
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Topotactic anion-exchange in thermoelectric nanostructured layered tin chalcogenides with reduced selenium content†
Guang
Han
a,
Srinivas R.
Popuri
b,
Heather F.
Greer
c,
Ruizhi
Zhang
d,
Lourdes
Ferre-Llin
e,
Jan-Willem G.
Bos
 b,
Wuzong
Zhou
c,
Michael J.
Reece
d,
Douglas J.
Paul
e,
Andrew R.
Knox
e and
Duncan H.
Gregory
*a
b,
Wuzong
Zhou
c,
Michael J.
Reece
d,
Douglas J.
Paul
e,
Andrew R.
Knox
e and
Duncan H.
Gregory
*a
aWestCHEM, School of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: Duncan.Gregory@glasgow.ac.uk
bInstitute of Chemical Sciences, Centre for Advanced Energy Storage and Recovery, School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK
cEaStCHEM, School of Chemistry, University of St Andrews, St Andrews, Fife KY16 9ST, UK
dSchool of Engineering & Materials Science, Queen Mary University of London, London, E1 4NS, UK
eSchool of Engineering, University of Glasgow, Glasgow, G12 8LT, UK
First published on 23rd March 2018
Abstract
Anion exchange has been performed with nanoplates of tin sulfide (SnS) via “soft chemical” organic-free solution syntheses to yield layered pseudo-ternary tin chalcogenides on a 10 g-scale. SnS undergoes a topotactic transformation to form a series of S-substituted tin selenide (SnSe) nano/micro-plates with tuneable chalcogenide composition. SnS0.1Se0.9 nanoplates were spark plasma sintered into phase-pure, textured, dense pellets, the ZT of which has been significantly enhanced to ≈1.16 from ≈0.74 at 923 K via microstructure texturing control. These approaches provide versatile, scalable and low-cost routes to p-type layered tin chalcogenides with controllable composition and competitive thermoelectric performance.
Introduction
Main group metal chalcogenides (MCs) are excellent candidates for thermoelectrics,1–4 (opto)electronics5 and photovoltaics6 due to their outstanding electronic, optical and thermal properties. Bottom-up solution syntheses afford energy-saving means of preparing MC nano/micro-structures with controllable morphology and size. Surface modification using organic surfactants can limit particle growth but such coatings can typically introduce impurities.7–10 By contrast, synthesis without organic surfactants, solvents or precursors can produce nanostructured MCs with impurity-free surfaces and enhanced electrical performance11–14 but require careful experiment design with appropriate synthesis parameters and reagents.Chemical transformations, including ion exchange, topotactic and pseudomorphic reactions, represent a versatile and effective means to produce new materials with control over crystal structure, composition and morphological complexity.15 Such transformations can realise prescribed materials that cannot be otherwise prepared.15–17 Indeed, doped compounds, multi-component composites or hetero-structures can be crafted by regulating the progress of transformations, leading to materials with engineered functional properties.18–21 When performed in solution, ion exchange can exploit the solubility difference between precursors and products to enable the rapid synthesis of nano/micro-structures (for example, MCs) with predetermined cation and/or anion compositions.15,18 Combining organic-free synthesis with ion exchange raises the prospect of producing MCs with compositions, crystal structures, morphologies and particle sizes that can be tailored towards delivering high electronic performance.
Thermoelectric materials can be utilised to convert thermal energy directly into electricity and vice versa, thus offering opportunities to refrigerate and to harvest electricity from waste heat via the Peltier and Seebeck effects, respectively.22,23 Layered tin chalcogenides (LTCs), including SnSe and SnS, have drawn much attention given a formidable combination of excellent thermoelectric conversion efficiency, relatively low toxicity and the Earth-abundance of their component elements.1,2,24–26 Notably, when p-type SnSe can be grown as a single crystal, it has demonstrated record high ZT values of 2.6 and 2.3 along the b and c crystallographic directions, respectively at 923 K.1 Polycrystalline SnSe and related doped materials have been prepared in an effort to improve mechanical properties, but ZT values cannot yet emulate those in the single crystalline material.27 The capacity to synthesise polycrystalline LTCs of premeditated composition to optimise thermoelectric performance is becoming gradually less elusive.28–50 For example, Ag-doped SnSe,28 alkali metal-doped SnSe,29–34 I-doped SnSe1−xSx (0 ≤ x ≤ 1),35 and Sn1−xPbxSe36 have demonstrated improvements in thermoelectric performance compared to undoped SnSe, pushing ZT to 1.2 at 773 K (Na, K co-doped SnSe)33 and ∼1.7 at 873 K (phase-separated Sn1−xPbxSe).36 Yet, polycrystalline LTCs are primarily fabricated by high-temperature, energy-intensive processes.27–29,31,32,35 Solution syntheses are an attractive alternative but generally involve using organics (solvents and/or surfactants, for example), can produce small sample yields and have offered little opportunity as yet to exert control over composition.51–60 For LTCs to be a practicable component of thermoelectric devices, a scalable and cost-effective organic-free synthesis approach to materials with tuneable composition and consistently excellent performance is essential.
In this study, we demonstrate how the combination of two organic-free aqueous solution strategies (anion exchange following direct precipitation) can be utilised to synthesise LTC nano/micro-plates with tuneable chalcogenide composition (e.g. >10 g SnS and SnS0.1Se0.9, respectively; Fig. S1 and S2†). The plates can be sintered into textured, dense pellets with competitive thermoelectric performance while partly replacing selenium with less toxic and more Earth-abundant sulfur.
Results and discussion
Characterisation and formation mechanism of anion-exchanged SnS1−xSex
The synthesis of phase-pure SnS (Fig. 1) involves injection of a Na2S aqueous solution into a Na2SnO2 solution that is subsequently boiled for 2 h (Fig. S1†). Powder X-ray diffraction (PXD) patterns (Fig. 1a) can be indexed exclusively to orthorhombic SnS (ICDD card no. 75-2115).61 Rietveld refinement against PXD data (Fig. S3; Tables S1 and S2†) confirms that the SnS product crystallises with orthorhombic space group Pnma; a = 11.2052(4) Å, b = 3.9877(2) Å and c = 4.3242(2) Å. Scanning electron microscopy (SEM) images (Fig. 1b and S4a–c†) reveal that the product predominantly forms flower-like nano/micro-structures that are composed of a series of plates, typically with a lateral size of 3–16 μm and a thickness of 80–650 nm. Imaging and electron diffraction of individual nanoplates were performed using transmission electron microscopy (TEM). Selected area electron diffraction (SAED) patterns collected with the incident beam perpendicular to an isolated plate (Fig. 1c) can be indexed to the [100] zone axis of SnS and confirm the single crystalline nature of the nanostructures. High resolution TEM (HRTEM) images (Fig. 1d) demonstrate a set of lattice spacings of 2.9 Å intersecting with an angle of 95(1)°, corresponding to the {011} planes of SnS. Energy dispersive X-ray spectroscopy (EDS) (Fig. 1e and S4d†) confirms Sn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S atomic ratios of 51(1):49(1). The organic-free SnS plates can be formed into dense pellets at 500 °C by either hot pressing or spark plasma sintering (SPS), each leading to strong texturing of the (h00) planes (Fig. S5 and S6†). The plate morphology, orthorhombic crystal structure and the overall Sn:S ratio are preserved on pressing (Fig. S7†). A pellet of the SnS material has an indirect optical bandgap of ca. 1.05 eV (Fig. S8†) and exhibits a negligible weight loss up to ca. 600 °C when heated under Ar gas, indicating excellent thermal stability (Fig. S9 and S10†).
:S atomic ratios of 51(1):49(1). The organic-free SnS plates can be formed into dense pellets at 500 °C by either hot pressing or spark plasma sintering (SPS), each leading to strong texturing of the (h00) planes (Fig. S5 and S6†). The plate morphology, orthorhombic crystal structure and the overall Sn:S ratio are preserved on pressing (Fig. S7†). A pellet of the SnS material has an indirect optical bandgap of ca. 1.05 eV (Fig. S8†) and exhibits a negligible weight loss up to ca. 600 °C when heated under Ar gas, indicating excellent thermal stability (Fig. S9 and S10†).
 | ||
| Fig. 1 Characterisation of SnS (a–e) and SnS0.1Se0.9 (a, e–h) nano/micro-plates: (a) PXD data; (b, f) SEM images; (c, g) TEM images of individual plates and corresponding SAED patterns along the [100] zone axis (inset); (d, h) HRTEM images of plates with d-spacings indicated, (e) EDS spectra collected from isolated plates. | ||
Thermoelectric measurements were performed on SnS pellets perpendicular to the pressing direction (Fig. 2). The pellet exhibits high electrical conductivity (σ), which rises from ca. 760 S m−1 at 323 K to ca. 2250 S m−1 at 523 K, gradually decreases to ca. 1475 S m−1 at 723 K and once again increases to ca. 1960 S m−1 at 773 K (Fig. 2a). These values are notably much higher than undoped SnS bulk counterparts (e.g. ∼5–32 S m−1 at 523 K) synthesised by mechanical alloying25 and high-temperature synthesis.26 Moreover, they also exceed those of pellets consolidated from solvothermally-synthesised SnS nanorods (e.g. ∼590 S m−1 at 523 K)62 and even Ag-doped SnS pellets (e.g. ∼530 S m−1 at 523 K).25 The high σ values of SnS pellets can be attributed to the organic-free surfaces of the plates, the orientation of the plates, high crystallinity and the high degree of densification (Fig. S5–S7†). The positive Seebeck coefficient (S) values for SnS pellets indicate p-type conducting behaviour (Fig. 2b). The Seebeck coefficient for SnS increases nearly linearly from ca. 285 μV K−1 at 323 K to ca. 455 μV K−1 at 723 K before decreasing to ca. 430 μV K−1 at 773 K. The combination of such high electrical conductivity and Seebeck coefficients leads to power factors (e.g. ∼0.33 mW m−1 K−2 at 573 K and ∼0.36 mW m−1 K−2 at 773 K) (Fig. 2c) that exceed those of previously reported undoped SnS pellets (e.g. ∼0.01–0.10 mW m−1 K−2 at 573 K).25,26,62 The value of thermal conductivity (κ) for SnS reduces from ≈1.918 W m−1 K−1 at 323 K to ≈0.786 W m−1 K−1 at 773 K (Fig. 2d), to which the lattice thermal conductivity (κL) is the main contributor (Fig. 2e). The ZT of a SnS pellet was thus calculated, increasing gradually from ca. 0.01 at 323 K to ca. 0.36 at 773 K (Fig. 2f). The ZT at 773 K is higher than the values reported for undoped SnS (e.g. 0.08–0.25 at 773 K) fabricated by various methods,25,26,62 indicating the significant potential of the organic-free method in synthesising high-performing metal chalcogenide thermoelectrics.
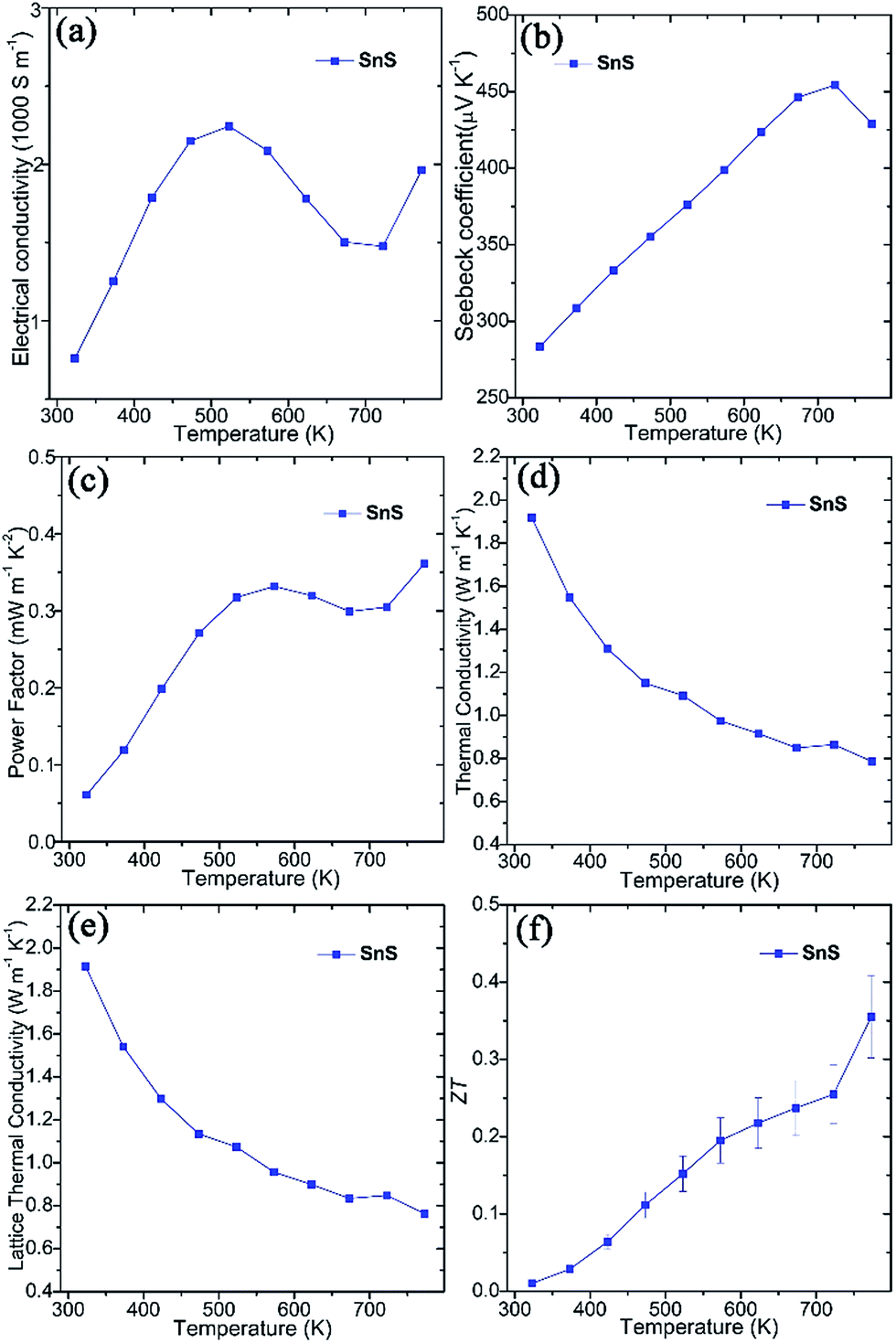 | ||
| Fig. 2 Thermoelectric properties of SnS pellets sintered using SPS, measured perpendicular to the pressing direction: (a) the electrical conductivity (σ), (b) the Seebeck coefficient (S), (c) the power factor (S2σ), (d) the thermal conductivity (κ), (e) the lattice κ (κL), and (f) ZT as a function of temperature. | ||
To demonstrate the utility of the organic-free strategy in obtaining a range of thermoelectrics with prescribed microstructure and tailored composition, we focused on the synthesis of substituted Sn(S,Se) materials, conscious that thermoelectric properties should improve at higher Se concentration.26,35 We achieved this via an organic-free anion exchange that involved injecting a NaHSe solution into the SnS suspension, followed by boiling for 2 h (Fig. S2†). Samples were prepared with NaHSe:Na2SnO2 in the appropriate molar ratios with the intention of preparing SnS1−xSex (0.5 ≤ x ≤ 1) (eqn (1) and (2)).
| Na2S + Na2SnO2 + 2H2O → SnS + 4NaOH | (1) |
| SnS + xNaHSe + xNaOH → SnS1−xSex + xNa2S + xH2O | (2) |
The anion exchange is likely driven by the lower solubility product (Ksp) of SnSe in water as compared to that of SnS.12,15 The PXD patterns of the anion-exchanged products ostensibly resemble that of orthorhombic SnS but with reflections shifted to lower 2θ (e.g. for x = 0.9 in Fig. 1a).61 SEM (Fig. 1f and S11a†) reveals that the product nano/micro- “flowers” consist of plates of approximately similar size (2–10 μm) and thickness (50–500 nm) to the original SnS plates. EDS spectra collected from both individual plates and clusters of plates in the 1:1, S:Se material consistently gave Sn:Se:S atomic ratios of 50(1):45(1):5(1) (Fig. 1e and S11b†). Rietveld refinement (Fig. S12; Tables S3 and S4†) confirms that the product crystallises with smaller cell parameters than SnSe (orthorhombic, Pnma; a = 11.4919(4) Å, b = 4.1507(2) Å, c = 4.4334(2) Å) with an anion site occupancy of 0.91(1):0.09(1) Se:S, corresponding to a composition close to SnS0.1Se0.9 and includes 1.4(1) wt% of SnS as a secondary phase. SAED patterns along the [100] zone axis (Fig. 1g) combined with HRTEM images (Fig. 1h) (which show lattice spacings of 3.0 Å with an intersection angle of 94(1)°, corresponding to the SnS0.1Se0.9 {011} plane spacings) identify preferred orientation along the 〈100〉 direction. Hence both the morphology and crystal structure before and after anion exchange are essentially unaltered and the process is topotactic (Fig. 3a).18
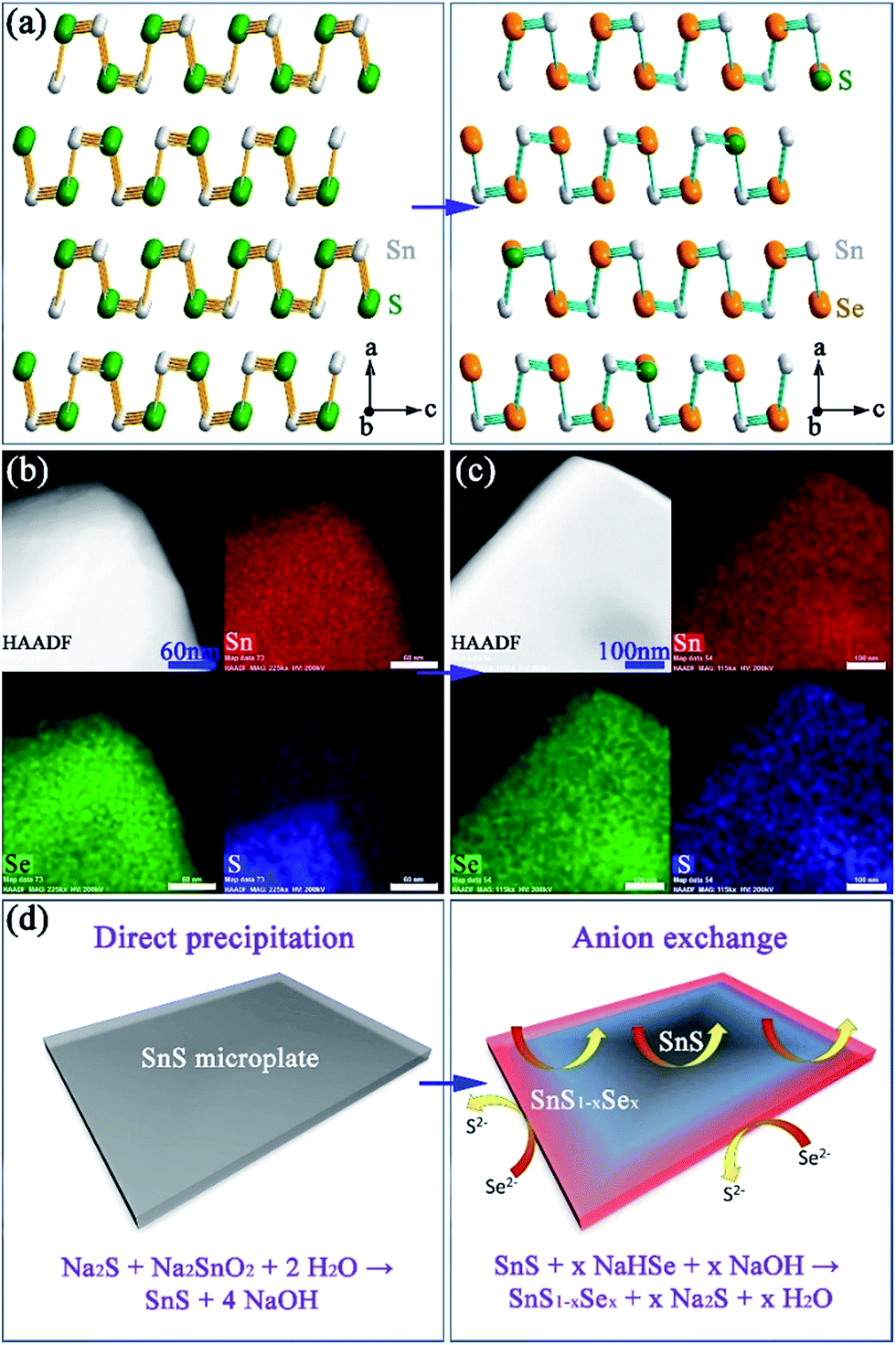 | ||
| Fig. 3 Structure, composition and formation mechanism of Sn(S,Se): (a) structural models of SnS and SnS0.1Se0.9; elemental mapping of isolated plates after anion exchange for (b) 1 min and (c) 2 h; (d) schematics of the initial precipitation and subsequent exchange processes. The four panels in (b) and (c) show the high angle annular dark field (HAADF) image of a plate and corresponding elemental maps for Sn (red), Se (green) and S (blue) respectively. | ||
In an effort to understand the formation mechanism of SnS0.1Se0.9, we investigated the outcome of an anion exchange reaction after only 1 min of boiling (using a Se:Sn molar ratio of 1). PXD shows that the product consists of both SnS1−xSex (x ≈ 0.9) and SnS (Fig. S13a†). SEM (Fig. S13b and c†) reveals clusters of nano/micro-plates, indicating again the retention of the SnS morphology. EDS (Fig. S13d†) gives an overall Sn:Se:S atomic ratio of 50(1):33(1):17(1) across the clusters, implying incomplete anion exchange. EDS element mapping (Fig. 3b) of an isolated plate from the 1 min synthesis reveals an uneven distribution of Se and S where the plate edges are much richer in Se than S (Se:S ratio of 49:1; Fig. S14†). Conversely the inner sections contain more S than Se (Se:S ratio of 23:27; Fig. S14†). By comparison, systematic elemental mapping of the product boiled for 2 h illustrates that ca. 50% of plates show an almost uniform distribution of Se and S (Fig. 3c), while the remainder demonstrate an uneven anion distribution. With Se:S ratios varying from 48(1):2(1) to 42(1):8(1) (Fig. S15†) the plates in the 2 h anion exchange sample are clearly much richer in Se than the 1 min product. Given the above observations, one can propose a formation process for the SnS1−xSex plates (Fig. 3d). The anion exchange should initiate at the edges and faces of plates. The periphery of a plate can access Se2− from both the edges and faces and therefore should be richer in Se than the centre. For thicker plates the contribution from anion exchange at the edges should become more pronounced. It is useful to note at this point that both the plate morphology and orthorhombic crystal structure are maintained after the subsequent high temperature sintering (e.g. at 500 °C) by either hot pressing or spark plasma sintering (SPS). Moreover, the overall Se:S ratio remains constant and the distribution of S and Se becomes more even across each plate (Fig. 4a–d).
 | ||
| Fig. 4 Characterisation of SnS0.1Se0.9 pellets: (a) TEM image of a SnS0.1Se0.9 plate peeled from a pellet and its corresponding SAED pattern along the [100] zone axis; (b) HRTEM image of the plate shown in (a) with d-spacings indicated; (c) EDS spectrum from the plate in (a); (d) elemental mapping of the same plate; (e, f) SEM images of the fractured cross sections of SnS0.1Se0.9-1 and SnS0.1Se0.9-2 with the viewing direction (parallel to pellet surface) indicated in the inset. The four panels in (d) are the HAADF image and corresponding elemental maps for Sn (blue), Se (green) and S (red) respectively. | ||
The NaHSe concentration is a crucial synthesis parameter for the synthesis of SnS1−xSex nano/micro-plates with tuneable S concentration. When the NaHSe:Na2SnO2 molar ratio is increased from 1 through 1.15 to 1.5, it is possible to obtain almost single-phase orthorhombic SnS1−xSex with very high Se concentrations (Fig. S16†). The Se site occupancy obtained from Rietveld refinement increases from 0.91(1) through 0.92(2) to 0.94(2) (Table S5†) (corresponding to x = 0.91, x = 0.92 and x = 0.94 respectively). On decreasing the Se:Sn molar ratio (and x) from 0.9 through 0.8 to 0.5, the SnS1−xSex products accordingly contain more sulphide together with rising phase fractions of SnS (Fig. S16; Table S6†). The lattice parameters and unit cell volumes of these products increase with increasing Se concentration (x) which follows Vegard's law (Fig. S17†). The products synthesised at various NaHSe concentrations universally take the form of nano/micro-flowers consisting of clusters of nano/micro-plates, reinforcing the premise that the anion exchange is (pseudomorphic and) topotactic (Fig. S18 and S19†). As the NaHSe:Na2SnO2 molar ratio is increased, EDS spectra show that the Se/(Se + S) ratio increases from ∼0.47, through ∼0.77, ∼0.85, ∼0.89, ∼0.92 to ∼0.93 (Fig. S18–S20†), which is consistent with the Rietveld refinement results.
Thermoelectric performance of anion-exchanged SnS1−xSex
The ability to prepare >10 g SnS1−xSex plates, with control over both structure and composition, facilitates the fabrication of textured, dense pellets. Of the various SnS1−xSex materials synthesised, SnS0.1Se0.9 was selected for thermoelectric measurements given previously documented electrical and thermal properties of the equivalent bulk material of the same composition.35 Pellets with ca. 98% of the SnS0.1Se0.9 theoretical density were consolidated from 2 h anion-exchanged SnS0.1Se0.9 plates via one-step (denoted SnS0.1Se0.9-1) and two-step (denoted SnS0.1Se0.9-2) SPS processes. The two-step SPS process can give rise to the superplastic flow of unconstrained samples in the transverse direction (i.e. perpendicular to the pressing direction), in turn producing textured samples.63 The orthorhombic structure of the materials persists after sintering and both pellets exhibit strong orientation in the (h00) plane (Fig. S21†). SnS0.1Se0.9-2 exhibits much stronger texturing than SnS0.1Se0.9-1 as evidenced by the higher relative diffraction intensities of the (h00) reflections. In fact, the orientation degree (F) for (h00), estimated by the Lotgering method64 and based on the PXD data (Fig. S21a†), is ca. 0.25 and ca. 0.46, for SnS0.1Se0.9-1 and SnS0.1Se0.9-2, respectively, demonstrating the greater texturing in the latter sample. SEM images of the cleavage surfaces (Fig. S22a–d†) and fractured cross-sections (Fig. 4e, f, S22e and f†) reveal that SnS0.1Se0.9-1 and SnS0.1Se0.9-2 consist of densely packed, stacked plates. In SnS0.1Se0.9-2 the plates are aligned almost parallel to the pellet surface (perpendicular to the pressing direction), while those in SnS0.1Se0.9-1 are more directionally disordered, which is consistent with PXD results. EDS (Fig. S22g and h†) reveals that the pellets conserve their composition post-sintering (Sn:Se:S = 50(1):45(1):5(1)). Both pellets have an indirect optical bandgap of ca. 0.86 eV (Fig. S23†) and exhibit a negligible weight loss (<0.5 wt%) below 700 °C when heated under Ar gas (Fig. S24 and S25†).
Thermoelectric measurements were performed on both SnS0.1Se0.9 pellets perpendicular to the pressing direction (Fig. 5). The electrical conductivity of SnS0.1Se0.9-1 (Fig. 5a) increases from ca. 1940 S m−1 at 300 K to ca. 3400 S m−1 at 423 K, gradually decreases to ca. 1740 S m−1 at 673 K and reaches a maximum of ca. 6200 S m−1 at 873 K. The value of σ subsequently subsides to ca. 5000 S m−1 at 923 K. This behaviour has been previously observed in SnSe polycrystalline materials. It has been suggested that the reduction in σ over the mid-temperature range followed by a subsequent increase could be related to a reduction in carrier mobility and a thermal excitation of carriers, respectively.55,56 SnS0.1Se0.9-2 exhibits very similar behaviour to SnS0.1Se0.9-1, but demonstrates higher electrical conductivity below ca. 823 K (and more notably at lower T). As with SnS samples, SnS0.1Se0.9-1 and SnS0.1Se0.9-2 demonstrate a combination of clean particle surfaces, pronounced plate orientation, high crystallinity and high density. Coupled with the reduced bandgap engendered by Se doping, the high σ values observed can be plausibly rationalised. That SnS0.1Se0.9-2 exhibits higher conductivity is likely due to the enhanced (h00) texturing induced by the two-step SPS process. Electrical conductivity within the Sn–Se layers (in the bc plane) of SnSe is expected to be much higher than that perpendicular to the layers (along a).1 Indeed, the room temperature Hall carrier mobility of SnS0.1Se0.9-2 (evidenced as more strongly textured) is approximately 20.6% higher than that of SnS0.1Se0.9-1 (Table 1).
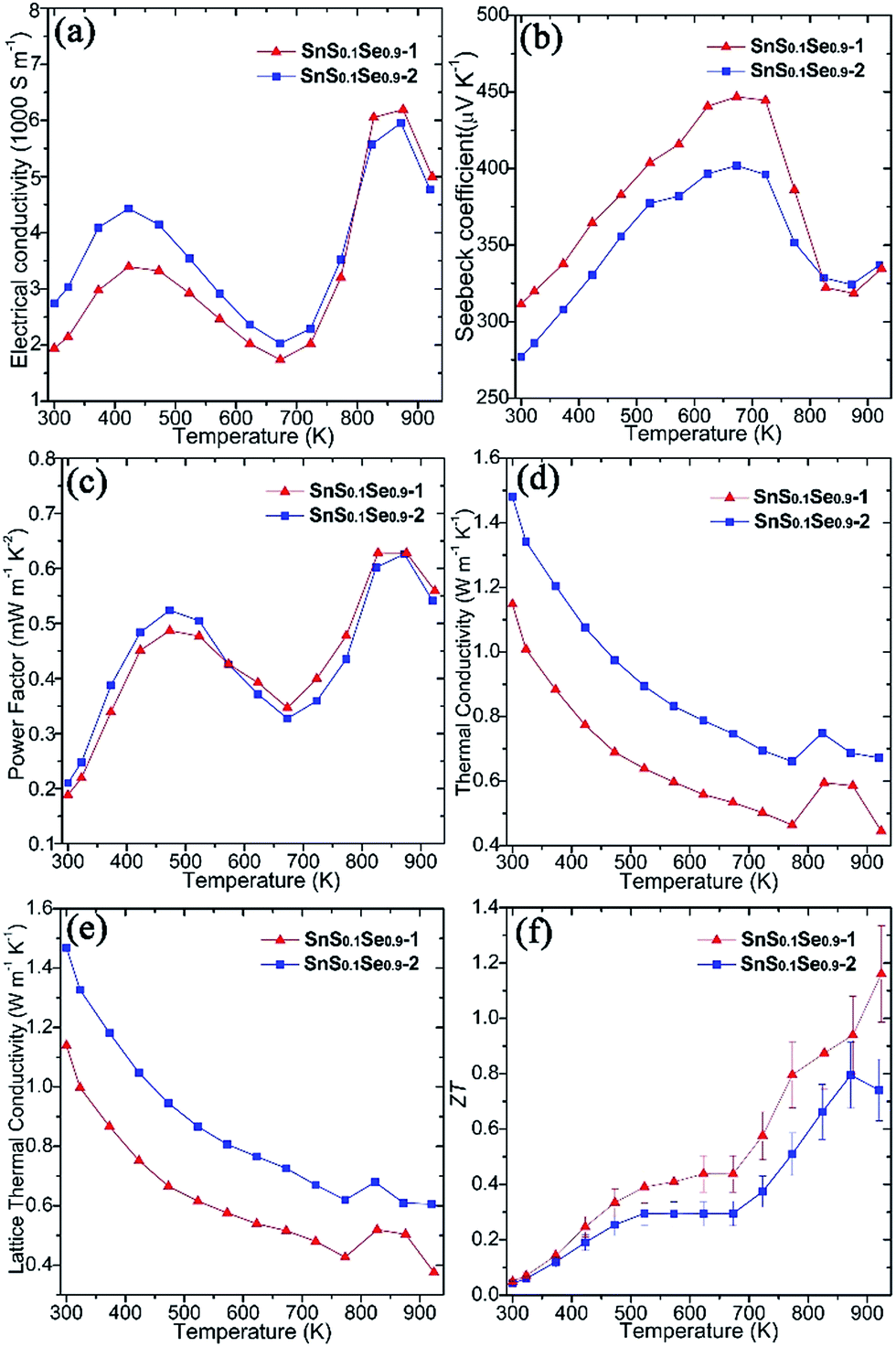 | ||
| Fig. 5 Thermoelectric properties of SnS0.1Se0.9 pellets SnS0.1Se0.9-1 and SnS0.1Se0.9-2 measured perpendicular to the pressing direction: (a) the electrical conductivity (σ), (b) the Seebeck coefficient (S), (c) the power factor (S2σ), (d) the thermal conductivity (κ), (e) the lattice κ (κL), and (f) ZT as a function of temperature. | ||
| Pellet | σ 300 K [S m−1] | S 300 K [μV K−1] | n H [1017 cm−3] | μ H [cm2 V−1 s−1] |
|---|---|---|---|---|
| SnS0.1Se0.9-1 | 1942 | 312 | 22.5 | 53.9 |
| SnS0.1Se0.9-2 | 2739 | 277 | 26.3 | 65.0 |
Both pellets exhibit p-type conducting behaviour, demonstrated by the positive Seebeck coefficient (Fig. 5b). S for SnS0.1Se0.9-1 increases almost linearly from ca. 310 μV K−1 at 300 K reaching a maximum at ca. 445 μV K−1 at 673 K. By 873 K, S drops to ca. 320 μV K−1 before a slight upturn to ca. 335 μV K−1 at 923 K. Below ca. 823 K, the Seebeck coefficient for SnS0.1Se0.9-2 is consistently slightly lower than that for SnS0.1Se0.9-1 but nevertheless follows the same trend with temperature. The decrease in S above 673 K could be related to the thermal excitation of minority carriers.55,56
The combination of large σ values coupled with high values of S results in exceptional power factors (S2σ) for SnS0.1Se0.9-1 and SnS0.1Se0.9-2 (Fig. 5c) which both show local maxima (of ca. 0.50 mW m−1 K−2) at 473 K and (of ca. 0.63 mW m−1 K−2) at 873 K. The S2σ vs. T behaviour hence broadly follows the temperature variation of σ, although the power factor for SnS0.1Se0.9-2 is marginally inferior to SnS0.1Se0.9-1 above 573 K. Interestingly SnS0.1Se0.9 and SnS show very similar trends in the variation of σ, S and S2σ with temperature (Fig. S26†), indicative of the broadly similar crystalline and electronic structures. Ultimately, SnS0.1Se0.9 reaches a higher value of σ (Fig. S26a†), probably primarily due to the reduction in the bandgap as Se replaces S.
Both SnS0.1Se0.9-1 and SnS0.1Se0.9-2 have higher power factors than SnSe pellets consolidated from surfactant-free nanoplates (increasing from ca. 0.05 mW m−1 K−1 at 300 K to ca. 0.40 mW m−1 K−1 at 550 K; Fig. S26c†),11 but perhaps particularly remarkable is that S2σ for SnS0.1Se0.9-1 at 773 K exceeds those of other SnS1−xSex (x < 1) materials (e.g. S2σ values at 773 K of ≈0.24 mW m−1 K−2 for p-type SnS0.2Se0.8 (ref. 26) and ≈0.40 mW m−1 K−2 for n-type SnS0.1Se0.87I0.03 (ref. 35)), and matches or surpasses those for the best examples of doped polycrystalline SnSe (e.g. 0.39–0.48 mW m−1 K−2 for p-type Na-doped SnSe at 773 K).29,31,32 Such materials require high-temperature methods to produce, so it is clearly possible to replace these synthesis methods by more efficient, energy-saving alternatives without sacrificing performance.
Microstructural texturing also influences the thermal conductivity (κ) of the pellets (Fig. 5d). The value of κ for SnS0.1Se0.9-1 is low, decreasing from ≈1.148 W m−1 K−1 at 300 K to ≈0.464 W m−1 K−1 at 773 K. The values for SnS0.1Se0.9-2 at the equivalent temperatures are approximately 29.0% and 42.4% larger respectively. The increase in κ at 823 K is most likely related to the second order displacive phase transformation of SnSe (from orthorhombic Pnma to orthorhombic Cmcm).65 As with the electrical conductivity, the thermal conductivity is anisotropic in SnS1−xSex and so the difference in κ can be attributed to the different degrees of texturing in SnS0.1Se0.9-1 and SnS0.1Se0.9-2. The thermal conductivity along the a-axis (perpendicular to the Sn–Se planes) is much lower than that along b or c.1 The main contribution to κ in the pellets is the lattice thermal conductivity (e.g. κL ≈ 0.427 W m−1 K−1 for SnS0.1Se0.9-1 and κL ≈ 0.620 W m−1 K−1 for SnS0.1Se0.9-2 at 773 K) (Fig. 5e) and the magnitude of κ and κL demonstrates the degree to which texturing can influence the thermal properties of polycrystalline materials such as SnSe. It should be noted that κL is still higher than the theoretical minimum for SnSe (κL ≈ 0.26 W m−1 K−1 at 770 K),35 so one might expect that by further texture control and/or by reducing the plate dimensions, κL could be driven further towards this theoretical minimum. It is also interesting to note that SnS (containing a lighter chalcogen), has a higher κL than SnS0.1Se0.9 across the whole measurement temperature range (Fig. S26e†).
ZT could be calculated for the pellets using the above electrical and thermal transport data (Fig. 5f). The ZT of SnS0.1Se0.9-1 is the higher across the whole T range, increasing from ca. 0.05 at 300 K to ca. 1.16 at 923 K; the ZT of SnS0.1Se0.9-2 reaches a maximum of ca. 0.80 at 873 K. The ZT of SnS0.1Se0.9-1 at 923 K is comparable to SnSe bulk materials with carbon inclusions measured at 903 K.37 Considering previous reports of the thermoelectric performance of SnSe up to 800 K, it is useful to compare the value of ZT close to this elevated temperature. The value for SnS0.1Se0.9-1 at 773 K is higher than that of sulfur-free p-type polycrystalline SnSe (ZT ≈ 0.39–0.66)27,66 and comparable to various metal-doped, p-type polycrystalline SnSe (ZT ≈ 0.5–1.2)26,28–32,36 (where all measurements were made perpendicular to the pressing direction at approximately 773 K; e.g. ≈0.6 for Ag-doped SnSe at 750 K,28 ≈0.8 for Na-doped SnSe at 800 K,31 ≈1.1 for K-doped SnSe at 773 K).30 Importantly, this indicates that tin chalcogenides can retain high thermoelectric performance when some of the more toxic Se is removed and replaced by less toxic S via anion exchange. Further systematic study of the thermoelectric performance of SnS1−xSex as a function of x should reveal the optimum materials and processing parameters in the system. Moreover, it may be possible to offset loss of performance with decreasing x by co-doping with appropriate low toxicity, Earth-abundant metals. Although homogeneity of the materials is realised in SPS treated pellets, we note that the extent of anion exchange during synthesis will be governed primarily by reaction kinetics. Both the kinetics and thermodynamics (Ksp) could be modified by replacing water with another solvent, but given the economic and environmental advantages in using aqueous chemistry, modifying reaction temperature (and/or autogenous pressure, hydrothermally) would be the obvious parameter(s) to investigate towards achieving complete topotactic conversion (Fig. 3d). There is thus considerable scope for polycrystalline SnSe materials to achieve (average) ZT values comparable to analogous single crystals. Given the materials design options to reduce κ (and notably κL) further as described above (and for example, via precipitates30) and to increase carrier concentration (to the magnitude of 1019 cm−3) and electrical conductivity, σ (e.g. through alkali ion doping2,24,29,31,33), it should be possible to propel ZT to still higher reaches in tin chalcogenides without sacrificing green chemistry principles.
Conclusions
In summary, organic-free, scalable, low-cost solution approaches have been developed to produce nanostructured layered tin chalcogenides with tuneable chalcogenide composition. This precipitation-anion exchange protocol yielded SnS0.1Se0.9 plates, which were consolidated into p-type textured pellets, achieving ZT ≈ 1.16 at 923 K through tuning the microstructural texturing. The precipitation-anion exchange route provides a versatile means to modify the microstructure and composition of layered tin chalcogenides directly from solution. Both variables have profound effects on the transport properties and thermoelectric performance of the resulting materials. The method should be extendable to the synthesis of a host of other p-block metal chalcogenides and provide scope for a wide-ranging strategy invoking nanostructuring and doping as fundamental parameters.Experimental
Full experimental details are provided in the ESI.†Materials synthesis
150 mmol NaOH and 10 mmol SnCl2·2H2O were added into 50 ml deionised water (DIW) to yield a transparent Na2SnO2 solution that was then heated to boil. 40 ml of Na2S(aq) (0.5 mol L−1) was promptly injected into the boiling solution, which was then further boiled for 2 h. The reaction was terminated at this point for the synthesis of SnS, while for the synthesis of SnS1−xSex materials such as SnS0.1Se0.9, used as an example here, 40 ml of freshly prepared NaHSe(aq) (0.25 mol L−1) was promptly injected into the SnS suspension that was boiled for another 2 h and then cooled to room temperature. Heating and cooling of the solution were performed under Ar(g) on a Schlenk line. The products were washed with DIW and ethanol and dried at 50 °C for 12 h. Scaled-up syntheses of SnS and SnS0.1Se0.9 were performed with eight-fold and six-fold precursor concentrations, respectively. SnS pellets (density of 5.17 g cm−3) were sintered in a graphite die (diameter: 15 mm) under vacuum by SPS (FCT HP D 25, FCT System GmbH; uniaxial pressure of 60 MPa; 500 °C; 5 min). SnS0.1Se0.9 pellets (density of 5.95 g cm−3) were sintered in a graphite die under vacuum by SPS in either a 1-step (uniaxial pressure of 60 MPa; 500 °C; 5 min; die diameter: 15 mm) or 2-step (1st step: 50 MPa; 450 °C; 5 min; die diameter: 15 mm. 2nd step: 50 MPa; 500 °C; 5 min; die diameter: 20 mm) process.63Materials characterisation and testing
PXD data were collected with a PANalytical X'pert Pro MPD diffractometer in Bragg–Brentano geometry (Cu Kα1 radiation, λ = 1.5406 Å). Rietveld refinement was performed against PXD data using the GSAS and EXPGUI software packages.67,68 Imaging and elemental analysis were conducted by SEM (Carl Zeiss Sigma, at 5 and 20 kV respectively) equipped with EDS (Oxford Instruments X-Max 80). Further imaging, elemental analysis and SAED were performed by TEM (FEI Titan Themis 200 equipped with Super-X windowless EDS detector, operated at 200 kV). Thermogravimetric-differential thermal analysis (TG-DTA) of the samples was performed using a Netzsch STA 409 thermal analyser under flowing Ar. Optical bandgaps were measured by DR-UV-Vis spectroscopy (Shimadzu, UV-2600). All thermoelectric measurements were made perpendicular to the pressing direction. Following prior determination of thermal stability and pre-heating treatments if necessary, the Seebeck coefficient (S) and electrical conductivity (σ) of pellets were measured simultaneously using a Linseis LSR-3 instrument from 300–923 K. The thermal conductivity, κ of the pellets was calculated by κ = DCpρ, where D, Cp and ρ are the thermal diffusivity coefficient, specific heat capacity and density, respectively. D of the SnS0.1Se0.9 pellets was measured using a Netzsch LFA 457 from 300–923 K (Fig. S27a†); Cp of SnS0.1Se0.9 (Fig. S27b†) was calculated from the weighted average26 of reported Cp of SnSe1 and SnS.25ρ of the pellets was measured by the Archimedes method. Electronic thermal conductivity (κe) was estimated by the Wiedemann–Franz law (κe = LσT, where L is the Lorentz number; L of 1.5 × 10−8 V2 K−2 was applied1), and lattice thermal conductivity (κL) was calculated by subtracting κe from κ. ZT was calculated via ZT = S2σT/κ. Hall measurements were performed on a nanometrics HL5500 Hall system using a van der Pauw configuration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the EPSRC (EP/K022156/1 and EP/P510968/1). The authors thank Mr Peter Chung for assistance with SEM. SRP and JWGB acknowledge the EPSRC for support (EP/N01717X/1). HFG and WZ acknowledge the EPSRC for the Equipment Grant to purchase a Titan Themis 200 microscope (EP/L017008/1). MJR and RZ would like to acknowledge the EPSRC for support under the DEFCOM grant (EP/N022726/1). RZ acknowledges the support from a Marie Curie International Incoming Fellowship within the 7th European Community Framework Programme (Contract No. PIIF-GA-2013-624474).References
- L. D. Zhao, S. H. Lo, Y. S. Zhang, H. Sun, G. J. Tan, C. Uher, C. Wolverton, V. P. Dravid and M. G. Kanatzidis, Nature, 2014, 508, 373–377 CrossRef CAS PubMed
.
- L.-D. Zhao, G. Tan, S. Hao, J. He, Y. Pei, H. Chi, H. Wang, S. Gong, H. Xu, V. P. Dravid, C. Uher, G. J. Snyder, C. Wolverton and M. G. Kanatzidis, Science, 2015, 351, 141–144 CrossRef PubMed
- G. Han, Z.-G. Chen, J. Drennan and J. Zou, Small, 2014, 10, 2747–2765 CrossRef CAS PubMed
- K. Biswas, J. Q. He, I. D. Blum, C. I. Wu, T. P. Hogan, D. N. Seidman, V. P. Dravid and M. G. Kanatzidis, Nature, 2012, 489, 414–418 CrossRef CAS PubMed
- S. J. Oh, C. Uswachoke, T. S. Zhao, J. H. Choi, B. T. Diroll, C. B. Murray and C. R. Kagan, ACS Nano, 2015, 9, 7536–7544 CrossRef CAS PubMed
- D. Zhitomirsky, M. Furukawa, J. Tang, P. Stadler, S. Hoogland, O. Voznyy, H. Liu and E. H. Sargent, Adv. Mater., 2012, 24, 6181–6185 CrossRef CAS PubMed
- M. Ibáñez, R. J. Korkosz, Z. S. Luo, P. Riba, D. Cadavid, S. Ortega, A. Cabot and M. G. Kanatzidis, J. Am. Chem. Soc., 2015, 137, 4046–4049 CrossRef PubMed
- D. Cadavid, M. Ibanez, A. Shavel, O. J. Dura, M. A. Lopez de la Torre and A. Cabot, J. Mater. Chem. A, 2013, 1, 4864–4870 CAS
- W. Zhou, W. Zhao, Z. Lu, J. Zhu, S. Fan, J. Ma, H. H. Hng and Q. Yan, Nanoscale, 2012, 4, 3926–3931 RSC
- R. J. Mehta, Y. Zhang, C. Karthik, B. Singh, R. W. Siegel, T. Borca-Tasciuc and G. Ramanath, Nat. Mater., 2012, 11, 233–240 CrossRef CAS PubMed
- G. Han, S. R. Popuri, H. F. Greer, J. W. G. Bos, W. Z. Zhou, A. R. Knox, A. Montecucco, J. Siviter, E. A. Man, M. Macauley, D. J. Paul, W. G. Li, M. C. Paul, M. Gao, T. Sweet, R. Freer, F. Azough, H. Baig, N. Sellami, T. K. Mallick and D. H. Gregory, Angew. Chem., Int. Ed., 2016, 55, 6433–6437 CrossRef CAS PubMed
- C. Han, Z. Li, G. Q. Lu and S. X. Dou, Nano Energy, 2015, 15, 193–204 CrossRef CAS
- G. Han, R. Z. Zhang, S. R. Popuri, H. F. Greer, M. J. Reece, J. W. G. Bos, W. Z. Zhou, A. R. Knox and D. H. Gregory, Materials, 2017, 10, 233 CrossRef PubMed
- G. Han, S. R. Popuri, H. F. Greer, L. F. Llin, J. W. G. Bos, W. Z. Zhou, D. J. Paul, H. Ménard, A. R. Knox, A. Montecucco, J. Siviter, E. A. Man, W.-g. Li, M. C. Paul, M. Gao, T. Sweet, R. Freer, F. Azough, H. Baig, T. K. Mallick and D. H. Gregory, Adv. Energy Mater., 2017, 7, 1602328 CrossRef
- G. D. Moon, S. Ko, Y. Min, J. Zeng, Y. N. Xia and U. Jeong, Nano Today, 2011, 6, 186–203 CrossRef CAS
- G. Han, Z.-G. Chen, L. Yang, M. Hong, J. Drennan and J. Zou, ACS Appl. Mater. Interfaces, 2015, 7, 989–995 CAS
- G. D. Moon, S. Ko, Y. N. Xia and U. Jeong, ACS Nano, 2010, 4, 2307–2319 CrossRef CAS PubMed
- G. Han, Z.-G. Chen, D. Ye, L. Yang, L. Wang, J. Drennan and J. Zou, J. Mater. Chem. A, 2014, 2, 7109–7116 CAS
- G. Han, Z.-G. Chen, D. Ye, B. Wang, L. Yang, Y. Zou, L. Wang, J. Drennan and J. Zou, J. Mater. Chem. A, 2015, 3, 7560–7567 CAS
- N. A. Moroz, A. Olvera, G. M. Willis and P. F. P. Poudeu, Nanoscale, 2015, 7, 9452–9456 RSC
- J. M. Pietryga, D. J. Werder, D. J. Williams, J. L. Casson, R. D. Schaller, V. I. Klimov and J. A. Hollingsworth, J. Am. Chem. Soc., 2008, 130, 4879–4885 CrossRef CAS PubMed
- G. J. Tan, L. D. Zhao and M. G. Kanatzidis, Chem. Rev., 2016, 116, 12123–12149 CrossRef CAS PubMed
- G. J. Snyder and E. S. Toberer, Nat. Mater., 2008, 7, 105–114 CrossRef CAS PubMed
- K. Peng, X. Lu, H. Zhan, S. Hui, X. Tang, G. Wang, J. Dai, C. Uher, G. Wang and X. Zhou, Energy Environ. Sci., 2016, 9, 454–460 CAS
- Q. Tan, L. D. Zhao, J. F. Li, C. F. Wu, T. R. Wei, Z. B. Xing and M. G. Kanatzidis, J. Mater. Chem. A, 2014, 2, 17302–17306 CAS
- Y.-M. Han, J. Zhao, M. Zhou, X.-X. Jiang, H.-Q. Leng and L.-F. Li, J. Mater. Chem. A, 2015, 3, 4555–4559 CAS
- S. Sassi, C. Candolfi, J. B. Vaney, V. Ohorodniichuk, P. Masschelein, A. Dauscher and B. Lenoir, Appl. Phys. Lett., 2014, 104, 212105 CrossRef
- C. L. Chen, H. Wang, Y. Y. Chen, T. Day and G. J. Snyder, J. Mater. Chem. A, 2014, 2, 11171–11176 CAS
- E. K. Chere, Q. Zhang, K. Dahal, F. Cao, J. Mao and Z. Ren, J. Mater. Chem. A, 2016, 4, 1848–1854 CAS
- Y.-X. Chen, Z.-H. Ge, M. Yin, D. Feng, X.-Q. Huang, W. Zhao and J. He, Adv. Funct. Mater., 2016, 26, 6836–6845 CrossRef CAS
- T.-R. Wei, G. Tan, X. Zhang, C.-F. Wu, J.-F. Li, V. P. Dravid, G. J. Snyder and M. G. Kanatzidis, J. Am. Chem. Soc., 2016, 138, 8875–8882 CrossRef CAS PubMed
- H.-Q. Leng, M. Zhou, J. Zhao, Y.-M. Han and L.-F. Li, RSC Adv., 2016, 6, 9112–9116 RSC
- Z.-H. Ge, D. Song, X. Chong, F. Zheng, L. Jin, X. Qian, L. Zheng, R. E. Dunin-Borkowski, P. Qin, J. Feng and L.-D. Zhao, J. Am. Chem. Soc., 2017, 139, 9714–9720 CrossRef CAS PubMed
- T. R. Wei, C. F. Wu, X. Z. Zhang, Q. Tan, L. Sun, Y. Pan and J. F. Li, Phys. Chem. Chem. Phys., 2015, 17, 30102–30109 RSC
- Q. Zhang, E. K. Chere, J. Y. Sun, F. Cao, K. Dahal, S. Chen, G. Chen and Z. F. Ren, Adv. Energy Mater., 2015, 5, 1500360 CrossRef
- G. Tang, W. Wei, J. Zhang, Y. Li, X. Wang, G. Xu, C. Chang, Z. Wang, Y. Du and L.-D. Zhao, J. Am. Chem. Soc., 2016, 138, 13647–13654 CrossRef CAS PubMed
- J. C. Li, D. Li, W. Xu, X. Y. Qin, Y. Y. Li and J. Zhang, Appl. Phys. Lett., 2016, 109, 4 Search PubMed
- X. Wang, J. T. Xu, G. Q. Liu, Y. J. Fu, Z. Liu, X. J. Tan, H. Z. Shao, H. C. Jiang, T. Y. Tan and J. Jiang, Appl. Phys. Lett., 2016, 108, 083902 CrossRef
- D. Li, J. C. Li, X. Y. Qin, J. Zhang, H. X. Xin, C. J. Song and L. Wang, Energy, 2016, 116, 861–866 CrossRef CAS
- Y. Fu, J. Xu, G.-Q. Liu, J. Yang, X. Tan, Z. Liu, H. Qin, H. Shao, H. Jiang, B. Liang and J. Jiang, J. Mater. Chem. C, 2016, 4, 1201–1207 RSC
- C. Chang, Q. Tan, Y. Pei, Y. Xiao, X. Zhang, Y.-X. Chen, L. Zheng, S. Gong, J.-F. Li, J. He and L.-D. Zhao, RSC Adv., 2016, 6, 98216–98220 RSC
- T. A. Wubieneh, C. L. Chen, P. C. Wei, S. Y. Chen and Y. Y. Chen, RSC Adv., 2016, 6, 114825–114829 RSC
- S. Lv, Z. H. Ge, Y. X. Chen, K. Y. Zhao, J. Feng and J. Q. He, RSC Adv., 2016, 6, 92335–92340 RSC
- L. Zhang, J. Wang, Q. Sun, P. Qin, Z. Cheng, Z. Ge, Z. Li and S. Dou, Adv. Energy Mater., 2017, 7, 1700573 CrossRef
- M. Hong, Z. G. Chen, L. Yang, T. C. Chasapis, S. D. Kang, Y. C. Zou, G. J. Auchterlonie, M. G. Kanatzidis, G. J. Snyder and J. Zou, J. Mater. Chem. A, 2017, 5, 10713–10721 CAS
- C. C. Lin, R. Lydia, J. H. Yun, H. S. Lee and J. S. Rhyee, Chem. Mater., 2017, 29, 5344–5352 CrossRef CAS
- K. L. Peng, H. Wu, Y. C. Yan, L. J. Guo, G. Y. Wang, X. Lu and X. Y. Zhou, J. Mater. Chem. A, 2017, 5, 14053–14060 CAS
- D. B. Li, X. J. Tan, J. T. Xu, G. Q. Liu, M. Jin, H. Z. Shao, H. J. Huang, J. F. Zhang and J. Jiang, RSC Adv., 2017, 7, 17906–17912 RSC
- F. Li, W. T. Wang, X. C. Qiu, Z. H. Zheng, P. Fan, J. T. Luo and B. Li, Inorg. Chem. Front., 2017, 4, 1721–1729 RSC
- Y. L. Li, X. Shi, D. D. Ren, J. K. Chen and L. D. Chen, Energies, 2015, 8, 6275–6285 CrossRef CAS
- W. J. Baumgardner, J. J. Choi, Y. F. Lim and T. Hanrath, J. Am. Chem. Soc., 2010, 132, 9519–9521 CrossRef CAS PubMed
- M. A. Franzman, C. W. Schlenker, M. E. Thompson and R. L. Brutchey, J. Am. Chem. Soc., 2010, 132, 4060–4061 CrossRef CAS PubMed
- L. Li, Z. Chen, Y. Hu, X. W. Wang, T. Zhang, W. Chen and Q. B. Wang, J. Am. Chem. Soc., 2013, 135, 1213–1216 CrossRef CAS PubMed
- D. D. Vaughn, S. I. In and R. E. Schaak, ACS Nano, 2011, 5, 8852–8860 CrossRef CAS PubMed
- Y. W. Li, F. Li, J. F. Dong, Z. H. Ge, F. Y. Kang, J. Q. He, H. D. Du, B. Li and J. F. Li, J. Mater. Chem. C, 2016, 4, 2047–2055 RSC
- D. Feng, Z.-H. Ge, D. Wu, Y.-X. Chen, T. Wu, J. Li and J. He, Phys. Chem. Chem. Phys., 2016, 18, 31821–31827 RSC
- S. G. Hickey, C. Waurisch, B. Rellinghaus and A. Eychmüller, J. Am. Chem. Soc., 2008, 130, 14978–14980 CrossRef CAS PubMed
- Z. Deng, D. Cao, J. He, S. Lin, S. M. Lindsay and Y. Liu, ACS Nano, 2012, 6, 6197–6207 CrossRef CAS PubMed
- A. J. Biacchi, D. D. Vaughn and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 11634–11644 CrossRef CAS PubMed
- A. de Kergommeaux, M. Lopez-Haro, S. Pouget, J.-M. Zuo, C. Lebrun, F. Chandezon, D. Aldakov and P. Reiss, J. Am. Chem. Soc., 2015, 137, 9943–9952 CrossRef CAS PubMed
- PDF-2 Release 2008, Joint Committee on Powder Diffraction Standards (JCPDS)-International Centre for Diffraction Data (ICDD), 2008.
- Q. Tan, C.-F. Wu, W. Sun and J.-F. Li, RSC Adv., 2016, 6, 43985–43988 RSC
- H. X. Yan, H. P. Ning, Y. M. Kan, P. L. Wang and M. J. Reece, J. Am. Ceram. Soc., 2009, 92, 2270–2275 CrossRef CAS
- F. K. Lotgering, J. Inorg. Nucl. Chem., 1959, 9, 113–123 CrossRef CAS
- T. Chattopadhyay, J. Pannetier and H. G. Von Schnering, J. Phys. Chem. Solids, 1986, 47, 879–885 CrossRef CAS
- S. R. Popuri, M. Pollet, R. Decourt, F. D. Morrison, N. S. Bennett and J. W. G. Bos, J. Mater. Chem. C, 2016, 4, 1685–1691 RSC
-
A. C. Larson and R. B. Von
Dreele, General Structure Analysis System (GSAS); Los Alamos National Laboratory Report LAUR 86-748, Los Alamos National Laboratory, 1994 Search PubMed
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc05190e |
| This journal is © The Royal Society of Chemistry 2018 |