
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn integrated mass spectrometry platform enables picomole-scale real-time electrosynthetic reaction screening and discovery†
Qiongqiong
Wan
 ,
Suming
Chen
and
Abraham K.
Badu-Tawiah
*
,
Suming
Chen
and
Abraham K.
Badu-Tawiah
*
Department of Chemistry and Biochemistry, The Ohio State University, Columbus, Ohio 43210, USA. E-mail: badu-tawiah.1@osu.edu
First published on 30th May 2018
Abstract
The identification of new electrosynthetic pathways enables environmentally friendly synthetic applications. However, the development of miniaturized screening procedures/platforms to expedite the discovery of electrooxidation reactions remains challenging. Herein, we developed an integrated system that serves as a reactor and ion source in a single experimental step using only picomole-scale reactants to monitor electrooxidation in real-time. This reaction screening platform utilizes the intrinsic electrochemical capabilities of nano-electrospray ionization mass spectrometry. We validated the feasibility of this method by reproducing three known electrochemical reactions. We also discovered two new electroorganic reaction pathways: (i) C–N dehydrodimerization of 8-methyl-1,2,3,4-tetrahydroquinoline to construct a novel quinoline skeleton, and (ii) TEMPO-mediated accelerated electrooxidative dehydrogenation of tetrahydroisoquinolines. Moreover, the radical cations and key intermediates captured by this screening platform provided direct evidence for the mechanism of these novel electrochemical reactions.
Introduction
Electroorganic synthesis is a promising modern synthetic tool and has attracted much attention. This is because electroorganic reactions not only represent a cost-efficient and sustainable “green chemistry” method, but more importantly, they provide outstanding selectivity and reactivity, allowing the formation of products that are sometimes not accessible via conventional methods.1 However, the insufficient number of reaction types when compared with traditional organic reactions has restricted the widespread application of electrosynthesis.Screening approaches have become the mainstay of discovery processes to identify new reactions. The establishment of efficient electrochemical reaction screening platforms is crucial for finding new electrosynthetic reactions. The general approach of electrochemical screening is based on the combinatorial measurement of cyclic voltammetry in microtiter plates or multichannel potentiostats,2 which could only provide electroanalytical data without structural information about the intermediates and final products involved. In addition, the electrolysis time required for each vial to reach complete conversion is long, making the whole screening method very time-consuming.1c Galvanostatic electrosynthetic screening was developed with the advantages of high efficiency and simple setup,3 but challenges of this method include extra analytical characterization steps needed to evaluate the reaction and large reagent consumption. Therefore, the development of a novel electrosynthetic screening method that could provide rapid results and accurate molecular information is necessary.
Mass spectrometry (MS) has the capacity to provide both qualitative and quantitative information on chemical substances.4 Although MS could be used to analyze products from off-line electrochemical cells, online reaction screening coupled with in situ MS will greatly expedite the discovery of new reaction pathways. Electrospray ionization (ESI) is not only an extensively applied ion source, but it has also been characterized as a controlled-current electrochemical cell, where redox chemistry occurs upon the application of direct current (DC) voltage to an analyte solution.5 However, no such approach has been developed or demonstrated for online electroorganic synthetic reaction screening; this is in part due to the low electrolysis efficiency owing to the dynamic nature of the system with high flow rates and ultralow electrical current. Some attempts were made to design special electrochemical cells coupled with an ESI MS to improve the electrolytic efficiency,6 but the necessity of an extra electrochemical system and relatively large sample consumption still present a challenge.
In the present study, we adapted and validated a simple nano-electrospray ionization (nESI) MS setup as a reactor/ion source integrated platform for the online screening and studying of electrochemical reactions (Fig. 1a). The electrochemical reactions occur in the glass capillary of the nESI emitter after applying a DC voltage and could be monitored in real time by MS. The features and advantages of this screening method are as follows: (1) the real-time MS monitoring capabilities provide qualitative and quantitative analysis, which offer a more convenient way to discover new reaction pathways; (2) the high sensitivity of nESI MS enables lower sample consumption on the order of picomolar levels. This ultralow amount also allows rapid product conversions (few minutes) under the nanoampere (∼150 nA) electric current reaction condition; (3) the ultra-low flow rate (∼100 nL min−1) offers extended reagent dwelling time, in a controllable manner, to obtain the desirable conversion yield, which is not available in traditional ESI; and (4) the accurate molecular information facilitates the study of the underlying mechanism of reaction pathways.
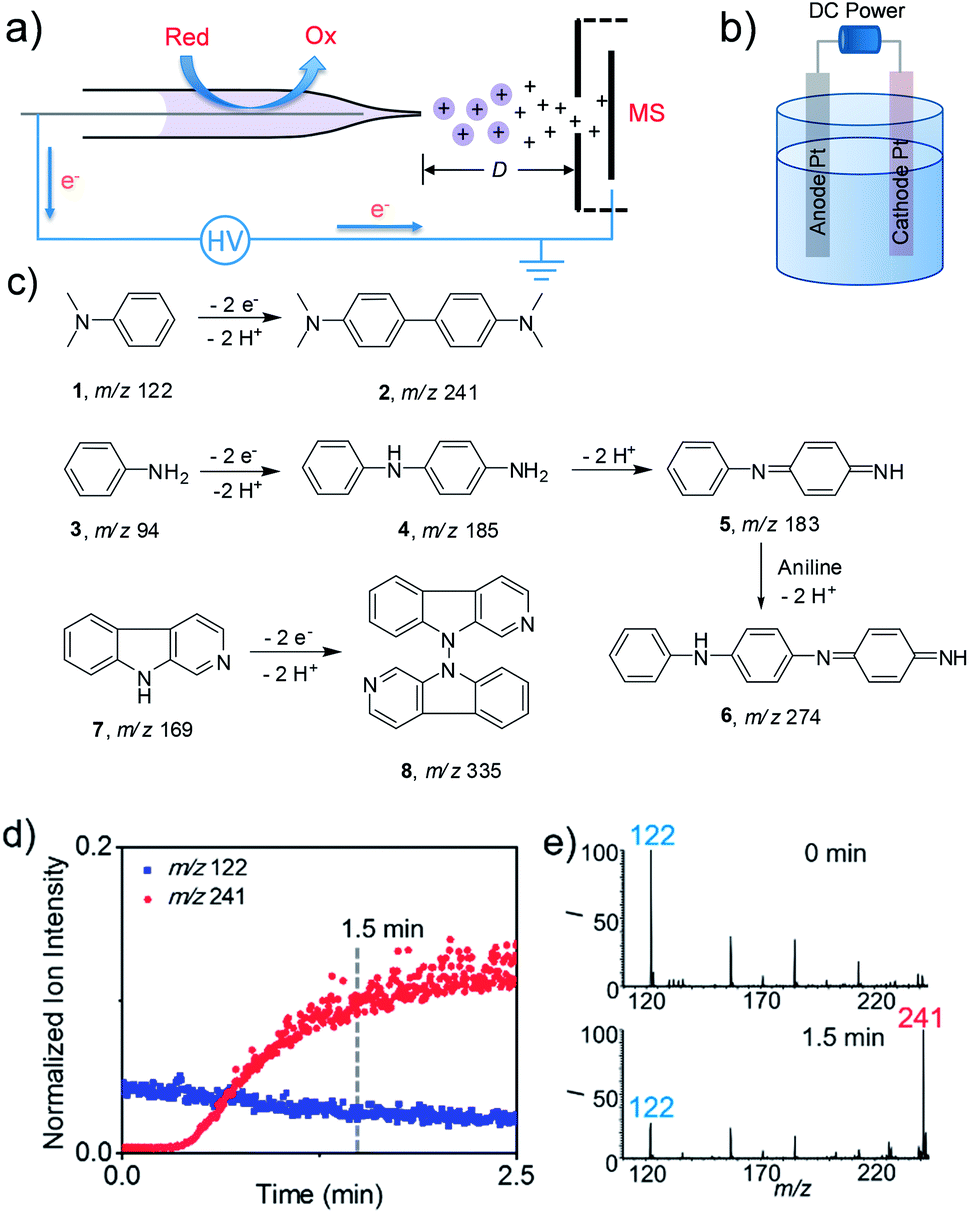 | ||
| Fig. 1 (a) Schematic of the real time electrochemical reaction platform. D is the distance between the capillary tip and the MS inlet. D was kept at 0.8 cm. The electrospray voltage is 2.0 kV with a Pt anode (diameter 0.2 mm, current density 1.2 μA cm−2). (b) Schematic of a classical controlled-current (40 mA, current density 0.1 A cm−2) undivided cell for electrolysis with Pt electrodes. (c) Chemical structures of electrochemical C,C-dehydrodimerization of N,N-dimethylaniline (DMA), C,N-polymerization of aniline and N,N-dehydrodimerization of β-carboline. (d) The real time normalized ion intensity of m/z 122 and m/z 241 to the total ion current during the DMA electrooxidation in CH3CN (10 μM) with nESI MS, and (e) mass spectra at spray times of 0 min and 1.5 min. m/z 122 and m/z 241 are assigned to protonated DMA (MW 121) and product N,N,N′,N′-tetramethylbenzidine (TMB, MW 240). The other peaks may correspond to background ions from solvents or the glass capillary. I represents the relative abundance (%) of detected ions. | ||
By using this nESI MS setup, we observed the electrooxidative conversions of different amines within a few minutes. The matched results of bulk reactions in an undivided electrolytic cell indicated the potential of this nESI MS apparatus as a direct and rapid screening platform for electrochemical transformations. With this screening platform, we discovered a new electrooxidative C–N coupling reaction from 8-methyl-1,2,3,4-tetrahydroquinoline, and TEMPO (2,2,6,6-tetramethyl-1-piperidine N-oxyl)-mediated electrooxidative dehydrogenation of 1,2,3,4-tetrahydroisoquinolines. These conversions were all validated by bulk reactions in the undivided electrolytic cell. Furthermore, this real-time MS method enabled the capture of radical cations and intermediates involved in the electrochemical reactions and provided direct evidence for the reaction mechanism.
Results and discussion
Design and validation of the reaction screening platform
The electrooxidation reaction screening platform employing nESI MS is shown in Fig. 1a and S1.† This nESI MS system could be considered as a controlled-current electrochemical cell consisting of two electrodes where the platinum electrode inside the nESI capillary acts as an anode and locus of oxidation reactions in positive ionization mode. The narrow capillary would facilitate the mass transfer of the reactants to the surface of electrodes that could expedite the reactions. The ultra-low flow rate (ca. 100 nL min−1) of nESI could also provide sufficient dwelling time for electrooxidation, which is beneficial for the conversion of compounds having relatively low reactivity. This MS detection method allowed both the qualitative and quantitative assessment of reaction progress. Like previous studies, the reaction progress was quantitatively evaluated by the parameter of the relative ion intensity (RI),4h,7 which is measured as the intensity of a specific ion (reactant or product) relative to the sum of intensities of the product, reactant and intermediates derived from this reactant. Platinum wire is used in this study because it is inert and durable (Fig. S2†). Furthermore, a traditional undivided electrolytic cell was created by using a DC power supply with two Pt electrodes as the anode and cathode (Fig. 1b) to test the feasibility of extending the screened small-scale reactions to a larger-scale synthesis.To validate the utility of this method, we first tested known electrochemical reactions (Fig. 1c) with the proposed screening platform. Electrooxidative reactions of amines like N,N-dimethylaniline (DMA),4g aniline8 and larger molecule β-carboline(9H-pyrido[3,4-b]indole) were studied. As shown in Fig. 1d and e, after applying DC voltage, nESI-MS analysis of DMA solution produced the predominant peak of protonated 1 at m/z 122 at the onset of the spray (i.e., 0 min). However, the relative intensity of this reactant ion decreased quickly with time, whereas the ion at m/z 241 corresponding to the expected dehydrodimerization product of N,N,N′,N′-tetramethylbenzidine (TMB) increased, which means the electrooxidative reaction indeed happened in this nESI process. The relative intensity of the product ion increased from 6% at 0 min to 78% at 1.5 min (Fig. 1e), which indicated a rapid conversion. Similarly, electrooxidative C–N oligomerization of aniline was studied using the nESI MS setup, and the results are shown in Fig. S3 in the ESI.† Initially, only the peak corresponding to protonated aniline at m/z 94 was observed. Then pronated dehydrodimeric intermediate (5, m/z 183) and dehydrotrimeric product (6, m/z 274) appeared after continuous application of the DC voltage for 1 min. We observed that the applied DC voltage has an influence on the electrochemical conversion of aniline in nESI. At a voltage of 1 kV, the relative intensities of the dimeric intermediate and trimeric product are relatively low (13% and 3%). However, when the voltage was increased to 2 kV, the relative intensity of intermediates and products increased dramatically to 19% and 27%, respectively. The continuous increase of the voltage to 3 kV didn't enhance the relative intensity of the intermediate (25%) and product (31%) significantly compared with 2 kV. In addition, the position of the Pt electrode inside the capillary also influenced the conversion of aniline (Fig. S4†). The obvious intermediates and product could be observed when the distance of the electrode from the capillary tip was kept at 2 mm. However, neither the intermediates nor the expected product could be detected when the electrode was pulled inwards with a concomitant increase of the distances to 4 mm and 6 mm.
Unlike the oligomerization of aromatic amines, the electrosynthesis of N,N-linkage compounds through oxidative coupling is very rare.1d The N,N dehydrodimerization of β-carboline has been reported to occur by anodic oxidation.9 Here, we also tested this reaction by nESI MS with tetraethylammonium perchloride as a supporting electrolyte. The expected product of N,N-dehydrodimerization was observed at m/z 335, corresponding to bis-β-carboline, albeit with small yields (Fig. S5†). The RI increased from 5% at 2 min to 10% at 5 min. This low conversion ratio of the product may be attributed to the relatively low reactivity of this compound, but the ability to reproduce this uncommon electrooxidative pathway validates the nESI MS approach.
To investigate if these reaction pathways match the results from bulk-phase reactions, all three reactions were performed on the undivided electrolytic cell (40 mA), and the reaction mixture was subsequently analyzed by a non-electrical sonic spray MS method to avoid any electrooxidation during detection. For the dehydrodimerization of DMA, the product ion of TMB at m/z 241 was observed and became evident (RI 22%) after 10 min of electrooxidation; the RI of the product ion reached 86% after 2 h reaction time (Fig. S6 and S7†). Likewise, the dehydrodimer and dehydrotrimer of aniline were also found in the bulk reaction (Fig. S8 and S9†), in which the RI of the trimeric product reached 25% and 77% at 30 min and 2 h, respectively. Interestingly, the ion signal corresponding to the dehydrodimer was very low in the electrolytic cell reaction, which suggests that a longer reaction time enabled the conversion from dehydrodimer to dehydrotrimer. For N,N-dehydrodimerization of β-carboline, the relatively low reaction rate was also observed in the bulk electrolytic cell. The RI of product 8 was detected at 8% yield after continuous electrooxidation for 3 h (Fig. S10c†). However, the RI of 8 increased dramatically to 85% after 30 h (Fig. S10d†). Collectively, the electrooxidative products of these selected reactions achieved using the nESI MS method are consistent with the scale-up synthesis in the undivided electrolytic cells, which indicated that this electrospray-based method could potentially be used as an electrosynthetic reaction screening platform to identify new reactions.
Comparing the results from the nESI MS screening method with those from the traditional undivided electrolytic cell revealed twofold advantages in favor of the nESI MS platform, which include rapid conversion rates and ultra-low sample consumption. For example, the relative intensity of the oxidative product TMB reaches 78% at only 1.5 min in the nESI MS setup (Fig. 1e) compared with a similar conversion ratio of 86% achieved after 2 h reaction time in the undivided electrolytic cell (Fig. S6f†). This represents approximately 80 times enhancement in reaction rate. Similarly, neither the intermediates nor the product could be detected within 5 min of electrochemical reaction of β-carboline in the undivided electrolytic cell (Fig. S11†). In contrast, evident conversion from β-carboline to bis-β-carboline was achieved in 5 min when the same concentration of reagents were reacted on the nESI mass spectrometric reaction platform (4%, Fig. S11c†). This conversion ratio is comparable to a 1.5 h reaction on the traditional electrolytic cell (4.5%, Fig. S10b†), representing approximately 18 times rate improvement in the nESI MS method for this challenging transformation. The rate of electrode reaction in solution generally relates to the current density and mass transport efficiency.10 The current density in the nESI (ca. 1.2 μA cm−2) is much less than the current density in the traditional electrolytic cell (ca. 0.1 A cm−2). But the absolute amount of reactant in the capillary of the nESI is about 2–3 orders of magnitude lower than that in the electrolytic cell. This may allow a higher product/reactant ratio in the nESI capillary for a comparable conversion rate in the traditional electrolytic cell. However, this effect may not fully account for the faster reaction rates recorded in the nESI capillary. A more important factor may relate to the mass transfer rate.11 Compared with the large volume (1.5 mL) of reaction solution in the electrolytic cell, the minuscule volume (<10 μL) of reactant solution in the smaller capillary (I.D. = 0.86 mm) is expected to facilitate efficient transfer of the reactant from the solution to the electrode surface due to the short diffusion distance and large surface-to-volume ratio.10b,c We investigated this possibility by employing two different sizes of glass capillaries (0.86 and 0.5 mm, I.D.) in the nESI. Using the electrooxidation of β-carbolines, the reactions rates recorded from the 0.5 mm glass capillary were about three times higher than those in our typical 0.86 mm capillary at the same reaction time of 5 min (Fig. S12†). We ascribed this increased conversion rate to more efficient mass transport in the smaller capillary. This unique feature found in the nESI MS approach enabled the rapid discovery and screening of new electrooxidative reactions.
The discovery and study of C–N dehydrodimerization of 8-methyl-1,2,3,4-tetrahydroquinoline
To apply the proposed method for the identification of new reactions, we first studied the electrochemical reaction of the heterocyclic compound 8-methyl-1,2,3,4-tetrahydroquinoline (9, Fig. 2a) as a proof-of-concept. This reaction was selected because the construction of larger quinoline scaffolds is important for the development of new drugs. Quinoline-based anticancer drugs have a strong position in modern medicinal chemistry.12 As shown in Fig. 2b, the protonated 9 at m/z 148 was the predominant peak at the beginning (0 min) of the spray process, after which new peaks at m/z 291 and 292 appeared when the DC voltage was continuously applied for 2 min. As an aromatic amine derivative, the dehydrodimerization of 9 is expected. Thus the peak at m/z 292 should be ascribed to the radical cation intermediate of the dehydrodimer (13). Interestingly, further dehydrogenations occurred to give products 14 (m/z 291) and 16 (m/z 289), which involved the removal of two and four hydrogen atoms, respectively. The successful capture of the important radical cation intermediate of 12 at m/z 293 provided more evidence for proposing the possible reaction mechanism in Fig. 2a. This reaction was also investigated at the same reagent conditions except that the Pt electrode was replaced with an Ag electrode (Fig. S13†). The intermediates 12, 13, 14 and 15, and the final product 16 were not observed when the DC voltage was continuously applied for 2 min. The production of characteristic isotopic peaks [Ag(CH3CN)2]+ at m/z 189 and 191 indicate that the sacrificial oxidation of the Ag anode may have limited the electrooxidation of 8-methyl-1,2,3,4-tetrahydroquinoline.5b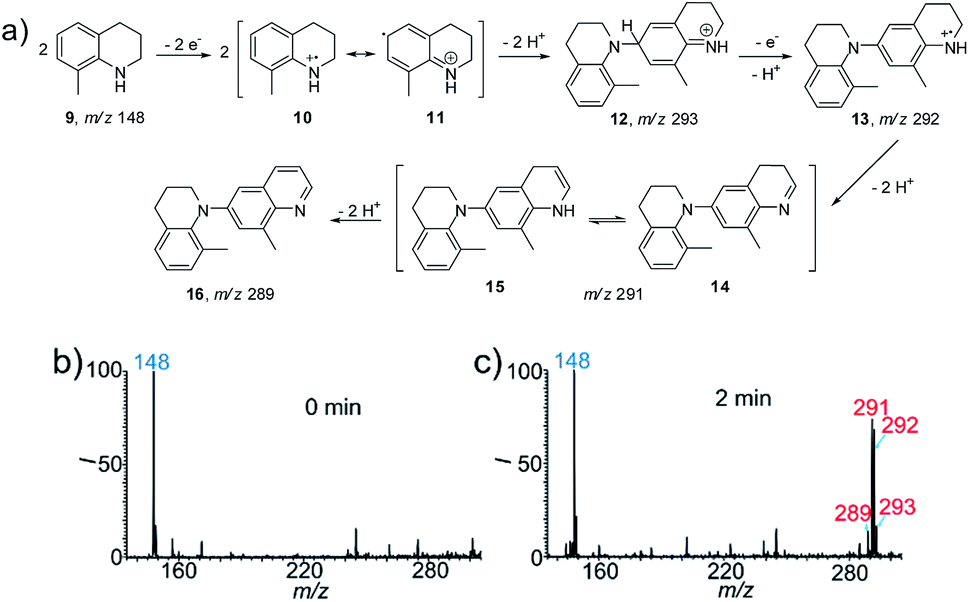 | ||
| Fig. 2 (a) The conversion and proposed mechanism for the electrochemical oxidation of 8-methyl-1,2,3,4-tetrahydroquinoline (9). (b and c) nESI MS spectra of 9 at the times (b) 0 min and (c) 2 min. The peaks at m/z 148, 289, 291, 292 and 293 correspond to compounds 9 (MW 147 g mol−1), 16 (MW 288 g mol−1), 14/15 (MW 290 g mol−1), 13 (MW 292 g mol−1), and 12 (MW 293 g mol−1), respectively. The reaction was conducted with 9 (10 μM) in acetonitrile. | ||
To further confirm and validate this reaction pathway, the electrochemical reaction of 9 was performed using the undivided electrolytic cell. A similar MS pattern of products peaks was observed when 9 was subjected to electrooxidation for 2 h (Fig. S14b†), i.e. the corresponding intermediates 12, 13, and 14/15 and final product 16 were formed. Note that a comparable conversion ratio in the nESI setup requires only 2 min, representing a >60 times increase in reaction speed. The intermediates further transformed into the final product 16 (RI 86%) when the reaction time was increased to 24 h (Fig. S14c†). The recorded 1H NMR, 13C NMR (Fig. S15 and S16†) and high resolution MS data further confirmed the special dehydrodimer structure of 16. Based on the above results, the proposed mechanism for the electrooxidation of 9 is summarized in Fig. 2a. First, anodic oxidation ionized 9via the removal of a single electron to yield radical cation 10. Tautomerization of intermediate 10 into 11 allows the formation of dehydrodimer cation intermediate 12 (m/z 293). A proton-coupled electron transfer reaction yields radical cation intermediate 13 (m/z 292). Then, dehydrogenation of 13 forms 14 (m/z 291) and an energetically equal tautomer, 15.13 Subsequently, the oxidative removal of two more hydrogen atoms leads to the formation of the final product, 16 (m/z 289). It is interesting to note that this product is a quinoline derivative, rather than a simple dehydrodimer or oligomer like other aromatic amines. This reaction provided a new route for the synthesis of the quinoline structure in a single step. The consistency of the reactivity and products observed for the nESI MS platform and the undivided electrolytic cell indicate the feasibility of using this method to screen other electrosynthetic reactions.
The discovery and study of TEMPO-mediated electrooxidative dehydrogenation of tetrahydroisoquinolines
To further extend the application of this screening platform, we investigated the reaction of tetrahydroisoquinolines and expected a new conversion pathway. The oxidative dehydrogenation of amine to imine with good selectivity has been achieved by various metal catalytic and photocatalytic pathways.14 However, the study of this transformation with the cleaner electrooxidative method is rare.15 Here 1,2,3,4-tetrahydroisoquinolines (17 and 19) were selected as examples (Fig. 3) because the corresponding dehydroaromatization products are prevalent in pharmaceuticals, agrochemicals, and functional organic materials.16 With the DC voltage applied on the reaction system inside the nESI capillary, the dehydrogenation products were indeed observed at m/z 132 (18) with an RI of 18% after 5 min (Fig. 3b). Nevertheless, the 4-methoxyphenyl-substituted 1,2,3,4-tetrahydroisoquinoline (19) (Fig. S17 and S18†) exhibited a much faster dehydrogenation rate (Fig. S19b and c†). The RI of product 20 was calculated to be 45% at 0.1 min (Fig. S19b†), which reached 80% in only 0.5 min (Fig. S19c†). Consistent results were also recorded from using the undivided electrolytic cell (Fig. S20a–c and S21a–c†).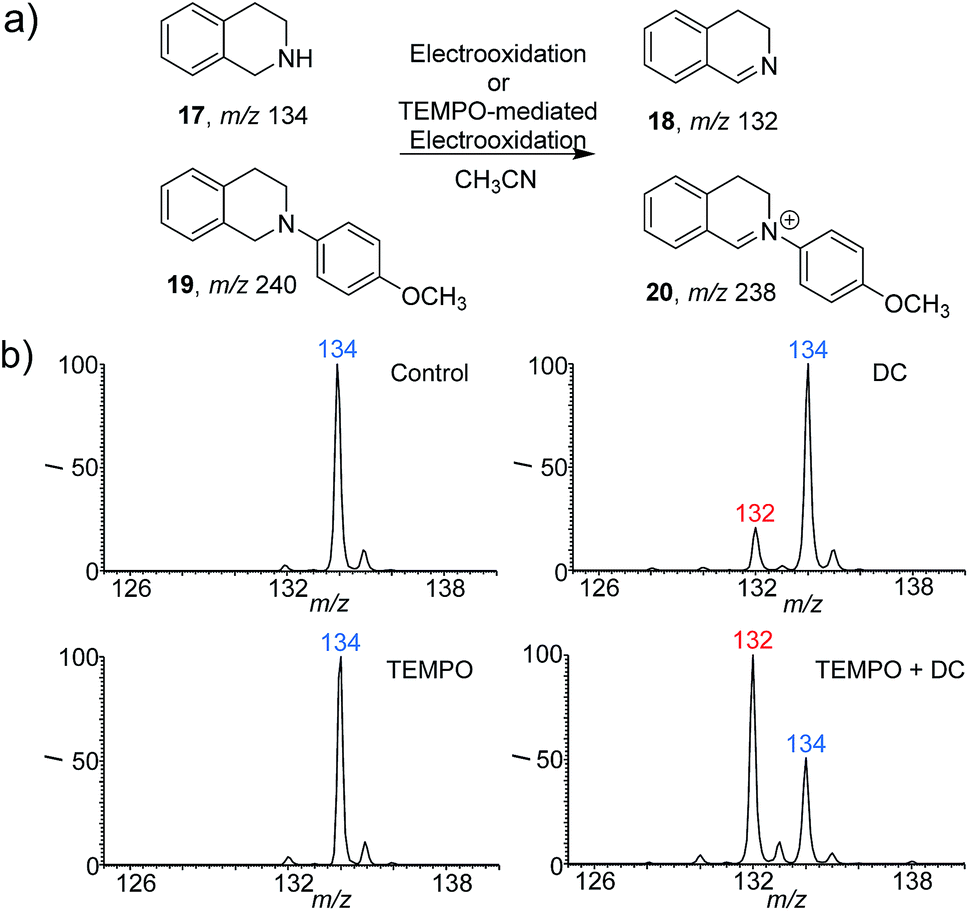 | ||
| Fig. 3 Electrooxidative dehydrogenation of 1,2,3,4-tetrahydroisoquinolines. (a) Electrooxidative reaction pathways of 1,2,3,4-tetrahydroisoquinoline (17) and 2-(4-methoxyphenyl)-1,2,3,4-tetrahydroisoquinoline (19). (b) Real time nESI mass spectra for the reaction of 17 (10 μM in ACN) under different conditions. Control, 17 only at 0 min; DC, 17 only at 5 min; TEMPO, 17 + TEMPO for 5 min and detected by non-electric sonic spray MS; TEMPO + DC, 17 + TEMPO with applying the DC voltage for 5 min. The concentration of TEMPO is 10 mol% (1 μM). The DC voltage is 2 kV. | ||
Given that a faster reaction rate would be more meaningful in chemical synthesis, we further explored the possibility of accelerating this new dehydrogenation pathway. As an efficient hydrogen abstraction reagent, 2,2,6,6-tetramethylpiperidinyl-1-oxy (TEMPO) has been used broadly as an electrocatalyst for alcohol oxidation,17 and we wondered whether it could be applied in the dehydrogenation of amines. Herein, TEMPO was introduced into this electrooxidative dehydrogenation reaction, and different reaction conditions were investigated using the nESI MS platform in detail. The reactions were conducted with TEMPO only and both TEMPO (10% mol) and DC applied. As can be seen from the results (Fig. 3b, and S20d–f and S21d–f†), TEMPO itself could not dehydrogenate these 1,2,3,4-tetrahydroisoquinolines (17/19). However, dehydrogenation reactions proceeded rapidly once electricity was applied. The products, 3,4-dihydroisoquinoline (18) at m/z 132 and 2-(4-methoxyphenyl)-3,4-dihydroisoquinolinium (20) at m/z 238, were clearly observed with higher reaction rates when compared to the systems with applied DC voltage without TEMPO (Fig. 3b and S19†). The RI of 18 increased to 66% at 5 min, which is about 3 times more than the yield recorded without TEMPO (Fig. 3b). This acceleration is also obvious for 19. The RI of product 20 even reached 90% within 0.1 min, which represents a 2 times increase compare to that without TEMPO (Fig. S19†). These results imply that TEMPO acts cooperatively in the presence of applied DC potential to accelerate these dehydrogenation reactions. Expectedly, the results obtained from undivided electrolytic cells are consistent with the reaction pathways screened from our nESI MS electrooxidation platform (Fig. S20 and S21†). The same products of 18 and 20 and acceleration effects were observed with TEMPO. The RIs of 18 and 20 were about 4 and 1.5 times higher than those without TEMPO after 30 min of electrooxidation. 1H NMR of products 18 and 20 further confirmed the structures and this new transformation (Fig. S22 and S23†). Again, a new TEMPO-mediated electrooxidative dehydrogenation reaction of 1,2,3,4-tetrahydroisoquinolines has been discovered by using this nESI MS electrochemical screening platform and validated by the scale-up synthesis in electrolytic cells. The results indicate that the introduction of TEMPO could greatly accelerate this oxidative dehydrogenation reaction.
Studies of the reaction mechanism of TEMPO-mediated electrooxidative dehydrogenation
As mentioned above, one of the advantages of this nESI MS platform is the convenience for mechanism study. Therefore, we further investigated the possible mechanism of this TEMPO-mediated reaction using this platform. TEMPO is known to be oxidized to a more oxidative TEMPO+ form by electrochemical oxidation, which is in turn reduced by a reductant to form TMEPOH.18 Both species (TEMPO+ and TEMPOH) were detected at m/z 156 and m/z 158 in the dehydrogenation reaction of 1,2,3,4-tetrahydroisoquinolines (Fig. 4a and S24†). The successful capture of these two key intermediates implies that the observed accelerated dehydrogenation reaction is TEMPO-mediated (Fig. 4b). In this case, TEMPO was oxidized at the anode to form reactive TEMPO+, which then oxidizes 1,2,3,4-tetrahydroisoquinolines to produce the imine products, accompanied by the reduction of TEMPO+ to TEMPOH. This reaction proceeds with a catalytic amount of TEMPO (10 mol%) implying that TEMPO was regenerated from TEMPOH at the anode by electrooxidation.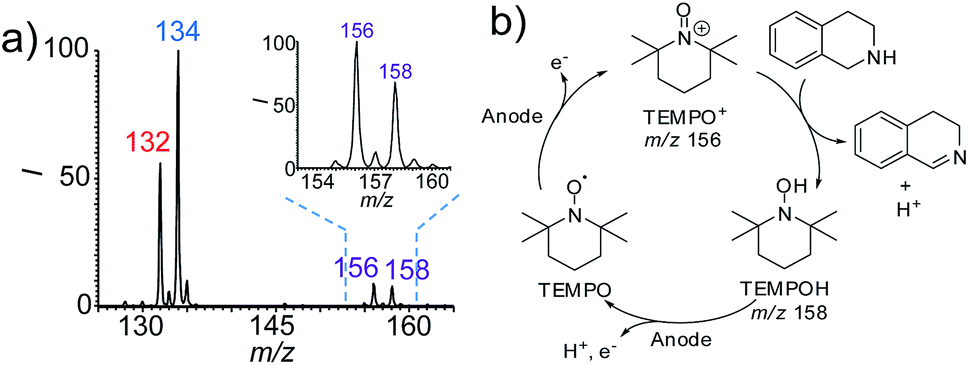 | ||
| Fig. 4 (a) nESI mass spectrum recorded from the electrooxidation of 1,2,3,4-tetrahydroisoquinoline (17, 10 μM in ACN) with TEMPO (10 mol%) at 2.5 min. The inset is the enlarged mass spectrum in the range of m/z 153 to 161. (b) The proposed mechanism of TEMPO-mediated oxidation of 17. | ||
As a whole, the above results of the discovery and study of the new electrochemical reactions demonstrated the unique advantages of the proposed integrated nESI MS platform. The rapid conversion in less than a few minutes could greatly save time and increase opportunities to discover new pathways, especially for reactants with low reactivity that need longer reaction times in traditional electrolytic cell. In addition, this nESI MS approach enables the real time observation of the reaction progress. This could prevent the escape of unexpected intermediates during interspersed sampling from electrolytic cells because some key intermediates for elucidating the reaction mechanism may disappear over short time periods.
Conclusions
In conclusion, by using the simple nESI MS apparatus, we have established a picomole-scale real-time electrooxidation screening platform for amines. The platform exhibits high efficiency and rapid electrosynthetic reaction screening capabilities. New chemical transformations such as the C–N dimerization and dehydrogenation of 8-methyl-1,2,3,4-tetrahydroquinoline and TEMPO-mediated electrocatalytic oxidative dehydrogenation of 1,2,3,4-tetrahydroisoquinoline and 2-(4-methoxyphenyl)-1,2,3,4-tetrahydroisoquinoline were discovered, and the corresponding intermediates were captured on this platform in real time. More importantly, the scale-up reactions confirm that these new pathways can be implemented under the traditional undivided electrolytic cell reaction conditions. The integration of the electrochemical reaction with nESI may be limited by the inability to independently optimize some reaction conditions. However, this integrated setup provided a rapid reaction screening platform to discover new electrochemical reaction pathways. After the verification of the feasibility of new reactions using this convenient and highly efficient platform, the individual reaction conditions could be optimized thereafter by other means. We believe that this platform may find more applications in the discovery and study of novel electrosynthetic reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Ohio State University start-up funds and U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Condensed Phase and Interfacial Molecular Science, under award number DE-SC0016044.Notes and references
- (a) J. i. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265 CrossRef PubMed; (b) J. B. Sperry and D. L. Wright, Chem. Soc. Rev., 2006, 35, 605 RSC; (c) C. Gütz, B. Klöckner and S. R. Waldvogel, Org. Process Res. Dev., 2016, 20, 26 CrossRef; (d) S. R. Waldvogel and B. Janza, Angew. Chem., Int. Ed., 2014, 53, 7122 CrossRef PubMed.
- (a) C. A. Briehn, M. S. Schiedel, E. M. Bonsen, W. Schuhmann and P. Bäuerle, Angew. Chem., Int. Ed., 2001, 40, 4680 CrossRef; (b) C. Leya, S. Z. Cekic, S. Kochius, K. M. Mangold, U. Schwaneberg, J. Schrader and D. Holtmann, Electrochim. Acta, 2013, 89, 98 CrossRef.
- T. Siu, W. Li and A. K. Yudin, J. Comb. Chem., 2000, 2, 545 CrossRef PubMed.
- (a) V. Prabhakaran, G. E. Johnson, B. Wang and J. Laskin, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13324 CrossRef PubMed; (b) M. R. Ligare, G. E. Johnson and J. Laskin, Phys. Chem. Chem. Phys., 2017, 19, 17187 RSC; (c) Y. Chen and M. T. Rodgers, J. Am. Chem. Soc., 2012, 134, 2313 CrossRef PubMed; (d) X. Ma, L. Chong, R. Tian, R. Shi, T. Y. Hu, Z. Ouyang and Y. Xia, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2573 CrossRef PubMed; (e) X. Ma and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 2592 CrossRef PubMed; (f) A. J. Ingram, C. L. Boeser and R. N. Zare, Chem. Sci., 2016, 7, 39 RSC; (g) T. A. Brown, H. Chen and R. N. Zare, Angew. Chem., Int. Ed., 2015, 54, 11183 CrossRef PubMed; (h) S. Chen, Q. Wan and A. K. Badu-Tawiah, Angew. Chem., Int. Ed., 2016, 55, 9345 CrossRef PubMed; (i) S. Chen, C. Xiong, H. Liu, Q. Wan, J. Hou, J. Wang, Q. He, A. Badu-Tawiah and Z. Nie, Nat. Nanotechnol., 2015, 10, 176 CrossRef PubMed; (j) R. M. Bain, C. J. Pulliam and R. G. Cooks, Chem. Sci., 2015, 6, 397 RSC; (k) T. Li, L. Fan, Y. Wang, X. Huang, J. Xu, J. Lu, M. Zhang and W. Xu, Anal. Chem., 2017, 89, 1453 CrossRef PubMed; (l) M. Wleklinski, B. P. Loren, C. R. Ferreira, Z. Jaman, L. Avramova, T. J. P. Sobreira, D. H. Thompson and R. G. Cooks, Chem. Sci., 2018, 9, 1647 RSC; (m) Y. Wang, M. Sun, J. Qiao, J. Ouyang and N. Na, Chem. Sci., 2018, 9, 594 RSC.
- (a) G. J. V. Berkel and F. Zhou, Anal. Chem., 1995, 67, 2916 CrossRef; (b) M. Abonnenc, L. Qiao, B. Liu and H. H. Girault, Annu. Rev. Anal. Chem., 2010, 3, 231 CrossRef PubMed; (c) J. F. d. l. Mora, G. J. V. Berkel, C. G. Enke, R. B. Cole, M. Martinez-Sanchez and J. B. Fenn, J. Mass Spectrom., 2000, 35, 939 CrossRef PubMed; (d) G. J. V. Berkel and V. Kertesz, Anal. Chem., 2007, 79, 5510 CrossRef PubMed.
- (a) A. H. Wonders, T. H. M. Housmans, V. Rosca and M. T. M. Koper, J. Appl. Electrochem., 2006, 36, 1215 CrossRef; (b) J. P. Grote, A. R. Zeradjanin, S. Cherevkon and K. J. Mayrhofer, Rev. Sci. Instrum., 2014, 85, 104101 CrossRef PubMed; (c) H. Oberacher, F. Pitterl, R. Erb and S. Plattner, Mass Spectrom. Rev., 2015, 34, 64 CrossRef PubMed; (d) F. M. Zhou and G. J. Vanberkel, Anal. Chem., 1995, 67, 3643 CrossRef.
- Z. Wei, M. Wleklinski, C. Ferreira and G. Cooks, Angew. Chem., Int. Ed., 2017, 56, 9386 CrossRef PubMed.
- H. Deng and G. J. V. Berkel, Anal. Chem., 1999, 71, 4284 CrossRef PubMed.
- B. R. Rosen, E. W. Werner, A. G. O'Brien and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 5571 CrossRef PubMed.
- (a) K. J. Vetter, Electrochemical Kinetics, ed. S. Bruckensein and B. Howard, Academic Press, New York, 1967 Search PubMed; (b) B. Ahmed, D. Barrow and T. Wirth, Adv. Synth. Catal., 2006, 348, 1043 CrossRef; (c) M. A. Modestino, D. Fernandez Rivas, S. M. H. Hashemi, J. G. E. Gardeniersb and D. Psaltis, Energy Environ. Sci., 2016, 9, 3381 RSC.
- T. Subbaiah, S. C. Das and R. P. Das, Hydrometallurgy, 1993, 33, 153 CrossRef.
- R. Musiol, Expert Opin. Drug Discovery, 2017, 12, 583 CrossRef PubMed.
- H. X. Li, J. L. Jiang, G. Lu, F. Huang and Z. X. Wang, Organometallics, 2011, 30, 3131 CrossRef.
- B. Chen, L. Y. Wang and S. Gao, ACS Catal., 2015, 5, 5851 CrossRef.
- M. Okimoto, Y. Takahashi, K. Numata, Y. Nagata and G. Sasaki, Synth. Commun., 2005, 35, 1989 CrossRef.
- G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef PubMed.
- (a) A. Badalyan and S. S. Stahl, Nature, 2016, 535, 406 CrossRef PubMed; (b) D. P. Hickey, M. S. McCammant, F. Giroud, M. S. Sigman and S. D. Minteer, J. Am. Chem. Soc., 2014, 136, 15917 CrossRef PubMed.
- M. F. Semmelhack and C. R. Schmid, J. Am. Chem. Soc., 1983, 105, 6732 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc00251g |
| This journal is © The Royal Society of Chemistry 2018 |