
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence[(Cp2M)2B9H11] (M = Zr or Hf): early transition metal ‘guarded’ heptaborane with strong covalent and electrostatic bonding†‡
Anangsha
De
a,
Qian-Fan
Zhang§
b,
Bijan
Mondal§
 a,
Ling Fung
Cheung
b,
Sourav
Kar
a,
Koushik
Saha
a,
Babu
Varghese
c,
Lai-Sheng
Wang
*b and
Sundargopal
Ghosh
*a
a,
Ling Fung
Cheung
b,
Sourav
Kar
a,
Koushik
Saha
a,
Babu
Varghese
c,
Lai-Sheng
Wang
*b and
Sundargopal
Ghosh
*a
aDepartment of Chemistry, Indian Institute of Technology Madras, Chennai 600036, India. E-mail: sghosh@iitm.ac.in
bDepartment of Chemistry, Brown University, Providence, Rhode Island 02912, USA. E-mail: Lai-Sheng_Wang@brown.edu
cSophisticated Analytical Instruments Facility, Indian Institute of Technology Madras, Chennai 600036, India
First published on 11th January 2018
Abstract
Among the series of stable closo-borate dianions, [BnHn]2−, the X-ray crystallographic structure of [B7H7]2− was determined only in 2011. To explore its chemistry and stability, we have isolated and structurally characterized two new transition metal complexes of the heptaborane, [(Cp2M)2B9H11] (Cp = η5-C5H5; M = Zr or Hf). The structures of [(Cp2M)2B9H11] contain a pentagonal bipyramidal B7 core, coordinated by two {Cp2M} and two {BH2} units equatorially. Structural and spectroscopic characterizations and DFT calculations show that [(Cp2M)2B9H11] complexes are substantially more stable than the parent dianion, in either [B7H7]2− or (nBu4N)2[B7H7]. Our theoretical study and chemical bonding analyses reveal that the surprising stability of the two new heptaborane metal complexes is due to multi-center covalent bonds related to the two exo-{Cp2M} units, as well as electrostatic interactions between the {Cp2M} units and the B7 core. The facile syntheses of the heptaborane metal-complexes will allow further exploration of their chemistry.
1. Introduction
Since the early work of Alfred Stock on borane chemistry,1 syntheses and characterizations of polyhedral borane compounds, especially with higher vertex closo geometries, have attracted significant interest.2,3 As a result, the syntheses and structural characterizations of closo-borate compounds with the general formula [BnHn]2− (n = 6–12) have been documented in detail.4,5 After the first isolation of the closo-borate dianion [B10H10]2− by Hawthorne in 1959, other members in the series [BnHn]2− (n = 6–12) were discovered successively.6 Interest in the closo-polyhedral borane compounds was stimulated by the first crystal structure report of icosahedral [K2(B12H12)] by Lipscomb in 1960.7 Interestingly, although extensive research had been carried out, X-ray crystallographic study of the borate dianion [B7H7]2− was still missing at that time, making the [BnHn]2− series incomplete. The only closo-heptaborate compound, [B7Br7]2−, was not known until the report of the crystal structure of [(C5H5N)2CH2][B7Br7].8 It should be noted that the parent [B7H7]2− was partially characterized in Cs2[B7H7] using 11B NMR and it was found to be the least stable polyhedral ion.9 The reported yield of [B7H7]2− synthesized from NaBH4 after several steps was only 0.008%. As a result, an improved synthesis and complete structural characterization of [B7H7]2− were of general interest. Although a number of theoretical studies on the structure and bonding of [B7H7]2− have been reported,10 its convenient synthesis and full structural characterization were established only recently by Bernhardt in 2011.11 In an effort to synthesize the neutral hypercloso-heptaborane analogue, Siebert reported [B7Cl5(PMe3)2], which was mainly characterized using 11B NMR.12The continuous efforts into the development of group 4 metalloboranes, both by the authors and others,14 allowed us to isolate and structurally characterize [(Cp2Zr)2B5H11] (I) from the reaction of [Cp2Zr(BH4)2] with [BH3 THF] recently. The single crystal X-ray structural analysis of I revealed an open geometry with a planar B3 ring. Structural and bonding analyses demonstrated that I indeed is a neutral triborane analogue. In addition, theoretical results revealed the intriguing role of early transition metal fragments {Cp2Zr} in the stabilization of the borane core. The synthesis of I paved the way for accessing group 4 metalloboranes readily utilizing the well-known borohydride complexes, [Cp2M(BH4)2] (M = Ti, Zr or Hf).15 These experimental results suggested that other boranes might also be stabilized synthetically by the {Cp2M} fragments of group 4 metals. This hypothesis motivated us to explore the group 4 metalloboranes extensively in a quest to synthesize higher boranes stabilized by the coordination sphere of early transition metals. In the current article, we present an efficient approach for the synthesis of the first neutral transition metal protected heptaboranes 1 and 2 by a simple borane condensation method utilizing group 4 metal borohydrides. In addition, theoretical calculations have been carried out to elucidate the stability and bonding of the two new metallo-borane clusters.
2. Experimental
The reaction of [Cp2Zr(BH4)2]15,16 with a large excess of [BH3 THF] was explored under vigorous conditions, yielding [(Cp2Zr)2B9H11] (1) with a novel B7 core (Fig. 1a and Scheme 1). Compound I was also isolated as a side product. The isolation of 1 tempted us to explore the Hf analogue, since structurally characterized hafnium–boron compounds are mostly unknown, except for one borohydride compound, [Hf(η3-BH4)4].17 Following this idea, we have investigated the reaction of [Cp2Hf(BH4)2]15 with [BH3 THF] under similar conditions. This indeed led to the formation of [(Cp2Hf)2B9H11] (2) (Fig. 1b and Scheme 1) and [(Cp2Hf)2B5H11] (II) as stable crystalline solids. Once compounds 1 and 2 were isolated as colourless solids, they were characterized using mass spectrometry, NMR, IR and single-crystal X-ray crystallography. Detailed syntheses and characterizations can be found in the ESI.‡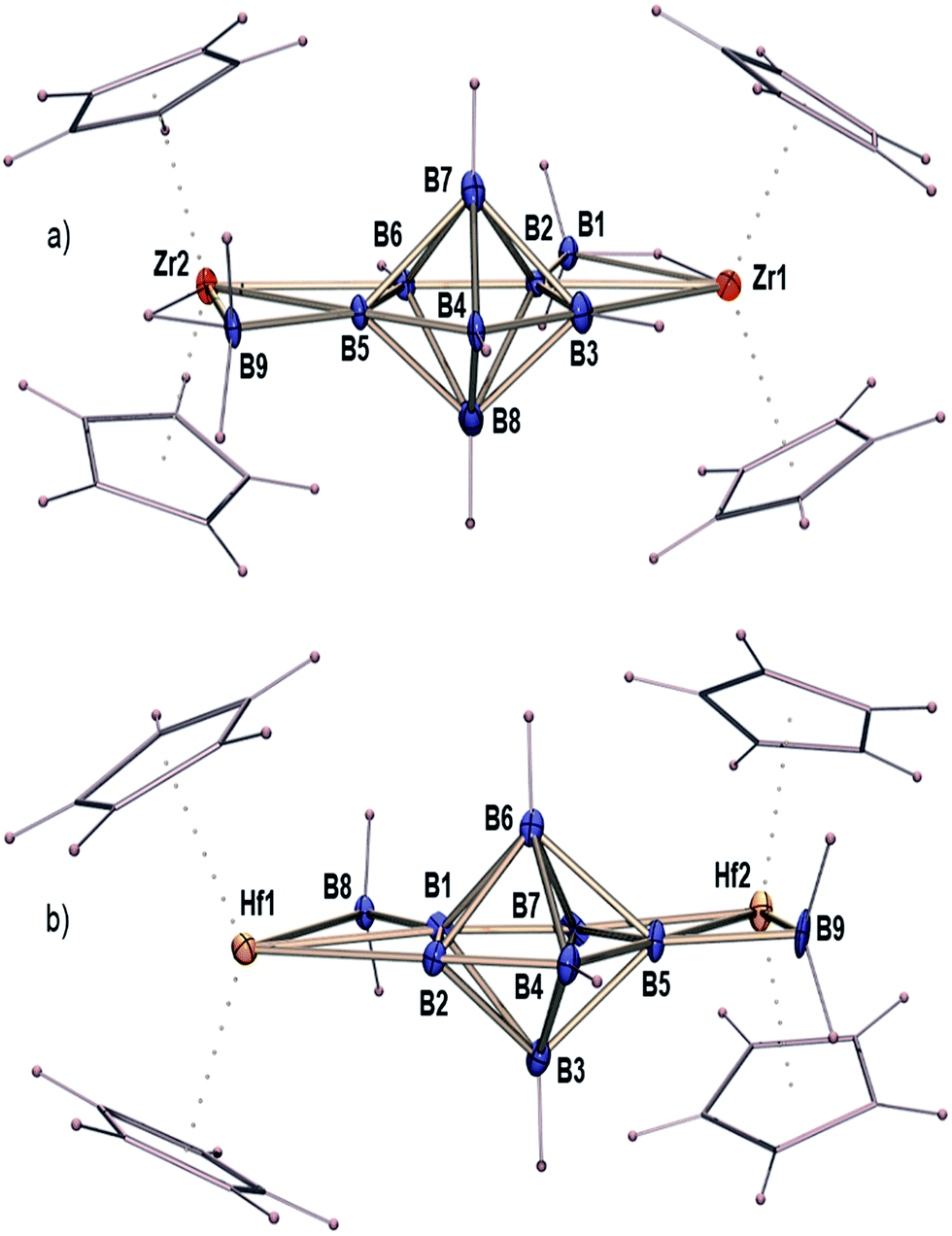 | ||
| Fig. 1 The crystal structures of [(Cp2M)2B9H11] [(a) M = Zr (1) and (b) M = Hf (2)] (some of the hydrogen atoms in 2 could not be located); details can be found in the ESI.‡ | ||
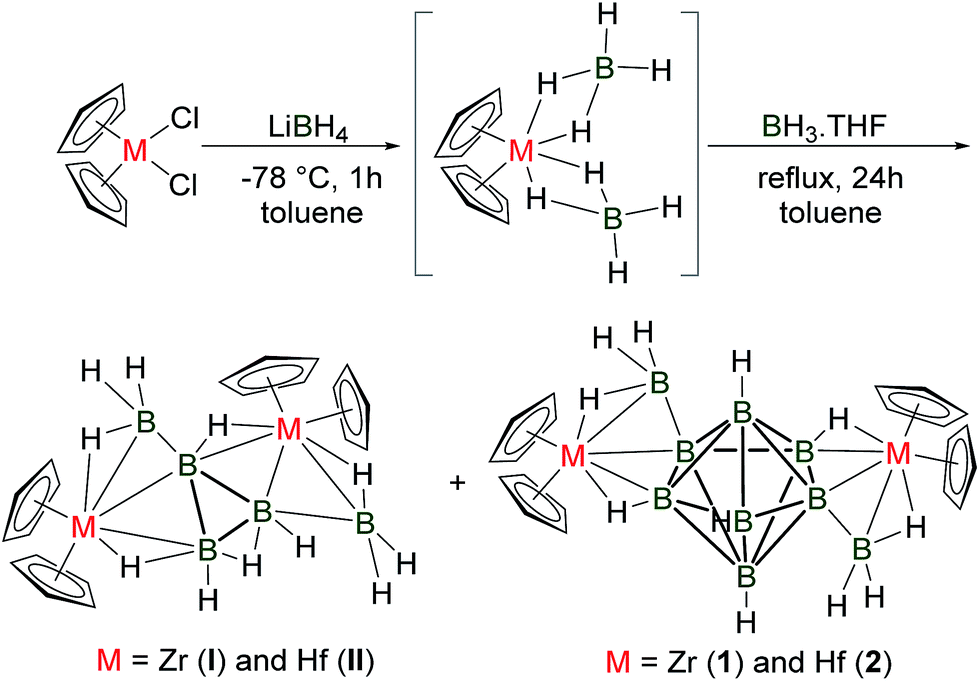 | ||
| Scheme 1 Synthesis of compounds 1 and 2. | ||
3. Results and discussion
3.1. Characterizations
The ESI mass spectrum of compound 1 in 100% MeCN (Fig. S1‡) shows the ion signal corresponding to the [M − (Cp2Zr)–BH2]− fragment (experimental m/z = 316.1341; calculated m/z = 316.1566). The isotopic distribution of the experimental data matches well with the calculated results. Furthermore, collision-induced dissociation (MS/MS) experiments show that the voltage threshold for the total fragmentation of [M − (Cp2Zr)–BH2]− is 25–30 V (Fig. S2‡). When the mobile phase in the ESI was changed to 95% MeCN + 5% H2O, the [M + Cl]– adduct was observed (experimental m/z = 585.1156; calculated m/z = 585.1369) with a good match in the isotopic distribution between the experiment and simulation (Fig. S3‡). The MS/MS experiments indicate that the major fragment is [M − (Cp2Zr)]−, with the voltage threshold of total fragmentation observed at 5–8 V (Fig. S4‡). Similarly, the mass spectrum of compound 2 in 100% MeCN (Fig. S5‡) shows the ion peak of [M − (Cp2Hf)BH2]− (experimental m/z = 404.1802; calculated m/z = 404.1919) with the voltage threshold of total fragmentation observed at 30–35 V (Fig. S6‡). When 95% MeCN + 5% H2O was used as the mobile phase, [M + Cl]− of 2 was observed again (experimental m/z = 761.1978; calculated m/z = 761.2076) (Fig. S7‡), which totally fragments to [M − (Cp2Hf)]− at 8–12 V (Fig. S8‡). All of the experimental isotopic distributions for compound 2 also match very well with the calculated results. The mass spectrometric analyses clearly show the structural similarity between 1 and 2. Both of them contain building units of {Cp2M} and {BH2} that can fragment easily. The only difference is the slightly higher voltage thresholds for 2.In order to confirm the spectroscopic assignments and determine the structures of 1 and 2, we did X-ray structure analyses (Fig. 1). The crystal structures of 1 and 2 both correspond to a 7-vertex pentagonal bipyramidal core consisting of a planar pentagonal B5 belt at the equatorial position (the average non-planar deviation of the B5 ring is 0.0006 Å for 1 and 0.019 Å for 2). The core geometries of 1 and 2 are virtually identical to the B7 structure in X2[B7H7] (X = PPh4 or nBu4N),11 Na3B20 and [(C5H5N)2CH2][B7Br7].8,18 The sum of the internal angles corroborates well with the ideal pentagon (540° for a pentagon, whereas it is 540.01° for 1 and 539.79° for 2). As expected, slight distortions are observed for individual internal angles, due to the asymmetry of the exo-{Cp2M} and exo-{BH2} moieties. The presence of the exo-{Cp2M} and exo-{BH2} moieties also supports the fragmentation channels observed in mass spectrometry.
Another interesting structural feature of compounds 1 and 2 is the presence of the {Cp2M} “wings”, which are anchored to the B7 core through M–B–B and M–H–B interactions. The crystallographic data show that the average B–B bond lengths in the B7 core of 1 and 2 are slightly shorter compared to [nBu4N]2[B7H7] and [B7H7]2−, indicating a more stable B7 core. Counting of the electrons becomes obvious when one realizes that the exo-{BH2} unit adds only one electron, like hydrogen.19 The core geometry turns out to be [B7H7]2−, which is protected by two 14 electron [(Cp2M)H]+ units. Alternatively, both compounds 1 and 2 can be considered as a fused cluster. As shown in Scheme S1,‡ cluster 1 or 2 may be described as a fusion20 of two arachno-MB3 {2[(14 + 12) + 6] = 64} moieties and a closo-B7 {(4 × 7 + 2) = 30} moiety through two B–B edges {2(6 × 2 + 2) = 28}. This arrangement of 1 or 2 generates an electron count of 66e which is equal to the total cluster valence electron (cve) number of 66 (the cve number of [(Cp2M)2B9H11] = 2 × 14 + 9 × 3 + 11 × 1 = 66). Although the isolation and structural characterization of 1 and 2 are closely connected to those of [(Cp*Re)2(B7H7)],21 the latter is highly electron deficient and shows an oblate-closo geometry that is inconsistent with prior theoretical findings.22 Furthermore, the metal atoms in both 1 and 2 are positioned in the same plane of B5. Interestingly, both the Zr and Hf atoms are in pseudo-tetrahedral geometries, contradictory to the theoretical finding which suggests that metal atoms with relatively diffuse frontier orbitals tend to occupy the higher-degree sites.22
The 1H and 11B{1H} NMR spectra of both 1 and 2 apparently suggest a lower symmetry than that in the solid-state structures. The hydrogen atoms in the compounds have been assigned using 1H–11B{1H} HSQC spectroscopy. The 11B{1H} chemical shift of 1 at δ = −3.6 ppm can be assigned to the axial boron atoms (B7 and B8 in Fig. 1), because the integration is twice that of the other boron signals and the HSQC result clearly indicates the attachment of hydrogen atom(s). To examine the fluxionality of 2, temperature-dependent 1H NMR spectroscopy was undertaken (Fig. S21‡). The 1H chemical shifts appearing at δ = −0.64 and −1.14 ppm can be assigned to two Hf–H–B protons and the broad peaks at δ = −3.26 and −3.42 ppm are assigned to the exo-{BH2} units. In addition, the experimental 11B{1H} NMR chemical shifts of 1 and 2 show reasonable agreement with the theoretical values (error range 3–7 ppm) (Table S1‡). The unusual negative 11B{1H} chemical shift for the axial boron atoms, both in 1 and 2, prompted us to apply the nucleus-independent chemical shift (NICS)23 criterion to investigate the magnetic shielding in 1 and 2. The value of the NICS(0.5) (−21.5 ppm) at the pentagonal plane of 1 is slightly more than that of the NICS(0) (−18.5 ppm). Interestingly, the NICS(3) of −6.2 ppm still shows a ring current indicating the presence of induced diatropic ring currents which is typical for a three-dimensional aromatic system (Fig. S35‡). Qualitatively, the NICS values observed for 2 (−22.2, −19.1 and −6.3 ppm; Fig. S36‡) are similar to those of 1 with slightly upfield chemical shifts. Therefore, the existence of the aromatic system plays a key role in the stabilization of 1 and 2.
3.2. Bonding analyses
An alternative description of compounds 1 and 2 is that they are metal-stabilized closo-compounds without the necessity of having any counter ions. In order to reveal the role of the metals in stabilizing the intriguing B7 core, bonding analyses of 1 and 2, as well as the naked dianion [B7H7]2−, were performed. However, due to the presence of highly delocalized electrons in such molecules, the bonding pictures established from the molecular orbital (MO) analysis (Fig. S30 and S31‡) cannot provide direct insight into the chemical bonding. Thus, adaptive natural density partitioning (AdNDP)24 analysis was employed to provide the intuitive bonding patterns. The bonding pattern obtained from the AdNDP analysis indicates the existence of both localized and delocalized bonds in compounds 1 and 2 (Fig. S32 and S33‡). Since the AdNDP results are similar for both compounds 1 and 2, only the case of Zr is discussed here for simplicity (Fig. 2). Seven 2c-2e B–H σ bonds (Fig. 2f) can be found in 1. Four of them are localized at the two exo-{BH2} units as expected. The other three, which are similar to the B–H σ bonds in [B7H7]2− (Fig. 2a), can be localized at the only equatorial non-bridged hydrogen and the two axial hydrogens. The remaining two equatorial bridged hydrogens have different bonding patterns, which will be discussed below. Although five 4c-2e σ bonds (delocalized between two adjacent equatorial boron atoms and two axial boron atoms) can be found in [B7H7]2− (Fig. 2b), only three of them are maintained in 1 (Fig. 2g). The other two are replaced by two 5c-2e σ bonds between the exo-{Cp2M}, the two axial and two equatorial borons (Fig. 2k). Such interactions provide strong stabilizing effects for the B7 core, which is also the main reason for the slightly shortened equatorial B–B bond lengths (Fig. S34‡) in the B7 core upon coordination by {Cp2Zr}. By forming 5c-2e σ bonds with the metal atoms, the high charge density in the naked [B7H7]2− can be lowered significantly. Therefore, the strong coulomb repulsion between the equatorial boron atoms is reduced, leading to shorter B–B bond lengths and the more stable B7 core.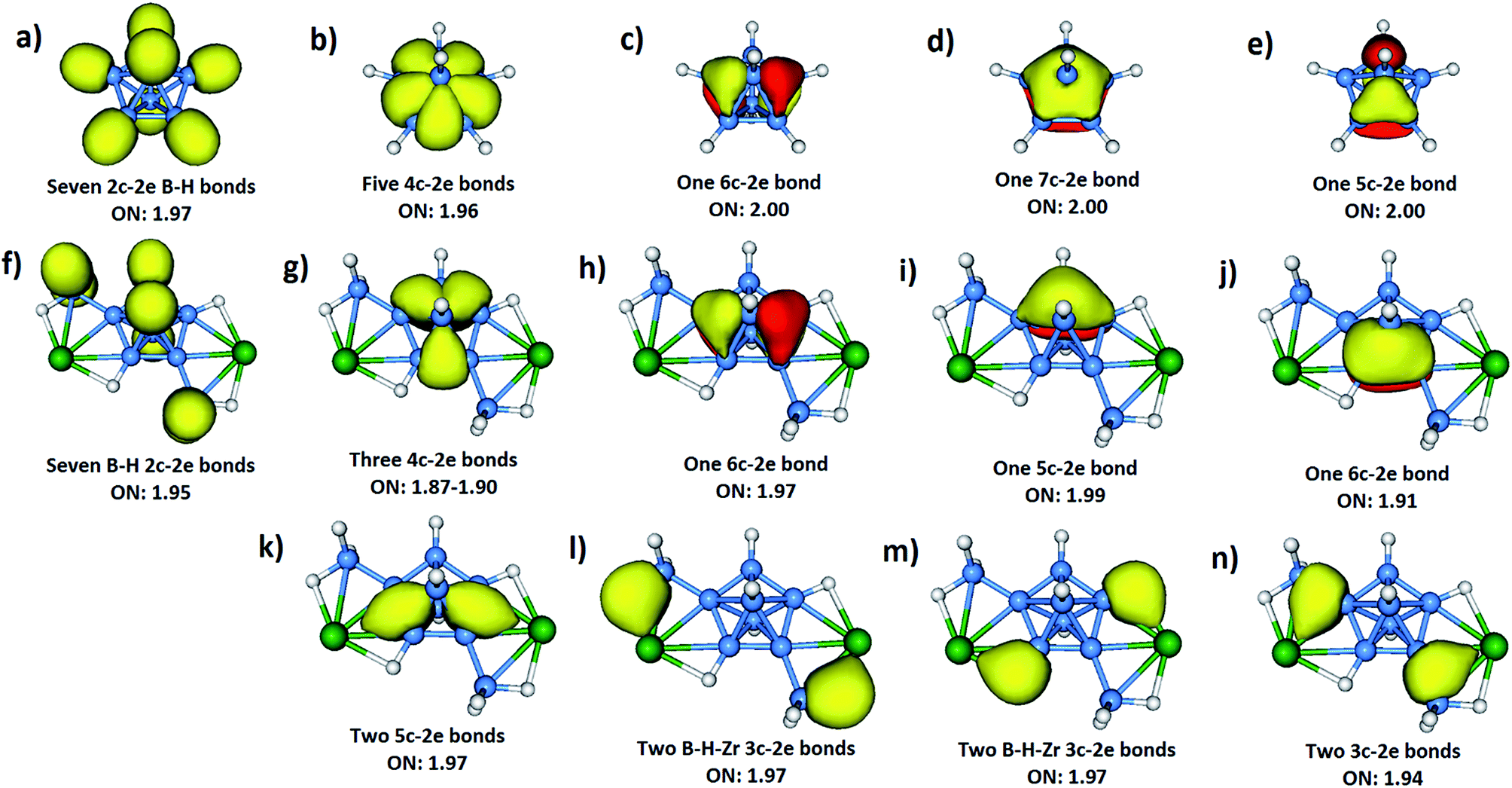 | ||
| Fig. 2 AdNDP analyses of [B7H7]2− (a–e) and 1 (f–n). The four Cp rings of 1 are omitted for clarity. | ||
Three delocalized π bonds (Fig. 2h–j) can be found in the B7 core of 1. Despite the similarity between them and the three π bonds in the naked [B7H7]2− (Fig. 2c–e), the π bonds in 1 have less symmetry due to the presence of exo-{Cp2M} and exo-{BH2} moieties. Besides the two 5c-2e σ bonds (Fig. 2k) discussed above, there are six more 3c-2e σ bonds (Fig. 2l–n) that contribute to the extra stability of 1. These bonds are absent from [B7H7]2−: (a) two 3c-2e σ bonds (Fig. 2l) between the exo-{Cp2M}, the exo boron and an exo bridged hydrogen; (b) two 3c-2e σ bonds (Fig. 2m) between the exo-{Cp2M}, the equatorial boron and hydrogen in the B7 unit; (c) two 3c-2e σ bonds (Fig. 2n) between the exo-{Cp2M}, the exo boron and the equatorial boron in B7. All of these multi-center bonds are crucial for keeping the integrity of compound 1, as also revealed by the large magnitudes of the NICS(0) values for these delocalized bonds (Fig. S35 and S36‡). The enhanced stability of the B7 core in 1 and 2 is also reflected in the large magnitudes of the NICS(0) values (−18.5 to −39.5 for 1; −19.1 to −39.8 for 2) in comparison to that of the bare [B7H7]2– (−21.3 in Fig. S37‡).
To further illustrate the strong stabilizing effect of the exo-{Cp2M} (M = Zr or Hf) units, we studied the binding energies of the system using DFT calculations. The binding energies were calculated as the energy difference between the parent [(Cp2M)2B9H11] molecule and the sum of two separate fragments of this compound at the PBE1PBE/Def2-TZVP level of theory. The single point energies of two schemes were calculated: (a) one of the exo-{Cp2M} units (charge state +2) was removed from the molecule, leaving the other part, [(Cp2M)B9H11], to have a −2 charge; and (b) similar to (a), but both fragments were kept neutral. The energies of these two fragments in each scheme were calculated separately with fixed geometries. The binding energies for these two schemes were 21.2 eV and 9.3 eV for compound 1, respectively. The difference between them, 11.9 eV, can be attributed to the stabilizing effect from electrostatic interaction or ionic bonding. The 9.3 eV value can be attributed to the stabilizing effect from the four multiple center σ bonds (three 3c-2e σ bonds and one 5c-2e σ bond) related to the exo-{Cp2Zr} unit or covalent bonding. As for 2, the electrostatic interaction was calculated to be 11.9 eV, while the effect of covalent bonding was 9.6 eV. The slightly higher binding energies of 2 also agree well with the slightly higher voltage thresholds observed in mass spectrometry. Therefore, the AdNDP analysis and binding energy calculations have revealed the crucial role of the exo-{Cp2M} and exo-BH2 moieties for stabilizing the whole (Cp2M)2B9H11 framework.
3.3. Characterization of the by-product
Compound II (Scheme 1) which was isolated along with the formation of 2 was separated using preparative thin-layer chromatography (TLC), allowing the characterization of the pure material. It was isolated as colourless crystals and characterized using 1H and 11B{1H} NMR and IR spectroscopy and single-crystal X-ray crystallography.25 In order to confirm the spectroscopic assignments and to determine the solid-state structure of II, X-ray structure analysis was also undertaken (Fig. S29‡). The crystal structure of II resembles that of [(Cp2Zr)2B5H11] (I).13 The core geometry can be described as arachno-triborane.26 It is worth noting that all of the B–B bond distances of the B3 core in II are significantly shorter as compared to the other [B3H8]− compounds, such as [Na(B3H8)], [Cs(B3H8)], [Cr(B3H8)2] and I.13,274. Conclusions
In summary, we have presented a facile synthetic method for two early-transition-metal-protected heptaborane compounds 1 and 2. They were synthesized using the borane condensation method utilizing group 4 metal borohydride compounds as the starting materials. Single crystal X-ray crystallography revealed the ubiquitous presence of a pentagonal bipyramidal B7 core. Our synthetic route successfully overcomes challenges faced previously in the continuing effort of isolating [B7H7]2− and its derivatives. The theoretical study revealed the intriguing role of the early transition metals in stabilizing the borane fragments via multicenter coordination bonding. The results described here lead to a new path towards the search for higher borane frameworks that go beyond the icosahedral topology in the coordination sphere of early transition metals. The current work may provide strategies for eventually synthesizing borospherenes and other novel boron clusters,28 for which CpM-type ligands have been suggested as ideal protecting ligands.29Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported financially by the Science and Engineering Research Board (SERB) (Project No. EMR/2015/001274), New Delhi, India, and the U. S. National Science Foundation (DMR-1655066). Computational facilities of IIT Madras and the Center for Computation and Visualization (CCV) of Brown University are gratefully acknowledged. The authors would also like to thank Dr Tun-Li Shen for help with the mass spectrometry experiments and Dr Abhishek Banerjee, IIT Madras for help with NMR spectroscopy. B. M. and S. K. thank IIT Madras for research fellowships. A. D. and K. S. thank UGC and CSIR, respectively, for research fellowships.Notes and references
- A. Stock, in Hydrides of Boron and Silicon, Cornell University Press, Ithaca, NY, 1933 Search PubMed.
- (a) J. D. Kennedy, Prog. Inorg. Chem., 1986, 36, 211 Search PubMed; (b) P. v. R. Schleyer, G. Subramanian, H. Jiao, K. Najafian and M. Hofmann, in Advances in Boron Chemistry, ed. W. Siebert, The Royal Society of Chemistry, Cambridge, U.K., 1997 Search PubMed; (c) E. D. Jemmis, M. M. Balakrishnarajan and P. D. Pancharatna, Chem. Rev., 2002, 102, 93–144 CrossRef CAS PubMed; (d) H. Nöth, in Homonuclear Boron Clusters in Molecular Clusters of the Main Group Elements, ed. M. Driess and H. Nöth, Wiley-VCH, Germany, 2005 Search PubMed.
- (a) G. B. Dunks, M. M. McKown and M. F. Hawthorne, J. Am. Chem. Soc., 1971, 93, 2541–2543 CrossRef; (b) A. S. F. Boyd, A. Burke, D. Ellis, D. Ferrer, B. T. Giles, M. A. Laguna, R. McIntosh, S. A. Macgregor, D. L. Ormsby, G. M. Rosair, F. Schmidt, N. M. M. Wilson and A. Welch, Pure Appl. Chem., 2003, 75, 1325–1333 CrossRef CAS; (c) G.-X. Jin, Coord. Chem. Rev., 2004, 248, 587–602 CrossRef CAS; (d) J. Zhang and Z. Xie, Chem.–Asian J., 2010, 5, 1742–1757 CrossRef CAS PubMed; (e) D. K. Roy, S. K. Bose, R. S. Anju, B. Mondal, V. Ramkumar and S. Ghosh, Angew. Chem., Int. Ed., 2013, 52, 3222–3226 CrossRef CAS PubMed.
- (a) W. L. Lipscomb, in Boron Hydrides, Benjamin, New York, 1963 Search PubMed; (b) J. L. Boone, J. Am. Chem. Soc., 1964, 86, 5036 CrossRef CAS; (c) Boron Hydride Chemistry, ed. E. L. Muetterties, Academic Press, New York, 1975 Search PubMed.
- (a) A. R. Pitochelli and F. M. Hawthorne, J. Am. Chem. Soc., 1960, 82, 3228–3229 CrossRef CAS; (b) F. Klanberg and E. L. Muetterties, Inorg. Chem., 1966, 5, 1955–1960 CrossRef CAS; (c) O. Volkov, N. P. Rath and L. Barton, J. Organomet. Chem., 2003, 680, 212–217 CrossRef CAS.
- W. N. Lipscomb, A. R. Pitochelli and M. F. Hawthorne, J. Am. Chem. Soc., 1959, 81, 5833–5834 CrossRef CAS.
- J. A. Wunderlich and W. N. Lipscomb, J. Am. Chem. Soc., 1960, 82, 4427–4428 CrossRef CAS.
- A. Franken, H. Thomsen and W. Preetz, Z. Naturforsch., B, 1996, 51, 744–746 CrossRef CAS.
- F. Klanberg, D. R. Eaton, L. J. Guggenberger and E. L. Muetterties, Inorg. Chem., 1967, 6, 1271–1281 CrossRef CAS.
- (a) C. E. Housecroft, R. Snaith, K. Moss, R. E. Mulvey, M. E. O’Neill and K. Wade, Polyhedron, 1985, 4, 1875–1881 CrossRef CAS; (b) Y.-F. Shen, C. Xu and L.-J. Cheng, RSC Adv., 2017, 7, 36755–36764 RSC.
- F. Schlüter and E. Bernhardt, Inorg. Chem., 2011, 50, 2580–2589 CrossRef PubMed.
- W. Einholz, in Advances in Boron Chemistry, ed. W. Siebert, The Royal Society of Chemistry, Cambridge, 1997, p. 44 Search PubMed.
- (a) R. S. Anju, D. K. Roy, B. Mondal, V. Ramkumar and S. Ghosh, Organometallics, 2013, 32, 4618–4623 CrossRef CAS; (b) D. K. Roy, B. Mondal, A. De, S. Panda and S. Ghosh, Organometallics, 2015, 34, 908–912 CrossRef CAS.
- (a) R. L. Thomas, N. P. Rath and L. Barton, J. Am. Chem. Soc., 1997, 119, 12358–12375 CrossRef CAS; (b) R. L. Thomas, N. P. Rath and L. Barton, Inorg. Chem., 2002, 41, 67–75 CrossRef CAS PubMed.
- T. J. Marks and J. R. Kolb, Chem. Rev., 1977, 77, 263–293 CrossRef CAS.
- R. K. Nanda and M. G. H. Wallbridge, Inorg. Chem., 1964, 3, 1798 CrossRef.
- R. W. Broach, I.-S. Chuang, T. J. Marks and J. M. Williams, Inorg. Chem., 1983, 22, 1081–1084 CrossRef CAS.
- B. Albert and K. Hofmann, Z. Anorg. Allg. Chem., 1999, 625, 709–713 CrossRef CAS.
- S. Ghosh, B. C. Noll and T. P. Fehlner, Angew. Chem., Int. Ed., 2005, 44, 2916–2918 CrossRef CAS PubMed.
- (a) D. M. P. Mingos, Nature (London), Phys. Sci., 1972, 236, 99–102 CrossRef CAS; (b) D. M. P. Mingos, Acc. Chem. Res., 1984, 17, 311–319 CrossRef CAS.
- B. L. Guennic, H. Jiao, S. Kahlal, J.-Y. Saillard, J.-F. Halet, S. Ghosh, M. Shang, A. M. Beatty, A. L. Rheingold and T. P. Fehlner, J. Am. Chem. Soc., 2004, 126, 3203–3217 CrossRef PubMed.
- E. D. Jemmis, J. Am. Chem. Soc., 1982, 104, 7017–7020 CrossRef CAS.
- (a) P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed; (b) Z. Chen, C. S. Wannere, C. Cominboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- D. Y. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207–5217 RSC.
- The mass spectrum of II showed a molecular ion peak at m/z 686.4 (Fig. S9‡). The 1H NMR spectrum displayed three signals at δ = 5.92, 5.90 and 5.89 ppm in a 2:1:1 ratio which rationalizes the presence of two types of Cp ligand. The presence of Zr–H–B (δ = −4.16 ppm) and B-H-B (δ = −2.74 ppm) was confirmed from 1H NMR. The 11B{1H} NMR spectrum showed three resonances at δ = 4.6, 2.0 and −3.8 ppm in a 2:1:2 ratio.
- Although the positions of the hydrogen atoms were not able to be fully located in the crystal structure, they were assigned based on the spectroscopic data, as shown in Scheme 1. The different B–B distances of the B3 unit in II are due to the presence of bridged hydrogen along the B2–B3 bond.
- (a) H. J. Deiseroth, O. Sommer, H. Binder, K. Wolfer and B. Frei, Z. Anorg. Allg. Chem., 1989, 571, 21–28 CrossRef CAS; (b) D. Y. Kim and G. S. Girolami, J. Am. Chem. Soc., 2006, 128, 10969–10977 CrossRef CAS PubMed; (c) A. C. Dunbar, J. A. Macor and G. S. Girolami, Inorg. Chem., 2014, 53, 822–826 CrossRef CAS PubMed.
- (a) H. J. Zhai, Y. F. Zhao, W. L. Li, Q. Chen, H. Bai, H. S. Hu, Z. A. Piazza, W. J. Tian, H. G. Lu, Y. B. Wu, Y. W. Mu, G. F. Wei, Z. P. Liu, J. Li, S. D. Li and L. S. Wang, Nat. Chem., 2014, 6, 727–731 CrossRef CAS PubMed; (b) Z. A. Piazza, H. S. Hu, W. L. Li, Y. F. Zhao, J. Li and L. S. Wang, Nat. Commun., 2014, 5, 3113 Search PubMed; (c) L. S. Wang, Int. Rev. Phys. Chem., 2016, 35, 69–142 CrossRef CAS; (d) W. L. Li, X. Chen, T. Jian, T. T. Chen, J. Li and L. S. Wang, Nat. Rev. Chem., 2017, 1, 0071 CrossRef.
- N. Karmodak and E. D. Jemmis, Chem.–Asian J., 2016, 11, 3350–3354 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Thomas P. Fehlner on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Detailed synthesis and characterizations; mass spectrometry details; NMR and IR spectral details; X-ray analysis details; bonding analyses and computational details. CCDC 1568096–1568098. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc05014c |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |