
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular hydride transfer onto arynes: redox-neutral and transition metal-free C(sp3)–H functionalization of amines†
Fahima I. M.
Idiris‡
 ,
Cécile E.
Majesté‡
,
Gregory B.
Craven
and
Christopher R.
Jones
*
,
Cécile E.
Majesté‡
,
Gregory B.
Craven
and
Christopher R.
Jones
*
School of Biological and Chemical Sciences, Queen Mary University of London, Mile End Road, London, E1 4NS, UK. E-mail: c.jones@qmul.ac.uk
First published on 8th February 2018
Abstract
Transition metal-free intramolecular hydride transfer onto arynes is reported for the first time. This unique transformation is utilized in redox-neutral intermolecular α-functionalization reactions of different tertiary amines, generating C(sp3)–C(sp3/sp2/sp) bonds in a single synthetic operation. Deuterium labeling studies support initial cleavage of the α-C–H bond via intramolecular 1,5-hydride transfer onto the aryne, which leads to activation of a range of integrated pronucleophiles and ultimately affords a new approach to cross-dehydrogenative coupling reactions which utilize aryne intermediates.
Introduction
The direct functionalization of C–H bonds offers a wealth of potential benefits to synthetic chemists,1 with significant progress having been made in recent decades.2 Activation of C(sp3)–H bonds α- to heteroatoms is particularly appealing,3 as evidenced by the burgeoning area of cross-dehydrogenative coupling (CDC) reactions,4 whereby certain substrates, especially amines, are functionalized using a sacrificial external oxidant and generally in the presence of a transition metal catalyst.5 α-Functionalization of amines has also received considerable recent interest through the development of redox-neutral processes6 that exploit the propensity of tertiary amines to undergo 1,5-hydride transfer onto a tethered acceptor,7 most commonly electron-deficient alkenes (Scheme 1a).8,9 Here, Lewis acid-catalyzed intramolecular hydride transfer results in a zwitterionic intermediate A that cyclizes to generate a new C–C bond. Maulide and co-workers elegantly extended this strategy to develop a redox-triggered approach to the C–H functionalization of cyclic amines (Scheme 1b).10 1,5-Hydride transfer onto an aldehyde acceptor – employed as a sacrificial oxidant – resulted in aminal B, which underwent nucleophilic attack upon addition of organometallic reagents.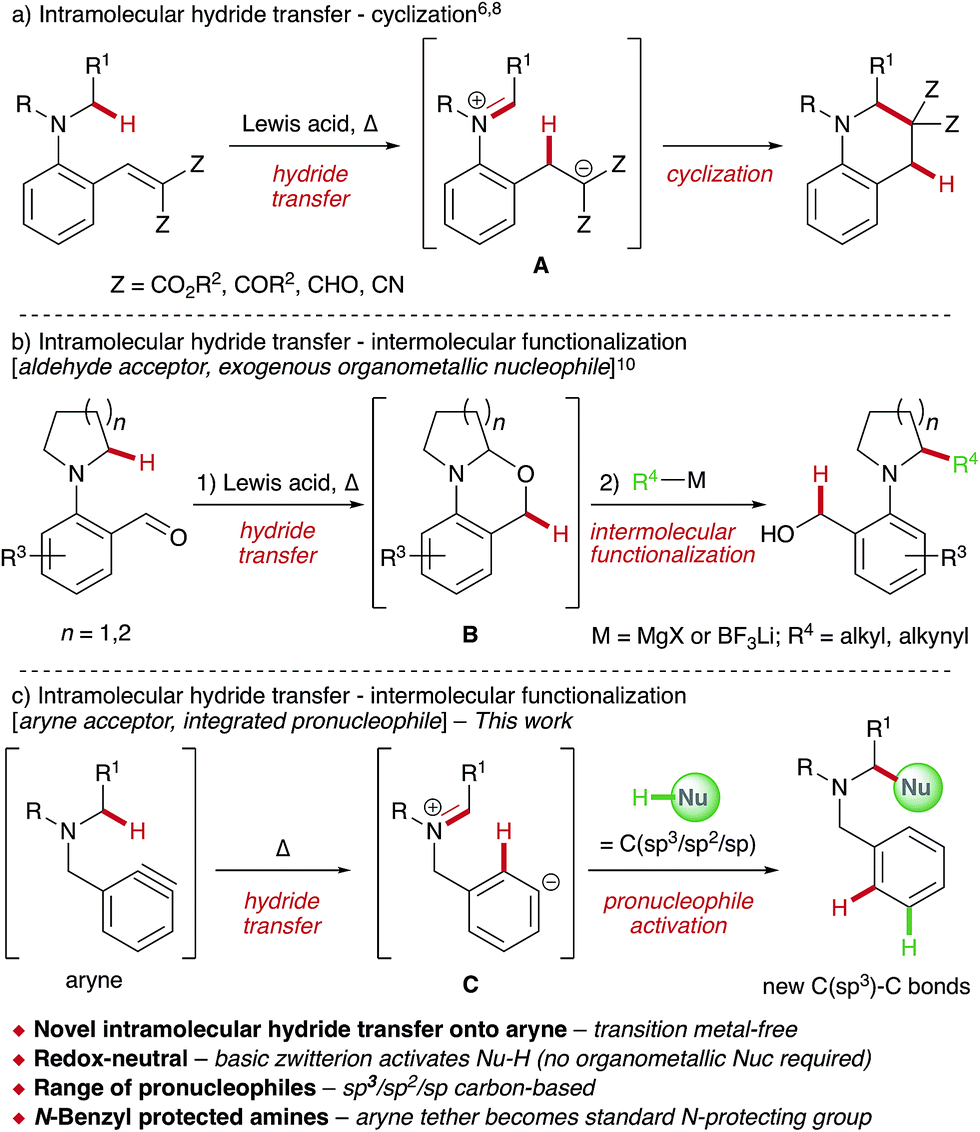 | ||
| Scheme 1 C(sp3)–H bond activation via intramolecular hydride transfer. | ||
Given our interest in aryne chemistry,11 we envisioned a related approach to a general α-functionalization of amines that employs arynes as internal hydride acceptors for the first time (Scheme 1c). This new transformation would reveal the unique zwitterionic intermediate C, prevented from undergoing intramolecular cyclization due to geometrical constraints. Significantly, zwitterion C contains a highly basic aryl anion that should be capable of activating a pronucleophile (Nu-H) within the reaction mixture,12 obviating the use of exogenous organometallic reagents in a second operation and rendering the overall process redox-neutral. Finally, the aryne tether would also operate as a latent N-benzyl protecting group; easily cleaved when desired.
Arynes are versatile reactive intermediates that have experienced a recent resurgence in interest13 due to the development of precursors that act under mild conditions, such as 2-(trimethylsilyl)aryl triflates14 and the hexadehydro-Diels–Alder reaction of polyalkynes.15 Despite undergoing myriad additions with an extensive range of nucleophiles,13 the reaction of arynes with C(sp3)–H bonds via ionic hydride transfer has yet to be realized.16–18 Herein we report this new transformation, which has enabled intermolecular α-functionalization of a range of tertiary amines with different carbon-based pronucleophiles, some of which are uncommon in CDC processes due to comparatively high pKa values. Deuterium labeling studies are described which support initial 1,5-hydride transfer onto the aryne, followed by activation of the integrated pronucleophile. Overall this furnishes new C(sp3)–C(sp3/sp2/sp) bonds in a single and operationally simple procedure via aryne-mediated CDC reactions.
Results and discussion
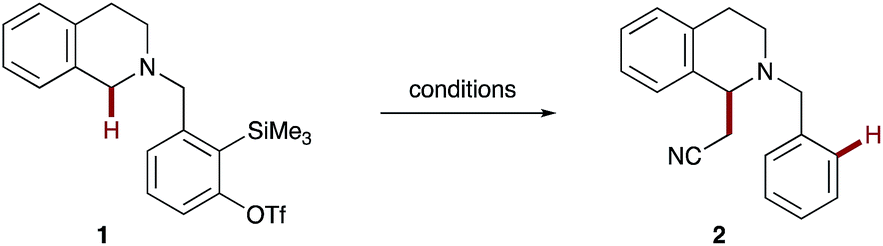
1,2,3,4-Tetrahydroisoquinoline (THIQ) was selected as the donor portion with which to initially evaluate our 1,5-hydride transfer hypothesis due to its biological activity and general synthetic utility.19,20 A suitable aryne acceptor was then tethered onto the amine donor using the 2-trimethylsilyl-3-trifluoromethanesulfonyl benzaldehyde precursor reported by Smith III and Kim.21 Acetonitrile was chosen as the initial pronucleophile as it is a common solvent for o-silylaryl triflate reactions and has been reported to undergo deprotonation by aryne-derived aryl anions.12 Selected optimization experiments, using THIQ scaffold 1 as a test system, are presented in Table 1. Evaluation of common o-silylaryl triflate activators (entries 1–7) identified KF/18-crown-6 and tetrabutylammonium triphenyldifluorosilicate (TBAT) as the most promising reagents. The introduction of toluene as a co-solvent (toluene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetonitrile, 3:1 by volume) enabled higher reaction temperatures which led to an increased yield with TBAT (entry 8). Both increasing and decreasing the reaction concentration resulted in lower yields (entries 9 & 10); most significantly at higher concentration due to competitive intermolecular amine arylation. At this stage we found that the α-cyanomethylated THIQ 2 could not be isolated cleanly during the reactions with TBAT, as the aryl silane by-product was a persistent contaminant, so we turned our attention to KF/18-crown-6 as the activator. A slight erosion in yield was observed for KF/18-crown-6 in the toluene–acetonitrile mixture (entry 11), presumably due to poorer solubility of fluoride. However, we were pleased to find that a solvent switch to 1,2-dimethoxyethane (DME) and acetonitrile (3:1 by volume) led to the complete consumption of aryne precursor 1 and the formation of the desired α-cyanomethylated THIQ 2a in 77% isolated yield (entry 12). The DME:acetonitrile ratio could be lowered to 19:1 by volume with a small drop-off in the yield of 2 (59%, entry 13). Encouragingly, further reduction of the pronucleophile loading to 150:1 (approx. 10 equivalents of acetonitrile, see entry 14) afforded 2 in a respectable 35% yield, which hinted at the potential to expand this method to more valuable pronucleophiles in the future. However, as the 3:1 volumetric ratio of DME:acetonitrile afforded the best yields and represented a good improvement in pronucleophile loading compared to the majority of CDC processes,4 especially those involving this less common pronucleophile,22 these conditions were selected for the study.
:acetonitrile, 3:1 by volume) enabled higher reaction temperatures which led to an increased yield with TBAT (entry 8). Both increasing and decreasing the reaction concentration resulted in lower yields (entries 9 & 10); most significantly at higher concentration due to competitive intermolecular amine arylation. At this stage we found that the α-cyanomethylated THIQ 2 could not be isolated cleanly during the reactions with TBAT, as the aryl silane by-product was a persistent contaminant, so we turned our attention to KF/18-crown-6 as the activator. A slight erosion in yield was observed for KF/18-crown-6 in the toluene–acetonitrile mixture (entry 11), presumably due to poorer solubility of fluoride. However, we were pleased to find that a solvent switch to 1,2-dimethoxyethane (DME) and acetonitrile (3:1 by volume) led to the complete consumption of aryne precursor 1 and the formation of the desired α-cyanomethylated THIQ 2a in 77% isolated yield (entry 12). The DME:acetonitrile ratio could be lowered to 19:1 by volume with a small drop-off in the yield of 2 (59%, entry 13). Encouragingly, further reduction of the pronucleophile loading to 150:1 (approx. 10 equivalents of acetonitrile, see entry 14) afforded 2 in a respectable 35% yield, which hinted at the potential to expand this method to more valuable pronucleophiles in the future. However, as the 3:1 volumetric ratio of DME:acetonitrile afforded the best yields and represented a good improvement in pronucleophile loading compared to the majority of CDC processes,4 especially those involving this less common pronucleophile,22 these conditions were selected for the study.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Activator | Additive | Solvent | T (°C) | Yieldb (%) |
| a Reaction conditions: activator (2.0 equiv.), additive (2.0 equiv.), solvent [0.01 M], 12 h. b 1H NMR yield vs. dibromomethane internal standard, isolated yield in parentheses, all reactions proceeded to full conversion after 12 h. c 1.0 M in THF. d 0.005 M. e 0.05 M. TBAT = tetrabutylammonium triphenyldifluorosilicate. | |||||
| 1 | CsF | — | CH3CN | 70 | 43 |
| 2 | CsF | 18-c-6 | CH3CN | 70 | 60 |
| 3 | KF | 18-c-6 | CH3CN | 70 | 70 |
| 4 | Cs2CO3 | 18-c-6 | CH3CN | 70 | 36 |
| 5 | TBAFc | — | CH3CN | 70 | 29 |
| 6 | TBAF·3H2O | — | CH3CN | 70 | 23 |
| 7 | TBAT | — | CH3CN | 70 | 66 |
| 8 | TBAT | — | 3:1 PhMe/CH3CN |
90 | 75 |
| 9 | TBAT | — | 3:1 PhMe/CH3CNd |
90 | 72 |
| 10 | TBAT | — | 3:1 PhMe/CH3CNe |
90 | 35 |
| 11 | KF | 18-c-6 | 3:1 PhMe/CH3CN |
90 | 64 |
| 12 | KF | 18-c-6 | 3:1 DME/CH3CN |
90 | 89 (77) |
| 13 | KF | 18-c-6 | 19:1 DME/CH3CN |
90 | 59 |
| 14 | KF | 18-c-6 | 150:1 DME/CH3CN |
90 | 35 |
A range of substituted THIQs 1b–i were found to be amenable to the optimized reaction conditions, affording the corresponding C1-cyanomethylated THIQs 2b–i (Scheme 2). Electron-donating substituents such as methoxy (1b) and hydroxy (1c) were excellent substrates, giving high yields of 2b and 2c. It is particularly interesting to note that 6-hydroxy THIQ precursor 2c was tolerated, as it demonstrated that the unprotected phenol did not cause significant quenching of the anion in zwitterionic intermediate C (see Scheme 1c). Halogens (Br, 1d and Cl, 1e) and moderately electron-withdrawing groups (SO2NEt2, 1f and CO2t-Bu, 1g) were also viable substrates, affording the corresponding products 2d–g in moderate yields; consistent with less hydridic C–H bonds and decreased carbocation stabilization in comparison to 1a–c. The incorporation of a strongly electron-withdrawing nitro group into the THIQ scaffold (1h) was found to almost completely inhibit hydride transfer, with only traces of 2h observed. Finally, 1-methyl THIQ 1i proved an effective substrate, generating the quaternary cyanomethylated THIQ 2i in a good yield. It is noteworthy that THIQs occupy a privileged position as benchmark substrates in CDC reactions, affording α-functionalized products that typically contain an N-aryl group.4 In comparison, the THIQs 2 produced here possess a synthetically practical N-benzyl protecting group.23
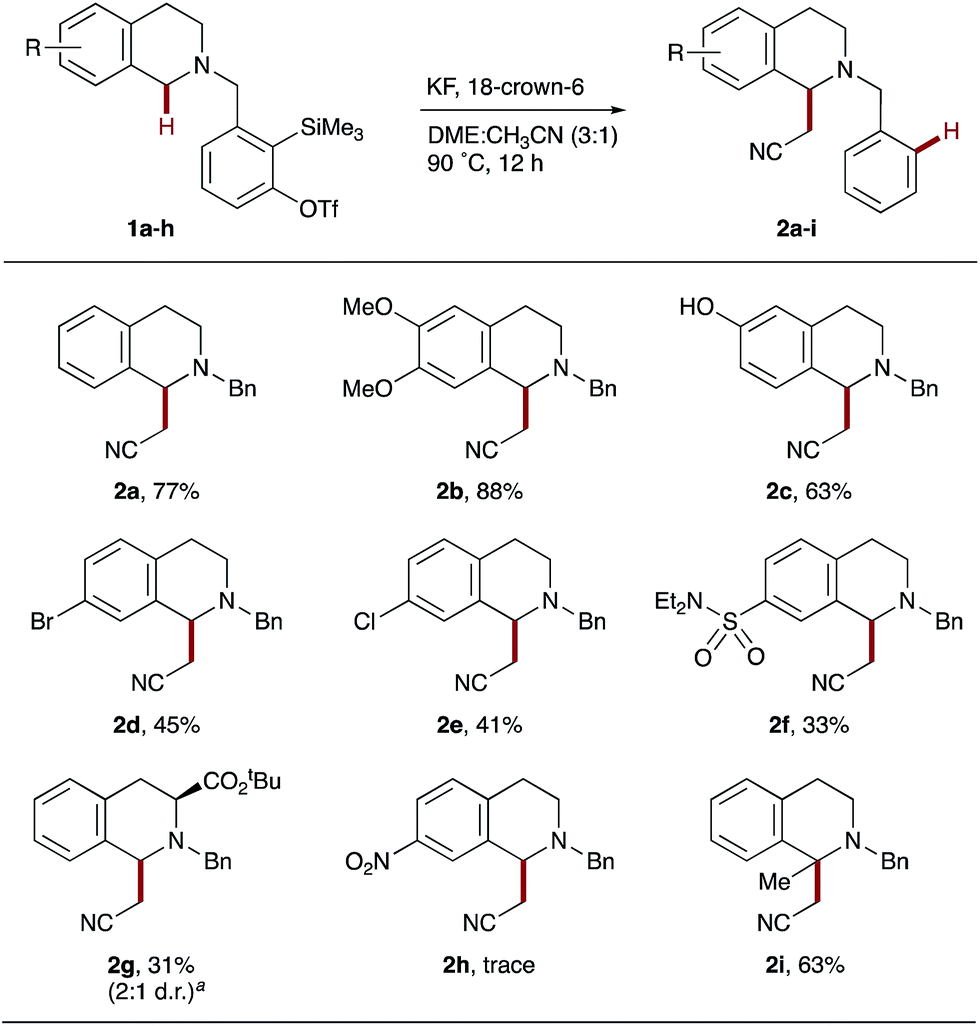 | ||
| Scheme 2 Intramolecular hydride transfer onto aryne with THIQ derivatives. Reaction conditions: amine 1 (1.0 equiv.), KF (2.0 equiv.), 18-crown-6 (2.0 equiv.), DME:CH3CN (3:1 by volume, 0.01 M), 90 °C, 12 h. Yields of isolated products throughout. aRatio determined by 1H NMR spectroscopy. | ||
Having established the feasibility of intramolecular hydride transfer onto arynes with a range of THIQ derivatives, we continued our investigations by varying the structure of the tertiary amine donor. Starting with dihydrophenanthridine derivative 3a, exposure to the established reaction conditions smoothly afforded 4a in 72% yield (Scheme 3). α-Phenylbenzylamine precursor 3b and α-methylbenzylamine 3c generated the quaternary products 4b and 4c in 53% and 32% yields respectively. Interestingly, no benzobarrelene products from a potentially competitive intramolecular Diels–Alder pathway were identified.24 Instead, the increase in conformational flexibility of the tether is proposed to account for the difference in reactivity between 3a and 3b/c.
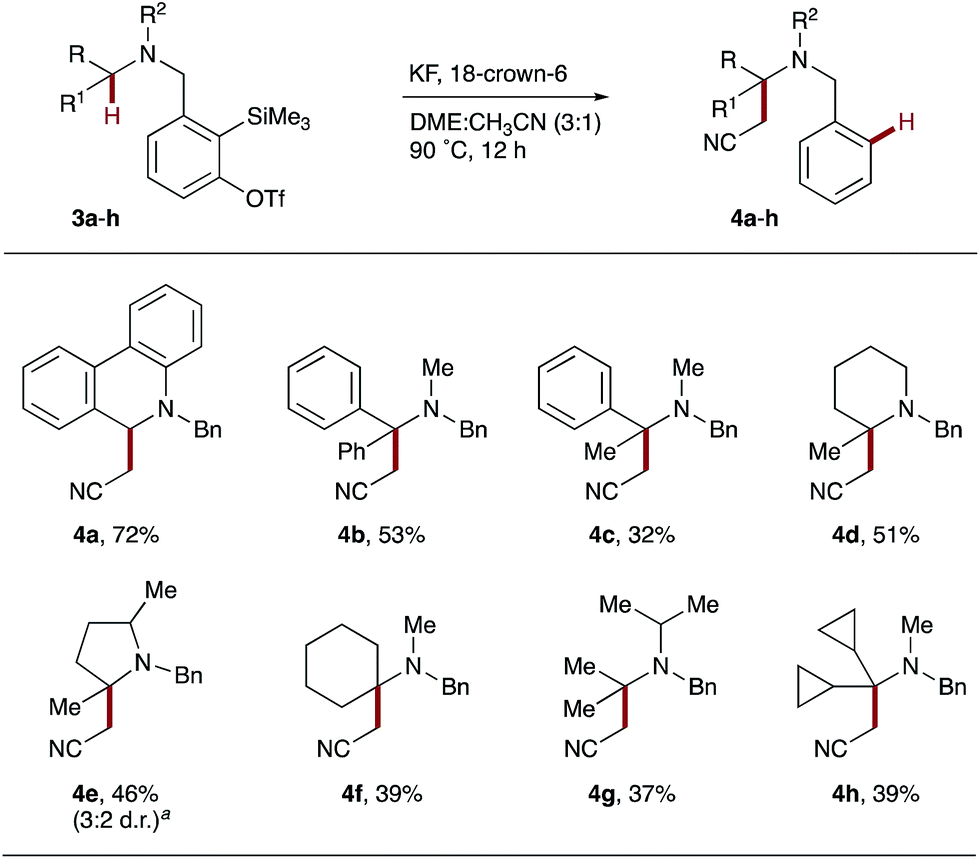 | ||
| Scheme 3 Hydride transfer from linear and cyclic amine derivatives. Reaction conditions are as shown in Scheme 2. Yields of isolated products throughout. aRatio determined by 1H NMR spectroscopy. | ||
Pleasingly, hydride transfer was not restricted to benzylic C–H bonds and proved equally effective with precursors 3d–h that each contained tertiary alkyl C–H bonds. Heterocyclic derivatives 2-methyl-piperidine 3d and 2,5-dimethyl-pyrrolidine 3e gave the corresponding α-quaternary heterocycles 4d and 4e in moderate yields. Similarly, the less conformationally-rigid amines 3f–h also promoted hydride transfer onto arynes, yielding spirocyclic and acyclic C–H functionalized amines, 4f and 4g/h, respectively.
Having applied the principle of aryne-mediated hydride transfer and subsequent co-solvent activation to the α-cyanomethylation of tertiary amines, we looked at introducing alternative pronucleophiles in this process. Pleasingly, when dimethoxy-THIQ derivative 1b was exposed to the standard reaction conditions, a range of different carbon-based coupling partners were found to be viable co-solvents (Scheme 4). For example, propionitrile, less acidic than acetonitrile (pKa = 32.5 in DMSO cf. 31.3 for MeCN),25 yielded β-substituted cyanoamine 5b as a 1:1 mixture of diastereoisomers in 66% yield. Nitromethane, a more commonly used solvent in CDC reactions4c due to significantly higher acidity (pKa = 17.2 in DMSO),25 produced nitromethylated THIQ 6b in a similarly good 70% yield. Interestingly, a more ‘inert’ solvent such as chloroform also operated as a coupling partner, producing trichloromethylated THIQ 7b in 41% yield. It is noteworthy that better yields were obtained here at a lower 9:1 DME:Nu-H ratio and no products from dichlorocarbene intermediates were detected. The use of pentafluorobenzene enabled access to new C(sp3)–C(sp2) bond formation (8b) in good yield, illustrating the potential for α-amino C(sp3)–H arylation with electron-deficient arenes in the absence of a transition metal catalyst. Lastly, C(sp3)–C(sp) coupling could be achieved with phenylacetylene, affording 9b in 48% yield; the addition of copper did not improve the outcome.26 THIQ derivatives 1a and 1i also proved amenable to these aryne-mediated CDC reactions, affording the corresponding α-functionalized THIQs in moderate to good yields.27
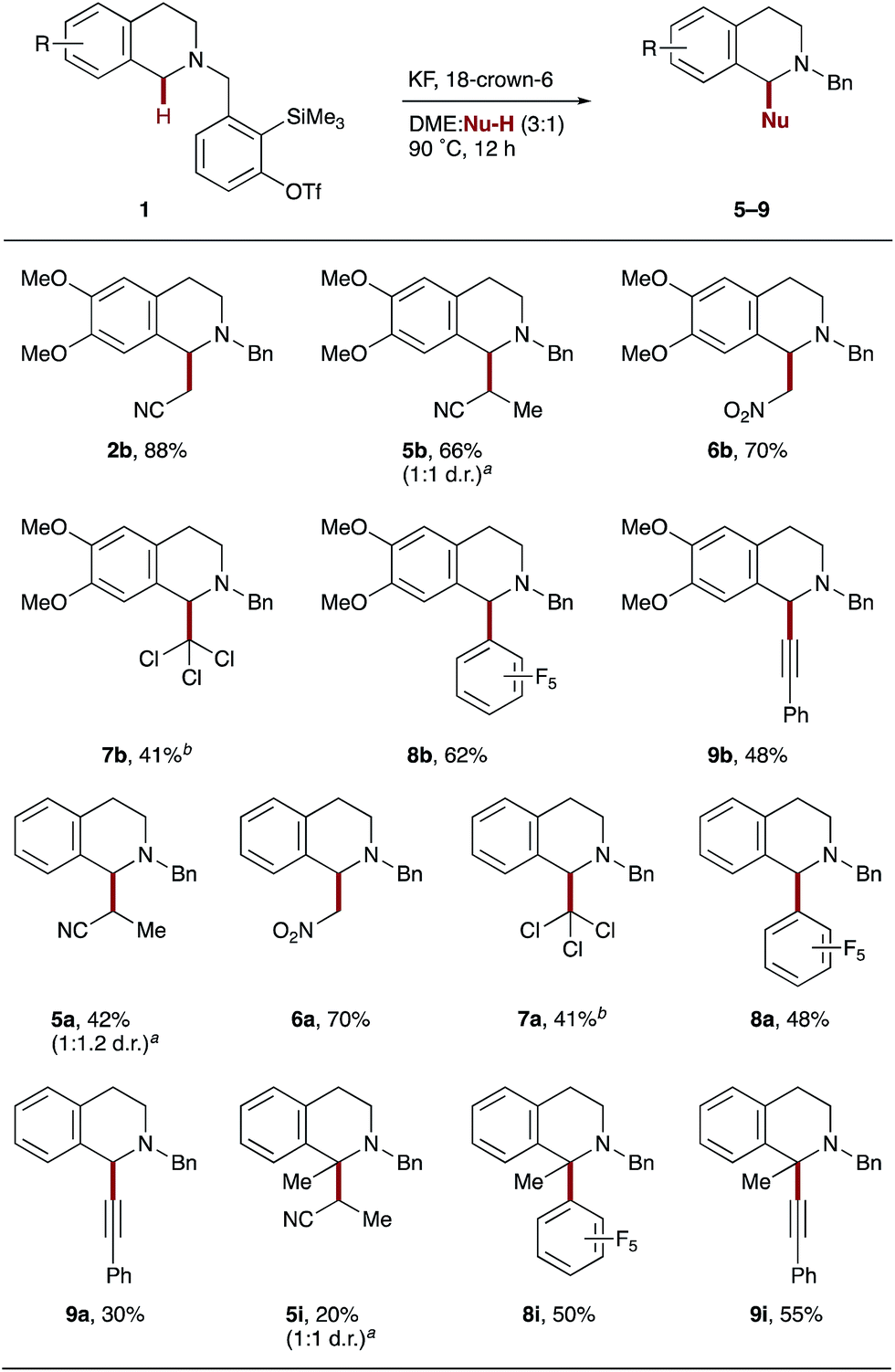 | ||
| Scheme 4 Alternative pronucleophiles. Reaction conditions are as shown in Scheme 2. Yields of isolated products throughout. aRatio determined by 1H NMR spectroscopy. bDME:Nu-H (9:1 by volume, 0.01 M). | ||
Finally, we sought to probe our mechanistic hypothesis. Support for an ionic hydride transfer process came from the retention of the cyclopropane rings in amine 4h (see Scheme 3), as it was reasoned that formation of a radical adjacent to nitrogen would result in rapid and irreversible cyclopropane ring-opening. Furthermore, conducting the cyanomethylation of 1a in the presence of a radical scavenger, TEMPO (2.0 equiv.), did not prohibit the reaction. Next we performed a series of deuterium labeling studies, starting with the reaction of THIQ precursor 1a in acetonitrile-d3 as co-solvent, which resulted in deuterium incorporation solely at the meta position of the benzene ring in THIQ 2a-d3 (Scheme 5a). Next, exposure of bisdeuterated precursor 1,1-d2-THIQ 1a-d2 to the reaction conditions afforded the corresponding product of 1,5-deuteride transfer, 2a-d2, with deuterium located at the ortho position of the benzene ring (Scheme 5b).28 Finally, a competition reaction between an equimolar amount of THIQ precursor 1a and the bisdeuterated isotopologue 1a-d2 supported the intramolecular nature of the hydride transfer, as the monodeuterated crossover products 2a-Hd and 2a-dH were not observed (Scheme 5c).
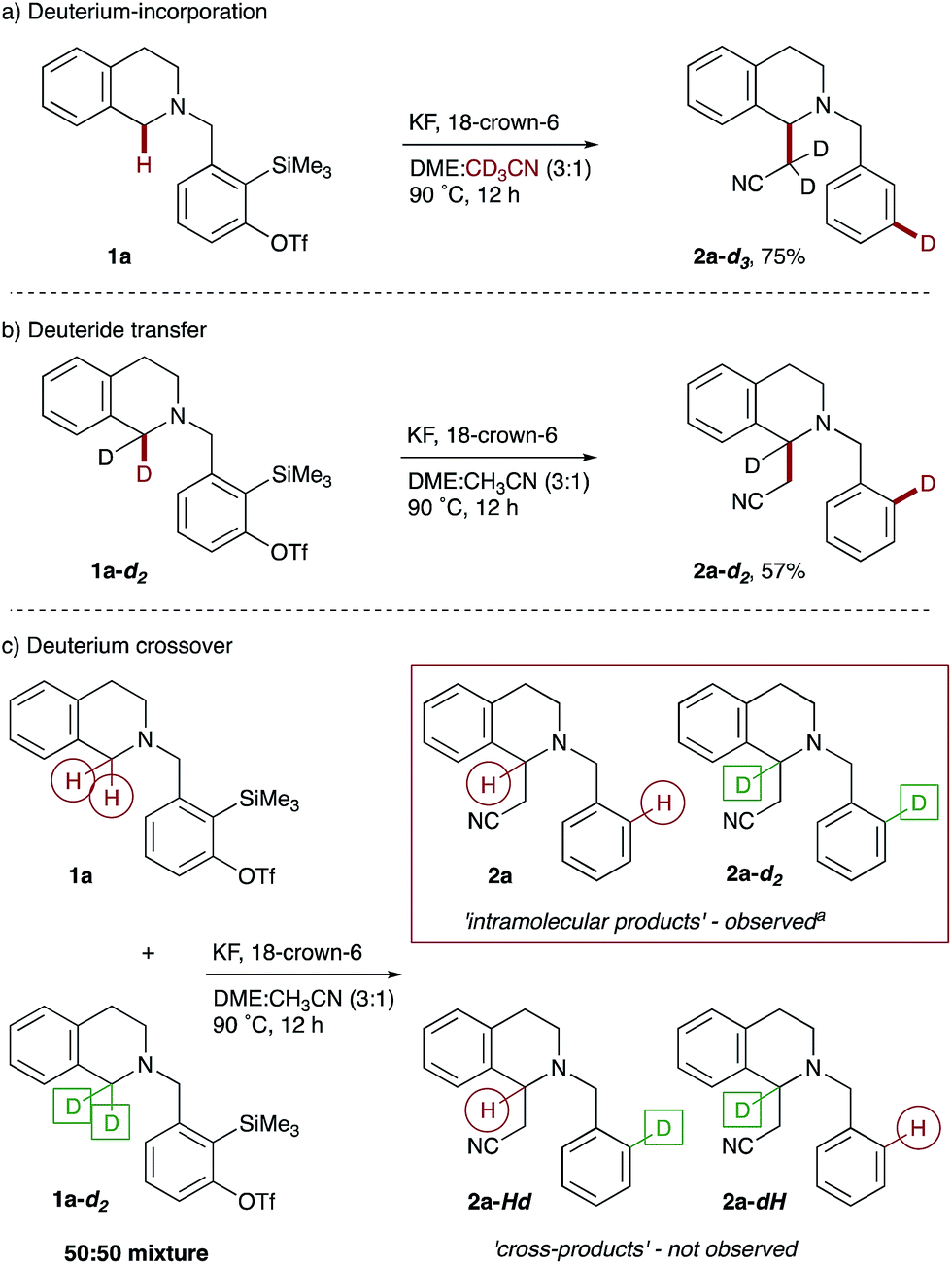 | ||
| Scheme 5 Mechanistic experiments. Reaction conditions are as shown in Scheme 2. aProducts 2a and 2a-d2 isolated as an inseparable mixture of isotopologues (5:3.5, as determined by 1H NMR spectroscopy) in a combined 70% yield. | ||
Considering the experimental evidence, the following mechanism is proposed (Scheme 6). Treatment of o-silylaryl triflate precursor 3 with fluoride reveals an aryne 10 that subsequently undergoes reduction via the intramolecular 1,5-hydride transfer of a C–H bond α- to nitrogen. The reactive zwitterionic intermediate 11 deprotonates the acetonitrile pronucleophile, which then adds to iminium ion 12 in a Mannich-type reaction to yield α-cyanomethylated amine 4.
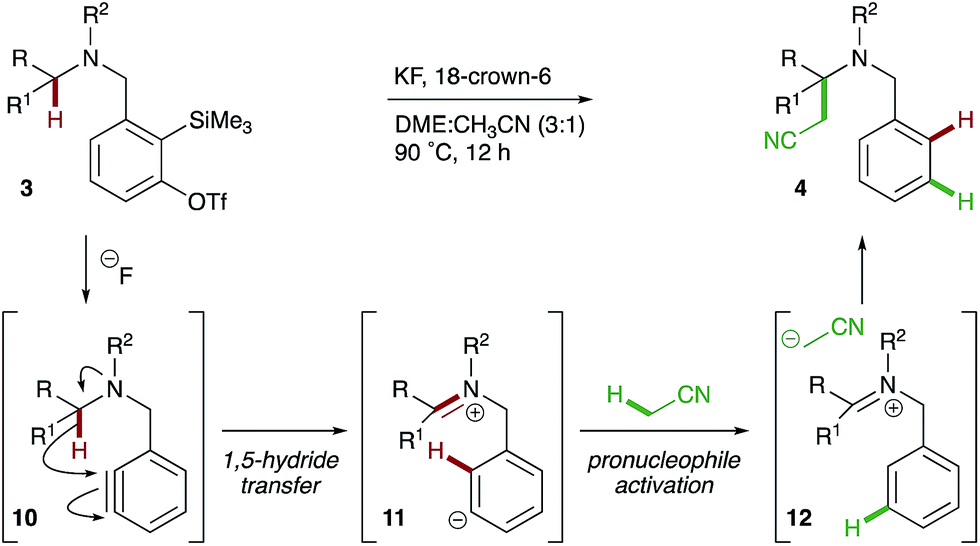 | ||
| Scheme 6 Proposed mechanism. | ||
Conclusions
In summary, we have described an intramolecular hydride transfer that uses arynes as acceptor moieties for the first time and exploited this in the development of aryne-mediated CDC reactions of heterocyclic and aliphatic tertiary amines. This is a transition metal-free and redox-neutral process that generates a new C(sp3)–C(sp3/sp2/sp) bond α- to nitrogen in a single synthetic operation. The approach is distinct from existing transition metal-free methods for C–H functionalization via hydride transfer as a Lewis acid is not required to activate the acceptor and intermediate zwitterion cyclization is geometrically inhibited. Furthermore, the highly basic aryl anion directly activates a number of diverse pronucleophiles – some of which are not often encountered in CDC processes due to high pKa values – which enables integration of an intermolecular coupling partner in the same reaction vessel. The reduced aryne tether also operates as a practical N-benzyl protecting group for the corresponding α-functionalized secondary amines. Finally, reactions conducted during the initial optimization studies revealed that lower pronucleophile loadings can be employed in these processes, hinting at the potential to expand to more valuable coupling partners, although the associated erosion in reaction yield means that further optimization would be required. To this end, work is currently underway in our laboratory to establish a full structure–activity profile for hydride transfer onto arynes.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to the EPSRC (EP/M026221/1, C. R. J.), Ramsay Memorial Trust (C. R. J.), QMUL (studentships to F. I. M. I. & C. E. M.) and the RSC Research Fund for financial support. We thank the EPSRC UK National Mass Spectrometry Facility at Swansea University.Notes and references
- J. Wencel-Delord and F. Glorious, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed.
- Selected reviews: (a) C. C. C. Johansson and T. J. Colacot, Angew. Chem., Int. Ed., 2010, 49, 676 CrossRef CAS PubMed; (b) O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902 RSC; (c) B.-J. Li and Z.-J. Shi, Chem. Soc. Rev., 2012, 41, 5588 RSC; (d) E. Geist, A. Kirschning and T. Schmidt, Nat. Prod. Rep., 2014, 31, 441 RSC.
- (a) W. Chen, L. Ma, A. Paul and D. Seidel, Nat. Chem., 2018, 10, 165 CrossRef CAS PubMed; (b) K. R. Campos, Chem. Soc. Rev., 2007, 36, 1069 RSC; (c) E. A. Mitchell, A. Peschiulli, N. Lefevre, L. Meerpoel and B. U. W. Maes, Chem.–Eur. J., 2012, 18, 10092 CrossRef CAS PubMed.
- Selected CDC reviews: (a) C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed; (b) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC; (c) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed; (d) B. V. Varun, J. Dhineshkumar, K. R. Bettadapur, Y. Siddaraju, K. Alagiri and K. R. Prabhu, Tetrahedron Lett., 2017, 58, 803 CrossRef CAS.
- Selected metal-free CDC: (a) Y. Zhang and C.-J. Li, J. Am. Chem. Soc., 2006, 128, 4242 CrossRef CAS PubMed; (b) H. Richter, R. Froehlich, C. G. Daniliuc and O. G. Mancheno, Angew. Chem., Int. Ed., 2012, 51, 8656 CrossRef CAS PubMed; (c) B. Schweitzer-Chaput and M. Klussmann, Eur. J. Org. Chem., 2013, 666 CrossRef CAS.
- Selected reviews: (a) B. Peng and N. Maulide, Chem.–Eur. J., 2013, 19, 13274 CrossRef CAS PubMed; (b) L. Wang and J. Xiao, Adv. Synth. Catal., 2014, 356, 1137 CrossRef CAS; (c) M. C. Haibach and D. Seidel, Angew. Chem., Int. Ed., 2014, 53, 5010 CrossRef CAS PubMed; (d) L. Wang and J. Xiao, Top. Curr. Chem., 2016, 374, 17 CrossRef PubMed.
- “tert-Amino effect”, see: (a) O. Meth-Cohn and H. Suschitzky, Adv. Heterocycl. Chem., 1972, 14, 211 CrossRef CAS; (b) O. Meth-Cohn, Adv. Heterocycl. Chem., 1996, 65, 1 CrossRef CAS.
- Selected examples with alkenes: (a) W. H. N. Nijhuis, W. Verboom and D. N. Reinhoudt, J. Am. Chem. Soc., 1987, 109, 3136 CrossRef CAS; (b) S. J. Pastine, K. M. McQuaid and D. Sames, J. Am. Chem. Soc., 2005, 127, 12180 CrossRef CAS PubMed; (c) M. C. Haibach, I. Deb, C. K. De and D. Seidel, J. Am. Chem. Soc., 2011, 133, 2100 CrossRef CAS PubMed; (d) K. Mori, K. Ehara, K. Kurihara and T. Akiyama, J. Am. Chem. Soc., 2011, 133, 6166 CrossRef CAS PubMed; (e) T. Yoshida and K. Mori, Chem. Commun., 2017, 53, 4319 RSC.
- Selected examples with (a) alkyne: P. A. Vadola and D. Sames, J. Am. Chem. Soc., 2009, 131, 16525 CrossRef CAS PubMed; (b) aldehyde: S. J. Pastine and D. Sames, Org. Lett., 2005, 7, 5429 CrossRef CAS PubMed; (c) iminium ion: C. Zhang, S. Murarka and D. Seidel, J. Org. Chem., 2009, 74, 419 CrossRef CAS PubMed; (d) allene: B. Bolte and F. Gagosz, J. Am. Chem. Soc., 2011, 133, 7696 CrossRef CAS PubMed; (e) metal carbenoid: F. Cambeiro, S. Lopez, J. A. Varela and C. Saa, Angew. Chem., Int. Ed., 2012, 51, 723 CrossRef CAS PubMed; (f) ketimine: K. Mori, N. Umehara and T. Akiyama, Adv. Synth. Catal., 2015, 357, 901 CrossRef CAS; (g) keteniminium ion: C. Theunissen, B. Metayer, N. Henry, G. Compain, J. Marrot, A. Martin-Mingot, S. Thibaudeau and G. Evano, J. Am. Chem. Soc., 2014, 136, 12528 CrossRef CAS PubMed.
- I. D. Jurberg, B. Peng, E. Wöstefeld, M. Wasserloos and N. Maulide, Angew. Chem., Int. Ed., 2012, 51, 1950 CrossRef CAS PubMed.
- P. Trinchera, W. Sun, J. E. Smith, D. Palomas, R. Crespo-Otero and C. R. Jones, Org. Lett., 2017, 19, 4644 CrossRef CAS PubMed.
- Arynes and solvent activation: (a) M. Jeganmohan and C.-H. Cheng, Chem. Commun., 2006, 2454 RSC; (b) D. Stephens, Y. Zhang, M. Cormier, G. Chavez, H. Arman and O. V. Larionov, Chem. Commun., 2013, 49, 6558 RSC; (c) S.-E. Suh and D. M. Chenoweth, Org. Lett., 2016, 18, 4080 CrossRef CAS PubMed.
- Selected reviews: (a) P. M. Tadross and B. M. Stoltz, Chem. Rev., 2012, 112, 3550 CrossRef CAS PubMed; (b) R. W. Hoffmann and K. Suzuki, Angew. Chem., Int. Ed., 2013, 52, 2655 CrossRef CAS PubMed; (c) C. Holden and M. F. Greaney, Angew. Chem., Int. Ed., 2014, 53, 5746 CrossRef CAS PubMed; (d) S. Yoshida and T. Hosoya, Chem. Lett., 2015, 44, 1450 CrossRef CAS; (e) J.-A. García-López and M. F. Greaney, Chem. Soc. Rev., 2016, 45, 6766 RSC; (f) S. S. Bhojgude, A. Bhunia and A. T. Biju, Acc. Chem. Res., 2016, 49, 1658 CrossRef CAS PubMed; (g) J. Shi, Y. Li and Y. Li, Chem. Soc. Rev., 2017, 46, 1707 RSC; (h) F. I. M. Idiris and C. R. Jones, Org. Biomol. Chem., 2017, 15, 9044 RSC.
- Y. Himeshima, T. Sonoda and H. Kobayashi, Chem. Lett., 1983, 12, 1211 CrossRef.
- (a) T. R. Hoye, B. Baire, D. Niu, P. H. Willoughby and B. P. Woods, Nature, 2012, 490, 208 CrossRef CAS PubMed; (b) D. Niu, P. H. Willoughby, B. P. Woods, B. Baire and T. R. Hoye, Nature, 2013, 501, 531 CrossRef CAS PubMed; (c) R. Karmakar and D. Lee, Chem. Soc. Rev., 2016, 45, 4459 RSC; (d) F. Xu, X. Xiao and T. R. Hoye, J. Am. Chem. Soc., 2017, 139, 8400 CrossRef CAS PubMed.
- Hydride transfer onto aryne proposed as rationale for unexpected by-product, however, no direct evidence presented: (a) E. J. Forbes and C. J. Gray, Tetrahedron, 1968, 24, 6223 CrossRef CAS; (b) R. Harrison, H. Heaney, J. M. Jablonski, K. G. Mason and J. M. Sketchley, J. Chem. Soc. C, 1969, 1684 RSC; (c) B. Jacques and R. G. Wallace, Tetrahedron, 1977, 33, 581 Search PubMed.
- Ag(I)-mediated hydride transfer onto aryl cation derived from aryne intermediate: P. Mamidipalli, S. Y. Yun, K.-P. Wang, T. Zhou, Y. Xia and D. Lee, Chem. Sci., 2014, 5, 2362 RSC.
- Formal aryne reduction via concerted double hydrogen atom transfer: see ref. 13b and P. H. Willoughby, D. Niu, T. Wang, M. K. Haj, C. J. Cramer and T. R. Hoye, J. Am. Chem. Soc., 2014, 136, 13657 CrossRef CAS PubMed and .
- Selected examples of THIQ bioactivity: (a) F. Crestey, A. A. Jensen, M. Borch, J. T. Andreasen, J. Andersen, T. Balle and J. L. Kristensen, J. Med. Chem., 2013, 56, 9673 CrossRef CAS PubMed; (b) J. N. Hanna, F. Ntie-Kang, M. Kaiser, R. Brun and S. M. N. Efange, RSC Adv., 2014, 4, 22856 RSC.
- Selected THIQ reviews: (a) J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669 CrossRef CAS PubMed; (b) K. W. Bentley, Nat. Prod. Rep., 2004, 21, 395 RSC; (c) W. Liu, S. Liu, R. Jin, H. Guo and J. Zhao, Org. Chem. Front., 2015, 2, 288 RSC.
- A. B. Smith III and W.-S. Kim, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6787 CrossRef PubMed ; see ESI† for full details of precursor preparation.
- W. Zhang, S. Yang and Z. Shen, Adv. Synth. Catal., 2016, 358, 2392 CrossRef CAS.
- P. D. Bailey, M. A. Beard, H. P. T. Dang, T. R. Phillips, R. A. Price and J. H. Whittaker, Tetrahedron Lett., 2008, 49, 2150 CrossRef CAS.
- V. D. Pogula, T. Wang and T. R. Hoye, Org. Lett., 2015, 17, 856 CrossRef CAS PubMed.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456 CrossRef CAS.
- Z. Li and C.-J. Li, J. Am. Chem. Soc., 2004, 126, 11810 CrossRef CAS PubMed.
- Nitromethyl and trichloromethyl derivatives of 1i could not be isolated, presumably due to dissociation being favoured by the increased stability of the C1-substituted iminium ion and the greater acidity of the corresponding co-solvents.
- Slower rate of deuterium transfer relative to hydride transfer enables competing and deleterious intermolecular amine arylation, resulting in slightly lower yields of cyanomethylated product 2a-d2.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc00181b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |