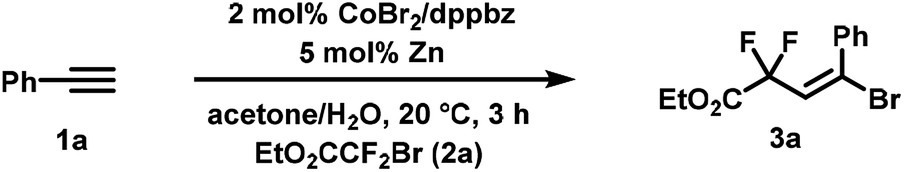DOI:
10.1039/C7SC04916A
(Edge Article)
Chem. Sci., 2018,
9, 1795-1802
Stereoselective cobalt-catalyzed halofluoroalkylation of alkynes†
Received
16th November 2017
, Accepted 3rd January 2018
First published on 5th January 2018
Abstract
Stereoselective additions of highly functionalized reagents to available unsaturated hydrocarbons are an attractive synthetic tool due to their high atom economy, modularity, and rapid generation of complexity. We report efficient cobalt-catalyzed (E)-halofluoroalkylations of alkynes/alkenes that enable the construction of densely functionalized, stereodefined fluorinated hydrocarbons. The mild conditions (2 mol% cat., 20 °C, acetone/water, 3 h) tolerate various functional groups, i.e. halides, alcohols, aldehydes, nitriles, esters, and heteroarenes. This reaction is the first example of a highly stereoselective cobalt-catalyzed halo-fluoroalkylation. Unlike related cobalt-catalyzed reductive couplings and Heck-type reactions, it operates via a radical chain mechanism involving terminal halogen atom transfer which obviates the need for a stoichiometric sacrificial reductant.
1 Introduction
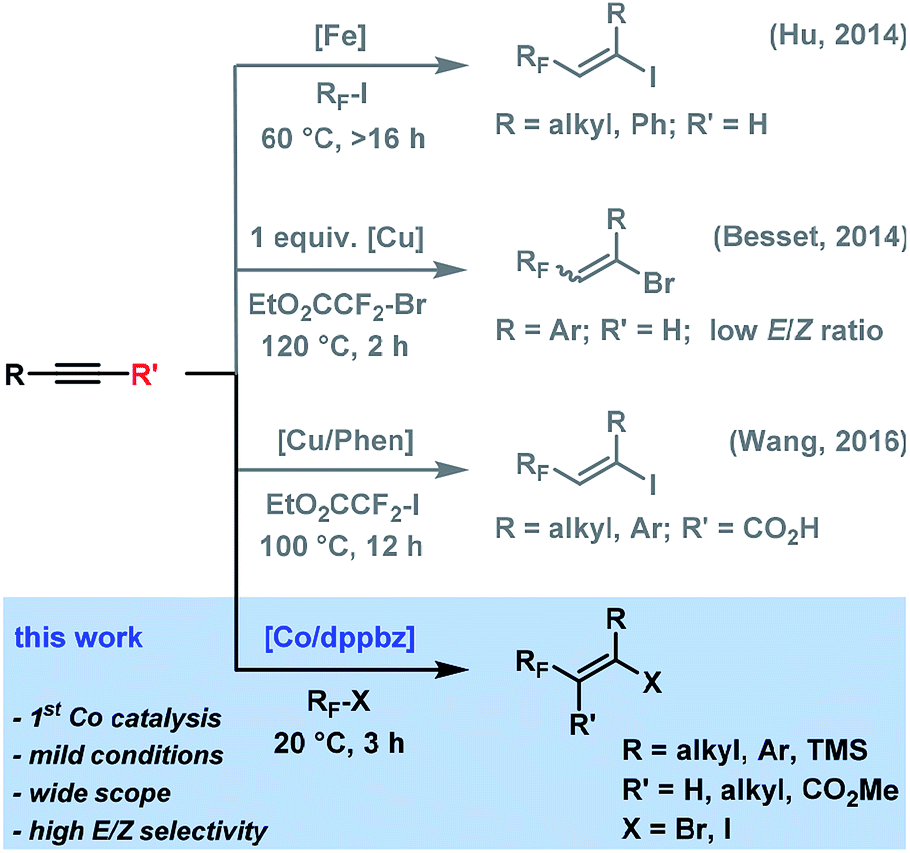
Fluorinated hydrocarbons constitute key structural motifs in many bioactive molecules, agrochemicals, and pharmaceuticals due to their high metabolic stability, lipophilicity, and bioavailability compared with the parent compounds.1 Fluoroalkylation methods of easily accessible precursors have therefore attracted great interest in the past years.2,3 While many protocols are substitution processes that require highly pre-functionalized starting materials and produce unwanted by-products, direct additions to unsaturated hydrocarbons exhibit higher modularity and atom-efficiency and provide ample opportunities of regio- and stereocontrol. The addition of halo-fluoroalkanes to alkynes is an especially attractive tool due to the easy availability of the reagents and the great synthetic versatility of the resultant halo-fluoroalkenes. Many methods operate via an atom transfer radical addition (ATRA) mechanism in the presence of radical initiators (e.g. BEt3, AIBN, Na2S2O3 or light)4 that showed narrow substrate scope and poor selectivity. Mechanistically closely related transition metal-mediated halo-fluoroalkylations have been recently reported, but with a narrow focus on iodofluoro-alkylations and/or moderate stereo-control (Scheme 1). Hu et al. devised an iron-catalyzed addition of perfluoroalkyl iodide to alkynes with moderate to good E/Z-selectivities in the presence of Cs2CO3. The radical reaction with alkyl-substituted alkynes required long reaction times at 60 °C and could not convert perfluoroalkyl bromides.5 Besset et al. postulated a different mechanism for the copper-mediated synthesis of difluoromethyl alkenes from BrCF2CO2Et and alkynes. However, significantly lower stereoselectivities were obtained and stoichiometric amounts of copper salt were employed.6 Very recently, Wang and co-workers reported a copper-catalyzed decarboxylative ATRA reaction between ICF2CO2Et and substituted propiolic acids.7
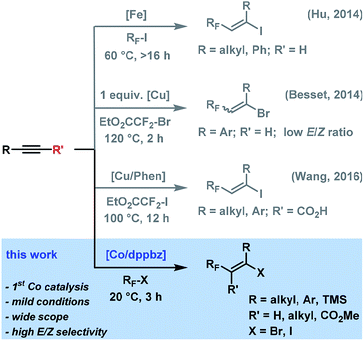 |
| | Scheme 1 Metal-mediated halo-fluoroalkylations of alkynes. | |
Despite the developments of novel iron- and copper-catalyzed procedures, the reactions generally utilize expensive fluoroalkyl iodides as starting materials, high catalyst loadings, long reaction times, and high reaction temperatures. An efficient and robust yet highly stereoselective method that operates at mild conditions and low catalyst loadings and that is applicable to various fluoroalkyl halides would constitute a significant advancement of the current technology and have considerable use in the synthesis of densely functionalized fluorinated building blocks. To the best of our knowledge, there are no literature reports of ATRA reaction between alkyl halides and alkynes with low-valent cobalt catalysts. Here, we report the cobalt-catalyzed halofluoro-alkylation of alkynes which enables the highly regioselective and stereoselective synthesis of a diverse set of halofluoroalkenes under unprecedentedly mild conditions (Scheme 1).
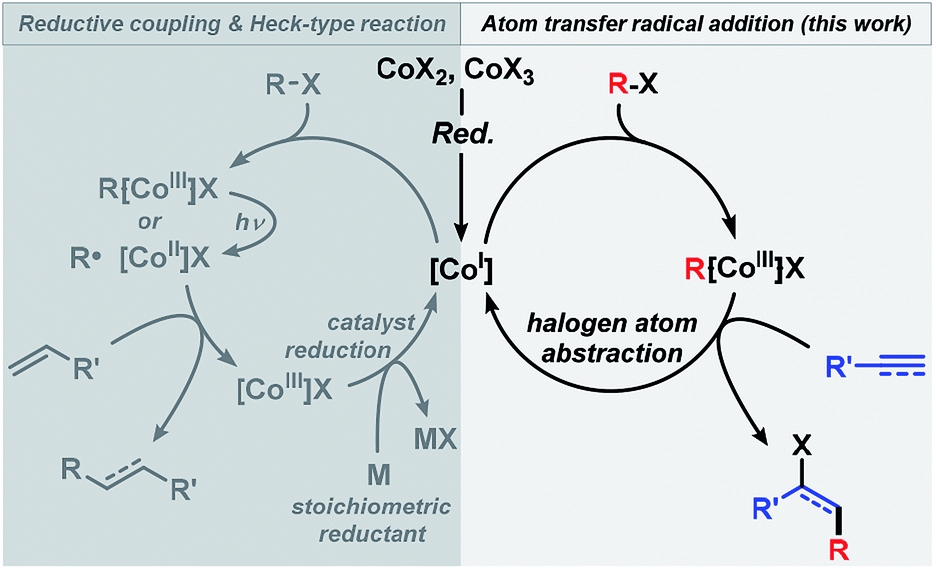
The utility of alkyl halides in cross-couplings and reductive additions has recently been greatly enhanced by the development of low-valent iron group metal catalysts (Fe, Co, Ni)8–10 that engage in facile alkyl-X activation. The high propensity of late 3d transition metals to undergo single-electron transfer (SET) processes often results in the intermediacy of carbon-centered radical species.8g,h,9h,10f The reductive formation of alkyl radicals from alkyl-X electrophiles is thermodynamically favoured when the formal electron septet-carbon is stabilized by heteroatoms, conjugation, hyperconjugation, or inductive effects. Efficient cobalt catalysts have been reported for several reductive coupling reactions between alkyl halides and Michael-type acceptors as well as for Heck-type reactions between alkyl halides and alkenes.11 These processes mostly follow the same mechanistic scenarios involving (i) initial formation of a low-valent Co(I) species from a Co(II,III) precursor in the presence of a reductant; (ii) reductive cleavage of the alkyl halide to give an alkyl radical R˙ and a Co(II) complex (by SET activation or homolysis of R–Co(III)), (iii) addition of R˙ to the olefin and formation of an organocobalt species that is subject to disproportionation (Heck-type reaction) or hydrolysis (reductive coupling) to release the Co(III) complex, (iv) regeneration of the Co(I) catalyst with a stoichiometric reductant (Scheme 2, left). We surmised that an ATRA reaction between alkyl halides and alkynes could follow a similar mechanism using low-valent Co catalysts but would require only catalytic amounts of a reductant (Scheme 2, right). Cobalt-catalyzed Heck-type and reductive coupling reactions were realized in the presence of stoichiometric reductants such as Zn, Mn, and Grignard reagents11 which effect the Co(III) → Co(I) reduction. Cobalt-catalyzed ATRA reactions of alkynes with alkyl halides have not been reported.
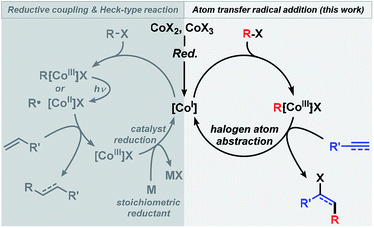 |
| | Scheme 2 Generalized mechanistic dichotomy of radical addition reactions under low-valent cobalt catalysis. | |
2 Results and discussion
2.1 Discovery of a cobalt-catalyzed ATRA reaction
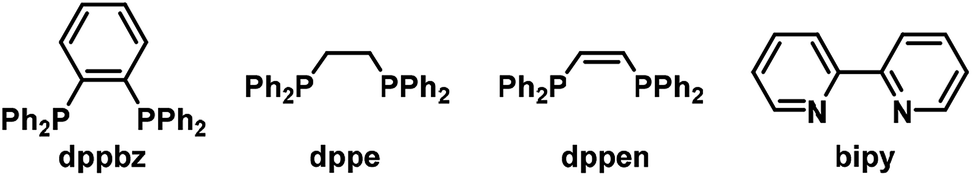
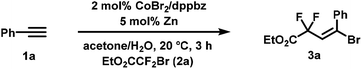
We commenced our investigations with the reaction of phenyl-acetylene (1a) and ethyl bromodifluoroacetate (2a). Variations of reaction conditions, solvents, and catalysts led to an optimized procedure that utilized a three-component catalyst comprising of CoBr2, dppbz (1,2-bis(diphenylphosphino)benzene) and zinc in acetone/water at 20 °C to furnish the synthesis of the desired adduct ethyl 4-bromo-2,2-difluoro-4-phenylbut-3-enoate (3a) in 83% yield (87% GC yield, Table 1, entry 1). Significantly lower yields were obtained when replacing dppbz with other bidentate phosphines or 2,2′-bipyridine (entries 5–8). Other transition metals were inactive (entries 9–12). CoCl2 and CoCl2·4H2O exhibited similar activity (entries 13, 14).
Table 1 Selected optimization experimentsa
|
|
| Entry |
Deviations from conditions above |
3a
[%] |
|
Conditions: 1a (0.3 mmol), 2a (0.45 mmol), CoBr2 (2 mol%), dppbz (2 mol%), Zn (5 mol%), 0.6 mL acetone/H2O, 20 °C, 3 h.
GC yield vs. internal n-dodecane.
1 mol% CoBr2, 1 mol% dppbz, 10 mol% Zn.
5 mol% CoBr2, 5 mol% dppbz, 20 mol% Zn.
|
|
1
|
None
|
87 (63)c |
| 2 |
Without H2O |
69 |
| 3 |
MeCN instead of acetone |
65 |
| 4 |
Without CoBr2 or Zn or dppbz |
0 |
| 5 |
dppe instead of dppbz |
29d |
| 6 |
dppen instead of dppbz |
31d |
| 7 |
dppf instead of dppbz |
0d |
| 8 |
bipy instead of dppbz |
0d |
| 9 |
FeBr2 instead of CoBr2 |
0d |
| 10 |
NiCl2 instead of CoBr2 |
0d |
| 11 |
CrCl3 instead of CoBr2 |
0d |
| 12 |
Cp2TiCl2 instead of CoBr2 |
0d |
| 13 |
CoCl2 instead of CoBr2 |
82d |
| 14 |
CoCl2·4H2O instead of CoBr2 |
83d |
|
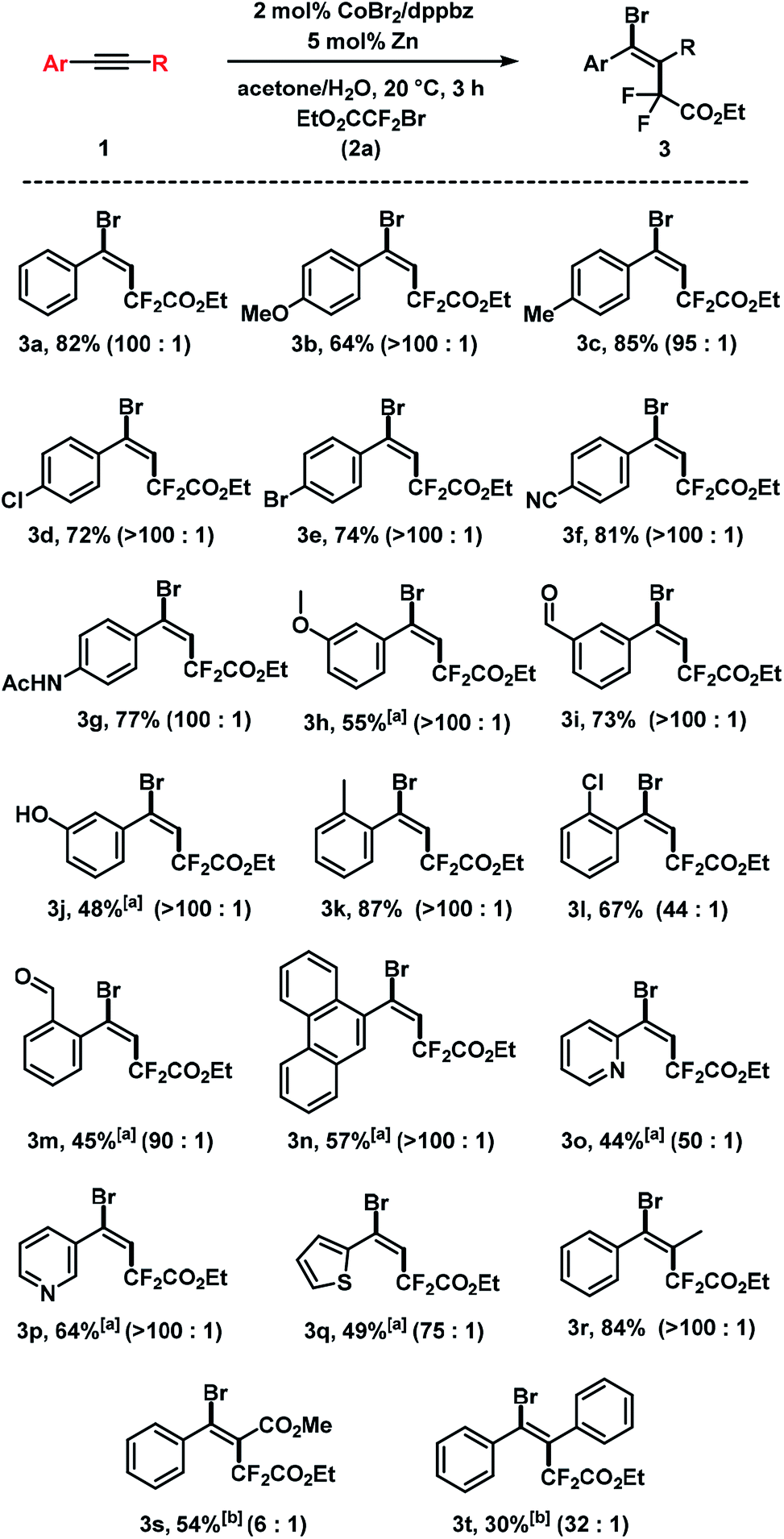
2.2 Substrate scope
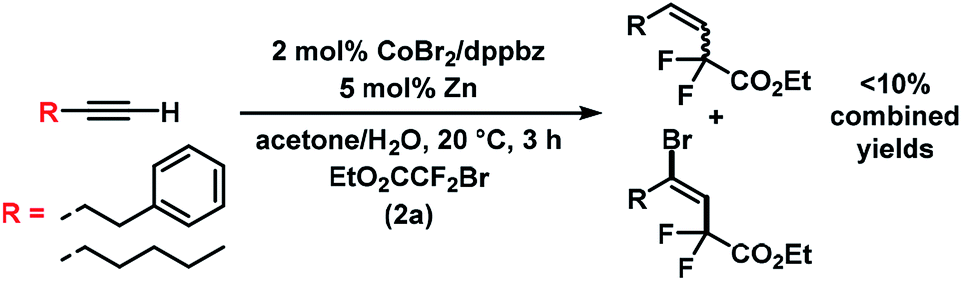
The substrate scope of reactions between terminal and internal aryl acetylenes and 2a (Scheme 3). Many substitution patterns were tolerated (ortho, meta, para, electron-withdrawing, electron-donating substituents). The reaction displayed remarkable compatibility with functional groups including aldehydes, halides, nitriles, amides, hydroxyl, pyridines, thiophenes. All products were obtained with perfect regiocontrol and high E/Z diastereo-selectivity (>50/1). However, alkyl-substituted terminal alkynes fared poorer (Scheme 4). Reactions of 2a with 1-heptyne and 4-phenyl-1-butyne, respectively, afforded mixtures of bromo-difluoro-alkylation and hydrodifluoroalkylation products in low yields.
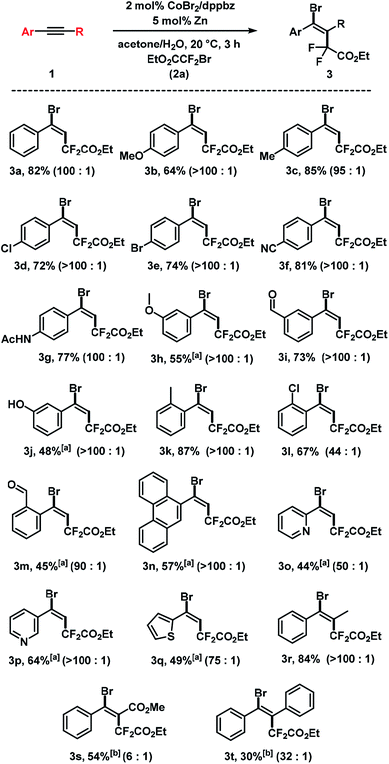 |
| | Scheme 3 Cobalt-catalyzed bromo-carboxydifluoromethylation of alkynes. Standard conditions: 1 (0.3 mmol), 2a (0.45 mmol), CoBr2 (2 mol%), dppbz (2 mol%) and Zn (5 mol%), 0.6 mL acetone/H2O, 20 °C, 3 h under N2. Isolated yields are given; E/Z ratios in parentheses (by 19F NMR). [a] 20 mol% Zn. [b] 40 mol% Zn, 8 h. | |
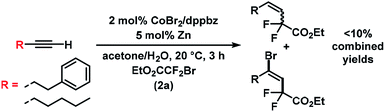 |
| | Scheme 4 Reactions with alkyl-substituted terminal alkynes. | |
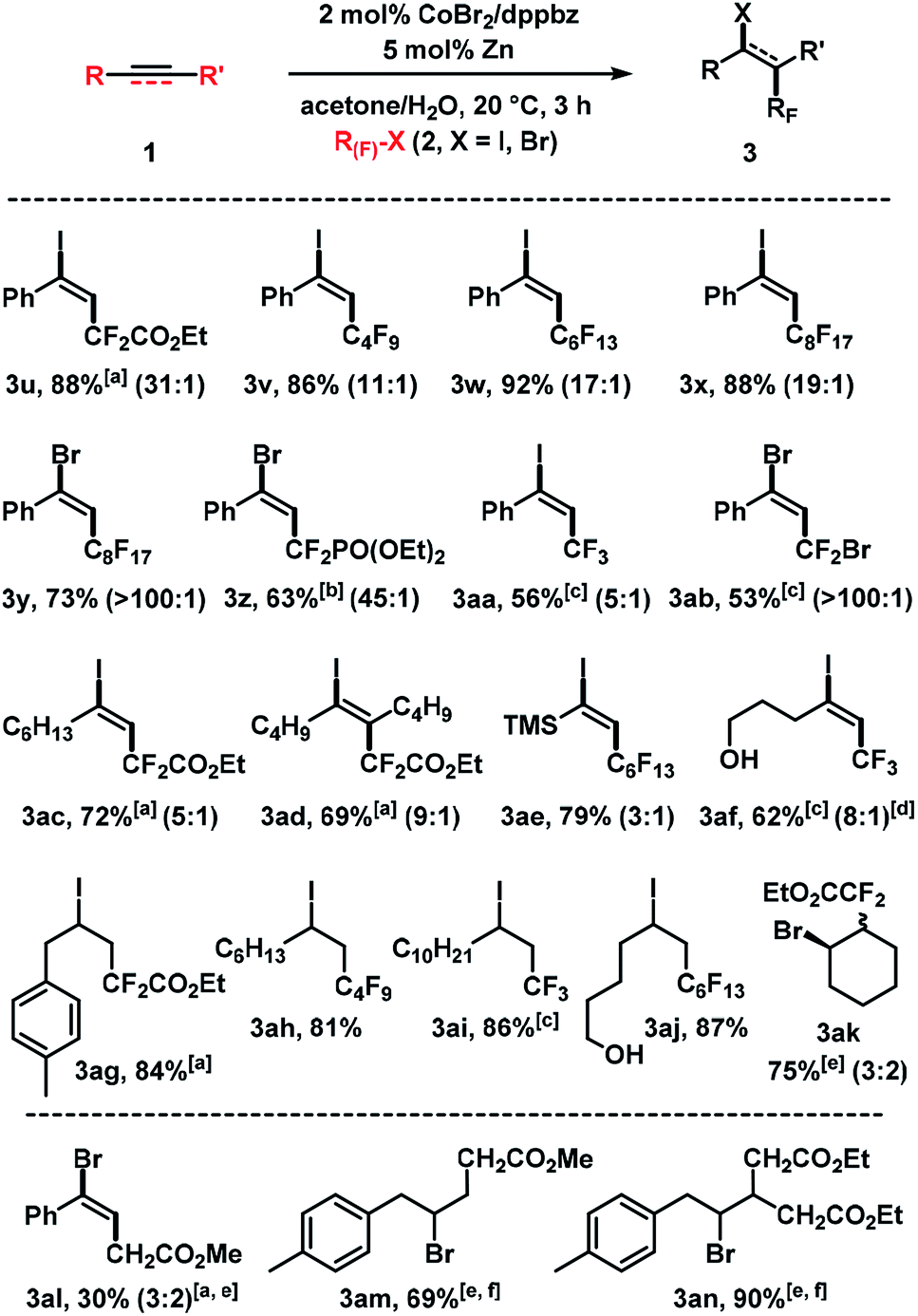
We then examined the cobalt-catalyzed halofluoroalkylation with different fluoroalkyl halides (Scheme 5). Iododifluoroacetate ICF2CO2Et, perfluoroalkyl iodides such as C4F9I, C6F13I and C8H17I, and the perfluoroalkyl bromide C8F17Br were competent electrophiles which afforded the desired adducts in good to excellent yields. The fluoroalkyl bromides gave generally better E/Z selectivities than the iodides. While this trend is in full agreement with the literature, it can now be harnessed at much milder conditions (room temp., 2 mol% catalyst, 3 h). BrCF2PO(OEt)2, CF3I, and CF2Br2 afforded slightly lower yields; the reaction with CF3I exhibited low stereocontrol. Reactions of alkyl-substituted alkynes with fluoroalkyl iodides gave good yields and moderate E/Z selectivities (3ac–3af). The reaction conditions were also applied to reactions of (cyclo)alkenes with halofluoroacetates (3ag–3ak). A method extension to reactions of simple bromo-acetates with alkenes gave the desired adducts 3al–3an.
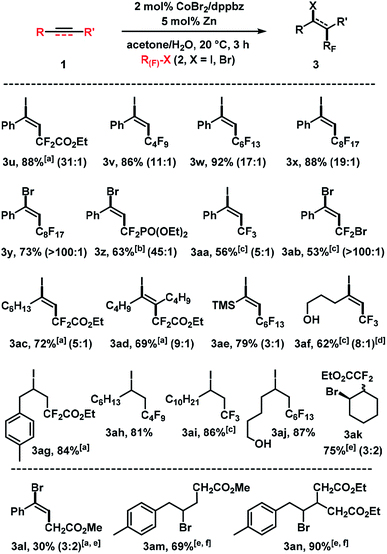 |
| | Scheme 5 Cobalt-catalyzed halo-fluoroalkylation of alkynes and alkenes. Standard conditions: 1 (0.3 mmol), 2 (0.45 mmol), CoBr2 (2 mol%), dppbz (2 mol%), Zn (5 mol%), 0.6 mL acetone/H2O, 20 °C, 3 h under N2. Isolated yields given; E/Z ratios in parentheses (by 19F NMR). [a] 20 mol% Zn. [b] RF–Br (1.0 equiv.), 10 mol% Zn. [c] RF–X (3.0 equiv.), 10 mol% Zn. [d]E/Z ratio of isolated products. [e] 10 mol% Zn. [f] 6 h. | |
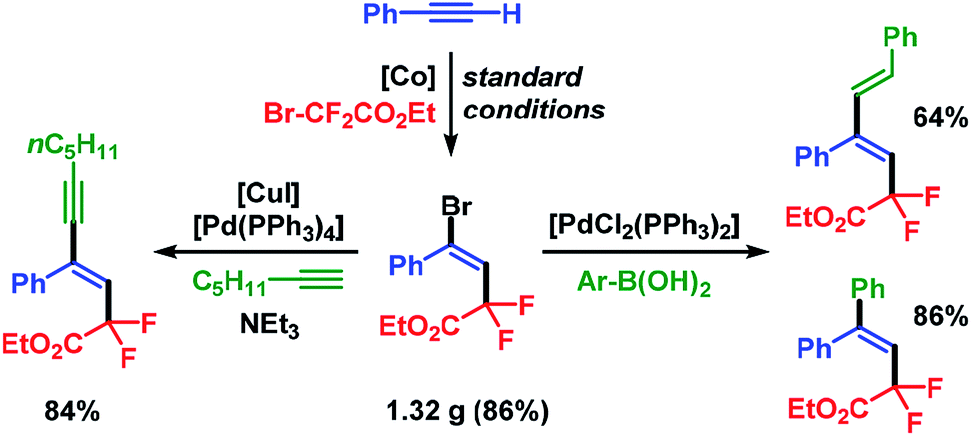
The synthetic utility of the stereoselective cobalt-catalyzed halo-fluoroalkylation protocol was demonstrated in a gram-scale setup which delivered pure ethyl (E)-4-bromo-2,2-difluoro-4-phenyl-but-3-enoate (3a) in 86% isolated yield (1.32 g) after 3 h. Substitution of the Br substituent in 3a by Sonogashira and Suzuki cross-coupling reactions, respectively, afforded the fluorinated alkenes in very good yields and with complete retention of stereochemistry (Scheme 6).
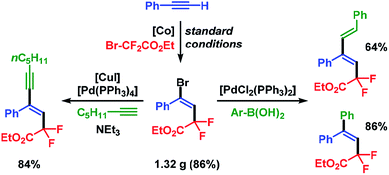 |
| | Scheme 6 Post-ATRA transformations by cross-coupling reactions. | |
2.3 Mechanistic studies
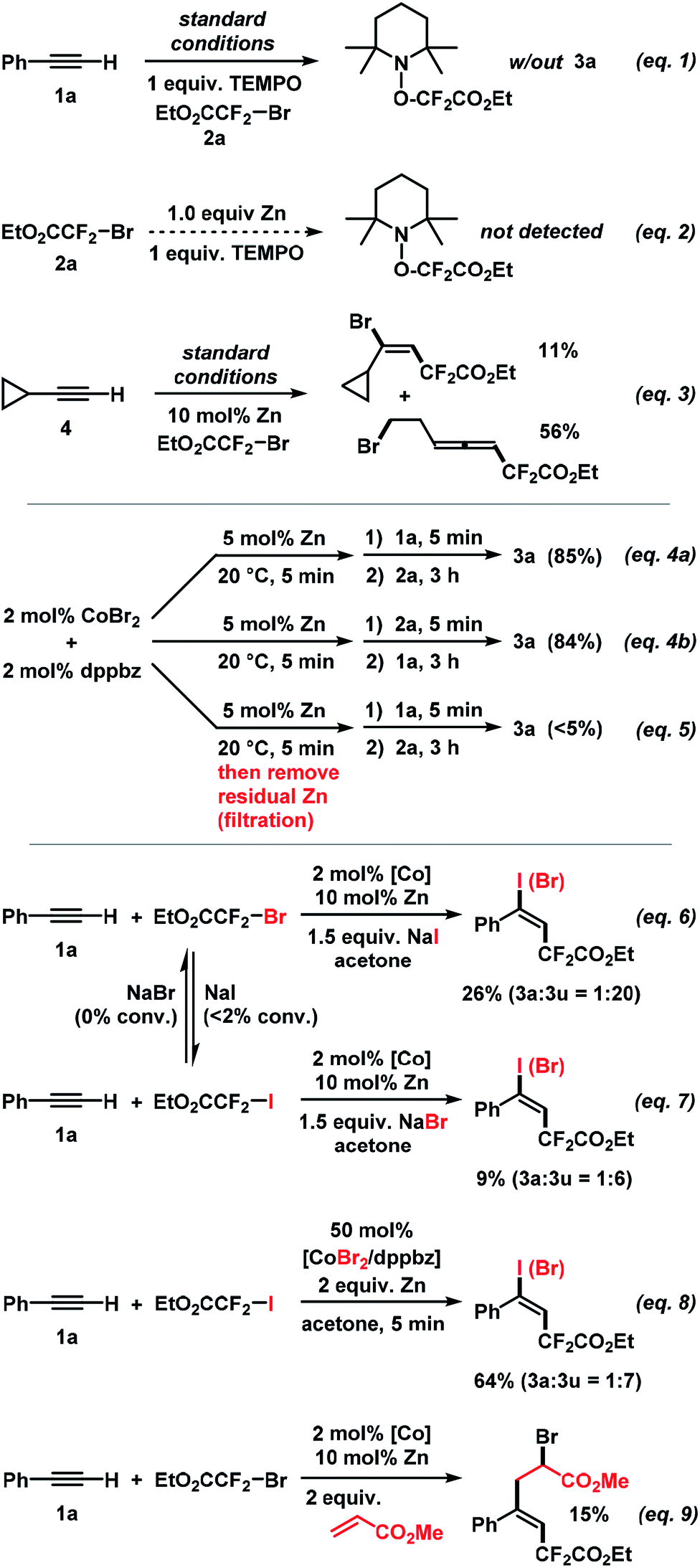
Further attention was devoted to the study of the reaction mechanism. In addition to the initial optimization reactions (Table 1), key mechanistic experiments were conducted. The model reaction between 1a and 2a was completely inhibited in the presence of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO). The TEMPO–CF2CO2Et adduct was observed by mass spectrometry (Scheme 7, eqn (1)). The TEMPO–CF2CO2Et adduct was not detected when treating 2a with equimolar Zn which supports the notion that the SET reduction of the alkyl halide is induced by the cobalt catalyst (Scheme 7, eqn (2)). Upon employment of cyclo-propylacetylene (4), the vinylcyclopropane product was formed in 11% yield while ring-opening to the 7-bromohepta-3,4-dienoate (56%) was the major pathway. This is in full agreement with the intermediacy of an internal vinyl radical formed by radical addition of EtO2CCF2˙ to the alkyne (Scheme 7, eqn (3)). Identical rates and yields were observed in reactions where 1a and 2a were successively added to the catalyst solution. The reverse order of addition (2a, then 1a) gave an identical result. Importantly, no product was formed when prior to the addition of 1a and 2a – the catalyst suspension (CoBr2, dppbz, Zn) was filtered (to remove residual zinc) or when the supernatant solution was decanted into a new reaction vessel (eqn (4)). These experiments suggest that the initially formed Co(I) species alone cannot catalyze the reaction but requires the presence of zinc, at least for the first turnover of the catalytic mechanism. Zn is employed only in catalytic amounts (5 mol%, i.e. 2.5 equiv. per Co)! The addition of sodium iodide and sodium bromide, respectively, shed light on the nature of the operating halogen transfer. 1a and 2a reacted with added NaI (1.5 equiv.) to give the iodo adduct 3u as major product (3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3u = 1:20, Scheme 7, eqn (6)). With NaBr added, the reaction between 1a and 2b gave 3a and 3u in a 1:6 ratio (Scheme 7, eqn (7)). Control experiments documented that no EtO2CCF2I was converted into EtO2CCF2Br using NaBr as additive; only minimal amounts of EtO2CCF2Br (<2%) were converted to EtO2CCF2I with NaI as additive under the same conditions. A similar outcome was observed when the standard reaction was performed with 50 mol% CoBr2/dppbz (3a:3u = 1:7, Scheme 7, eqn (8)). These experiments document that the halogen atom X in the product does not originate from the electrophilic RFX via a direct radical chain transfer but is transferred from the cobalt catalyst. This is a fine but important distinction from previously reported ATRA reactions that all involved halogen transfer from RFX to the vinyl radical. This has great implications for catalyst design and reaction development as the thermodynamics and kinetics of the halogen atom transfer step are no longer depending on the nature of the employed substrates but can be finely tuned through the stereoelectronic properties of the catalyst. We further believe that halogen atom transfer to a vinyl radical intermediate (rather than a vinyl cation) is operative: (i) the addition of water (as a nucleophile) resulted in no product bearing oxo functions; (ii) the presence of methyl acrylate as a radical acceptor led to the formation of the heptene-1,7-dioate via radical insertion of the acrylate (eqn (9)). A cationic intermediate would not add to this Michael acceptor.
:3u = 1:20, Scheme 7, eqn (6)). With NaBr added, the reaction between 1a and 2b gave 3a and 3u in a 1:6 ratio (Scheme 7, eqn (7)). Control experiments documented that no EtO2CCF2I was converted into EtO2CCF2Br using NaBr as additive; only minimal amounts of EtO2CCF2Br (<2%) were converted to EtO2CCF2I with NaI as additive under the same conditions. A similar outcome was observed when the standard reaction was performed with 50 mol% CoBr2/dppbz (3a:3u = 1:7, Scheme 7, eqn (8)). These experiments document that the halogen atom X in the product does not originate from the electrophilic RFX via a direct radical chain transfer but is transferred from the cobalt catalyst. This is a fine but important distinction from previously reported ATRA reactions that all involved halogen transfer from RFX to the vinyl radical. This has great implications for catalyst design and reaction development as the thermodynamics and kinetics of the halogen atom transfer step are no longer depending on the nature of the employed substrates but can be finely tuned through the stereoelectronic properties of the catalyst. We further believe that halogen atom transfer to a vinyl radical intermediate (rather than a vinyl cation) is operative: (i) the addition of water (as a nucleophile) resulted in no product bearing oxo functions; (ii) the presence of methyl acrylate as a radical acceptor led to the formation of the heptene-1,7-dioate via radical insertion of the acrylate (eqn (9)). A cationic intermediate would not add to this Michael acceptor.
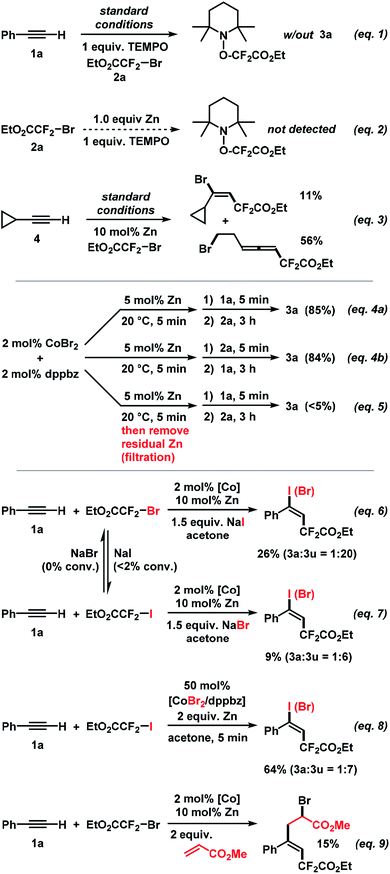 |
| | Scheme 7 Key mechanistic experiments. | |
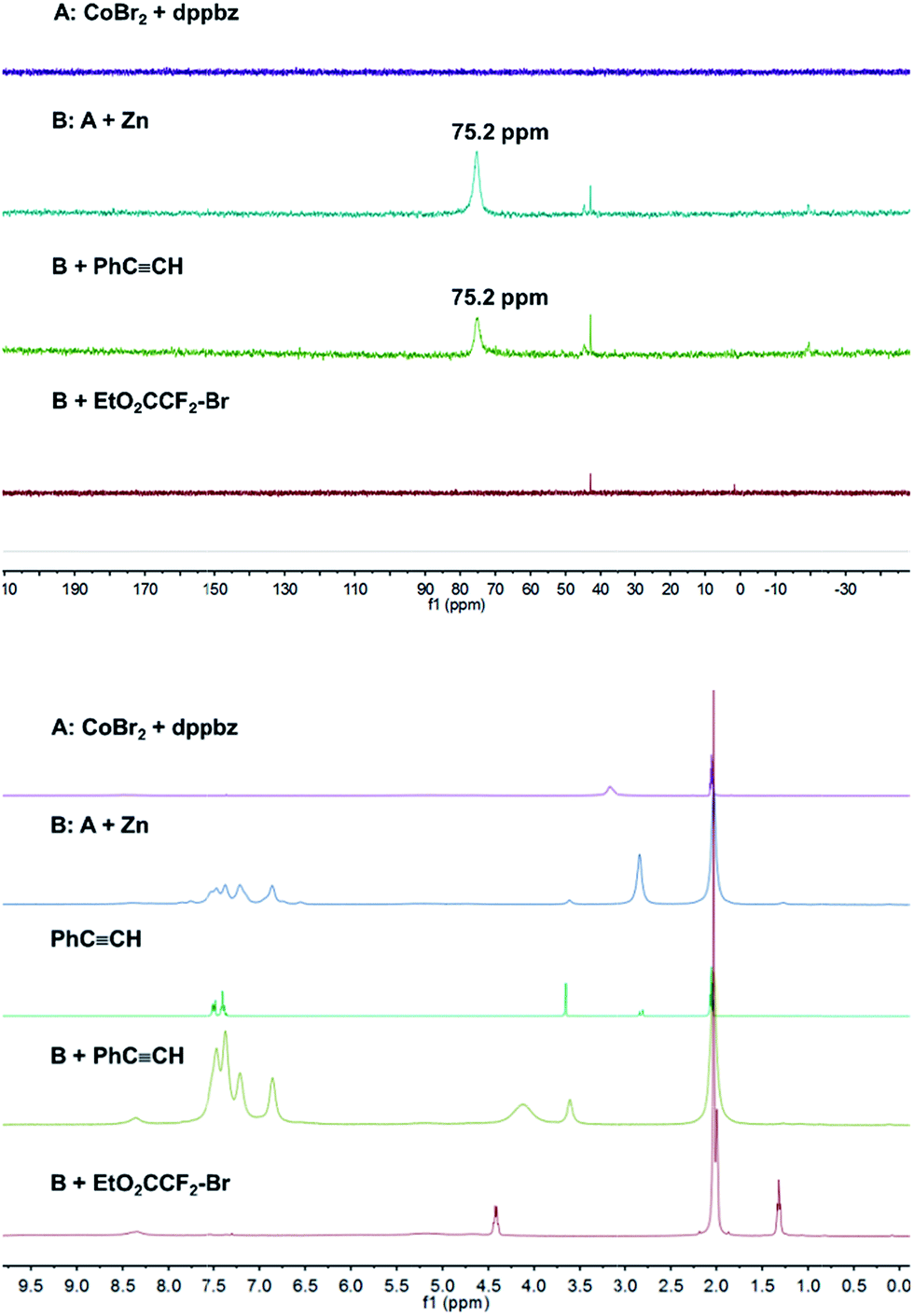
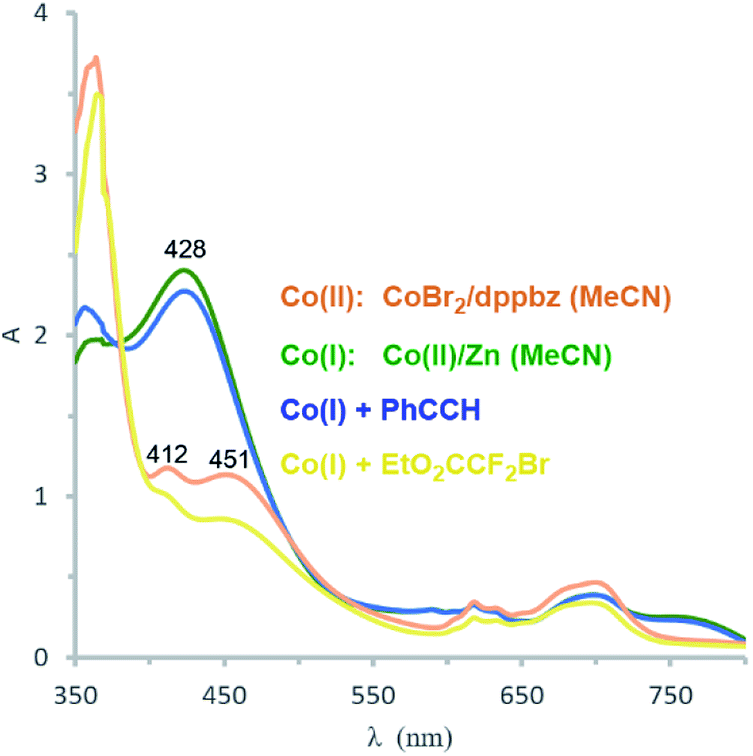
Catalyst formation and substrate additions were monitored by 31P NMR and 1H NMR spectroscopy (Fig. 1). The reduction of the (NMR silent) CoBr2/dppbz mixture with Zn resulted in a Co(I) species with a 31P resonance at 75.2 ppm. The 1H NMR spectrum of this low-spin Co(I) complex gave signals 7–8 ppm. No changes were observed in 31P and 1H NMR spectra when phenyl acetylene (1a) was added to Co(I) which suggests the absence of significant alkyne-catalyst coordination. On the other hand, complete disappearance of the 31P (75.2 ppm) and 1H (7–8 ppm) signals was observed upon addition of EtO2CCF2Br (2a). This is a direct consequence of the reductive activation of 2a which leads to a paramagnetic Co(II,III) species and the carbon-centered radical.12 These results are consistent with the UV-vis spectra (Fig. 2). Reduction of Co(II) with Zn (and removal of residual Zn) resulted in an intense absorption of the Co(I) complex at 428 nm (green curve). Addition of 1a to this solution gave no change of the absorption in this region (blue curve), whereas the addition of 2a to Co(I) led to immediate colour change and the appearance of two weak bands at 412 and 451 nm (yellow curve).
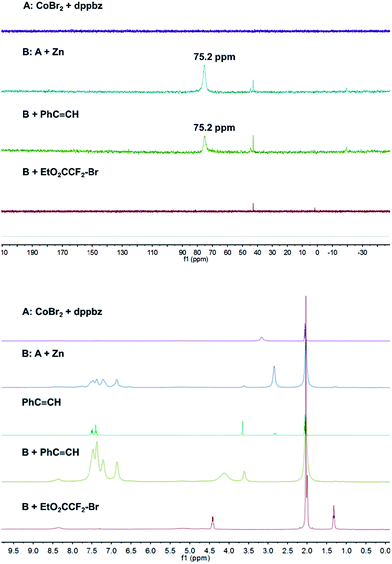 |
| | Fig. 1
31P NMR (top) and 1H NMR (bottom) monitoring of catalyst formation and reductive electrophile activation. | |
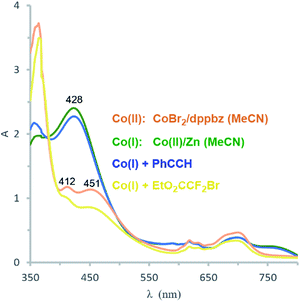 |
| | Fig. 2 UV-vis absorption spectra: CoBr2/dppbz (orange); after reduction of Co(II) with Zn to Co(I) (green); Co(I) with 1 equiv. 1a (blue); Co(I) with 1 equiv. of 2a (yellow). | |
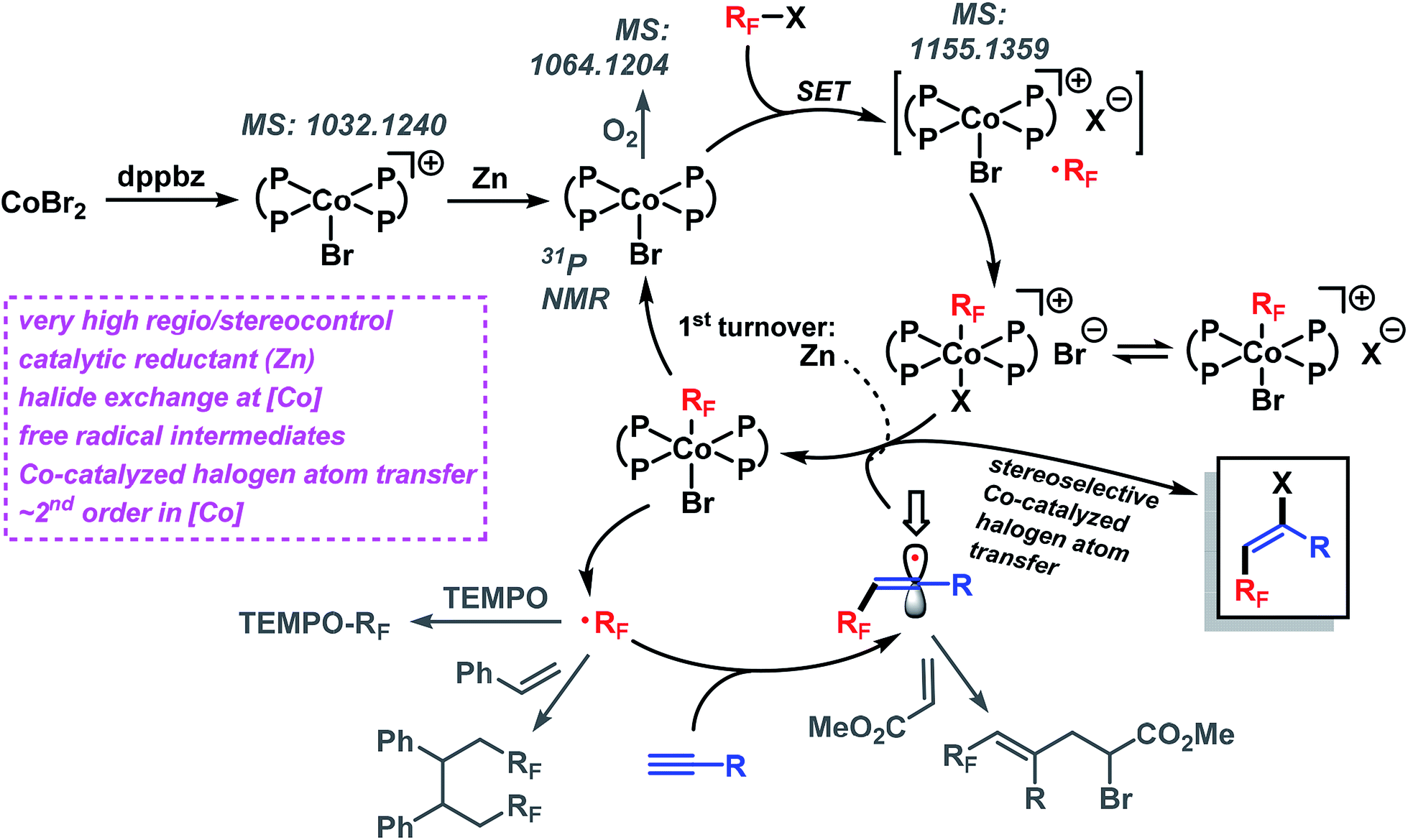
The standard reaction between 1a and 2a went to completion within 90 min (with 2 mol% catalyst) and 12 min (4 mol% catalyst), respectively. Analysis of the initial rates (0.5–8 min, 1–4 mol% catalyst) displayed a near-2nd order behavior of the catalyst concentration. We postulate the following reaction mechanism (Scheme 8). Complexation of dppbz with CoBr2 leads to the formation of [CoII(dppbz)2Br]+ as observed by the soft and inert mass spectrometric technique for sensitive organometallics LIFDI-MS (liquid injection field desorption ionization mass spectrometry). Reduction of the Co(II) complex with equimolar Zn generates the catalytically active [CoI(dppbz)2Br].12d Exposure to oxygen/air gives [CoIII(dppbz)2Br(O2)] (LIFDI-MS). [CoI(dppbz)2Br] effects the reductive single-electron activation of RFBr to give a [RFCoIII(dppbz)2Br]+ species (LIFDI-MS)12 which is presumably a direct result of rapid combination of the intermediate Co(II) complex and free radical RF˙.14 We have demonstrated that in the absence of residual zinc [RFCoIII(dppbz)2Br]+ is not a catalyst (vide supra). Therefore, we propose – in contrast to the light-induced Co–R homolysis11g,13 – the reduction of [RFCoIII(dppbz)2Br]+ by Zn in the first catalytic turnover to form the unstable complex [RFCoII(dppbz)2Br]. Dissociation of the fluoroalkyl radical RF˙ regenerates [CoI(dppbz)2Br]12h–j which can undergo another reductive activation of RFX to give [RFCoIII(dppbz)2Br]+. The catalytic amounts of Zn present in the reaction dictate that another mechanism operates from the 2nd turnover on, most likely an ATRA reaction involving halogen atom transfer from the cobalt complex [RFCoIII(dppbz)2Br]+. Accordingly, the addition of RF˙ to the alkyne results in the formation of a vinyl radical intermediate which undergoes rapid halogen atom abstraction from [CoIII(dppbz)2Br]+ to form the catalytically active Co(I) complex and RF˙.15 The high E-selectivity of the radical addition is a direct consequence of the steric hindrance by the RF group in the vinyl radical.16 The higher E/Z stereoselectivity of the bromoalkylation over the iodoalkylation reactions can be explained by the shorter Co–Br bond (vs. Co–I) in the key catalytic Co(III) species which effects an enhanced facial differentiation of the vinyl radical. The facile operation of this halogen atom transfer step with the intermediate vinyl radical is the key to the realization of an overall process that is catalytic in both metals, Co and Zn.
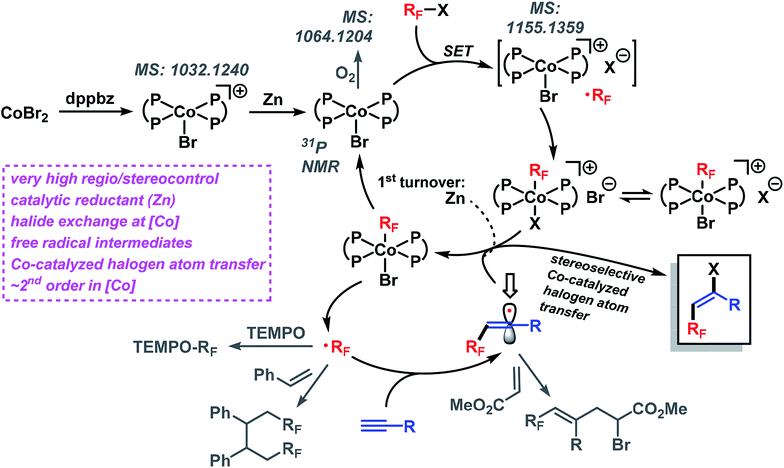 |
| | Scheme 8 Proposed reaction mechanism. | |
3 Conclusions
We have developed a convenient cobalt-catalyzed halofluoro-alkylation that exhibits wide substrate scope including terminal and internal alkynes, alkenes, and various fluoroalkyl and alkyl bromides and iodides. The protocol enables the highly regio-selective and stereoselective synthesis of densely functionalized halogenated (E)-alkenes under very mild reaction conditions (2 mol% catalyst, 5 mol% Zn, acetone/water, 20 °C, 3 h). Contrary to literature reports, mechanistic studies documented for the first time that the halogen atom transfer is a cobalt-mediated process. The RFCoIIIX complex is the key catalytic intermediate which generates the free RF˙ radical and mediates the halogen atom transfer to the terminal vinyl radical. This mechanistic deviation from substrate control to catalyst control may provide the basis for the development of related halogen atom transfer reactions through catalyst design. Further, this ATRA reaction operates without a stoichiometric reductant for the regeneration of the low-valent Co(I) catalyst. The high functional group tolerance and mild reaction conditions make this protocol highly attractive in the context of complex molecule synthesis with potential utility for medicinal chemistry endeavours.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was generously supported by the Deutsche Forschungs-gemeinschaft (DFG, JA 1107/6-1) and the European Research Council (ERC, CoG 683150). We thank Dr Michal Majek and Dr Uttam Chakraborty for insightful discussions and technical support.
Notes and references
-
(a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed;
(b) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed;
(c) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC;
(d) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC;
(e)
V. Gouverneur and K. Müller, Fluorine in Pharmaceutical and Medicinal Chemistry: From Biophysical Aspects to Clinical Applications, Imperial College Press, London, 2012 Search PubMed.
- Trifluoromethylations:
(a) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed;
(b) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed;
(c) T. Besset, C. Schneider and D. Cahard, Angew. Chem., Int. Ed., 2012, 51, 5048 CrossRef CAS PubMed;
(d) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed;
(e) L. Chu and F.-L. Qing, Acc. Chem. Res., 2014, 47, 1513 CrossRef CAS PubMed;
(f) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598 RSC;
(g) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294 CrossRef CAS PubMed;
(h) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed;
(i) X. Liu, C. Xu, M. Wang and Q. Liu, Chem. Rev., 2015, 115, 683 CrossRef CAS PubMed;
(j) C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765 CrossRef CAS PubMed;
(k) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef CAS PubMed;
(l) C. Alonso, E. M. Marigotra, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847 CrossRef CAS PubMed;
(m) P. Gao, X.-R. Song, X.-Y. Liu and Y.-M. Liang, Chem.–Eur. J., 2015, 21, 7648 CrossRef CAS PubMed.
- Difluoroalkylations:
(a) K. Fujikawa, Y. Fujioka, A. Kobayashi and H. Amii, Org. Lett., 2011, 13, 5560 CrossRef CAS PubMed;
(b) P. S. Fier and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 5524 CrossRef CAS PubMed;
(c) Z. He, T. Luo, M. Hu, Y. Cao and J. Hu, Angew. Chem., Int. Ed., 2012, 51, 3944 CrossRef CAS PubMed;
(d) Q. Qi, Q. Shen and L. Lu, J. Am. Chem. Soc., 2012, 134, 6548 CrossRef CAS PubMed;
(e) Z. Feng, F. Chen and X. Zhang, Org. Lett., 2012, 14, 1938 CrossRef CAS PubMed;
(f) X. Jiang, L. Liu and F.-L. Qing, Org. Lett., 2012, 14, 2870 CrossRef CAS PubMed;
(g) Z. He, M. Hu, T. Luo, L. Li and J. Hu, Angew. Chem., Int. Ed., 2012, 51, 11545 CrossRef CAS PubMed;
(h) G. K. S. Prakash, S. K. Ganesh, J.-P. Jones, A. Kulkarni, K. Masood, J. K. Swabeck and G. A. Olah, Angew. Chem., Int. Ed., 2012, 51, 12090 CrossRef CAS PubMed;
(i) Z. Li, Z. Cui and Z.-Q. Liu, Org. Lett., 2013, 15, 406 CrossRef CAS PubMed;
(j) Q.-Q. Min, Z. Yin, Z. Feng, W.-H. Guo and X. Zhang, J. Am. Chem. Soc., 2014, 136, 1230 CrossRef CAS PubMed;
(k) Z. Feng, Y.-L. Xiao and X. Zhang, Org. Chem. Front., 2014, 1, 11 RSC;
(l) Y.-L. Xiao, W.-H. Guo, G.-Z. He, Q. Pan and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 9909 CrossRef CAS PubMed;
(m) S. Ge, S. I. Arlow, M. G. Mormino and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 14401 CrossRef CAS PubMed;
(n) Z. Feng, Q.-Q. Min, H.-Y. Zhao, J.-W. Gu and X. Zhang, Angew. Chem., Int. Ed., 2015, 54, 1270 CrossRef CAS PubMed;
(o) C. Shao, G. Shi, Y. Zhang, S. Pan and X. Guan, Org. Lett., 2015, 17, 2652 CrossRef CAS PubMed;
(p) L. An, Y.-L. Xiao, Q.-Q. Min and X. Zhang, Angew. Chem., Int. Ed., 2015, 54, 9079 CrossRef CAS PubMed;
(q) Z. Li, A. Garcia-Domínguez and C. Nevado, J. Am. Chem. Soc., 2015, 137, 11610 CrossRef CAS PubMed;
(r) G. Li, T. Wang, F. Fei, Y.-M. Su, Y. Li, Q. Lan and X.-S. Wang, Angew. Chem., Int. Ed., 2016, 55, 3491 CrossRef CAS PubMed;
(s) Y.-L. Xiao, Q.-Q. Min, C. Xu, R.-W. Wang and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 5837 CrossRef CAS PubMed;
(t) M. V. Ivanova, A. Bayle, T. Besset, T. Poisson and X. Pannecoucke, Angew. Chem., Int. Ed., 2015, 54, 13406 CrossRef CAS PubMed;
(u) X. Wang and A. Studer, Org. Lett., 2017, 19, 2977 CrossRef CAS PubMed. For reviews, see ref. 2e and
(v) T. Besset, T. Poisson and X. Pannecoucke, Eur. J. Org. Chem., 2015, 2765 CrossRef CAS;
(w) M.-C. Belhomme, T. Besset, T. Poisson and X. Pannecoucke, Chem.–Eur. J., 2015, 21, 12836 CrossRef CAS PubMed.
-
(a) Y. Takeyama, Y. Ichinose, K. Oshima and K. Utimato, Tetrahedron Lett., 1989, 30, 3159 CrossRef CAS;
(b) Y. Li, H. Li and J. Hu, Tetrahedron, 2009, 65, 478 CrossRef CAS;
(c) X. Fang, X. Yang, X. Yang, S. Mao, Y. Wang, G. Chen and F. Wu, Tetrahedron, 2007, 63, 10684 CrossRef CAS;
(d) W. Huang, L. Lv and Y. Zhang, Chin. J. Chem., 1990, 4, 350 Search PubMed;
(e) G.-B. Rong and R. Keese, Tetrahedron Lett., 1990, 31, 5615 CrossRef CAS;
(f) K. Tsuchii, M. Imura, N. Kamada, T. Hirao and A. Ogawa, J. Org. Chem., 2004, 69, 6658 CrossRef CAS PubMed. For selected other methods, see:
(g) T. Ishihara, M. Kuroboshi and Y. Okada, Chem. Lett., 1986, 1895 CrossRef CAS;
(h) S. Ma and X. Lu, Tetrahedron, 1990, 46, 357 CrossRef CAS;
(i) M. P. Jennings, E. A. Cork and P. V. Ramachandran, J. Org. Chem., 2000, 65, 8763 CrossRef CAS PubMed;
(j) D. Motoda, H. Kinoshita, H. Shinokubo and K. Oshima, Adv. Synth. Catal., 2002, 344, 261 CrossRef CAS.
- T. Xu, C. Cheung and X. Hu, Angew. Chem., Int. Ed., 2014, 53, 4910 CrossRef CAS PubMed.
- M.-C. Belhomme, D. Dru, H.-Y. Xiong, D. Cahard, T. Besset, T. Poisson and X. Pannecoucke, Synthesis, 2014, 46, 1859 CrossRef.
- G. Li, Y.-X. Cao, C.-G. Luo, Y.-M. Su, Y. Li, Q. Lan and X.-S. Wang, Org. Lett., 2016, 18, 4806 CrossRef CAS PubMed.
-
(a) B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500 CrossRef CAS PubMed;
(b) E. Nakamura and N. Yoshikai, J. Org. Chem., 2010, 75, 6061 CrossRef CAS PubMed;
(c) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed;
(d) R. B. Bedford and P. B. Brenner, Top. Organomet. Chem., 2015, 50, 19 CrossRef CAS;
(e) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170 CrossRef CAS PubMed;
(f) A. Fürstner, ACS Cent. Sci., 2016, 2, 778 CrossRef PubMed;
(g) T. L. Mako and J. A. Byers, Inorg. Chem. Front., 2016, 3, 766 RSC;
(h) A. Guérinot and J. Cossy, Top. Curr. Chem., 2016, 374, 49 CrossRef PubMed.
- For selected examples, see:
(a) L. Nicolas, P. Angibaud, I. Stanfield, P. Bonnet, L. Meerpoel, S. Reymond and J. Cossy, Angew. Chem., Int. Ed., 2012, 51, 11101 CrossRef CAS PubMed;
(b) J. Mao, F. Liu, M. Wang, L. Wu, B. Zhang, S. Liu, J. Zhong, Q. Bian and P. J. Walsh, J. Am. Chem. Soc., 2014, 136, 17662 CrossRef CAS PubMed;
(c) J. M. Hammann, D. Hass and P. Knochel, Angew. Chem., Int. Ed., 2015, 54, 4478 CrossRef CAS PubMed;
(d) J. M. Hammann, D. Hass, C.-P. Tüllmann, K. Karaghiosoff and P. Knochel, Org. Lett., 2016, 18, 4778 CrossRef CAS PubMed;
(e) M. S. Hofmayer, J. M. Hammann, D. Hiana and P. Knochel, Org. Lett., 2016, 18, 6456 CrossRef CAS PubMed, and references therein. For selected reviews, see:
(f) H. Yorimitsu and K. Oshima, Pure Appl. Chem., 2006, 78, 441 CrossRef CAS;
(g) C. Gosmini, J.-M. Bégouin and A. Moncomble, Chem. Commun., 2008, 3221 RSC;
(h) G. Cahiez and A. Moyeux, Chem. Rev., 2010, 110, 1435 CrossRef CAS PubMed.
-
(a) J. Zhou and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 14726 CrossRef CAS PubMed;
(b) G. D. Jones, J. L. Martin, C. McFarland, O. R. Allen, R. E. Hall, A. D. Haley, R. J. Brandon, T. Konovalova, P. J. Desrochers, P. Pulay and D. A. Vicic, J. Am. Chem. Soc., 2006, 128, 1317 Search PubMed;
(c) J. Terao and N. Kambe, Acc. Chem. Res., 2008, 41, 1545 CrossRef CAS PubMed;
(d) V. B. Phapale and D. J. Cárdenas, Chem. Soc. Rev., 2009, 38, 1598 RSC;
(e) A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656 CrossRef CAS PubMed;
(f) X. Hu, Chem. Sci., 2011, 2, 1867 RSC;
(g) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed;
(h) T. Iwasaki and N. Kambe, Top. Curr. Chem., 2016, 374, 66 CrossRef PubMed.
- S. Torii, T. Inokuchi and T. Yukawa, J. Org. Chem., 1985, 50, 5875 CrossRef CAS; C.-M. Hu and Y.-L. Qiu, J. Fluorine Chem., 1991, 55, 113 CrossRef;
(a) C.-M. Hu and Y.-L. Qiu, Tetrahedron Lett., 1991, 32, 4001 Search PubMed;
(b) B. P. Branchaud and W. D. Detlefsen, Tetrahedron Lett., 1991, 32, 6273 Search PubMedC.-M. Hu and Y.-L. Qiu, J. Org. Chem., 1992, 57, 3339 Search PubMed;
(c) Y. Ikeda, T. Nakamura, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2002, 124, 6514 Search PubMed;
(d) W. Affo, H. Ohmiya, T. Fujioka, Y. Ikeda, T. Nakamura, H. Yorimitsu, K. Oshima, Y. Imamura, T. Mizuta and K. Miyoshi, J. Am. Chem. Soc., 2006, 128, 8068 Search PubMed;
(e) P. Shukla, Y.-C. Hsu and C.-H. Cheng, J. Org. Chem., 2006, 71, 655 Search PubMed;
(f) M. E. Weiss, L. M. Kreis, A. Lauber and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 11125 Search PubMed;
(g) S. Lu, T. Jin, M. Bao and Y. Yamamoto, J. Am. Chem. Soc., 2011, 133, 12842 Search PubMed.
-
(a) A. A. Isse, A. Gennaro and E. Vianello, J. Electroanal. Chem., 1998, 444, 241 CrossRef CAS;
(b) A. J. Moad, L. J. Klein, D. G. Peters, J. A. Karty and J. P. Reilly, J. Electroanal. Chem., 2002, 531, 163 CrossRef CAS;
(c) J. D. Persinger, J. L. Hayes, L. J. Klein, D. G. Peters, J. A. Karty and J. P. Reilly, J. Electroanal. Chem., 2004, 568, 157 CrossRef CAS;
(d) P. Vanalabhpatana, D. G. Peters and J. A. Karty, J. Electroanal. Chem., 2005, 580, 300 CrossRef CAS;
(e) P. C. Gach, J. A. Karty and D. G. Peters, J. Electroanal. Chem., 2008, 612, 22 CrossRef CAS;
(f) L. Pan, H. Shimakoshi, T. Masuko and Y. Hisaeda, Dalton Trans., 2009, 38, 9898 RSC;
(g) T. Ueda, N. Inazuma, D. Komatsu, H. Yasuzawa, A. Onda, S.-X. Guo and A. M. Bond, Dalton Trans., 2013, 42, 11146 RSC;
(h) M. Giedyk, K. Goliszewska and D. Gryko, Chem. Soc. Rev., 2015, 44, 3391 RSC;
(i) M. Giedyk, H. Shimakoshi, K. Goliszewska, D. Gryko and Y. Hisaeda, Dalton Trans., 2016, 45, 8340 RSC;
(j) H. Shimakoshi, Z. Luo, T. Inaba and Y. Hisaeda, Dalton Trans., 2016, 45, 10173 RSC.
-
(a) D. N. Ramakrishna, R. Symons and M. Symons, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 423 RSC;
(b) B. D. Gupta and S. Roy, Inorg. Chim. Acta, 1988, 146, 209 CrossRef CAS.
-
(a) R. Breslow and P. L. Khanna, J. Am. Chem. Soc., 1976, 98, 1297 CrossRef CAS PubMed;
(b) H. Eckert, D. Lenoir and I. Ugi, J. Organomet. Chem., 1997, 141, C23 CrossRef;
(c) J. Schafler and J. Retey, Angew. Chem., Int. Ed. Engl., 1978, 17, 845 CrossRef;
(d) A. I. Scott, J. B. Hansen and S. K. Chung, J. Chem. Soc., Chem. Commun., 1980, 388 RSC;
(e) B. D. Gupta and S. Roy, Inorg. Chim. Acta, 1988, 146, 209 CrossRef CAS.
- D. B. Bagal, G. Kachkovskyi, M. Knorn, T. Rawner, B. M. Bhanage and O. Reiser, Angew. Chem., Int. Ed., 2015, 54, 6999 CrossRef CAS PubMed.
-
(a) Y. Yauchi, M. Ide, R. Shiogai, T. Chikugo and T. Iwasawa, Eur. J. Org. Chem., 2015, 938 CrossRef CAS;
(b) K. C. Sproul and W. A. Chalifoux, Org. Lett., 2015, 17, 3334 CrossRef CAS PubMed;
(c) X. Wang and A. Studer, J. Am. Chem. Soc., 2016, 138, 2977 CrossRef CAS PubMed;
(d) X. Wang and A. Studer, Org. Lett., 2017, 19, 2977 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04916a |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab
*ab