
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unusual mechanisms in Claisen rearrangements: an ionic fragmentation leading to a meta-selective rearrangement†
Boris
Maryasin
 *ab,
Dainis
Kaldre‡
a,
Renan
Galaverna‡
c,
Immo
Klose
a,
Stefan
Ruider
ab,
Martina
Drescher
a,
Hanspeter
Kählig
a,
Leticia
González
b,
Marcos N.
Eberlin
c,
Igor D.
Jurberg
*d and
Nuno
Maulide
*a
*ab,
Dainis
Kaldre‡
a,
Renan
Galaverna‡
c,
Immo
Klose
a,
Stefan
Ruider
ab,
Martina
Drescher
a,
Hanspeter
Kählig
a,
Leticia
González
b,
Marcos N.
Eberlin
c,
Igor D.
Jurberg
*d and
Nuno
Maulide
*a
aUniversity of Vienna, Faculty of Chemistry, Institute of Organic Chemistry, Währinger Strasse 38, 1090 Vienna, Austria. E-mail: nuno.maulide@univie.ac.at; boris.maryasin@univie.ac.at
bUniversity of Vienna, Faculty of Chemistry, Institute of Theoretical Chemistry, Währinger Strasse 17, 1090 Vienna, Austria
cState University of Campinas, ThoMSon Mass Spectrometry Laboratory, Institute of Chemistry, Rua Monteiro Lobato 270, 13083-970, Campinas, Brazil
dState University of Campinas, Institute of Chemistry, Rua Monteiro Lobato 270, 13083-970, Campinas, Brazil. E-mail: idjurberg@iqm.unicamp.br
First published on 10th April 2018
Abstract
A mechanistic investigation of the acid-catalysed redox-neutral oxoarylation reaction of ynamides using electrospray ionisation mass-spectrometry (ESI-MS) and quantum chemical calculations (DFT and MP2) is presented. This study reveals the diversity of pathways and products available from an otherwise deceptively simple-looking, classical transformation: fragmentation, an unusual meta-arylation and competing α-carbonyl cation pathways are some of the alternatives unveiled by ESI-MS and mechanistic experiments. Detailed calculations explain the observed trends and rationalise the results.
Introduction
The [3,3]-sigmatropic rearrangement of vinyl(allyl)ethers was first introduced in 1912 by the seminal work of L. Claisen, whereby O-allyl phenols were thermally converted into 2-allyl phenols (Scheme 1).1 Inspired by this contribution, in the following years several studies were reported expanding the scope of this transformation into a powerful and versatile C–C bond forming strategy.2 Indeed, a list of well-known examples of such variants now hold “textbook status” in the field of organic chemistry, including those developed by Ireland,3 Johnson,4 Eschenmoser,5 Overman,6 and Ficini,7 among others.8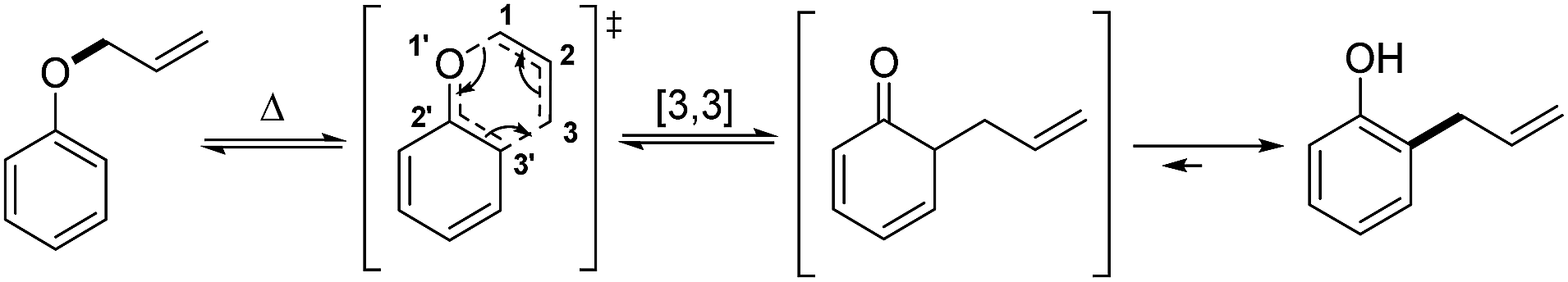 | ||
| Scheme 1 The Claisen rearrangement originally reported in 1912. | ||
More recently, one of us9 has been interested in the development of novel variants of this venerable reaction that can be deployed in alternative contexts. Maulide and co-workers thus described a TfOH-catalysed oxoarylation of ynamides 1 using sulfoxides 2. Mechanistic analysis assumed the involvement of a [3,3]-sigmatropic shift analogous to the Claisen rearrangement, most likely taking place on cationic intermediate 5+, en route to the corresponding final α-arylated acyloxazolidinone 7 (Scheme 2, top).10
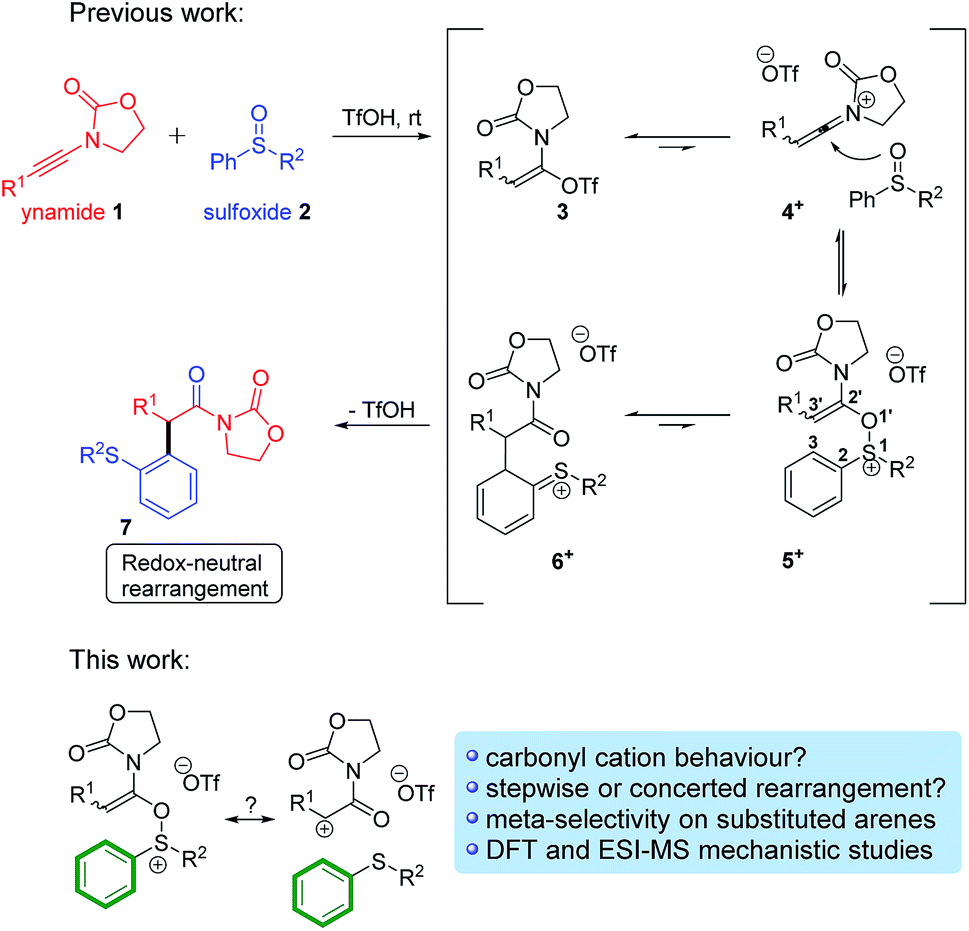 | ||
| Scheme 2 The redox-neutral oxoarylation reaction previously reported10 and mechanistic questions addressed in this manuscript. | ||
Interestingly, whereas the latter rearrangement proceeds at temperatures below and at room temperature, typically in no more than 30 minutes reaction time, and in high yields, the analogous Ficini–Claisen rearrangement generally requires higher temperatures, longer reaction times and generally produces lower yields of the corresponding final alkylated products.7
Intrigued by this great difference in reactivity and in order to gather more information concerning the mechanism of this formal α-arylation reaction, we became interested in monitoring this transformation online. Given the abundance of (postulated) positively charged reaction intermediates along the reaction pathway, ESI-MS11 emerged as a promising tool to observe and intercept those species directly. Additionally, we aimed to complement those studies with quantum chemical calculations and additional experiments. Herein, we report the results of this study, culminating in a unified mechanistic view of this transformation. On the way to that goal, several unusual, unexpected alternative mechanistic pathways (Scheme 2, bottom) emerged showcasing the complex reactivity network in which this system is embedded.
Discussion of results
Analysis of reaction intermediates by online ESI(+)-MS
Based on the putative mechanism outlined in Scheme 2, we were eager to verify whether certain key charged intermediates [3 + Y]+ (Y = H, Na), 4+, 5+ and 6+ (Scheme 2) could be intercepted and characterised via electrospray ionisation MS operated in positive-ion mode (ESI(+)-MS).12Initially, we were curious to verify whether, in the absence of sulfoxide 2, a cationic form of the enamine-triflate [3 + Y]+ (Y = H, Na) could be detected. Nevertheless, initial attempts employing ynamide 1a (cf.1, with R1 = n-C6H13) failed. As the corresponding protonated amide [8a + H]+, of m/z 214, was typically the main fragment observed, we rationalised that the timeframe between sample preparation and injection in the mass spectrometer might be too long for the detection of transient [3a + Y]+. Thus, we switched to DESI(+)-MS,13 in order to achieve quasi simultaneous sample preparation and MS analysis. For this purpose, a solution of triflic acid (1 mmol) in DCM (500 μL) was used for the DESI spray, whereas ynamide 1a (1 μL) was deposited on a glass surface. While the spray droplets slowly mixed the reagents on the glass surface (10 μL min−1), the resulting intermediate [3a + Y]+ was simultaneously transferred to the gas phase. Using this approach, we were indeed able to detect intermediate [3a + H]+, as well as 4a+ at room temperature. The low abundances obtained for these ions are presumably due to their transient nature (Fig. 1).
 | ||
| Fig. 1 ESI(+)-MS monitoring of the reaction solution containing ynamide 1a and TfOH in DCM. (a) DESI-MS(+) spectrum. (b) DESI(+)-MS/MS of the ion of m/z 346.09, attributed to [3a + H]+. | ||
Being able to detect the key intermediates [3a + H]+ and 4a+ in the presence of TfOH, we then turned our attention to the action of other common Brønsted acids: PhCO2H, pTSA and (PhO)2P(O)OH. Interestingly, whereas PhCO2H barely reacts with either 1a or 1b (cf.1, with R1 = Ph), with the major fraction of the ynamides remaining unreacted, extensive addition of pTSA and (PhO)2P(O)OH to 1a and 1b is observed. As a consequence, these three acids are not capable of promoting this Claisen rearrangement (see ESI† for details).
Next, we investigated the TfOH-catalysed arylation of ynamide 1a with diphenyl sulfoxide 2a by ESI(+)-MS. The MS-spectra recorded immediately after the preparation of the reaction mixture already exhibited the key intermediate 5a+, and the final sodiated aryl amide [7a + Na]+ (Fig. 2).
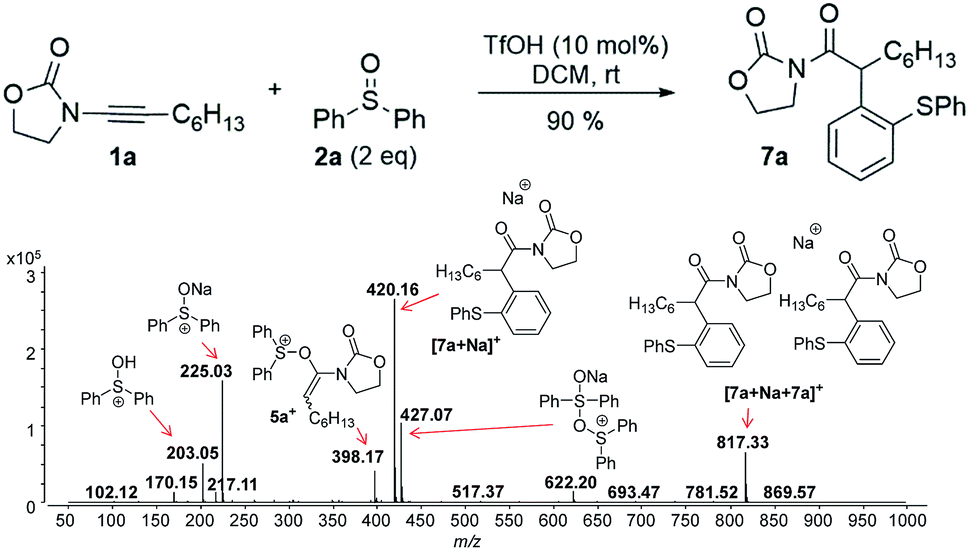 | ||
| Fig. 2 ESI(+)-MS recorded immediately after ynamide 1a and diphenyl sulfoxide 2a are mixed in the presence of a catalytic amount of TfOH in DCM. | ||
In this context, an important observation is that 7a binds preferentially to Na+, rather than to H+ (due to the absence of strong Brønsted basic sites), and is largely detected as [7a + Na]+ of m/z 420. Furthermore, although 5a+ and [7a + H]+ are isomers of m/z 398, they can be distinguished by comparing the ESI(+)-MS/MS spectra obtained from isolated 7a and intermediate 5a+ detected from the reaction mixture. Indeed, the fragmentation patterns are found to be remarkably different. The ESI(+)-MS/MS of [7a + H]+ of m/z 398 (acquired from the isolated product) shows major fragment ions of m/z 283, 311 and 398, whereas the MS-spectrum of 5a+, acquired from the reaction mixture, mainly contains the fragment ions of m/z 88, 125, 170, 212, 283, 311 and 398 (Fig. 3). Furthermore, the reaction of ynamide 1a (R1 = n-C6H13) with a different sulfoxide 2b (R2 = Me) was also studied, providing similar observations (see ESI† for the corresponding spectra). Under the same experimental conditions, a second reaction involving ynamide 1b (R1 = Ph) was studied. This time, not only intermediate 5b+ and the final product [7b + Na]+ can be observed in the full MS spectrum, but the prominent detection of a carbenium intermediate 9b+ (m/z 204) is a remarkable observation (Fig. 4). In contrast to the previous analyses of 7a (Fig. 2 and 3), the analogous 9a+ was only visible in the MS/MS spectrum of 5a+ (Fig. 3b). In the same manner as before, the comparison of MS/MS spectra associated to m/z 390, assigned to intermediate 5b+ and the protonated compound [7b + H]+ (spectra also acquired from isolated 7b), allows for the discrimination among them. Indeed, fragments of m/z 105, 132, 160, 176 and 204 are only found for 5b+, while the fragment of m/z 197 is unique for [7b + H]+ (Fig. 5).
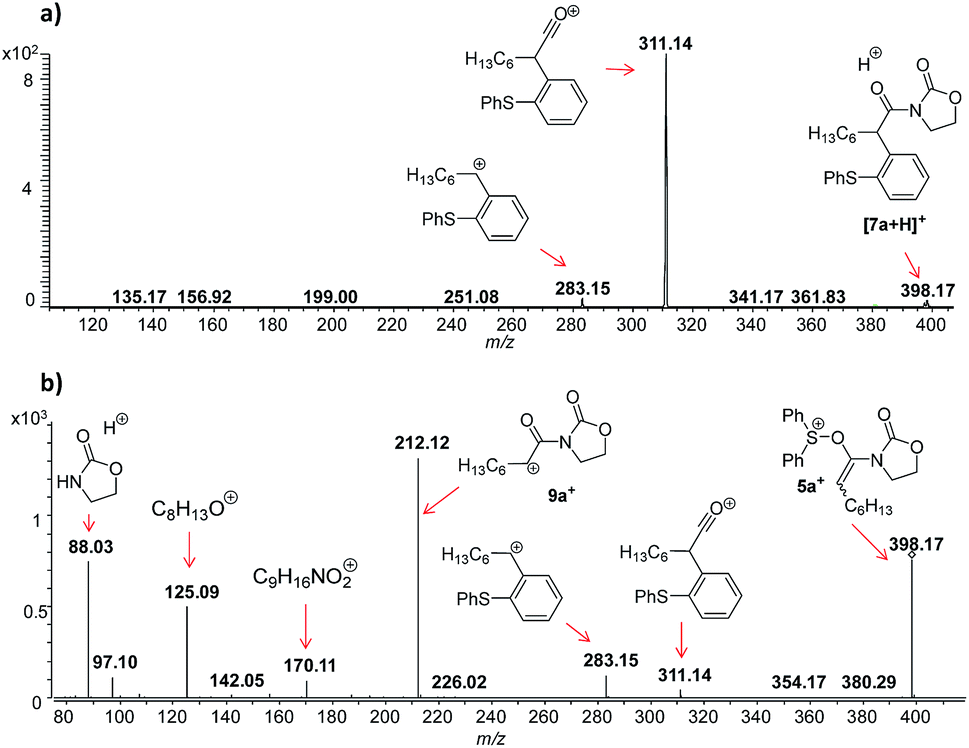 | ||
| Fig. 3 ESI(+)-MS/MS of the ion of m/z 398 analyzed from (a) isolated [7a + H]+, and (b) from the reaction mixture, which is clearly not identical with (a) and can be assigned to 5a+. | ||
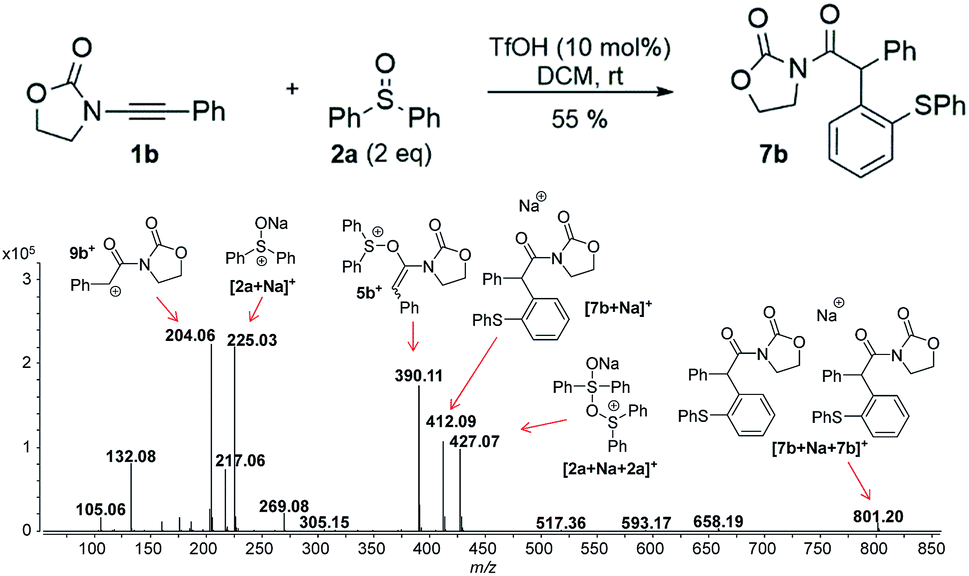 | ||
| Fig. 4 Full ESI-MS(+) immediately recorded after ynamide 1b and diphenyl sulfoxide 2a are mixed in the presence of a catalytic amount of TfOH in DCM. | ||
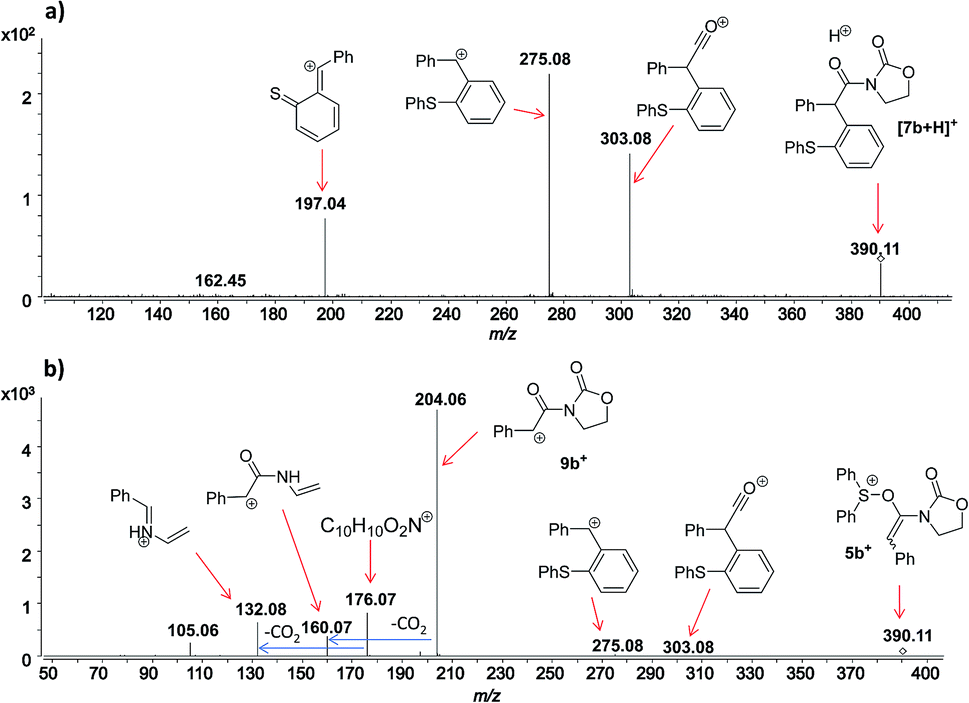 | ||
| Fig. 5 ESI(+)-MS/MS of the ion of m/z 390 analyzed from (a) isolated [7b + H]+, and (b) from the reaction mixture, which corresponds to 5b+. | ||
Theoretical study and auxiliary experiments
In order to explain the dependence of experimentally observed intermediates on reaction substrate, namely the marked presence of the cation 9b+ in the case of a phenyl-substituted ynamide, and to clarify the mechanism of the considered reactions, we have carried out quantum chemical calculations at the DFT (B3LYP-D3-SMD/6-31+G(d,p)) and MP2//DFT (RI-MP2-COSMO/def2-TZVP//B3LYP-D3-SMD/6-31+G(d,p)) levels of theory (see the ESI† for computational details).14Fig. 6 shows the computed reaction profile for conversion of the ynamide (A) into the cationic intermediate D with a characteristic loss of aromaticity.
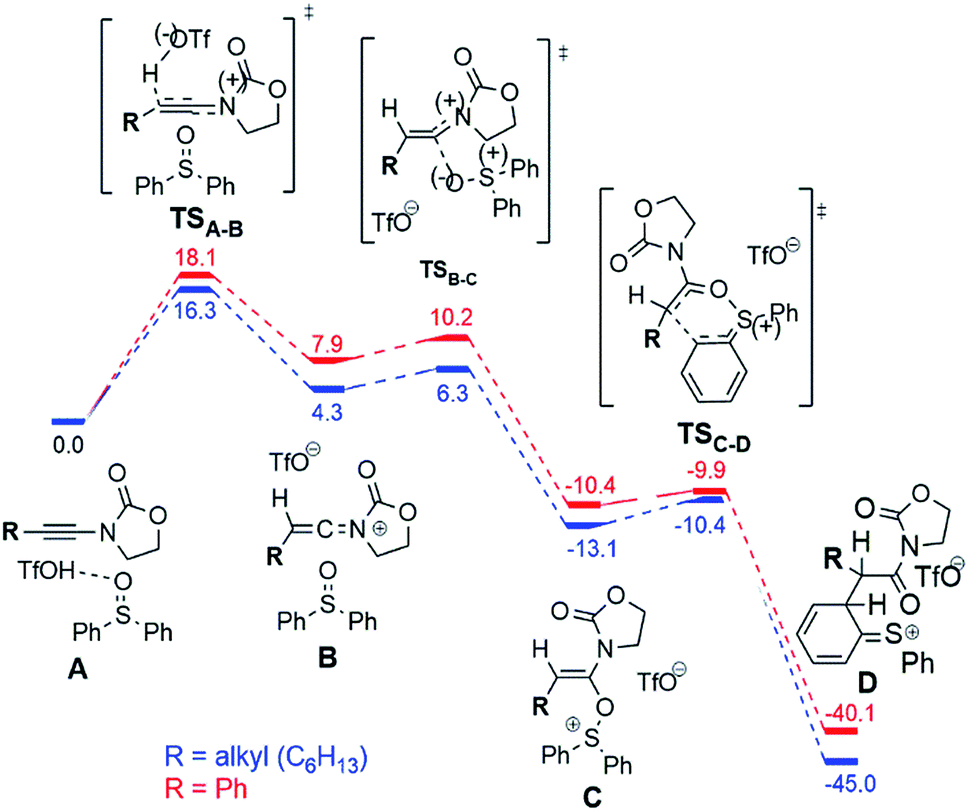 | ||
| Fig. 6 Computed reaction profile (DFT, ΔG298,DCM) for the conversion of ynamide (reactant complex A) into the intermediate D for the phenyl (red) and the alkyl (blue) substrates. The energy of the reactant complex A is taken as a reference (0.0 kcal mol−1). | ||
The first step is formation of the transient keteniminium triflate B through protonation by TfOH. In the sequence, reaction with the sulfoxide yields the unusual N,O-ketene acetal C, that is further converted to intermediate D.
As can be seen, the aryl- and the alkyl ynamide substrates show similar behaviour in the formation of the intermediate D from the ynamide A. The reaction is exergonic for both substrates, and the barriers are relatively low, in agreement with the mild reaction conditions.
The situation changes substantially for the next steps, where intermediate D undergoes a conversion to the experimentally observed products. Two possible pathways were found and are presented in Fig. 7. The first scenario leads to the formation of the main product F (Fig. 7, left), while an alternative is the formation of the bicyclic cation E (Fig. 7, right). This cation was detected experimentally by ESI(+)-MS, but only for the phenyl-substituted ynamide (E is effectively the more stable form of the cation 9b+ shown in Fig. 5).15 Analysis of the relative Gibbs free energies presented in Fig. 7 provides a rationale. One can see that for both phenyl- and alkyl substituted intermediates D, the formation of the product F is energetically favourable, and therefore is experimentally observed for both systems. However, the barriers of the formation of the product E are very different for different substrates ΔΔG‡298,DCM = ΔG‡298,DCM(alkyl) − ΔG‡298,DCM(phenyl) = 9.9 kcal mol−1 at the DFT or even 16.4 kcal mol−1 at the higher (MP2//DFT) level of theory. This substantial gap between the barriers and the large absolute value of the barrier for the alkyl-substituted system (21.8 kcal mol−1 at the DFT or 26.7 kcal mol−1 at the MP2//DFT level) is in good agreement with the experimental evidence, namely that the carbenium intermediate 9b+ is detected only in the case of the phenyl substituent.
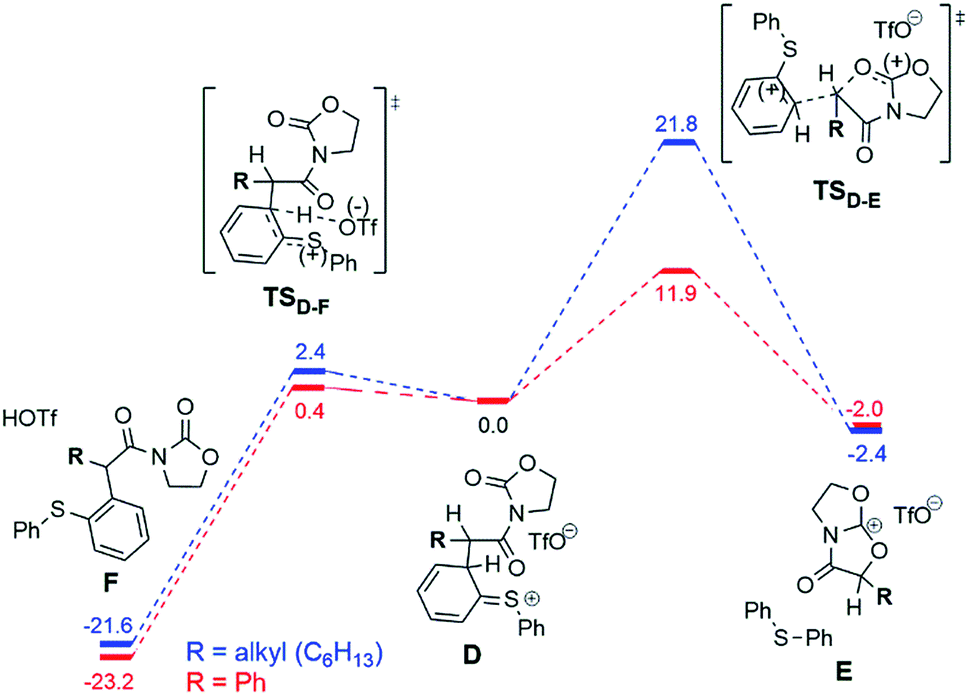 | ||
| Fig. 7 Computed reaction profile (DFT, ΔG298,DCM) for the conversion of the intermediate D into the main product F or the bicyclic cation E for the phenyl (red) and the alkyl (blue) substrates. The energy of the intermediate D is taken as a reference (0.0 kcal mol−1). | ||
The conversion of intermediate D into the bicycle E is an intramolecular SN2-type process. Fig. 8 shows the computed structures of the transition states TSD–E for phenyl (bottom) and alkyl (top) substituents, from which the typical trigonal bipyramidal structure of the SN2 transition state can be recognised.
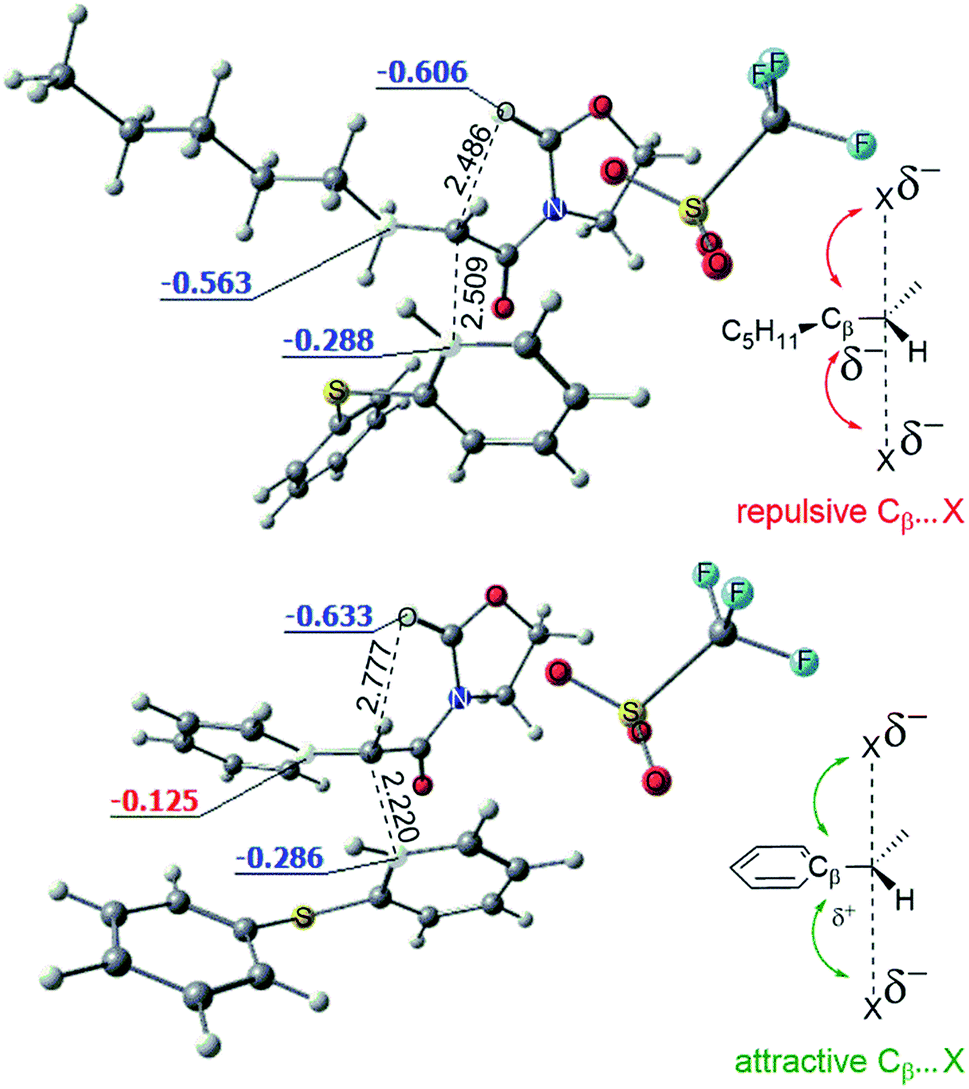 | ||
| Fig. 8 Optimized structures of the transition states TSD–E for the phenyl (bottom) and alkyl (top) substituted systems. Computed NBO charges of the selected atoms reveal the attractive and repulsive interactions in the transition states of the intramolecular SN2 reactions. The breaking and forming bonds are shown (Å). | ||
Fig. 8 also depicts the calculated NBO (Natural Bond Orbital) charges for the atoms involved in the SN2 transformation. Electrostatic interactions within the transition state stabilise the phenyl variant of the TSD–E as compared to the alkyl TSD–E. As shown, attractive Cβ(δ+)–X(δ−) interactions in the phenyl-substituted structure stabilise the transition state (leading to a higher rate for the SN2 process). The importance of such electrostatic interactions for classical SN2 reactions was previously explored by Wu et al.16
Alternatively, the bicyclic cation E can form directly from the intermediate C (before the sigmatropic rearrangement C → D) by a SN1-type reaction (Fig. 9).
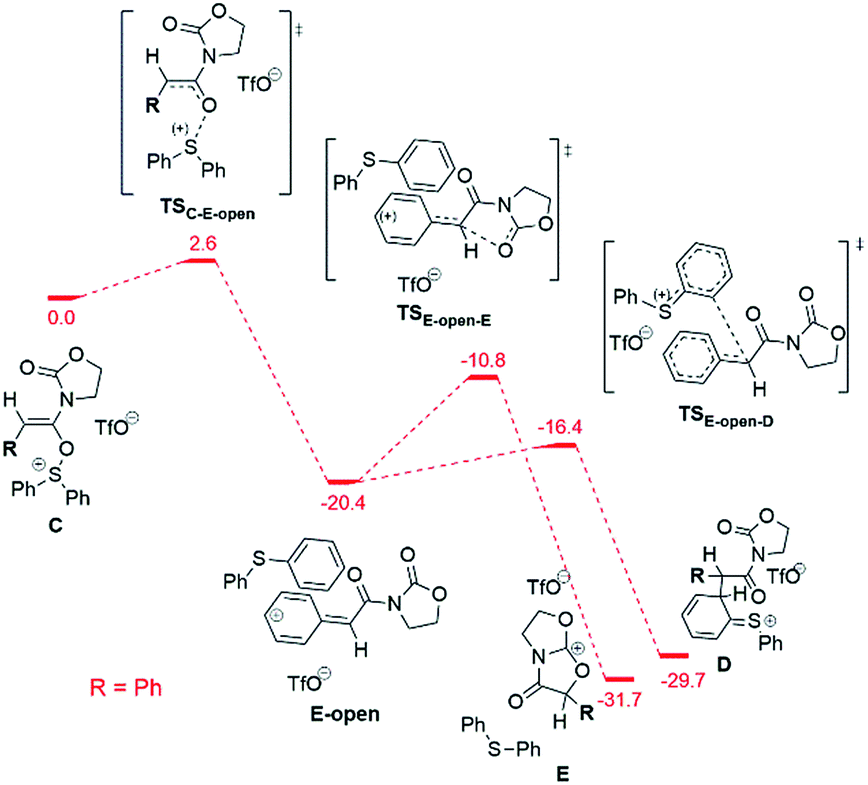 | ||
| Fig. 9 Computed reaction profile (DFT, ΔG298,DCM) for the conversion of the intermediate C into the bicyclic cation E for the phenyl substrate. The possible conversion of the “open” intermediate E-open to the intermediate D is also shown. The energy of the intermediate C is taken as a reference (0.0 kcal mol−1). | ||
This C → E conversion proceeds via an open form of the intermediate 9b+ (E-open), which undergoes annulation to E on the last step. The phenyl substituent stabilises the E-open cationic intermediate by hyperconjugation in a fashion not accessible to an alkyl group. Therefore, the SN1-like process C → E happens exclusively for the phenyl-substituted system, again in agreement with the experimental results (vide supra).
The barriers of the rate limiting steps of the SN2 and the SN1 processes for the phenyl substituted system are comparable with each other (11.9 kcal mol−1vs. 9.6 kcal mol−1). Thus, both pathways can be realised in the case of the phenyl substituent. However, the SN1 mechanism is more probable due to the low barrier of the D → F step competing to the SN2 pathway. In the case of the alkyl substituent both SN1 and SN2 pathways are unlikely, since the barrier of the SN2 pathway is too high (21.8 kcal mol−1) and the SN1 mechanism is forbidden (the necessary E-open intermediate is not formed).
Intrigued by the plausible intermediacy of a cyclic cationic structure such as E as inferred from both experimental and theoretical evidence, we set out to experimentally probe its intermediacy in these redox-neutral arylation reactions. In particular, we were aware that, if formed, E would necessarily coexist with an equivalent of the Friedel–Crafts-competent nucleophile diphenyl sulfide.
As shown in Scheme 3a, treatment of the readily available α-bromoimide 10 with 1.5 equivalents of AgSbF6 in the presence of an excess of diphenylsulfide indeed led to a C–C coupled product of the same molecular weight as 12. Detailed NMR analysis and X-ray single crystal analysis, however, revealed this to be the para-substituted product and not the ortho-adduct that was exclusively obtained in the previously described catalytic transformation.10 Thus, while electrophilic aromatic substitution of the cationic intermediate E is possible, its outcome is predictably governed by sterics.
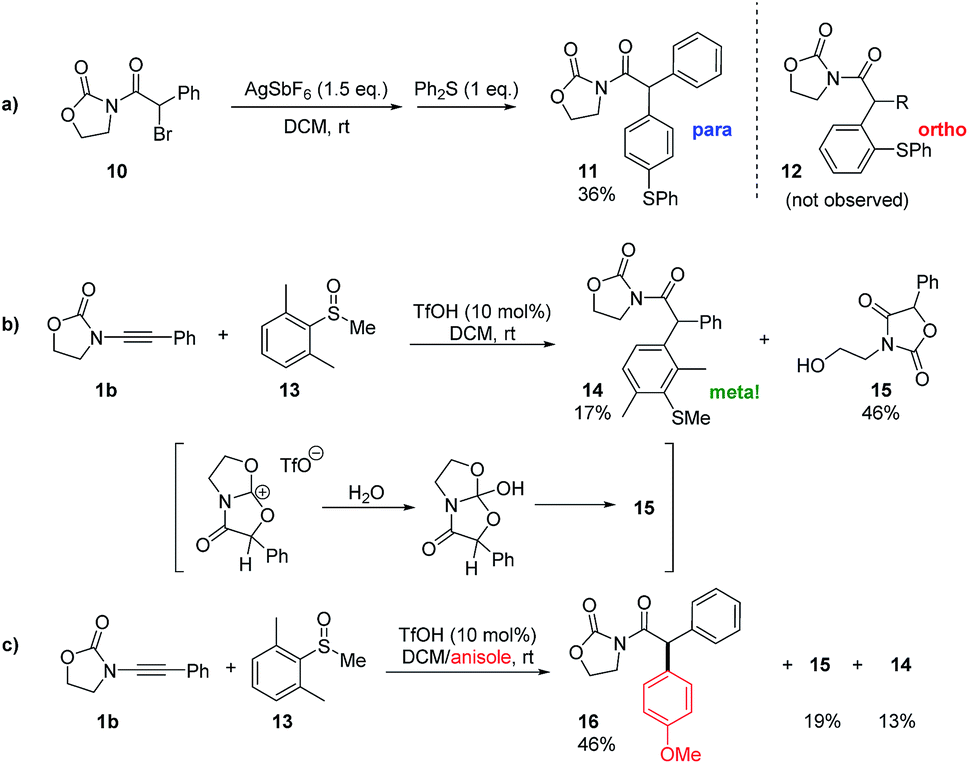 | ||
| Scheme 3 Mechanistic experiments. | ||
The absence of para-products akin to 11 in any of the redox arylation transformations previously reported by Maulide and co-workers suggests that this is not an operative pathway in the originally reported catalytic reaction.10
Nonetheless, this result intriguingly suggested a catalytic experiment in which a sulfoxide 13, where both ortho-positions are blocked, is employed as a nucleophile (Scheme 3b). Two products were observed: the alcohol 15, which appears to be the product of hydrolysis of E (Scheme 3b), and the unanticipated meta-substituted, α-arylated product 14 in a low yield.17 The result of this experiment suggests that E might indeed be an energy minimum in cases where evolution to product F is blocked, and we hypothesised that it might be possible to intercept E if a more powerful aromatic nucleophile were present. We were delighted to see that the repetition of the experiment shown in Scheme 3b, in this instance in the presence of a large excess of anisole, led to a respectable 46% yield of the intermolecular C–C bond captured product 16 along with 19% of the hydrolysis product 15 and 13% of the meta-arylated compound 14 (Scheme 3c).
The observation of meta-substituted products on the rearrangement of ortho-substituted sulfoxides is an unusual result.18 We hypothesised that strongly electron-donating moieties located ortho to the sulfoxide might be able to render this an even more efficient process and thus we decided to probe the reaction with (2-alkoxy)aryl sulfoxides. The results of our ensuing investigation are compiled in Scheme 4 (for the optimisation of reaction conditions see the ESI†).
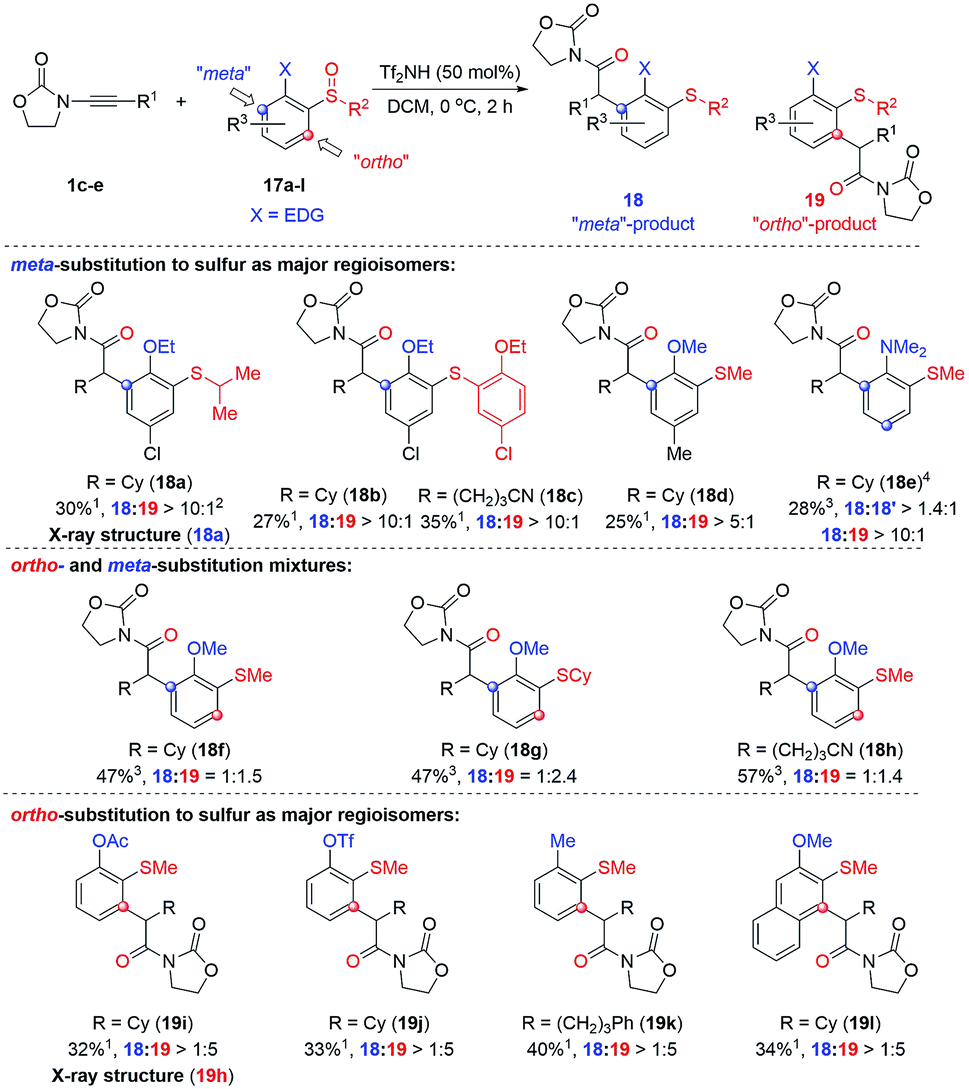 | ||
Scheme 4 Acid-catalysed reaction of ynamides with (ortho-donor-arylsulfoxides). 1Isolated yields of the product; 2Ratio of product 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19 determined from by crude 1H NMR analysis; 3Overall isolated yield of 18 + 19. 4120 mol% of Tf2NH was used. :19 determined from by crude 1H NMR analysis; 3Overall isolated yield of 18 + 19. 4120 mol% of Tf2NH was used. | ||
As can be seen, two regioisomeric products compete: the unexpected “meta”-arylated product 18 and the “ortho”-arylated compound 19. Even though yields are modest,19 high regioselectivities were observed in many cases which led us to divide the substrates into three subgroups dependent on the ratio in which the respective isomers were formed. As shown, strongly electron-donating groups, such as dimethyl amino, ethoxy- or methoxy-groups, in combination with an additional substituent, clearly favour the formation of the “meta” products 18 (cf.18a–18e; the structure of 18a was confirmed by X-ray analysis, see ESI† for details).20 However, removing the sterically biasing substituent (18f–18hcf.Fig. 10 and discussion thereafter to rationalise the effect of the –Cl substituent) or changing to less powerful electron-donating groups such as –OAc or –OTf or even –Me (18f–18hvs.19g–19i) led to increased formation of the “ortho” arylated products 19i–k.21 Counterintuitively, in case of the naphthalene derivative 19l the “ortho”-isomer was the major product suggesting the fused aromatic system to favour an ortho arylation in this case.22
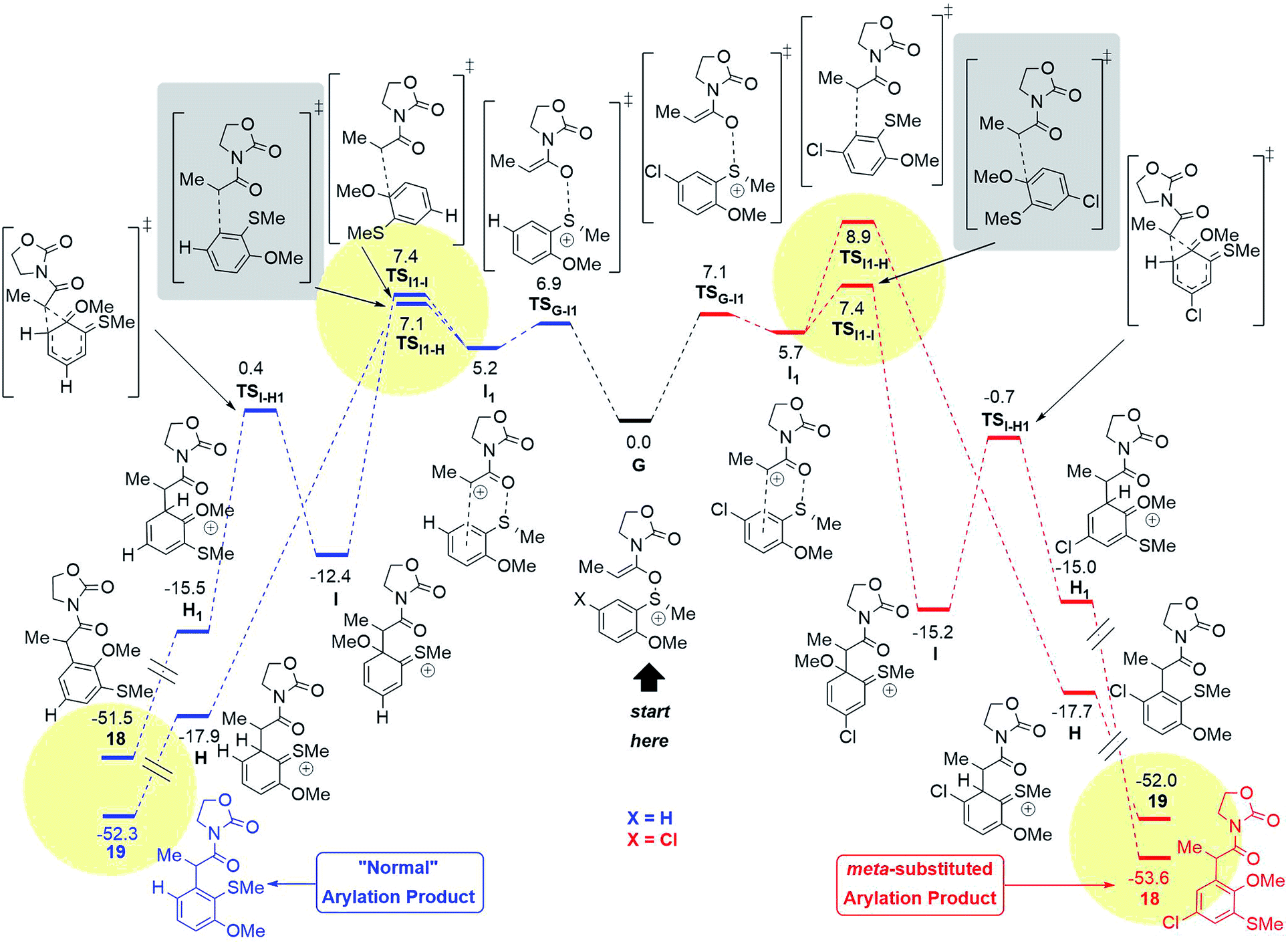 | ||
| Fig. 10 Computed reaction profile (SCS-RI-MP2-COSMO/def2-QZVP//RI-MP2/def2-TZVP,24 ΔG298,DCM) for the conversion of the intermediate G into both possible products for the X: H (blue) and the X: Cl (red) systems. The summarised energies of the intermediate G and the counterion NTf2− are taken as a reference (0.0 kcal mol−1). The yellow circles highlight the kinetic and thermodynamic factors, responsible for the regioselectivity. The grey boxes highlight the key transition states responsible for the proposed pathways. | ||
Unsure whether the meta-substituted products 18 arise by a direct process akin to a sigmatropic rearrangement, we carried out the experiment shown in Scheme 5. As depicted, the use of the commercially available, chiral enantioenriched sulfoxide 17g in conjunction with ynamide 1c led to both rearranged products 18g and 19g in good to excellent levels of chirality transfer.23 This suggests a high degree of concertedness, that is not readily compatible with a Friedel Crafts-type mechanism.
 | ||
| Scheme 5 Asymmetric rearrangement with chiral enantiopure sulfoxides. | ||
Quantum chemical calculations have been performed in order to clarify the observed regioselectivity. The reaction profile in Fig. 10 represents both possible pathways for the formation of products with the general structure 18 and 19 for two different systems: the aromatic ring with a chloro-substituent in the para-position (X = Cl, shown in red) and the unsubstituted system (X = H, shown in blue).
The initial N,O-ketene acetal intermediate G (Fig. 10, middle) can undergo two types of reactions (vide supra): (1) the classical concerted rearrangement and (2) the fragmentation. In the case of the concerted mechanism, the intermediate G must be a divergence point for the reaction to form one of the products 18 or 19. However, the DFT results and moreover the calculations at the higher level of theory (ab initio) RI-MP2/def2-TZVP support the stepwise mechanism,24 where the S–O bond of the intermediate G breaks viaTSG–I1 leading to the intermediate I1. The formation of the intermediate I1 is energetically similar for both systems (X = H or X = Cl) as shown in Fig. 10.
After the intermediate I1, two possibilities exist. One of them proceeds viaTSI1–H and forms the intermediate H, which rearomatises to the “normal” product with general structure 19. Alternatively, the reaction can proceed via the TSI1–I leading to the intermediate I, where a C–C bond is formed to the methoxy-substituted atom of the aromatic system. Intermediate I can undergo a 1,2-shift18 to form the intermediate H1, which finally rearomatises analogously to the intermediate H to form the meta-product with the general structure 18.
In the case of X = H (blue), the pathway to form 19 is preferable compared to the formation of 18 (see 18d–fScheme 4), while the situation is changed for X = Cl with the “abnormal” product 18 as a main product (see 18a–cScheme 4). This switch of regioselectivity is in good agreement with the experimental results. The calculations show that the observed regioselectivity is controlled by both kinetic and thermodynamic factors. Indeed, for the case of X = H, the barrier associated with the TSI1–I is 0.3 kcal mol−1 larger as compared to that of TSI1–H, while for X = Cl, the I1–I barrier is vice versa 1.5 kcal mol−1 smaller than the I1–H barrier. Thermodynamically, the “abnormal” pathway (formation of the product 18) is also less favourable than the “normal” rearrangement for X = H: ΔG(G → 18) = −51.5 kcal mol−1, while ΔG(G → 19) = −52.3 kcal mol−1. However, for the chloro-substituent, the formation of the meta-product 18 becomes more probable: ΔG(G(Cl) → 18(Cl)) = −53.6 kcal mol−1vs. ΔG(G(Cl) → 19(Cl)) = −52.0 kcal mol−1. In any event, it is remarkable that a chiral sulfoxide is able to mediate efficient chirality transfer from sulfur to carbon over a sequence of transformations, as depicted in Fig. 10 and Scheme 5.
Conclusions
In summary, we have presented a combined ESI-MS/quantum chemical mechanistic analysis of the redox-neutral arylation of ynamides and sulfoxides. Several noteworthy observations result from this study. First, ESI(+)-MS suggested the unusual presence of α-carbonyl cations whenever aryl substitution is present. Second, these were substantiated by computations, which outline the existence of a fine borderline between sigmatropy and fragmentation that can be crossed for particularly stabilised systems. Third, key mechanistic experiments showcased in practice how particularly “difficult” (or “sluggish”) rearrangements may easily cross the aforementioned borderline between sigmatropy and fragmentation. Fourth, additional work shows how electronically biased systems can provide meta-arylated products by a combination of unusual [3,3]- and [1,2]-shifts. Together, these observations reveal a rich panorama of mechanistic pathways available to an otherwise apparently “innocent”, classical Claisen-type [3,3]-sigmatropic rearrangement.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. M. acknowledges the generous support of this research by the ERC (StG FLATOUT and CoG VINCAT) and the University of Vienna. IDJ acknowledges the support of Fapesp (2015/20809-4) and CNPq (458416/2014-2). Calculations were partially performed at the Vienna Scientific Cluster (VSC). Invaluable support by Ing. A. Roller (U. Vienna) with X-ray structure analysis is acknowledged.Notes and references
- (a) L. Claisen, Chem. Ber., 1912, 45, 3157–3166 CrossRef CAS , see also: ; (b) L. Claisen and E. Tietze, Chem. Ber., 1925, 58, 275–281 CrossRef; (c) L. Claisen and E. Tietze, Chem. Ber., 1926, 58, 2344–2351 CrossRef.
- Selected reviews: (a) M. Hiersemann and U. Nubbemeyer, The Claisen Rearrangement, 2007, Wiley-VCH Search PubMed; (b) S. J. Rhoads and R. Raulins, Org. React., 1975, 22, 1–252 CAS; (c) F. E. Ziegler, Acc. Chem. Res., 1977, 10, 227–232 CrossRef CAS; (d) F. E. Ziegler, Chem. Rev., 1988, 88, 1423–1452 CrossRef CAS; (e) H. Ito and T. Taguchi, Chem. Soc. Rev., 1999, 28, 43–50 RSC; (f) U. Nubbemeyer, Synthesis, 2003, 961–1008 CAS; (g) A. M. M. Castro, Chem. Rev., 2004, 104, 2939–3002 CrossRef CAS PubMed.
- Selected references: (a) R. E. Ireland and R. H. Mueller, J. Am. Chem. Soc., 1972, 94, 5897–5898 CrossRef CAS; (b) R. E. Ireland and A. K. Willard, Tetrahedron Lett., 1975, 16, 3975–3978 CrossRef; (c) R. E. Ireland, R. H. Mueller and A. K. Willard, J. Am. Chem. Soc., 1976, 98, 2868–2877 CrossRef CAS.
- Selected references: (a) W. S. Johnson, L. Werthemann, W. R. Bartlett, T. J. Brocksom, T.-T. Li, D. J. Faulkner and M. R. Petersen, J. Am. Chem. Soc., 1970, 92, 741–743 CrossRef CAS; (b) R. A. Fernandes, A. K. Chowdhury and P. Kattanguru, Eur. J. Org. Chem., 2014, 2833–2871 CrossRef CAS.
- Selected references: (a) A. E. Wick, D. Felix, K. Steen and A. Eschenmoser, Helv. Chim. Acta, 1964, 47, 2425–2429 CrossRef CAS; (b) A. E. Wick, D. Felix, K. Gschwend-Steen and A. Eschenmoser, Helv. Chim. Acta, 1969, 52, 1030–1042 CrossRef.
- Selected references: (a) L. E. Overman, J. Am. Chem. Soc., 1974, 96, 597–599 CrossRef CAS; (b) L. E. Overman, J. Am. Chem. Soc., 1976, 98, 2901–2910 CrossRef CAS; (c) C. E. Anderson and L. E. Overman, J. Am. Chem. Soc., 2003, 125, 12412–12413 CrossRef CAS PubMed , see also: ; (d) O. Mumm and F. Moller, Chem. Ber., 1937, 708, 2214–2227 CrossRef.
- (a) J. Ficini and C. Barbara, Tetrahedron Lett., 1966, 52, 6425–6429 CrossRef; (b) J. A. Mulder, R. P. Hsung, M. O. Frederick, M. R. Tracey and C. A. Zificsak, Org. Lett., 2002, 4, 1383–1386 CrossRef CAS PubMed.
- See for instance: (a) B. Chen and A. Mapp, J. Am. Chem. Soc., 2005, 127, 6712–6718 CrossRef CAS PubMed; (b) R. Malherbe and D. Bellus, Helv. Chim. Acta, 1978, 61, 3096–3099 CrossRef CAS; (c) R. Malherbe, G. Rist and D. Bellus, J. Org. Chem., 1983, 48, 860–869 CrossRef CAS; (d) D. A. Mundal, C. T. Avetta Jr and R. J. Thomson, Nat. Chem., 2010, 2, 294–297 CrossRef CAS PubMed.
- (a) B. Peng, D. Geerdink, C. Farès and N. Maulide, Angew. Chem., Int. Ed., 2014, 53, 5462–5466 CrossRef CAS PubMed; (b) B. Peng, D. Geerdink and N. Maulide, J. Am. Chem. Soc., 2013, 135, 14968–14971 CrossRef CAS PubMed; (c) V. Valerio, D. Petkova and N. Maulide, Chem.–Eur. J., 2013, 19, 2606–2610 CrossRef CAS PubMed; (d) B. Peng, D. O'Donovan, I. D. Jurberg and N. Maulide, Chem.–Eur. J., 2012, 18, 16292–16296 CrossRef CAS PubMed; (e) V. Valerio, C. Madelaine and N. Maulide, Chem.–Eur. J., 2011, 17, 4742–4745 CrossRef CAS PubMed; (f) X. Huang and N. Maulide, J. Am. Chem. Soc., 2011, 133, 8510–8513 CrossRef CAS PubMed , for related reactions, see: ; (g) A. B. Cuenca, S. Montserrat, K. M. Hossain, G. Mancha, A. Lledós, M. Medio-Simón, G. Ujaque and G. Asensio, Org. Lett., 2009, 11, 4906–4909 CrossRef CAS PubMed; (h) B. Lu, Y. Li, Y. Wang, D. H. Aue, Y. Luo and L. Zhang, J. Am. Chem. Soc., 2013, 135, 8512–8524 CrossRef CAS PubMed; (i) N. D. Shapiro and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 4160–4161 CrossRef CAS PubMed; (j) A. J. Eberhart, C. Cicoira and D. J. Procter, Org. Lett., 2013, 15, 3994–3997 CrossRef CAS PubMed; (k) A. J. Eberhart and D. J. Procter, Angew. Chem., Int. Ed., 2013, 52, 4008–4011 CrossRef CAS PubMed , see also: ; (l) Z. Jia, E. Gálvez, R. M. Sebastián, R. Pleixats, A. Álvarez-Larena, E. Martin, A. Vallribera and A. Shafir, Angew. Chem., Int. Ed., 2014, 53, 11298–11301 CrossRef CAS PubMed , for a recent review on the use of sulfoxides for the functionalization of C–H bonds, see: ; (m) A. P. Pulis and D. J. Procter, Angew. Chem., Int. Ed., 2016, 55, 9842–9860 CrossRef CAS PubMed.
- B. Peng, X. Huang, L.-G. Xie and N. Maulide, Angew. Chem., Int. Ed., 2014, 53, 8718–8721 CrossRef CAS PubMed.
- For some recent contributions of the Eberlin and Jurberg groups to the monitoring of organic reactions using ESI-MS, see: (a) R. Y. Souza, G. A. Bataglion, D. A. C. Ferreira, C. C. Gatto, M. N. Eberlin and B. A. D. Neto, RSC Adv., 2015, 5, 76337–76341 RSC; (b) C. A. Oliveira, M. V. Marques, M. N. Godoi, T. Regiani, V. G. Santos, E. A. F. dos Santos, M. N. Eberlin, M. M. Sá and C. R. D. Correia, Org. Lett., 2014, 16, 5180–5183 CrossRef CAS PubMed; (c) S. T. Rodrigues, V. H. C. Silva, P. M. Lalli, H. C. B. de Oliveira, W. A. da Silva, F. Coelho, M. N. Eberlin and B. A. D. Neto, J. Org. Chem., 2014, 79, 5239–5248 CrossRef PubMed; (d) M. L. Stivanin, M. Duarte, C. Sartori, N. M. R. Capreti, C. F. F. Angolini and I. D. Jurberg, J. Org. Chem., 2017, 82, 10319–10330 CrossRef CAS PubMed.
- V. Tona, S. Ruider, M. Berger, S. Shaaban, M. Padmanaban, L.-G. Xie, L. González and N. Maulide, Chem. Sci., 2016, 7, 6032–6040 RSC.
- In agreement with the low binding affinity of 7a to H+, the acquirement of the ESI(+)-MS/MS of m/z 398, associated to intermediate [7a + H]+, was only possible by employing a 7.2T LTQ-FT Ultra mass spectrometer (ThermoScientific, Bremen, Germany) of increased sensibility. Other spectra were acquired using a 6500 Series Accurate-Mass Quadrupole Time-of-Flight (Q-TOF) LC/MS (Agilent technologies, California, USA). See ESI† for more information.
- Tables S11 and S12 of the ESI† contain the total energies and the Gibbs free energies for all found stationary points.
- For a recent observation of α-carbonyl cation behaviour in unactivated alkyne chemistry, see: T. Stopka, M. Niggemann and N. Maulide, Angew. Chem., Int. Ed., 2017, 56, 13270–13274 CrossRef CAS PubMed.
- (a) C.-H. Wu, B. Galabov, J. I.-C. Wu, S. Ilieva, P. v. R. Schleyer and W. D. Allen, J. Am. Chem. Soc., 2014, 136, 3118–3126 CrossRef CAS PubMed; (b) The charge at Cβ is calculated to be negative for both phenyl and alkyl substituents. However, the absolute values of the charges Cβ@Ph and Cβ@Alk are very different, and the Cβ@Ph is positive relative to the Cβ@Alk as well as to the interacting atoms “X”: Cβ@Ph − Cβ@Alk = −0.125 − (−0.563) = +0.438 a.u..
- When a 2,4,6-trimethylated arylsulfoxide was employed, the alcohol 15 and the highly congested meta-substituted arene 14b were obtained in low combined yield. See ESI† for details.
- T. Yangi, S. Otsuka, Y. Kasuga, K. Fujimoto, K. Murakami, K. Nogi, H. Yorimitsu and A. Osuka, J. Am. Chem. Soc., 2016, 138, 14582–14585 CrossRef PubMed.
- Several side products are formed. The major ones derive from hydration of the keteniminium ion 4+, thus leading to amide 8, and also addition products to cation E, such as 18 (cf.Scheme 4).
- However, when the sulfoxide is flanked by two methoxy-groups on the arene, no meta-arylated product is observed.
- Trace amounts of meta-arylated product 18 were observed in the crude 1H NMR.
- This result is in agreement with the mechanism presented as a “meta”-arylated product would require an intermediate in which the aromaticity of the naphthalene system is broken whereas the intermediate leading to 19l does leave the fused arene intact.
- The Maulide group has recently shown that chiral enantiopure sulfoxides are amenable to chirality transfer in sigmatropic rearrangement chemistry: D. Kaldre, B. Maryasin, D. Kaiser, O. Gajsek, L. González and N. Maulide, Angew. Chem., Int. Ed., 2017, 56, 2212–2215 CrossRef CAS PubMed.
- The described system is highly “sensitive” to the applied level of theory. Thus, among the tested DFT functionals (PBE0, M062X, B3LYP) only the B3LYP is able to predict the intermediate I1. However, the higher level calculations at the MP2 level of theory are in agreement with the B3LYP. Calculations suggest that the reaction shown in Fig. 10 goes exclusively via the stepwise mechanism. See ESI† for details.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, full characterization for new compounds, spectral data and computational details. CCDC 1540803, 1540804 and 1568395. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04736c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |