicate C2v symmetry in solution. Structural confirmation was obtained by single crystal X-ray diffraction revealing square-pyramidal (τ = 0.182) metal coordination with an oxygen atom of the bidentate carbamyldiide ligand in apical position (Fig. 8). This immediate product of CO2 addition rearranges at r.t. in solution and in the solid state to mainly the cyclometalated carbamate complex 13 upon proton transfer to the basic carbamyldiide ligand (Scheme 5). The C–H activation product 13 was fully characterized including single crystal X-ray diffraction (Fig. 8).
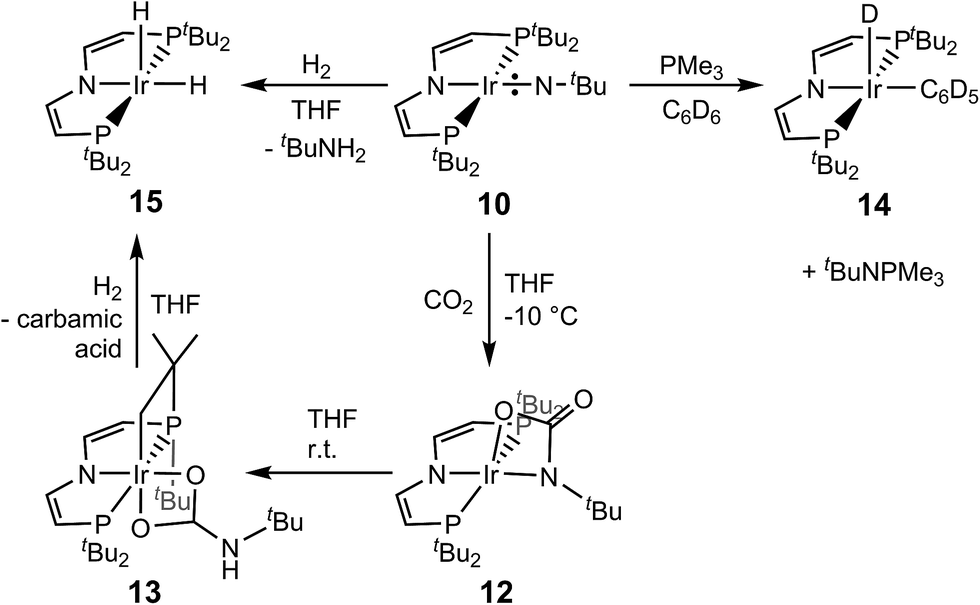 | ||
| Scheme 5 Reactivity of neutral imido complex 10 with PMe3, CO2 and H2. | ||
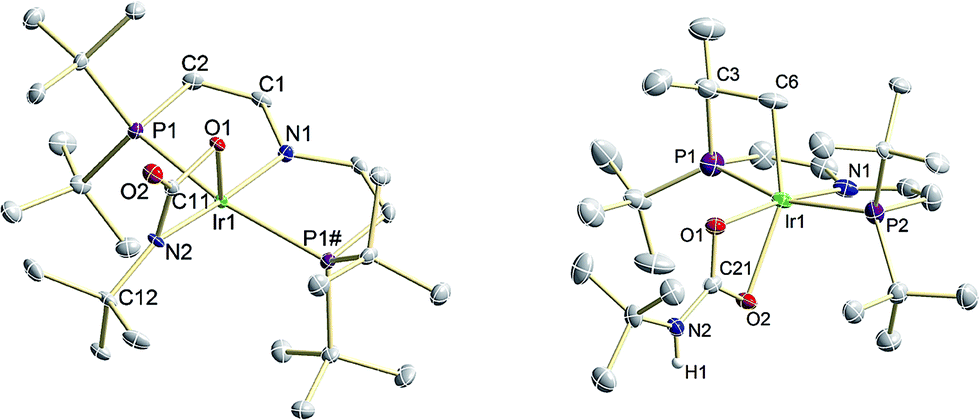 | ||
| Fig. 8 Molecular structures of [Ir{μ-κOC(O)κN(tBu)}(PNP)] (12, left) and [Ir(O2CNHtBu)2{(CH2CMe2)P(tBu)CHCHNCHCHPtBu2}] (13, right), derived by single X-ray diffraction. ORTEP plots with anisotropic displacement parameters set at 50% probability. Solvent molecules and hydrogen atoms except for N–H and C–H2 are omitted for clarity. Selected bond lengths [Å] and angles [°]: 12: Ir1–N1 2.021(3), Ir1–N2 2.112(3), Ir1–O1 2.013(2), N2–C11 1.327(4), C11–O2 1.237(4), C11–O1 1.366(4), P1–Ir1–P1# 162.60(3), N1–Ir1–N2 173.5(2), Ir1–N2–C12 143.7(2); 13: Ir1–N1 2.022(4), Ir1–C6 2.098(6), Ir1–O1 2.129(3), Ir1–O2 2.262(3), C21–O1 1.290(5), C21–O2 1.275(5), C21–N2 1.334(6), P1–Ir1–P2 161.49(5), N1–Ir1–O1 172.0(2), C6–Ir1–O2 155.2(2), C21–N2–C22 127.2(4). | ||
Interestingly, 10 also undergoes nitrene transfer to PMe3 at r.t. in C6D6 over the course of several days (Scheme 5). Formation of the free phosphoraneimine tBuN = PMe3 is evidenced by NMR spectroscopy. The Ir(PNP) fragment is trapped by solvent oxidative addition to the previously reported phenyl hydride complex [Ir(D)(C6D5)(PNP)] (14).24 This result suggests that the lack of nitrene transfer with 9 is not attributable to steric reasons. However, the reaction with 10 might be driven by the C–H oxidative addition rather than an electrophilic nitrene transfer step, which would be expected to be more rapid for cationic 9. Furthermore, it was shown for nitride transfer that the potential ambiphilicity of phosphines can make the rationalization of phosphine imination ambiguous.25,26
Chirik and co-workers reported a square planar iron phenylimido complex with an S = 1 ground-state that forms aniline upon hydrogenolysis.27 In analogy, complex 10 reacts with H2 (1 bar) in THF to give free tert-butylamine and iridium(III) dihydride [IrH2(PNP)] (15) (Scheme 5). The molecular structure of complex 15 in the solid state exhibits square-pyramidal metal coordination with an apical hydride ligand (τ = 0.097). However, on the 1H NMR timescale at room temperature C2v-symmetry is observed due to dynamic behavior. Compound 15 is also obtained from hydrogenolysis of the carbamate 13 (Scheme 5).
Conclusions
In conclusion, the proton coupled oxidation of metal-bound tert-butylamine provides a versatile nitrene source for the synthesis of iridium imido complexes. The spectroscopic, magnetic, crystallographic and computational characterization of the iridium(III/IV/V) imido redox series 9–11 is in line with highly covalent Ir–NtBu bonding as expressed by the simple MO model shown in Fig. 6. The data supports an electronic doublet ground state for 9 with cylindrical delocalization of the spin density perpendicular to the Ir–NtBu bond as a result of SOMO/LUMO mixing through SOC. The reduced, neutral imide 10 is a highly unusual complex well beyond the ‘oxo wall’ with a triplet ground-state, i.e. unprecedented for iridium. It features a (π*1/π*2)2 state with near degeneracy of these orbitals which results in ambiphilic nitrene transfer reactivity with both CO2 and PMe3. In comparison, the chemical inertness of 9 is striking given its ‘imidyl’ radical character. We attribute this finding to stabilizing relativistic contributions by SOC-induced spin delocalization, which moderates the reactivity of this class of electron-rich heavy metal imido complexes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. S. acknowledges to the European Research Council (ERC Grant Agreement 646747) for funding. B. d. B. gratefully acknowledges financial support from NWO-CW (VICI project 016122613) and the University of Amsterdam (RPA Sustainable Chemistry). J. v. S. acknowledges funding for the MCD-spectrometer from the DFG (INST41/864). Quantum-chemical calculations of the Frankfurt group were performed at the Center for Scientific Computing (CSC) Frankfurt on the LOEWE-CSC computer cluster.Notes and references
- (a) A. Gunay and K. H. Theopold, Chem. Rev., 2010, 110, 1060 CrossRef CAS PubMed; (b) G. Dequirez, V. Pons and P. Dauban, Angew. Chem., Int. Ed., 2012, 51, 7384 CrossRef CAS PubMed; (c) J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911 CrossRef CAS PubMed.
- J. R. Winkler and H. B. Gray, Struct. Bonding, 2012, 142, 17 CrossRef CAS.
- J. Abbenseth, S. C. Bete, C. Volkmann, C. Würtele and S. Schneider, Organometallics, 2018, 37, 802 CrossRef CAS.
- Representative examples: (a) C. G. Espino and J. Du Bois, Angew. Chem., Int. Ed., 2001, 40, 598 CrossRef CAS; (b) V. Lyaskovskyy, A. I. Olivos Suárez, H. Lu, H. Jiang, X. P. Zhang and B. de Bruin, J. Am. Chem. Soc., 2011, 133, 12264 CrossRef CAS PubMed; (c) U. Hintermair, S. W. Sheehan, A. R. Parent, D. H. Ess, D. T. Richens, P. H. Vaccaro, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2013, 135, 10837 CrossRef CAS PubMed; (d) K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040 CrossRef CAS PubMed.
- P. Kuijpers, J. I. van der Vlugt, S. Schneider and B. de Bruin, Chem.–Eur. J., 2017, 23, 13819 CrossRef CAS PubMed.
- Recent review articles: (a) J. F. Berry, Comments Inorg. Chem., 2009, 30, 28 CrossRef CAS; (b) A. I. O. Suarez, V. Lyaskovskyy, J. N. H. Reek, J. I. van ver Vlugt and B. de Bruin, Angew. Chem., Int. Ed., 2013, 52, 12510 CrossRef CAS PubMed; (c) M. G. Scheibel, J. Abbenseth, M. Kinauer, F. W. Heinemann, C. Würtele, B. de Bruin and S. Schneider, Inorg. Chem., 2015, 54, 9290 CrossRef CAS PubMed.
- (a) P. L. Larsen, T. J. Parolin, D. R. Powell, M. P. Hendrich and A. S. Borovik, Angew. Chem., Int. Ed., 2003, 42(1), 85 CrossRef CAS; (b) F. F. Pfaff, S. Kundu, M. Risch, S. Pandian, F. Heims, I. Pryjomska-Ray, P. Haack, R. Metzinger, E. Bill, H. Dau, P. Comba and K. Ray, Angew. Chem., Int. Ed., 2011, 50, 1711 CrossRef CAS PubMed; (c) D. C. Lacy, Y. L. Park, J. W. Ziller, J. Yano and A. S. Borovik, J. Am. Chem. Soc., 2012, 134, 17526 CrossRef CAS PubMed; (d) S. Kundu, E. Miceli, E. R. Farquhar, F. F. Pfaff, U. Kuhlmann, P. Hildebrandt, B. Braun, C. Greco and K. Ray, J. Am. Chem. Soc., 2012, 134, 14710 CrossRef CAS PubMed; (e) S. Hong, F. F. Pfaff, E. Kwon, Y. Wang, M.-S. Seo, E. Bill, K. Ray and W. Nam, Angew. Chem., Int. Ed., 2014, 53, 10403 CrossRef CAS PubMed.
- R. S. Hay-Motherwell, G. Wilkinson, B. Hussain-Bates and M. B. Hursthouse, Polyhedron, 1993, 12, 2009 CrossRef CAS.
- E. Poverenov, I. Efremenko, A. I. Frenkel, Y. Ben-David, L. J. W. Shimon, G. Leitus, L. Konstantinovski, J. M. L. Martin and D. Milstein, Nature, 2008, 455, 1093 CrossRef CAS.
- C. A. Laskowski, A. J. M. Miller, G. L. Hillhouse and T. R. Cundari, J. Am. Chem. Soc., 2011, 133, 771 CrossRef CAS PubMed.
- (a) D. S. Glueck, J. Wu, F. J. Hollander and R. G. Bergman, J. Am. Chem. Soc., 1991, 113, 2041 CrossRef CAS; (b) D. M. Jenkins, T. A. Betley and J. C. Peters, J. Am. Chem. Soc., 2002, 124, 11238 CrossRef CAS PubMed; (c) D. T. Shay, G. P. A. Yap, L. N. Zakharov, A. L. Rheingold and K. H. Theopold, Angew. Chem., Int. Ed., 2005, 44, 1508 CrossRef CAS PubMed; (d) R. E. Cowley, R. P. Bontchev, J. Sorrell, O. Sarracino, Y. Feng, H. Wang and J. M. Smith, J. Am. Chem. Soc., 2007, 129, 2424 CrossRef CAS PubMed; (e) E. R. King, G. T. Sazama and T. A. Betley, J. Appl. Chem. Sci., 2012, 134, 17858 CrossRef CAS PubMed; (f) A. M. Geer, C. Tejel, J. A. Lõpez and M. A. Ciriano, Angew. Chem., Int. Ed., 2014, 53, 5614 CrossRef CAS PubMed; (g) J. Du, L. Wang, M. Xie and L. Deng, Angew. Chem., Int. Ed., 2015, 54, 12640 CrossRef CAS PubMed.
- Representative examples: (a) B. L. Tran, M. P. Washington, D. A. Henckel, X. Gao, H. Park, M. Pink and D. J. Mindiola, Chem. Commun., 2012, 48, 1529 RSC; (b) D. L. J. Broere, B. de Bruin, J. N. H. Reek, M. Lutz, S. Dechert and J. I. van der Vlugt, J. Am. Chem. Soc., 2014, 136, 11574 CrossRef CAS PubMed; (c) V. Vreeken, M. A. Siegler, B. de Bruin, J. N. H. Reek, M. Lutz and J. I. van der Vlugt, Angew. Chem., Int. Ed., 2015, 54, 7055 CrossRef CAS PubMed; (d) Y. Park, J. Heo, M.-H. Baik and S. Chang, J. Am. Chem. Soc., 2016, 138, 14020 CrossRef CAS PubMed; (e) B. Bagh, D. L. J. Broere, M. A. Siegler and J. l. van der Vlugt, Angew. Chem., Int. Ed., 2016, 55, 8381 CrossRef CAS PubMed; (f) L. Nurdin, D. M. Spasyuk, W. E. Piers and L. Maron, Inorg. Chem., 2017, 56, 4157 CrossRef CAS PubMed; (g) L. Hu and H. Chen, ACS Catal., 2017, 7, 285 CrossRef CAS.
- (a) M. Goswami, V. Lyaskovskyy, S. R. Domingos, W. J. Buma, S. Woutersen, O. Troeppner, I. Ivanović-Burmazović, H. Lu, X. Cui, X. P. Zhang, E. J. Reijerse, S. DeBeer, M. M. Van Schooneveld, F. F. Pfaff, K. Ray and B. de Bruin, J. Am. Chem. Soc., 2015, 137, 5468 CrossRef CAS PubMed; (b) P. F. Kuijpers, M. J. Tiekink, W. B. Breukelaar, D. L. J. Broere, N. P. van Leest, J. I. van der Vlugt, J. N. H. Reek and B. de Bruin, Chem.–Eur. J., 2017, 23(33), 7945 CrossRef CAS PubMed.
- (a) J. D. Blakemore, N. D. Schley, D. Balcells, J. F. Hull, G. W. Olack, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2010, 132, 16017 CrossRef CAS PubMed; (b) M. Zhou, D. Balcells, A. R. Parent, R. H. Crabtree and O. Eisenstein, ACS Catal., 2012, 2, 208 CrossRef CAS; (c) M. C. Lehman, D. R. Pahls, J. M. Meredith, R. D. Sommer, D. M. Heinekey, T. R. Cundari and E. A. Ison, J. Am. Chem. Soc., 2015, 137, 3574 CrossRef CAS PubMed; (d) B. Bagh, D. L. J. Broere, V. Sinha, P. F. Kuijpers, N. P. van Leest, B. de Bruin, S. Demeshko, M. A. Siegler and J. I. van der Vlugt, J. Am. Chem. Soc., 2017, 139, 5117 CrossRef CAS PubMed.
- (a) M. G. Scheibel, B. Askevold, F. W. Heinemann, E. J. Reijerse, B. de Bruin and S. Schneider, Nat. Chem., 2012, 4, 552 CrossRef CAS PubMed; (b) M. G. Scheibel, Y. Wu, A. C. Stückl, L. Krause, E. Carl, D. Stalke, B. de Bruin and S. Schneider, J. Am. Chem. Soc., 2013, 135, 17719 CrossRef CAS PubMed; (c) Y. Gloaguen, C. Rebreyend, M. Lutz, P. Kumar, M. Huber, J. I. van der Vlugt, S. Schneider and B. de Bruin, Angew. Chem., Int. Ed., 2014, 53, 6814 CrossRef CAS PubMed.
- (a) J. Meiners, M. G. Scheibel, M. H. Lemée-Cailleau, S. A. Mason, M. B. Boeddinghaus, T. F. Fässler, E. Herdtweck, M. M. Khusniyarov and S. Schneider, Angew. Chem., Int. Ed., 2011, 50, 8184 CrossRef CAS PubMed; (b) M. Kinauer, M. G. Scheibel, J. Abbenseth, F. W. Heinemann, P. Stollberg, C. Würtele and S. Schneider, Dalton Trans., 2014, 43, 4506 RSC.
- J. Schöffel, A. Y. Rogachev, S. DeBeer and P. Burger, Angew. Chem., Int. Ed., 2009, 48, 4734 CrossRef PubMed.
- J. Abbenseth, M. Diefenbach, S. C. Bete, C. Würtele, C. Volkmann, S. Demeshko, M. C. Holthausen and S. Schneider, Chem. Commun., 2017, 53, 5511 RSC.
- L. A. Watson, O. V. Ozerov, M. Pink and K. G. Caulton, J. Am. Chem. Soc., 2003, 125, 8426 CrossRef CAS PubMed.
- B. Askevold, M. M. Khusnivarov, W. Kroener, K. Gieb, P. Müller, E. Herdtweck, F. W. Heinemann, M. Diefenbach, M. C. Holthausen, V. Vieru, L. F. Chibotaru and S. Schneider, Chem.–Eur. J., 2015, 21, 579 CrossRef CAS PubMed.
- M. Atanasov, D. Aravena, E. Suturina, E. Bill, D. Maganas and F. Neese, Coord. Chem. Rev., 2015, 289, 177 CrossRef.
- R. A. Eikey and M. M. Abu-Omar, Coord. Chem. Rev., 2003, 243, 83 CrossRef CAS.
- B. A. Arndtsen, H. F. Sleiman, A. K. Chang and L. McElwee-White, J. Am. Chem. Soc., 1991, 113, 4871 CrossRef CAS.
- M. Kinauer, M. G. Scheibel, J. Abbenseth, F. W. Heinemann, P. Stollberg, C. Würtele and S. Schneider, Dalton Trans., 2014, 43, 4506 RSC.
- J. Scepaniak, C. G. Margarit, J. N. Harvey and J. M. Smith, Inorg. Chem., 2011, 50, 9508 CrossRef CAS PubMed.
- F. S. Schendzielorz, M. Finger, C. Volkmann, C. Würtele and S. Schneider, Angew. Chem., Int. Ed., 2016, 55, 11417 CrossRef CAS PubMed.
- S. C. Bart, E. Lobkovsky, E. Bill and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 5302 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1546382–1546385. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01113c |
| ‡ For synthetic, spectroscopic, electrochemical, crystallographic and computational data see ESI. |
| § Oxidation state assignments refer to formal oxidation states throughout this paper unless otherwise noted. |
| ¶ Another irreversible reduction wave was found at very low potential (Epc = −2.7 V vs. Fc+/Fc). |
| This journal is © The Royal Society of Chemistry 2018 |