
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of histidine-containing hydrocarbon stapled cell penetrating peptides for in vitro and in vivo delivery of siRNAs†
Soonsil
Hyun
 a,
Yoonhwa
Choi
b,
Ha Neul
Lee
c,
Changki
Lee
c,
Donghoon
Oh
c,
Dong-Ki
Lee
d,
Changjin
Lee
*c,
Yan
Lee
*e and
Jaehoon
Yu
*ab
a,
Yoonhwa
Choi
b,
Ha Neul
Lee
c,
Changki
Lee
c,
Donghoon
Oh
c,
Dong-Ki
Lee
d,
Changjin
Lee
*c,
Yan
Lee
*e and
Jaehoon
Yu
*ab
aInstitute of Molecular Biology and Genetics, Seoul National University, Seoul 08826, Korea
bDepartment of Chemistry & Education, Seoul National University, Seoul 08826, Korea. E-mail: jhoonyu@snu.ac.kr
cHugel, Inc., Chuncheon 24206, Korea. E-mail: senovia@hugel.co.kr
dDepartment of Chemistry, Sungkyunkwan University, Suwon, Korea
eDepartment of Chemistry, Seoul National University, Seoul 08826, Korea. E-mail: gacn@snu.ac.kr
First published on 3rd April 2018
Abstract
A hydrocarbon stapled peptide based strategy was used to develop an optimized cell penetrating peptide for siRNA delivery. Various stapled peptides, having amphipathic Leu- and Lys-rich regions, were prepared and their cell penetrating potentials were evaluated. One peptide, stEK, was found to have high cell penetration and siRNA delivery abilities at low nanomolar concentrations. In order to improve its ability to promote gene silencing, stEK was modified by replacing several Lys residues with His moieties. The modified peptide, LKH-stEK, was found to facilitate endosomal escape and to display >90% knock-down with 50 nM of a siRNA targeting cyclophilin B in HeLa cells. The results of an in vivo animal wound healing model study demonstrate that LKH-stEK promotes delivery of an siRNA, which targets the connective tissue growth factor, and that this process leads to efficient gene silencing by the siRNA at a nanomolar level in mouse skin.
Introduction
The discovery of small noncoding RNAs was a highly significant advance in the area of RNA biology.1 Among small RNAs, short interfering RNAs (siRNAs) act as defenders of genome integrity in response to foreign nucleic acids. Through a silencing mechanism, siRNA therapeutics have the potential to regulate abnormal gene expression such as that taking place in genomic diseases or tumorigenic disorders.2However, numerous optimization processes and related mechanistic studies have been performed to develop therapeutically important siRNAs.3–5 For example, chemical modification is necessary in order to improve the stability of siRNA.6 Another approach to improve the potency of siRNA therapeutics involves the use of delivery tools,7,8 such as lipid nanoparticle or cell penetrating peptides (CPPs).9–11 However, accumulation in specific organs, immunity activation, and the associated toxicity hamper the use of lipid–siRNA complexes for delivery.12 Thus, a variety of studies have probed the use of covalent conjugates13,14 and non-covalent complexes of CPPs15,16 for delivery of siRNA. In the former approach, CPPs are linked to the surfaces of liposomes or polymer complexes of siRNA13 or directly conjugated to siRNA through cleavable or non-cleavable chains.14 In the latter method, formation of non-covalent complexes between CPPs and siRNA is driven by ionic interactions. Because it employs simple mixing, this delivery strategy has become more ready to use than those that utilize covalently linked CPPs. Therefore, the search for new CPPs, which have improved efficacy for noncovalent siRNA delivery and reduced cytotoxicity, is in high demand.
Our recent efforts have focused on using systematic modification to develop amphipathic α-helical Lys- and Leu-rich RNA binding peptides for specific RNA targets.17 We postulated that insertion of hydrocarbon cross-links would be a useful strategy in designing CPPs for the delivery of siRNA due to the increased stability, cell penetrating abilities, and appropriate binding affinity to RNA (Fig. 1a).18,19 Moreover, stapled LK-peptides could be further modified to incorporate His moieties (Fig. 1d). This change could facilitate escape of the siRNA from the CPP/siRNA complex entrapped in endosomes, because side chains of His have near endosomal pKa values and, consequently, undergo changes in their protonation states in endosomal compartments (Fig. 1c (3)).20
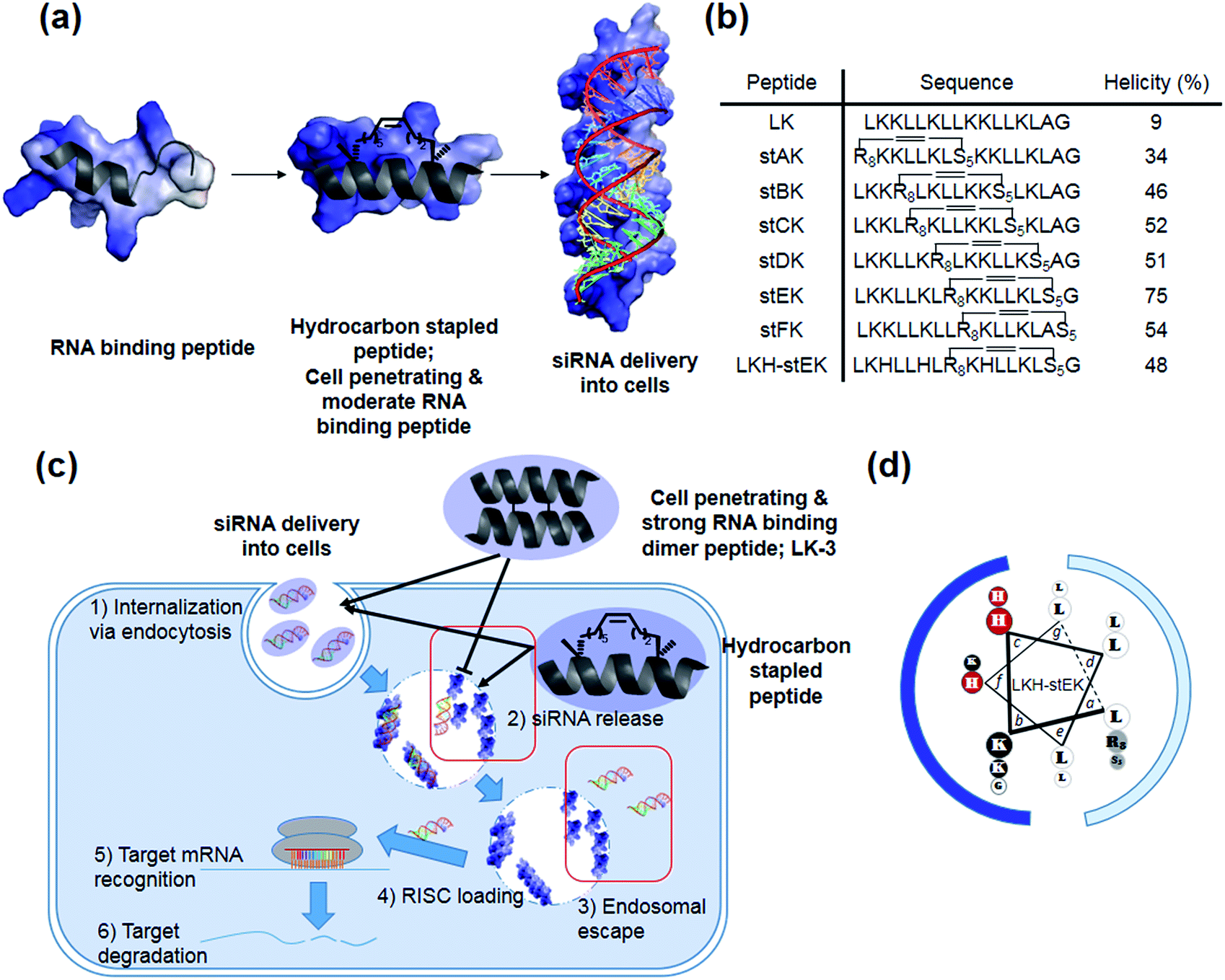 | ||
| Fig. 1 (a) Schematic representation of hydrocarbon stapled peptides for siRNA delivery. (b) Sequences and helicities of LK peptide and their stapled derivatives. (c) Design of peptides for siRNA delivery. Although both the dimer (LK-3) and stapled peptides can be delivered into cells, the latter peptide has advantages over the former owing to facile release and endosomal escape of siRNA. (d) LKH-stEK. Lowercase letters indicate positions in the heptad repeat (abcdefg), with hydrophobic residues at a, d, e, and g sites. Polar Lys, nonpolar Leu residues, alkene-bearing non-natural amino acids, and His residues are shown in black, white, gray, and red circles, respectively. Cationic hydrophilic face and hydrophobic face are marked as blue and pale blue. | ||
In the investigation described below, we prepared and evaluated a small library of CPPs (Fig. 1b), which is comprised of positional hydrocarbon stapled peptide isomers. An assessment of the siRNA delivery potentials of these peptides led to the observation that the stapled peptide, stEK, displays the highest cell penetrating ability in the group as well as the potential for efficient delivery of the siRNA targeting endogenous gene, cyclophilin B (CypB). Partial substitution of Lys by His moieties in stEK produced the descendent peptide, LKH-stEK, which is able to deliver low nM concentrations of the siRNA at a concentration of 2.5 μM and to promote a >90% gene knock-down of the target gene in HeLa cells. To demonstrate its use in an in vivo application, LKH-stEK was employed for delivery of siRNA to silence the activity of connective tissue growth factor (CTGF), a promising target for fibrotic skin disorders such as keloids.21 The results show that the use of this hydrocarbon stapled peptide in conjunction with the siRNA leads to efficient promotion of gene silencing in the mouse skin wound healing model.
Results and discussion
Design and synthesis of peptides for siRNA delivery
Recently, we demonstrated that the disulfide dimer peptide, LK-3 (Fig. 1c and S2a†), which consists of mainly Lys and Leu residues, has an efficient cell penetrating ability.22 Owing to its pico-molar affinity for hairpin RNA targets,23 LK-3 was explored as an agent for non-covalent delivery of siRNAs. Even though a fluorescence labeled siRNA is efficiently delivered into the cells (Fig. S2b†), we found that LK-3 does not display a high ability to promote gene silencing (only 23%) by an siRNA (Fig. S2c†). We reasoned that this result is likely a consequence of the extremely strong binding ability of LK-3, which prevents the efficient release of the siRNA (Fig. 1c (2)). This hypothesis, if correct, suggests that the efficient nucleic acid delivery potential of LK peptides17 would only be favorable if the level of release of “free” siRNA were increased.Incorporation of monomeric stapled peptides possessing positively charged moieties should lead RNA binding not only to make stable complexes outside cells but also to be able to release siRNA in cytosol (Fig. 1a and c (2)), while the cell penetrating abilities of peptides can be improved by utilizing the stapling technique. To assess this proposal, we prepared the positional specific stapled peptides (Fig. 1b and S3†), having amphipathic Leu- and Lys-rich regions. Unnatural amino acids, (R)-2-(7′octenyl)alanine (R8) and (S)-2-(4′pentenyl)-alanine (S5), were introduced on the hydrophobic face by replacing Leu at the i and i + 7 positions.24 For example, stAK has R8 at position 1 and S5 at position 8. Then, the second stapled peptide, stBK, which contains R8 at position 4 and S5 at position 11, is introduced. This pattern is used repeatedly to create other members of the series of isomeric stapled peptides. Moreover, stFK, containing R5 and S5 at Lys 9 and Gly 16, respectively, was prepared so that the effects of replacing one Lys residue for stapling on hydrophobicity can be assessed. Each of the required bis-alkene linked peptides employed in the preparative sequences was synthesized using solid phase techniques and then subjected to on-resin ring-closing olefin metathesis (RCM) using the previously described procedure.24
Firstly, the cell penetrating abilities of 5-TAMRA-labeled derivatives of the stapled peptides were determined using flow cytometry. Analysis of cellular fluorescence intensities revealed that the cell penetrating abilities are dependent on the position of the hydrocarbon cross-links. Among the stapled peptides, stEK displays the highest tendency for cell penetration as reflected in the observation that >70% of HeLa cells are fluorescent positive when incubated with only 10 nM 5-TAMRA-labeled stEK for 24 h (Fig. 2a). The stEK has a cell penetrating efficiency that is similar to that of the previously reported bis-disulfide dimer peptide, LK-3, and showed much higher cell penetration ability than that of the unstapled monomeric LK peptide.22
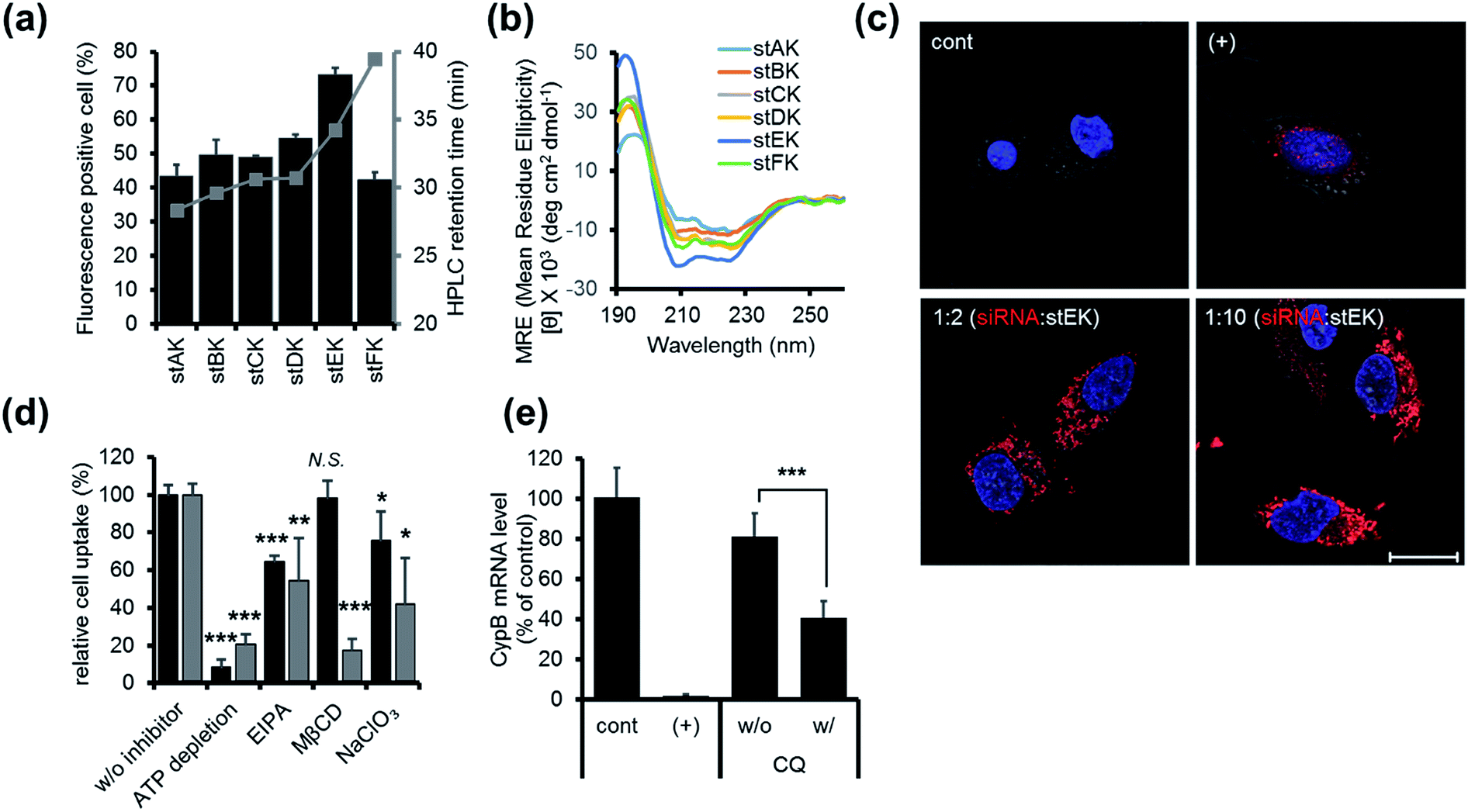 | ||
| Fig. 2 (a) Cell penetrating abilities of 10 nM of stapled peptides (mean ± s.d., n = 3) and their HPLC retention times in gray. (b) CD spectra of 20 μM of stapled peptides in 10 mM sodium phosphate (pH 7.4). (c) Confocal laser scanning microscopy (CLSM) images of HeLa cells transfected with 50 nM of DY-547 labeled siRNA (siGLO™) (red) for 24 h. Cell nuclei were stained with Hoechst 33342 (blue). DharmaFECT™ 1 was used as a positive (+) transfecting agent (scale bar, 20 μm). (d) Relative cell penetration of fluorescence labeled stEK (black) and stEK-siGLO™ complex (gray) in the presence and absence of endocytosis inhibitors (mean ± s.d., n = 3, N.S. not significant, *P < 0.5, **P < 0.01, ***P < 0.001 compared with the control w/o inhibitor). ATP depletion, 5-(N-ethyl-N-isopropyl)amiloride (EIPA) and sodium chlorate (NaClO3) were used to block the ATP-dependent pathway, macropinocytosis and proteoglycan-dependent endocytosis, respectively. Methyl-β-cyclodextrin (MβCD) was used to inhibit cholesterol associated membrane processes, including caveloin, lipid raft, and macropinocytosis. (e) In vitro gene silencing efficiency of siRNA–stEK complexes in HeLa cells. Cells were incubated with 50 nM of siRNA and 500 nM of stEK in the presence and absence of 120 μM of chloroquine (CQ). Relative mRNA expression levels were determined by RT-qPCR (mean ± s.d., n = 3, ***P < 0.001 compared with the control w/o CQ). | ||
Secondly, relative hydrophobicities of the peptides were determined by using HPLC retention times on a C18 reverse phase column (Fig. 2a). Interestingly, the hydrophobicities of these peptides are governed by the position of the hydrocarbon cross-link. Specifically, the closer the cross-link is to the C-terminus the more hydrophobic is the stapled peptide. Furthermore, the more hydrophobic peptides penetrate into cells more efficiently. However, replacement of Lys by an unnatural hydrophobic amino acid linker, as in the case of stFK, does not lead to an increase in the cell penetrating efficiency even though it does promote an increase in hydrophobicity.
Thirdly, the α-helicity of each stapled peptide was evaluated using circular dichroism (CD). The results show that stEK has the greatest α-helical propensity (Fig. 1b and 2b). In agreement with previous results,25 the combined results demonstrate that hydrophobicity and α-helicity, all of which are governed by the position of hydrophobic cross-links, are important factors determining cell penetrating efficiency. In addition, stEK, which has the highest hydrophobicity and helical propensity, displays the best cell penetrating ability in the series of stapled peptides evaluated in this effort.
In vitro siRNA delivery with hydrocarbon stapled peptides
The intracellular delivery of siRNA by stEK was examined by confocal microscopy. HeLa cells were treated with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and 1:10 molar complexes comprised of fluorescence-labeled siRNA and stEK. Fluorescence microscopy images confirmed that only a two-fold molar excess of stEK promotes siRNA delivery into cells.
:2 and 1:10 molar complexes comprised of fluorescence-labeled siRNA and stEK. Fluorescence microscopy images confirmed that only a two-fold molar excess of stEK promotes siRNA delivery into cells.
Initially, we explored the mechanism of cell penetrating peptide alone (Fig. 2d). The mechanistic probes of the best cell penetrating stEK showed that stEK gets into cells through both macropinocytosis (36% inhibition in the presence of EIPA) and sulfate proteoglycan-mediated endocytosis (25% inhibition in the presence of NaClO3). Based on previous studies26,27 we tested if CPP mediated nucleic acid internalization proceeds by proteoglycan-dependent macropinocytosis and lipid rafts. Indeed, delivery of the siRNA–stEK complex is still dependent on macropinocytosis (45% inhibition) as shown in stEK delivery. Surprisingly, some differences between the cell invasion features of stEK and siRNA–stEK complex do exist. Specifically, intracellular delivery of siRNA promoted by the siRNA–stEK complex is decreased by 58% and 82% using NaClO3 and MβCD, respectively. These findings suggest that, in contrast to that of stEK, siRNA delivery by stEK is more dependent on binding to sulfated cell surface proteoglycans and cholesterol associated membrane deformation pathways including macropinocytosis.
Next, the ability of stEK to promote gene silencing by an siRNA was examined by using RT-qPCR (Fig. 2e). We have chosen the validated siRNA targeting CypB, an abundant gene but it is known that its knockdown has no effect on cell survival and growth. Disappointingly, the siRNA–stEK complex brings about only 20% gene silencing at 1:10 molar ratio of siRNA:stEK, in spite of its excellent cell penetrating ability. To determine if endosomal entrapment of the complex is the source of the low efficiency, we tested whether co-treatment with the endosomal destabilization reagent, chloroquine (CQ), increases the level of gene knock-down.28 We found that the expression of the target gene is reduced significantly (60% inhibition) by co-treatment with CQ. This result shows that endosome escape of the siRNA complex by different stapled peptides should be a feasible approach to overcoming the inefficiency of gene silencing (Fig. 1c (3)).
Preparation and evaluation of second-generation stapled peptides for siRNA delivery
Because the partial replacement of Lys by His residues leads to higher gene knock-down efficacy in cationic polymers,20,29 the His containing derivatives of stEK, LKH-stEK, LKH-stEK-1, and LKH-stEK-2 (Fig. 3a and S4a†), were prepared. The respective His residues in LKH-stEK and LKH-stEK-1 are located at i and i + 3 (or i + 4) and i and i + 7 (or i + 4) positions that reside on the same face of the helix, respectively, while all the His residues in LKH-stEK-2 are located on the N-terminus. In order to gain information about the influence of His on the enhancement of siRNA gene silencing, two non-canonical amino acids were incorporated into stEK. First, 3-pyridyl-ala was introduced at the same positions as His residues of LKH-stEK to generate LKpyr-stEK (Fig. 3a). Because its conjugate acid has a pKa of 3.28, the pyridine group in LKpyr-stEK should be deprotonated even in the maturated acidic late endosomes (pH ∼ 5.5).30 In addition, acridinylated Lys moieties (pKa of 9.03)31 were incorporated in place of His to form LKacr-stEK, which enables us to determine if intercalating effects enhance siRNA gene silencing through increasing the stability of the CPP/siRNA complex.32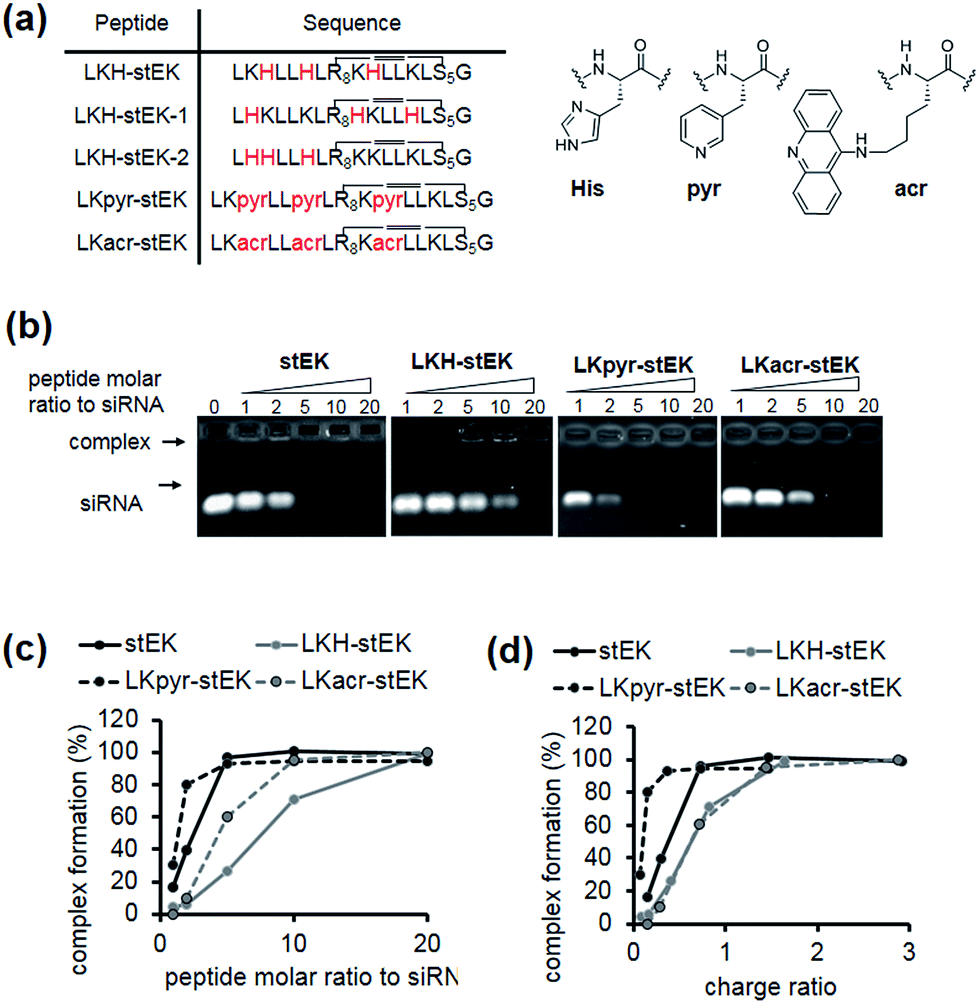 | ||
| Fig. 3 (a) Sequences of aromatic amino acids containing stapled peptides. (b) Gel retardation assay of siRNA/stEK or aromatic amino acid derivative complexes. The preformed siRNA/CPP complexes were analyzed by electrophoresis on 1% of agarose gel stained with ethidium bromide. siRNA was pre-incubated with peptides for 30 min in 0.5× PBS buffer. (c) Dose-dependent complex formation was determined by assessing unbound band intensities. Plot of siRNA complex formation against the molar ratio of stEK and LKH-stEK peptide shown in (b). (d) Plot of siRNA complex formation against charge ratio (+/−) shown in (b). The respective net charge of each peptide at pH 7.4 was calculated to be 5.99, 3.37, 3.01, and 5.97 for stEK, LKH-stEK, LKpyr-stEK, and LKacr-stEK, respectively using MarvinView software package (free software from ChemAxon). | ||
The abilities of these peptides to form stable complexes with siRNA were evaluated by using a gel retardation assay (Fig. 3b). The results show that a 5 molar excess of stEK brings about complete complexation of siRNA whereas 20 molar excess of LKH-stEK is necessary to promote complete complexation (Fig. 3b and c). However, the (+/−) charge ratio on the peptide to siRNA is only 1.6 in the latter complex because the His containing peptide LKH-stEK has a low net charge (N = 3.37) at pH 7.4 (Fig. 3d). In contrast, stEK undergoes complete complexation with siRNA with a charge ratio of less than one. Thus, this peptide has a strong RNA binding affinity that is not only derived from charge–charge interactions but also from specific hydrogen bonding and other interactions.17 LKpyr-stEK shows the highest propensity for complex formation, followed by stEK and LKacr-stEK, and the His containing LKH-stEK displays the lowest tendency for complex formation.
Next, we determined the siRNA–peptide complex uptake efficiency of these peptides using flow cytometry (Fig. 4a). The data, in the form of plots of siRNA uptake efficiency vs. complex formation efficiency (Fig. 3b and c), show that siRNA uptake efficiency is directly proportional to the efficiency of complex formation (Fig. 4b).
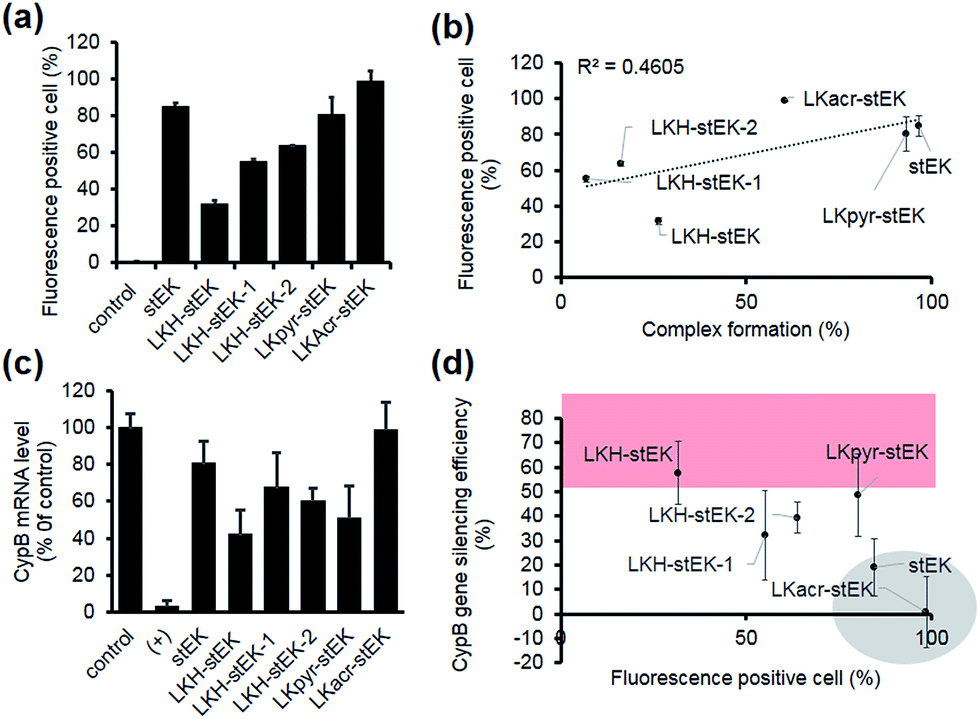 | ||
| Fig. 4 (a) siRNA cell uptake efficiency. Fluorescence positive cell (%) was measured using flow cytometry. HeLa cells were incubated with siRNA/stapled peptide complexes formed with a 1:10 molar ratio (50 nM of siGLO, mean ± s.d., n = 3). (b) Relationship between the siRNA cell uptake and complex formation efficiency at 1:10 molar ratio. The dotted line is a trend line. R2 (squared correlation coefficient) and the linear regression formula are shown. (c) Relative mRNA expression of the target gene transfected using siRNA/other stapled peptide complexes formed with a 1:10 molar ratio (50 nM of siRNA). HeLa cells were transfected and incubated for 24 h (mean ± s.d., n = 3). (d) Relationship between siRNA cell uptake and in vitro gene silencing efficiency of siRNA-peptide complexes in HeLa cells. Data for LKacr-stEK and stEK and those for a group showing gene silencing efficiency with over 50% are indicated in a gray circle and a red box, respectively. | ||
Target gene silencing was then used to determine the transfection efficiencies of complexes comprised of siRNA and each peptide (Fig. 4c). The results show that LKH-stEK promotes the most efficient silencing (>60% knock-down of the target gene) determined by using RT-qPCR. LKpyr-stEK, which most efficiently forms a complex with siRNA (Fig. 3b–d), displays the next highest silencing efficiency. All His containing derivatives of stEK, LKH-stEK, LKH-stEK-1, and LKH-stEK-2, have greater abilities to promote target gene silencing relative to stEK. Among these isomeric peptides, LKH-stEK displays higher efficiencies than LKH-stEK-1 and LKH-stEK-2 do (Fig. 4c and S4c†). This finding demonstrates that the introduction of His promotes functional siRNA delivery and that the effect on gene silencing efficiency is dependent on the positions of the His residue.
In contrast, the gene knock-down efficiency of siRNA does not correlate with its uptake efficiency (Fig. 4d). For example, LKacr-stEK and stEK display higher siRNA uptake efficiencies but lower gene knock-down efficiencies than other peptides (Fig. 4d, shown in a gray circle). A quantitative analysis of gene silencing showed that target gene expression does not decrease as a function of a decrease in the molar ratio of LKacr-stEK (Fig. S5a†), irrespective of the strong ability of acridine moieties to intercalate with nucleic acids.31 Also, LKpyr-stEK does not have a molar ratio dependent siRNA transfection efficiency, although its gene silencing efficiency is maximized at 1:5 molar ratio (Fig. S5b†). The results indicate that these peptides bind with siRNA too strongly to release siRNA to promote target gene knock-down. Therefore, although it is important to optimize cell penetrating efficiencies and complex formation abilities of CPPs needed for the required amount of intracellular siRNA transport, intracellular release of siRNA is the key determinant in functional siRNA delivery.
Finally, we chose LKH-stEK as the best siRNA delivery peptide as a result of the gene silencing efficiency. Studies of the gene silencing as a function of molar ratio and concentration show that >90% knock-down is achieved by using a 1:50 molar ratio (Fig. 5a) obtained by mixing 50 nM of siRNA and a 50 molar excess of LKH-stEK (Fig. 5b). Western blot analysis confirmed that LKH-stEK delivered siRNA targeting CypB and that target protein expression decreases (Fig. 5c). The data also showed that the transfection efficiency of siRNA delivered using LKH-stEK is comparable to that of siRNA delivered using the cationic lipid, DharmaFECT™ 1. Mechanism study proves that delivery of the siRNA–LKH–stEK complex is still dependent on macropinocytosis (18% inhibition in the presence of EIPA), proteoglycan-dependent endocytosis (50% inhibition in the presence of NaClO3), and cholesterol associated processes (75% inhibition in the presence of MβCD) (Fig. 5d), which is similar to that of siRNA–stEK. Furthermore, the knock-down efficiency promoted by LKH-stEK is not significantly altered by the presence or absence of CQ (Fig. 5e). These results confirm that LKH-stEK not only efficiently delivers siRNA into cells through the same cell uptake mechanisms with the best cell penetrating stEK but also promotes endosomal escape for functional siRNA delivery.
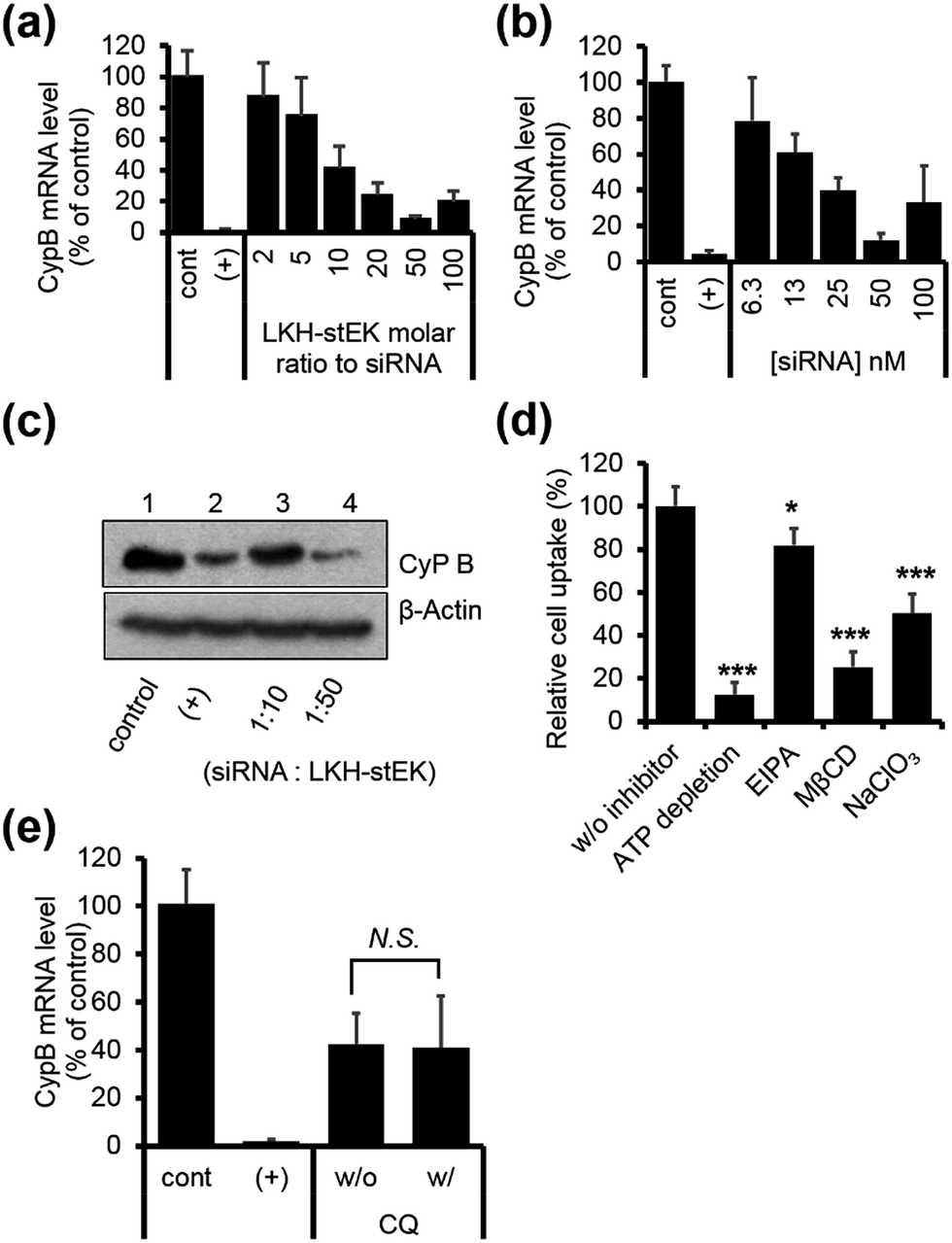 | ||
| Fig. 5 (a) HeLa cells were transfected using siRNA and LKH-stEK complexes formed with different molar ratios from 1:2 to 1:100 (50 nM of siRNA) and incubated for 24 h (mean ± s.d., n = 3). (b) Concentration dependent target mRNA expression in a ratio of 1:50 (siRNA:LKH-stEK, mean ± s.d., n = 3). (c) Target protein repression was confirmed by using western blot. (d) Relative cell penetration of LKH-stEK-siGLO™ complex in the presence and absence of endocytosis inhibitors (mean ± s.d., n = 3, *P < 0.5, **P < 0.01, ***P < 0.001 compared with the control w/o inhibitor). (e) In vitro gene silencing efficiency of siRNA/LKH-stEK complex in HeLa cells in the presence and absence of 120 μM of CQ. Cells were incubated with 50 nM of siRNA and 500 nM of LKH-stEK for 24 h. The mRNA expression levels of CypB were measured using qRT-PCR (mean ± s.d., n = 3, N.S., not significant). | ||
The combined observations suggest that the efficiency of peptide promoted siRNA gene silencing is affected by the complex formation efficiency, by siRNA release utilized via moderate binding with siRNA, and by the efficiency of siRNA endosomal escape. Although strong complex formation efficiency might affect gene silencing in the sense that it induces cell penetration at lower concentrations, its effect is less than that of releasing siRNA from the complex and facile endosomal escape of siRNA.
In vivo application of LKH-stEK to deliver asiCTGF
Considerable efforts have been made recently to develop cell permeable siRNAs using a nucleotide modification strategy. As part of the efforts, we developed the cell-permeable siRNA, cp-asiCTGF.21 Even though several studies including our previous report have demonstrated the viability of intradermal injection of naked oligonucleotides,34–36 observations made in these investigations show that high doses in the range of μg to mg are required to elicit observable target repression by cell permeable siRNAs.21,37,38 A basic goal of the current investigation is to show that cell penetrating peptides would enhance the delivery potential of target siRNAs.The findings summarized above led to the selection of LKH-stEK as an optimized peptide to probe this proposal. The results of preliminary cell viability studies showed that LKH-stEK is not cytotoxic in the range of concentrations employed in this effort (Fig. S6†) and also applicable to various other genes (Fig. S7†). Because the long range aim of this in vivo animal study was to develop a therapeutic agent for treatment of tissue fibrosis, the stapled peptide, LKH-stEK, was utilized to deliver the siRNA targeting CTGF, asiCTGF. Based on the observations, which show that a 1:50 molar ratio of siRNA to LKH-stEK brings about maximum gene silencing, a 50-fold molar excess of LKH-stEK and only 10 nM and 100 nM siRNA were employed (Fig. 6).39 These amounts of siRNA correspond to 6 and 60 ng per injection, which are much lower amounts than those of normally administered siRNAs for in vivo applications.21,34,36–38 Comparing the results with the 100 nM of cell permeable siRNA (cp-asiCTGF), 10 nM of cell impermeable siRNA was efficiently delivered by LKH-stEK. After 1 and 3 d following intradermal injection of asiCTGF in complexes with LKH-stEK, skin biopsy samples were subjected to RT-qPCR analysis. The results show that gene silencing of asiCTGF promoted by LKH-stEK peptide displayed 49–58% repression of CTGF. In addition, the gene silencing effect of asiCTGF with LKH-stEK was retained up to 3 d after injection.
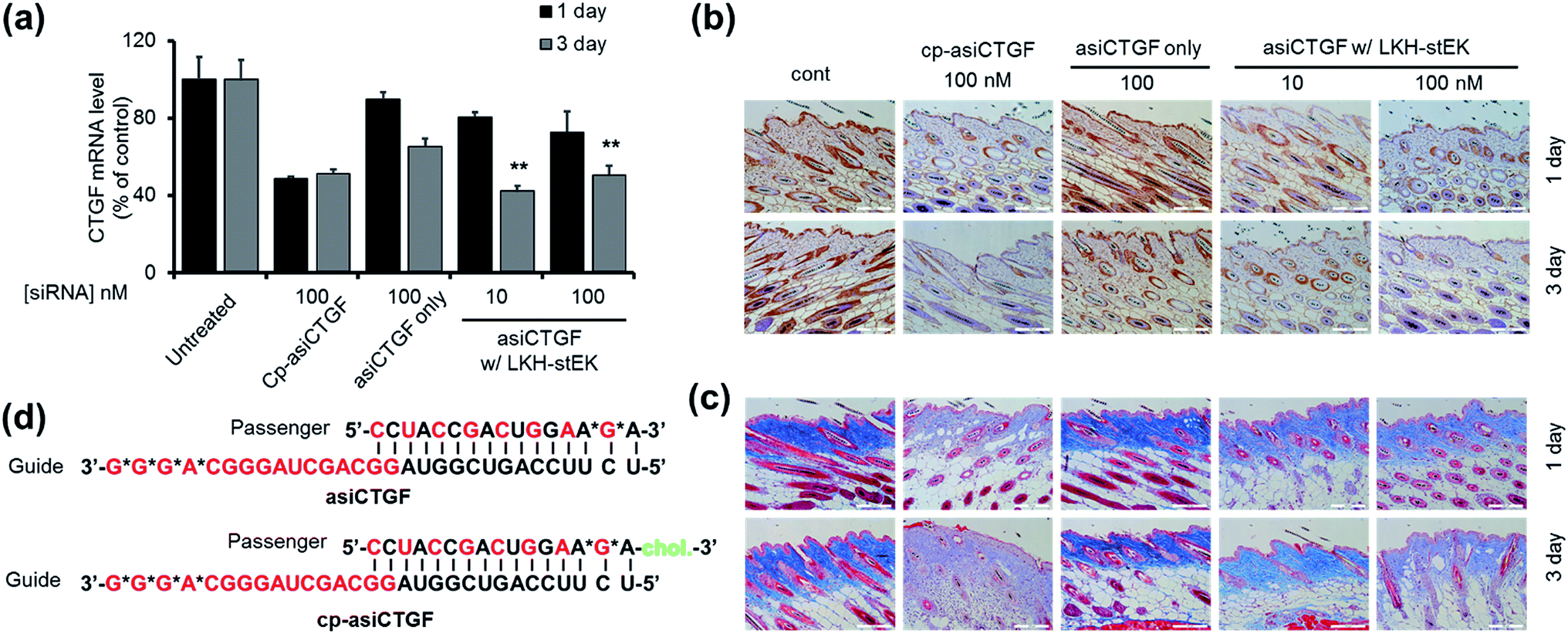 | ||
| Fig. 6 In vivo analysis of asiCTGF using LKH-stEK. (a) Repression of target mRNA by asiCTGF delivered by LKH-stEK in mouse skin through intradermal injection, 0.4 pmoles (10 nM) and 4 pmoles (100 nM) per injection. After 1 or 3 days, total RNA extracted from the skin of treated mice (n = 5/group) was reverse transcribed, and CTGF mRNA levels were quantified by using qRT-PCR. Mouse CTGF levels were normalized to GAPDH levels (mean ± s.e.). The marker (**) indicates P < 0.01 compared with the siRNA only groups. (b) Repression of CTGF protein expression was examined by immunohistochemistry on day 3. CTGF protein was stained with brown color. Magnification ×200. (c) Collagen deposition represented through Masson's Trichrome staining. Magnification ×200. (d) Schematic presentation of duplex asiCTGF and cp-asi-CTGF. Red characters indicate 2′-O-methyl modification and asterisks indicate phosphorothioate modification. Cholesterol modification is shown in green. Animal studies were carried out in accordance with the 8th edition of the Guide for the Care and Use of Laboratory Animals (National Research Council, 2011), and the protocol was approved by the Institutional Animal Care and Use Committee (IACUC) at the Hugel, Inc. | ||
Immunohistochemistry and Masson's Trichrom staining experiments also demonstrate that LKH-stEK efficiently delivers asiCTGF and that consequently repression of both CTGF protein and collagen deposition takes place (Fig. 6b and c). Because CTGF plays a role in promoting collagen synthesis,40 we showed that asiCTGF delivered at nano-molar concentrations using a 50-fold excess of LKH-stEK significantly reduces keloid disorder. The use of siRNA and LKH-stEK complexes in vivo still needs to be optimized before the full potential of therapeutic siRNA is realized. However, the study described above has demonstrated the possibility of using cell penetrating peptides in vivo for this purpose.
Conclusions
In the study described above, we demonstrated that the hydrocarbon stapling technology can be employed to enhance delivery of an siRNA by an amphipathic α-helical peptide, which binds to RNA double strands. To prepare the stapled peptides, two alkene-bearing non-natural amino acids were introduced into the amphipathic LK peptide by replacement of two hydrophobic Leu residues. Ring closure metathesis reaction was then used to construct hydrocarbon cross-links. The stapled amphipathic α-helical peptides were observed to have higher cell penetrating abilities as compared to the unstapled LK peptide. Among the stapled peptides, stEK was found to display the highest cell penetrating ability, which is comparable to that of the previously studied dimeric bundle LK-3 peptide. However, the siRNA silencing efficiency of the target gene promoted by stEK is low (20%). This observation suggested that even though stEK delivers siRNA into cells via endocytosis, the delivered siRNA is entrapped in the endosome. In contrast, LKH-stEK, the His-containing descendent of stEK, facilitates endosomal escape of the siRNA and, consequently, promotes >90% gene silencing. Taken together, these results indicate that in designing cell penetrating peptides for siRNA delivery, one needs to consider not only the cell penetrating ability of the peptide but also the functional siRNA delivery of the siRNA–peptide complex. In other words, the peptide needs to have a moderate binding affinity to the siRNA, so that the peptide–siRNA complex is able to release the siRNA in the endosome, and the siRNA can undergo endosomal escape so that it can function in the cytosol. Finally, the results of an in vivo study demonstrated that LKH-stEK efficiently assists asiCTGF promoted knock-down in an animal model by enabling nanomolar dose of asiCTGF. This is the first observation of a His containing stapled peptide that is capable of efficiently delivering siRNA in vitro and in vivo. The findings suggest that LKH-stEK is a promising candidate to promote siRNA gene knock-down for therapeutic purposes. Considering the tunable molar ratio of siRNA by simple mixing with LKH-stEK to optimize the efficiency, it is expected that LKH-stEK is easily applicable to various unmodified cell impermeable siRNAs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported financially by the National Research Foundation of Korea (NRF-2015R1C1A1A02037640 to S. H. and NRF-2017R1A2B3003750 and NRF-2017M3A9E4077444 to J. Y.) and by the Ministry of Health & Welfare (grants of the Korea Health Technology R&D Project, HI15C2878 to Y. L. and HI17C2052 to J. Y.).References
- R. W. Carthew and E. J. Sontheimer, Cell, 2009, 136, 642–655 CrossRef CAS PubMed.
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS PubMed.
- D. R. Corey, J. Clin. Invest., 2007, 117, 3615–3622 CrossRef CAS PubMed.
- D. M. Kenski, G. Butora, A. T. Willingham, A. J. Cooper, W. Fu, N. Qi, F. Soriano, I. W. Davies and W. M. Flanagan, Mol. Ther.–Nucleic Acids, 2012, 1, e5 CrossRef PubMed.
- N. T. Schirle, G. A. Kinberger, H. F. Murray, W. F. Lima, T. P. Prakash and I. J. MacRae, J. Am. Chem. Soc., 2016, 138, 8694–8697 CrossRef CAS PubMed.
- G. F. Deleavey, J. K. Watts and M. J. Damha, Curr Protoc Nucleic Acid Chem, 2009, ch. 16, Unit 16 13 Search PubMed.
- A. de Fougerolles, H. P. Vornlocher, J. Maraganore and J. Lieberman, Nat. Rev. Drug Discovery, 2007, 6, 443–453 CrossRef CAS PubMed.
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Nat. Mater., 2013, 12, 967–977 CrossRef CAS PubMed.
- I. Nakase, H. Akita, K. Kogure, A. Graslund, U. Langel, H. Harashima and S. Futaki, Acc. Chem. Res., 2012, 45, 1132–1139 CrossRef CAS PubMed.
- A. van den Berg and S. F. Dowdy, Curr. Opin. Biotechnol., 2011, 22, 888–893 CrossRef CAS PubMed.
- R. H. Mo, J. L. Zaro and W. C. Shen, Mol. Pharm., 2012, 9, 299–309 CrossRef CAS PubMed.
- J. G. van den Boorn, M. Schlee, C. Coch and G. Hartmann, Nat. Biotechnol., 2011, 29, 325–326 CrossRef CAS PubMed.
- C. Zhang, N. Tang, X. Liu, W. Liang, W. Xu and V. P. Torchilin, J. Controlled Release, 2006, 112, 229–239 CrossRef CAS PubMed.
- J. Wang, Z. Lu, M. G. Wientjes and J. L. Au, AAPS J., 2010, 12, 492–503 CrossRef CAS PubMed.
- M. Gooding, L. P. Browne, F. M. Quinteiro and D. L. Selwood, Chem. Biol. Drug Des., 2012, 80, 787–809 CAS.
- H. Margus, K. Padari and M. Pooga, Mol. Ther., 2012, 20, 525–533 CrossRef CAS PubMed.
- S. J. Lee, S. Hyun, J. S. Kieft and J. Yu, J. Am. Chem. Soc., 2009, 131, 2224–2230 CrossRef CAS PubMed.
- G. L. Verdine and G. J. Hilinski, Methods Enzymol., 2012, 503, 3–33 CAS.
- L. D. Walensky and G. H. Bird, J. Med. Chem., 2014, 57, 6275–6288 CrossRef CAS PubMed.
- P. Midoux, C. Pichon, J. J. Yaouanc and P. A. Jaffres, Br. J. Pharmacol., 2009, 157, 166–178 CrossRef CAS PubMed.
- J. Hwang, C. Chang, J. H. Kim, C. T. Oh, H. N. Lee, C. Lee, D. Oh, C. Lee, B. Kim, S. W. Hong and D. K. Lee, J. Invest. Dermatol., 2016, 136, 2305–2313 CrossRef CAS PubMed.
- S. Jang, S. Hyun, S. Kim, S. Lee, I. S. Lee, M. Baba, Y. Lee and J. Yu, Angew. Chem., Int. Ed., 2014, 53, 10086–10089 CrossRef CAS PubMed.
- S. Hyun, J. Na, S. J. Lee, S. Park and J. Yu, ChemBioChem, 2010, 11, 767–770 CrossRef CAS PubMed.
- Y. W. Kim, T. N. Grossmann and G. L. Verdine, Nat. Protoc., 2011, 6, 761–771 CrossRef CAS PubMed.
- G. H. Bird, E. Mazzola, K. Opoku-Nsiah, M. A. Lammert, M. Godes, D. S. Neuberg and L. D. Walensky, Nat. Chem. Biol., 2016, 12, 845–852 CrossRef CAS PubMed.
- K. A. Mislick and J. D. Baldeschwieler, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 12349–12354 CrossRef CAS.
- S. Sandgren, A. Wittrup, F. Cheng, M. Jonsson, E. Eklund, S. Busch and M. Belting, J. Biol. Chem., 2004, 279, 17951–17956 CrossRef CAS PubMed.
- R. M. Steinman, I. S. Mellman, W. A. Muller and Z. A. Cohn, J. Cell Biol., 1983, 96, 1–27 CrossRef CAS PubMed.
- A. Erazo-Oliveras, N. Muthukrishnan, R. Baker, T. Y. Wang and J. P. Pellois, Pharmaceuticals, 2012, 5, 1177–1209 CrossRef CAS PubMed.
- J. Huotari and A. Helenius, EMBO J., 2011, 30, 3481–3500 CrossRef CAS PubMed.
- Y. Lee, S. Hyun, H. J. Kim and J. Yu, Angew. Chem., Int. Ed., 2008, 47, 134–137 CrossRef CAS PubMed.
- K. Zhou, P. Kos, Y. Yan, H. Xiong, Y. L. Min, K. A. Kinghorn, J. T. Minnig, J. B. Miller and D. J. Siegwart, Chem. Commun., 2016, 52, 12155–12158 RSC.
- S. M. Liao, Q. S. Du, J. Z. Meng, Z. W. Pang and R. B. Huang, Chem. Cent. J., 2013, 7, 44 CrossRef PubMed.
- V. Hegde, R. P. Hickerson, S. Nainamalai, P. A. Campbell, F. J. Smith, W. H. McLean and D. M. Pedrioli, J. Controlled Release, 2014, 196, 355–362 CrossRef CAS PubMed.
- R. P. Hickerson, M. A. Flores, D. Leake, M. F. Lara, C. H. Contag, S. A. Leachman and R. L. Kaspar, J. Invest. Dermatol., 2011, 131, 1037–1044 CrossRef CAS PubMed.
- E. Gonzalez-Gonzalez, H. Ra, R. P. Hickerson, Q. Wang, W. Piyawattanametha, M. J. Mandella, G. S. Kino, D. Leake, A. A. Avilion, O. Solgaard, T. C. Doyle, C. H. Contag and R. L. Kaspar, Gene Ther., 2009, 16, 963–972 CrossRef CAS PubMed.
- C. M. Lin, K. Huang, Y. Zeng, X. C. Chen, S. Wang and Y. Li, Arch. Dermatol. Res., 2012, 304, 139–144 CrossRef CAS PubMed.
- Y. Deng, J. Chen, Y. Zhao, X. Yan, L. Zhang, K. Choy, J. Hu, H. J. Sant, B. K. Gale and T. Tang, Sci. Rep., 2016, 6, 21422 CrossRef CAS PubMed.
- The discrepancy of the sequence from the previous paper (ref. 21) occurs from the species, human and mouse siRNA targeting CTGF.
- Y. T. Khoo, C. T. Ong, A. Mukhopadhyay, H. C. Han, D. V. Do, I. J. Lim and T. T. Phan, J. Cell. Physiol., 2006, 208, 336–343 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, peptide synthesis and characterization, siRNA delivery of LK-3, helical wheel diagrams of stapled peptides, siRNA complex formation & delivery efficiency of stapled peptides, cytotoxicity test results, and various siRNA delivery efficiencies of LKH-stEK. See DOI: 10.1039/c8sc00074c |
| This journal is © The Royal Society of Chemistry 2018 |