
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel-catalyzed coupling reaction of alkyl halides with aryl Grignard reagents in the presence of 1,3-butadiene: mechanistic studies of four-component coupling and competing cross-coupling reactions†
Takanori
Iwasaki
 *a,
Asuka
Fukuoka
a,
Wataru
Yokoyama
a,
Xin
Min
a,
Ichiro
Hisaki
b,
Tao
Yang
*cde,
Masahiro
Ehara
*cd,
Hitoshi
Kuniyasu
a and
Nobuaki
Kambe
*a
*a,
Asuka
Fukuoka
a,
Wataru
Yokoyama
a,
Xin
Min
a,
Ichiro
Hisaki
b,
Tao
Yang
*cde,
Masahiro
Ehara
*cd,
Hitoshi
Kuniyasu
a and
Nobuaki
Kambe
*a
aDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: iwasaki@chem.eng.osaka-u.ac.jp; kambe@chem.eng.osaka-u.ac.jp
bDepartment of Material and Life Science, Graduate School of Engineering, Osaka University, Suita, Osaka 565-0871, Japan
cDepartment of Theoretical and Computational Molecular Science, Institute for Molecular Science, 38 Nishigo-Naka, Myodaiji, Okazaki, Aichi 444-8585, Japan. E-mail: ehara@ims.ac.jp
dElements Strategy Initiative for Catalysts and Batteries (ESICB), Kyoto University, Katsura, Kyoto 615-8510, Japan
eFachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Strasse, Marburg 35032, Germany. E-mail: yangt@staff.uni-marburg.de
First published on 5th January 2018
Abstract
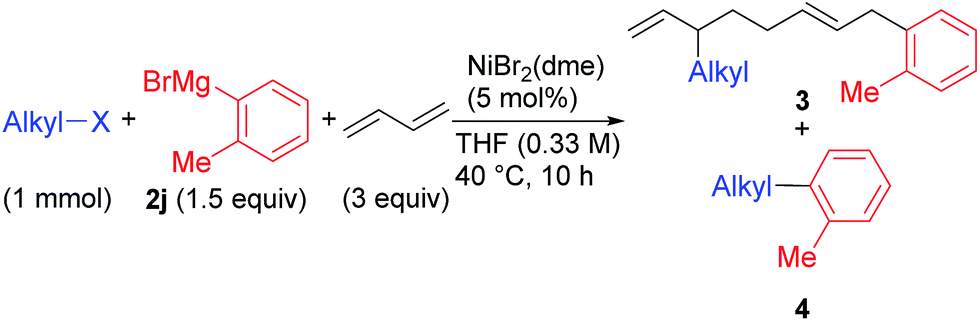
We describe the mechanism, substituent effects, and origins of the selectivity of the nickel-catalyzed four-component coupling reactions of alkyl fluorides, aryl Grignard reagents, and two molecules of 1,3-butadiene that affords a 1,6-octadiene carbon framework bearing alkyl and aryl groups at the 3- and 8-positions, respectively, and the competing cross-coupling reaction. Both the four-component coupling reaction and the cross-coupling reaction are triggered by the formation of anionic nickel complexes, which are generated by the oxidative dimerization of two molecules of 1,3-butadiene on Ni(0) and the subsequent complexation with the aryl Grignard reagents. The C–C bond formation of the alkyl fluorides with the γ-carbon of the anionic nickel complexes leads to the four-component coupling product, whereas the cross-coupling product is yielded via nucleophilic attack of the Ni center toward the alkyl fluorides. These steps are found to be the rate-determining and selectivity-determining steps of the whole catalytic cycle, in which the C–F bond of the alkyl fluorides is activated by the Mg cation rather than a Li or Zn cation. ortho-Substituents of the aryl Grignard reagents suppressed the cross-coupling reaction leading to the selective formation of the four-component products. Such steric effects of the ortho-substituents were clearly demonstrated by crystal structure characterizations of ate complexes and DFT calculations. The electronic effects of the para-substituent of the aryl Grignard reagents on both the selectivity and reaction rates are thoroughly discussed. The present mechanistic study offers new insight into anionic complexes, which are proposed as the key intermediates in catalytic transformations even though detailed mechanisms are not established in many cases, and demonstrates their synthetic utility as promising intermediates for C–C bond forming reactions, providing useful information for developing efficient and straightforward multicomponent reactions.
1 Introduction
Metal-catalyzed multicomponent coupling reactions are useful and straightforward synthetic methods, because they can construct complex carbon frameworks from relatively simple organic molecules through the concomitant formation of multiple new bonds.1 In these transformations, the oxidative dimerization of 1,3-butadiene on low valent group 10 metals is a fundamental process, being often used to produce a variety of C8 carbon frameworks.2 A well-known application is telomerization,3 in which bis(π-allyl)metal complexes of group 10 metals (Ni and Pd) generated by the oxidative dimerization of two molecules of 1,3-butadiene on the metals react with heteroatom nucleophiles such as water, alcohols, and amines. This process has been used for the industrial production of C8 chemicals including 1-octanol and 1-octene.4 In this context, pioneering studies of the Ni-catalyzed multicomponent reaction of two molecules of 1,3-butadiene with carbonyl compounds5a,b and imines5b–d as well as CO and CO2 (ref. 6) were reported in the 1970s. Kimura and Tamaru have expanded these multicomponent coupling reactions of 1,3-butadiene to the bifunctionalization of the C8 carbon framework through the combined use of ketones, aldehydes, or imines with organozinc or organoaluminium reagents.7,8 This reaction could introduce two different carbon moieties into the 2,6-octadiene framework. However, in some cases, three component coupling of these reagents in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio without dimerization can occur as a competing or preferred reaction7a,c,9 due to the competing oxidative cyclization of 1,3-butadiene with carbonyl compounds to form oxanickelacycle intermediates.10
:1:1 ratio without dimerization can occur as a competing or preferred reaction7a,c,9 due to the competing oxidative cyclization of 1,3-butadiene with carbonyl compounds to form oxanickelacycle intermediates.10
We have reported the dimerization and silylation of 1,3-butadiene as another application of these transformations.11 In addition, aiming at the construction of carbon skeletons, we recently developed the multicomponent coupling reaction of two molecules of 1,3-butadiene, aryl Grignard reagents, and alkyl fluorides to give a 1,6-octadiene bearing an alkyl group arising from the alkyl fluorides at the 3-position, and an aryl group arising from the aryl Grignard reagents at the 8-position (Scheme 1).12 It was also revealed that a similar multicomponent coupling reaction using perfluoroarenes instead of alkyl fluorides proceeds smoothly to yield the corresponding 1,6-octadienes with perfluoroaryl and aryl groups.13 In these reactions, three-component coupling products consisting of organo fluorides, aryl Grignard reagents, and 1,3-butadiene in a 1:1:1 ratio were not produced due to the intermediacy of anionic bis(allyl)nickel intermediates. In the alkylation reaction mentioned above, the use of ortho-substituted aryl Grignard reagents is essential for selective formation of four-component coupling products. When unsubstituted aryl Grignard reagents were employed instead, the competing reaction of direct cross-coupling of alkyl fluorides with aryl Grignard reagents occurred, resulting in a mixture of cross-coupling and four-component coupling products (Scheme 1).14 In these reactions as well as our previously reported cross-coupling reactions,15 an anionic nickel complex B generated by the reaction of the bis(π-allyl)nickel complex A with the Grignard reagent is likely the key intermediate, which reacts with electrophiles at the Ni center to promote the cross-coupling reaction (via complex D) or at the γ-carbon of the σ-allyl giving rise to four-component coupling products (via complex C).
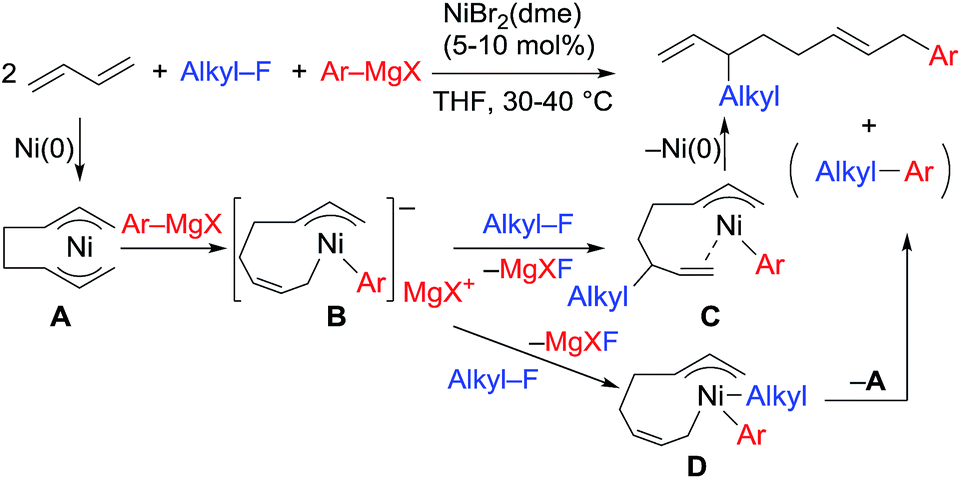 | ||
| Scheme 1 Ni-catalyzed dimerization and alkylarylation of 1,3-dienes. | ||
Herein, to understand the detailed mechanism of the four-component coupling reaction of alkyl fluorides, aryl Grignard reagents, and two molecules of 1,3-butadiene, we performed mechanistic studies and theoretical calculations to clarify the structures and chemical behavior of the anionic nickel intermediate B. In addition, we performed the reaction using various aryl Grignard reagents including ortho-unsubstituted ones to reveal not only the substrate scope of the reaction but also the origin of the selectivity of the four-component coupling reaction over the direct cross-coupling reaction. Steric and electronic effects on the selectivity were also investigated by theoretical calculations.
Notably, the structures and chemical characteristics of anionic complexes of transition metals (the so-called ate complexes16) have rarely been studied, except for the case of cuprates,17 even though ate complexes have been proposed as key intermediates in catalytic C–C bond formations.18–21 In addition, theoretical calculations of ate complexes have not been well-established because of a lack of structural information of ate complexes (especially the counter cation). Therefore, another challenge in this study is to develop a means for theoretical calculations of ate complex-mediated transformations.
2 Results and discussion
2.1. Reaction using aryl Grignard reagents
We first examined the reaction using n-OctF (1a) and p-fluorophenylmagnesium bromide (2a), and the representative results are summarized in Table 1. When the reaction was conducted in the presence of NiBr2(dme) (20 mol%) and 1,3-butadiene (3 equiv.) at 40 °C for 24 h, the cross-coupling product 4aa was obtained in 33% yield along with 3aa incorporating two 1,3-butadiene molecules in 50% yield (entry 1). The structure of compound 3aa was fully assigned by NMR. The alkyl and aryl groups were introduced site-selectively onto the 3- and 8-positions of the 1,6-octadiene framework, respectively, with an E-configuration of the internal alkene moiety. The amount of Ni catalyst could be reduced to 10 mol% (entry 2); however, a further reduction to 5 mol% led to incomplete reaction within 24 h (79% conversion of 1a). We found that the selectivity of the four-component coupling versus cross-coupling reactions is temperature-dependent. When the reaction temperature was increased to 50 °C, the selectivity decreased to almost 1:1 (entry 3). In contrast, a lower reaction temperature increased the selectivity to 1.7:1 (entry 4), although the reaction was sluggish at 0 °C, and 41% of 1a remained after 24 h (entry 5).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | X | T (°C) | 3aa (%)b | 4aa (%)b |
| a Reaction conditions: catalyst, n-OctF (1.0 mmol), p-F-C6H4MgBr (2.0 mmol), and 1,3-butadiene (3.0 mmol) in THF (3 mL) for 24 h. b Yields were determined by GC. Isolated yields are in parentheses. | |||||
| 1 | NiBr2(dme) (20) | F | 40 | 50 | 33 |
| 2 | NiBr2(dme) (10) | F | 40 | 48 | 36 |
| 3 | NiBr2(dme) (10) | F | 50 | 49 | 49 |
| 4 | NiBr2(dme) (10) | F | 30 | 63 (48) | 37 (37) |
| 5 | NiBr2(dme) (10) | F | 0 | 31 | 19 |
| 6 | NiBr2(dme) (10) | Cl | 30 | 2 | 10 |
| 7 | NiBr2(dme) (10) | Br | 30 | 15 | 52 |
| 8 | NiBr2(dme) (10) | I | 30 | 6 | 58 |
| 9 | NiBr2(dme) (10) | OTs | 30 | 17 | 68 |
| 10 | None | F | 25 | n.d. | n.d. |
| 11 | Pd(acac)2 (20) | F | 25 | n.d. | 52 |
| 12 | PtCl2 (20) | F | 25 | n.d. | 3 |
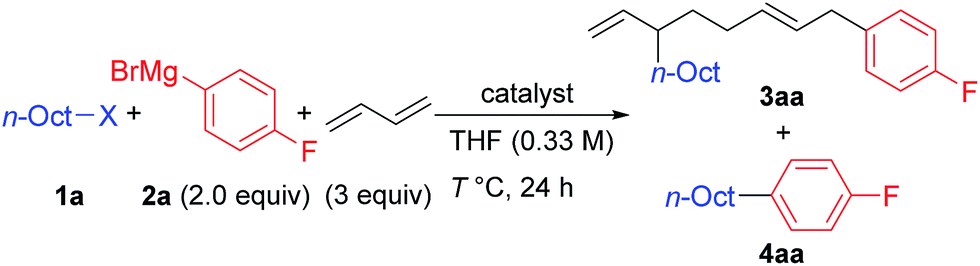
Next, we investigated the effect of the leaving group (entries 6–9). The reaction of n-OctCl was sluggish (entry 6). Alkyl bromides, iodides, and tosylates (which are good coupling partners in alkyl–alkyl cross-coupling reactions)15,20a–c gave 4aa as the major product in 52 to 68% yields along with small amounts of 3aa (entries 7–9), where the product ratios 3aa/4aa were 0.20:1 (Cl), 0.29:1 (Br), 0.10:1 (I), and 0.25:1 (OTs). These results imply the usefulness of alkyl fluorides as promising alkylating reagents toward 1,3-butadiene.22,23 Neither four-component coupling nor cross-coupling reactions could proceed without the Ni catalyst (entry 10). It is known that other group 10 metals also promote the oxidative dimerization of 1,3-dienes.3,20a However, no desired four-component coupling product 3aa was yielded by Pd and Pt catalysts, while the cross-coupling product 4aa was obtained in 52% and 3% yields, respectively (entries 11 and 12).
In order to improve the selectivity of 3aa over 4aa, we tested the reaction using various ligands, additives, and solvents. However, these attempts were not fruitful. For instance, the addition of PPh3 slightly affected the reaction efficiency without appreciable change in the selectivity. The addition of Mg and Li salts, on the other hand, decreased the selectivity to 1:1 and 0.6:1, respectively. The reactions in mixed solvents of toluene-THF or hexane-THF depressed both the yields and selectivity.24
Scheme 2 summarizes the results of the four-component coupling reaction with various aryl Grignard reagents and alkyl fluorides under the same conditions as entry 4 in Table 1, where the yields of the direct cross-coupling product 4 are given in parentheses. Alkyl fluorides carrying various functional groups produced mixtures of 3 and 4 in a 1.5:1 to 1:1 ratio. It should be noted that the aromatic C–F and C–Cl bonds remained intact, and the aliphatic C–F bonds were cleaved exclusively. The reaction of γ-branched alkyl fluoride 1g resulted in better selectivity (3ga:4ga = 5.2:1) although the reaction was slow and did not complete within 24 h (80% conv.). This result may suggest that sterically hindered alkyl fluorides prefer the four-component coupling reaction to give 3 rather than the cross-coupling reaction. However, the reaction of (fluoromethyl)cyclohexane (1h) resulted in a low yield, and no reaction took place with fluorocyclohexane (1i). Next, we ran the reactions using different aryl Grignard reagents (2b–i). PhMgBr (2b) afforded the corresponding products 3ab and 4ab in 40% and 55% yields, respectively (95% total yield with a 0.7:1 ratio). The introduction of electron-donating groups such as Me, MeO, and Me2N led to the preferential formation of the cross-coupling product 4. On the other hand, the para-chlorophenyl Grignard reagent (2e) gave a 1:1 mixture of 3ae and 4ae in 44% total yield. Substituents at the meta-position showed a similar tendency. The reaction of 1g with phenyl and para- and meta-tolyl Grignard reagents afforded four-component coupling products 3gb, 3gc, and 3gg as the major product. The thienyl Grignard reagent 2i underwent a cross-coupling reaction exclusively to give 4gi in 23% yield.
 | ||
| Scheme 2 The Ni-catalyzed four-component coupling reaction of alkyl fluorides, aryl Grignard reagents, and 1,3-butadiene. Isolated yields. The yields of cross-coupling products 4 are shown in parentheses. aca. 20% of 1g was recovered. bThe reaction time was 42 h. cn.i. = not isolated. d44% of 1g was recovered. | ||
2.2. Reaction using ortho-substituted aryl Grignard reagents
We found a remarkable effect of the ortho-substituent in the aryl Grignard reagents, as shown in eqn (1).12 When o-tolylmagnesium bromide (2j) was employed in the reaction with 1g, the four-component coupling product 3gj was obtained as the sole product in 85% yield, and no 4gj was observed.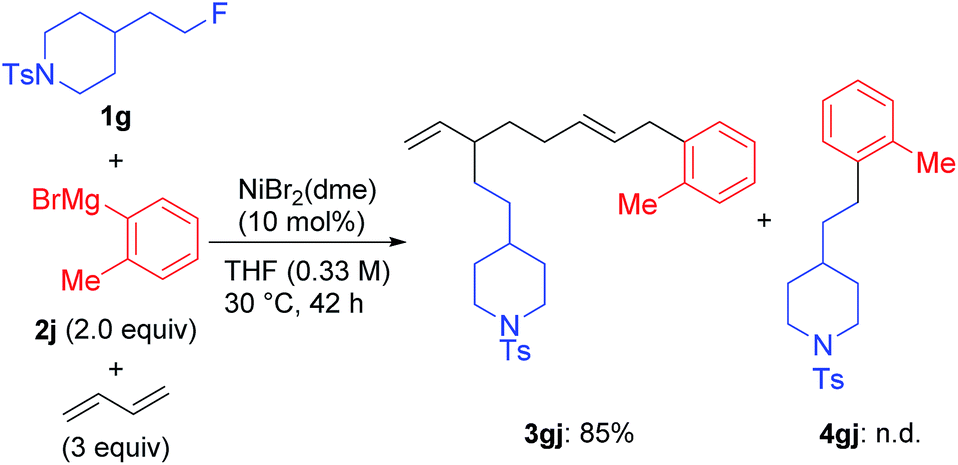 | (1) |
Using 1a and 2j under the previously optimized conditions (Table 1, entry 4), the reaction proceeded smoothly to give 3aj in 92% GC yield within 10 h (Table 2, entry 1). The addition of PPh3 or a bidentate phosphine (dppf) hindered the reaction (entries 2 and 3). Other Ni salts such as NiCl2 and Ni(acac)2 showed a comparable reactivity (entries 4 and 5). The amount of Grignard reagent 2j could be reduced to 1.5 equiv. without significant loss of yield (entry 6). Although the reaction did not complete within 10 h with 5 mol% of NiBr2(dme) (entry 7, 88% conv. of 1a), the four-component coupling product 3aj was obtained in 87% yield by elevating the reaction temperature to 40 °C (entry 8).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | 2j | Temp. (°C) | 3aj (%)a |
| a Determined by GC (isolated yield is in parentheses). In all cases, ca. 5% of 4aj was observed in the GC analysis. | ||||
| 1 | NiBr2(dme) (10) | 2.0 equiv. | 30 | 92 |
| 2 | NiCl2(PPh3)2 (10) | 2.0 equiv. | 30 | 43 |
| 3 | NiCl2(dppf) (10) | 2.0 equiv. | 30 | 8 |
| 4 | NiCl2 (10) | 2.0 equiv. | 30 | 90 |
| 5 | Ni(acac)2 (10) | 2.0 equiv. | 30 | 95 |
| 6 | NiBr2(dme) (10) | 1.5 equiv. | 30 | 88 |
| 7 | NiBr2(dme) (5) | 1.5 equiv. | 30 | 79 |
| 8 | NiBr2(dme) (5) | 1.5 equiv. | 40 | 87 (86) |
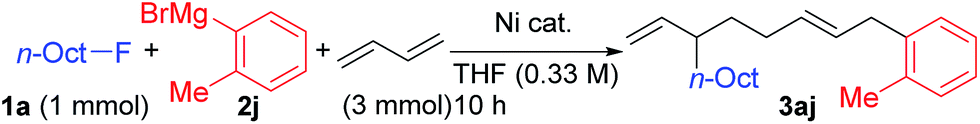
The results using various alkyl (pseudo)halides are summarized in Table 3. While the C(sp3)–Cl bond is weaker than the C(sp3)–F bond, n-OctCl was less reactive and mostly recovered unreacted (entry 2). Although alkyl bromides and iodides afforded the cross-coupling product predominantly with the p-fluorophenyl Grignard reagent 2a, as shown in Table 1, introducing an ortho-substituent into ArMgBr largely suppressed the cross-coupling, giving rise to the selective formation of the four-component coupling product albeit only in moderate yields (entries 3 and 4). Alkyl mesylate was found to be ineffective as an alkylating reagent in the present reaction (entry 5).
Under the optimized conditions shown in entry 8 of Table 2, the scope and limitations of the aryl Grignard reagents and alkyl fluorides were further examined, and the results are summarized in Scheme 3.12 Sterically hindered aryl Grignard reagents such as 2-ethyl-, 2-isopropyl-, and 2,6-dimethylphenyl Grignard reagents (2k–m) underwent the four-component coupling reaction to produce 3ak–am in good yields. On the other hand, the introduction of a methoxy group at the ortho-position (2n) completely suppressed the reaction (3an). When o-tolyl Grignard reagents bearing electron-withdrawing groups at the para-position (2o and 2p) were employed, the yields were slightly reduced (3ao and 3ap). The reaction of the 2,4-dimethylphenyl Grignard reagent (2q) resulted in an excellent yield (3aq) although the p-methoxy substituent led to a somewhat lower yield (3ar). The thienyl Grignard reagent (2t) was less effective, and the naphthyl Grignard reagent (2s) afforded 3as in 81% yield.
 | ||
| Scheme 3 The scope and limitations of the four-component coupling reaction. aThe reaction was conducted in a 0.1 M concentration of 1 for 20 h. | ||
Alkyl fluorides (2b–m) carrying various functionalities including acetal, silyl ether, N-tosyl, and N-Boc protecting groups afforded the four-component coupling products in good yields. A terminal olefin moiety in 1j remained intact to give 3jm in 91% yield under the selective oxidative dimerization conditions of 1,3-dienes. The alkyl fluoride 1d bearing an acidic hydrogen at the α-position of thiophene also gave 3dm in an excellent yield (93%). It is well known that low-valent Ni intermediates can cleave C(sp2)–X bonds including the C–F bond.22 However, under the reaction conditions, both the C(sp2)–Cl and C(sp2)–F bonds remained unchanged, and the C(sp3)–F bond was selectively cleaved to give 3em, 3eo, 3fm, and 3fs. In addition, 1-chloro-6-fluorohexane (1m) reacted at the C(sp3)–F bond rather than the C(sp3)–Cl bond to exclusively give 3mm and 3mo.
When isoprene was employed as the conjugated diene under the optimized conditions, the corresponding four-component coupling product 3aj′ was obtained as a mixture of four inseparable regioisomers in a 1:1.3:1.4:1.5 ratio in 76% total yield (eqn (2)). In contrast, the reaction of 1,3-pentadiene was sluggish, and no desired four-component coupling products were observed.
 | (2) |
2.3. Mechanistic studies
The mechanistic studies include the observation and isolation of catalytic intermediates, kinetic parameters, electronic effects of the Grignard reagents, and theoretical calculations. Based on the results, plausible catalytic cycles of the Ni-catalyzed coupling reactions of alkyl fluorides with aryl Grignard reagents in the presence of 1,3-butadiene are proposed (Scheme 4). These reactions are triggered by the oxidative dimerization of 1,3-butadiene on Ni(0), generated in situ by the reduction of Ni salt with Grignard reagents, to form the bis(π-allyl)nickel A (step A). The reaction of A with aryl Grignard reagents yields the anionic Ni complex B, which has higher nucleophilicity towards alkyl fluorides. Although the four potent nucleophilic centers exist in complex B, namely the Ni center, the ipso carbon of the Ar group, and the α- and γ-positions of the σ-allyl group, two reaction pathways operate. When the γ-carbon of the σ-allyl group attacks the alkyl fluoride (step C), the four-component coupling product 3 is yielded through the reductive elimination of the thus-formed complex C (step D). Alternatively, the Ni center in complex B can react with alkyl fluorides to give the cross-coupling product 4 through complex D (steps E and F).25 Therefore, the reaction courses of four-component coupling vs. cross-coupling are determined by the reaction of alkyl fluorides with complex B (step C vs. step E).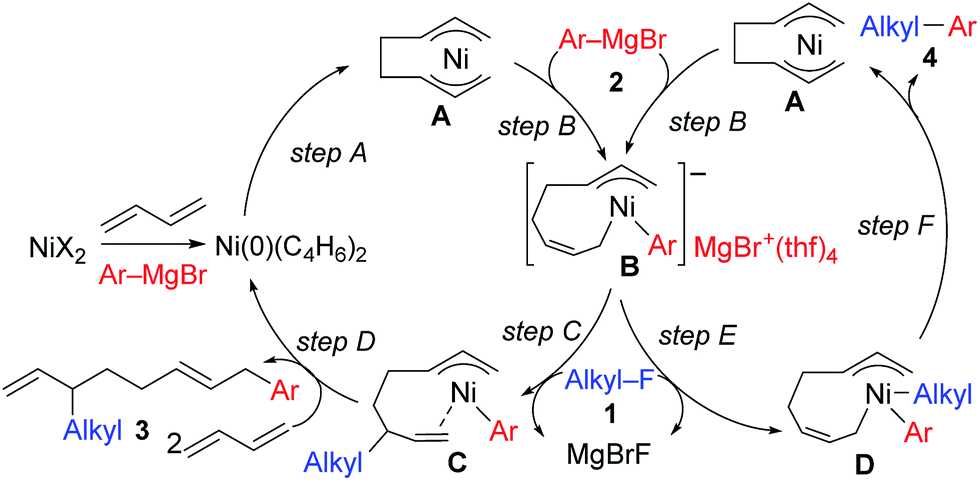 | ||
| Scheme 4 Proposed catalytic cycles of the Ni-catalyzed four-component coupling and cross-coupling reactions. | ||
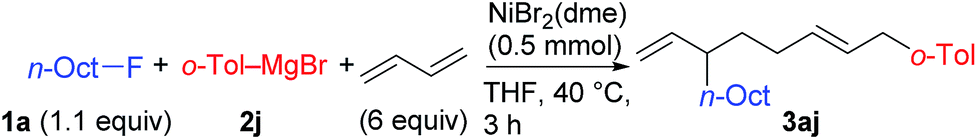
 ) and one 2,6-dimethylphenyl group (
) and one 2,6-dimethylphenyl group ( ) (Fig. 1c), which could be assigned to the corresponding nickelate complex 5,28 along with signals of free 1,3-butadiene (
) (Fig. 1c), which could be assigned to the corresponding nickelate complex 5,28 along with signals of free 1,3-butadiene ( ). The formed nickelate complex 5 was stable in THF for a day even at room temperature.
). The formed nickelate complex 5 was stable in THF for a day even at room temperature.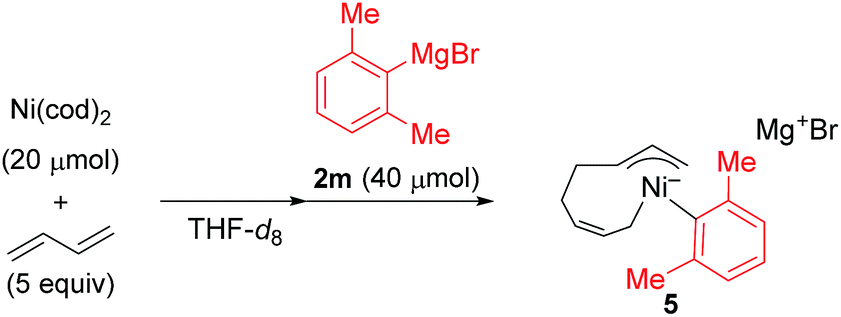 | (3) |
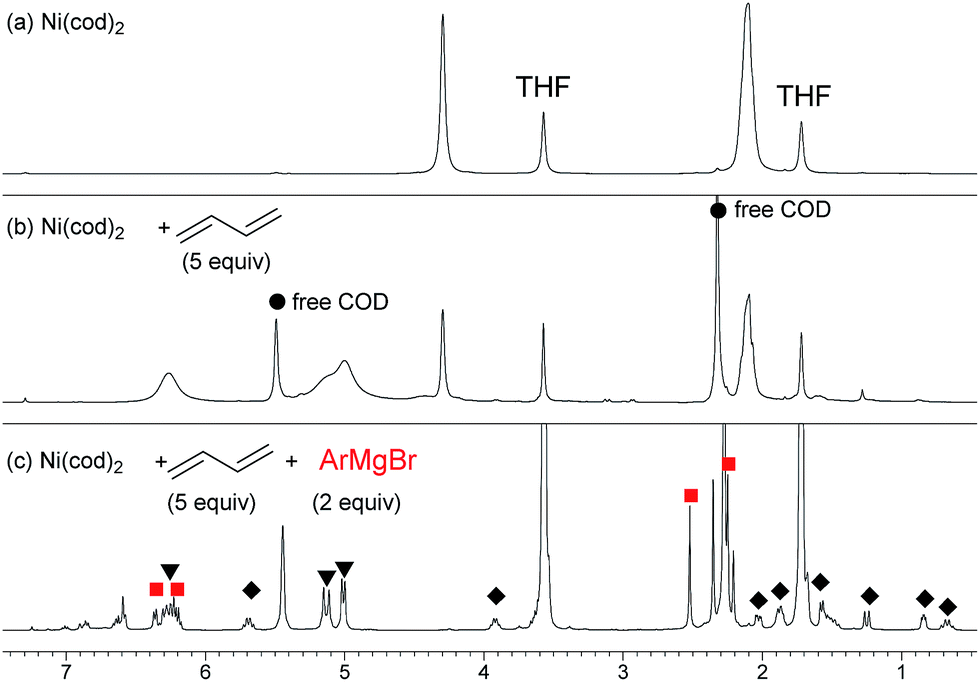 | ||
| Fig. 1 1H NMR spectra for the reaction of Ni(0) with 1,3-butadiene and 2m in THF-d8 at 20 °C. (a) Ni(cod)2, (b) Ni(cod)2 and 5 equiv. of 1,3-butadiene, and (c) Ni(cod)2, 1,3-butadiene (5 equiv.), and 2m (2 equiv.). | ||
In order to isolate the nickelate complex, we tested various organometallic reagents and ligands for the counter cation. Lithium bearing polyether ligands such as 1,2-dimethoxyethane (DME) and 12-crown-4 ether were found to be a suitable counter cation. For instance, when Ni(cod)2 was treated with 2,6-dimethylphenyllithium (6) and 1,3-butadiene in Et2O and then DME was added, nickelate complex 7 was obtained as an orange semi-solid in 94% yield (Scheme 5). The 1H NMR spectrum of thus formed nickelate 7 is in good agreement with the above-mentioned NMR spectra when using the corresponding Grignard reagent 2m (Fig. 1c).
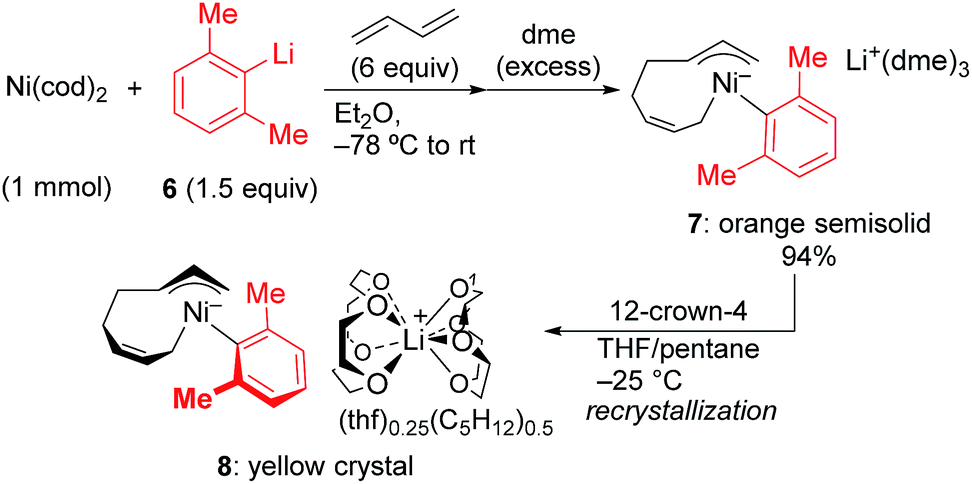 | ||
| Scheme 5 Isolation of anionic Ni complexes. | ||
Recrystallization of the nickelate complex 7 to obtain crystals suitable for X-ray was not successful. However, fine crystals of 8 were obtained by changing DME to 12-crown-4 (Scheme 5). As shown in Fig. 2,24,29 the crystal structure of 8 contains anionic Ni and the Li cation, where the octadiene moiety, generated by the oxidative dimerization of 1,3-butadiene, binds to the Ni center in η1,η3-fashion as expected. It is also revealed that the C–C double bond (C6![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C7) of the σ-allyl group has the Z-configuration. The Ni center carries π-allyl, σ-allyl, and aryl groups in a square planar geometry.
C7) of the σ-allyl group has the Z-configuration. The Ni center carries π-allyl, σ-allyl, and aryl groups in a square planar geometry.
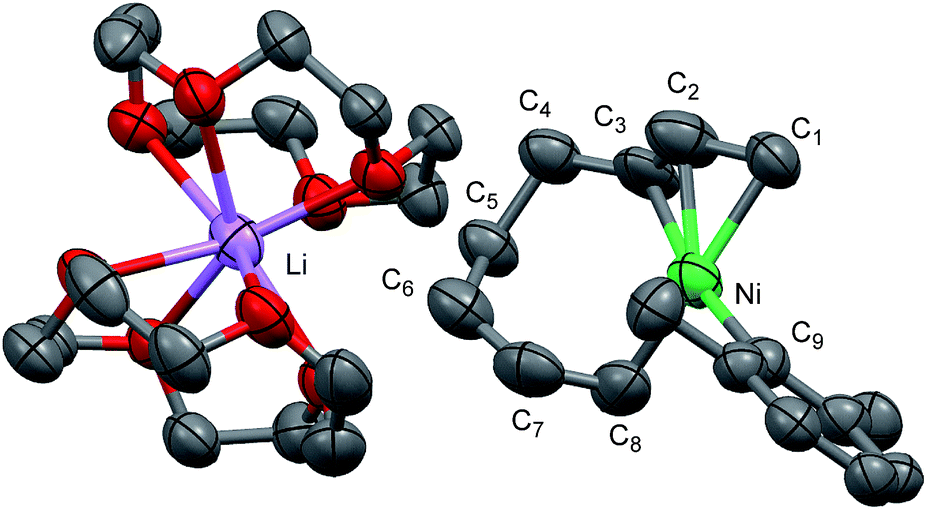 | ||
| Fig. 2 An ORTEP drawing of one of the four asymmetric units of [Ni(C8H9)(C8H12)·Li(12-crown-4)2]4(thf)(C5H12)2 (8) with thermal ellipsoids at the 50% probability level. All hydrogen atoms and solvent molecules are omitted for clarity. | ||
 | (4) |
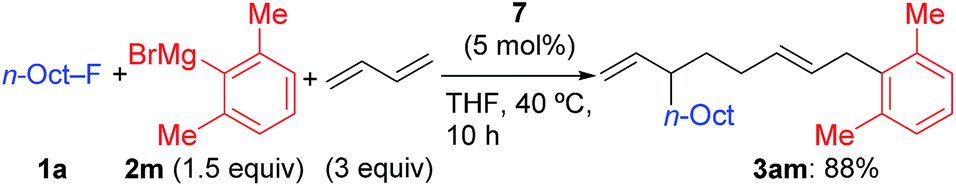
In contrast, when 7 was treated with 2 equiv. of n-OctF (1a) in THF-d8 at 40 °C for 4 h, no change was observed in its 1H NMR spectrum. The addition of MgCl2 to the reaction mixture promoted the reaction to give 3am in 10% yield (Scheme 6).12 As shown in Scheme 7, when PhZnI (9) and PhLi (10) were used instead of Grignard reagents, neither four-component coupling product 3ab nor cross-coupling product 4ab was yielded.12 PhMgCl (2b′) also gave a mixture of 3ab and 4ab albeit in somewhat lower total yields compared to PhMgBr (2b) (Scheme 7).30 These results imply that the Mg cation contributes to the reaction of the nickelate intermediate B with alkyl fluorides to activate the C–F bond by coordination to F.30
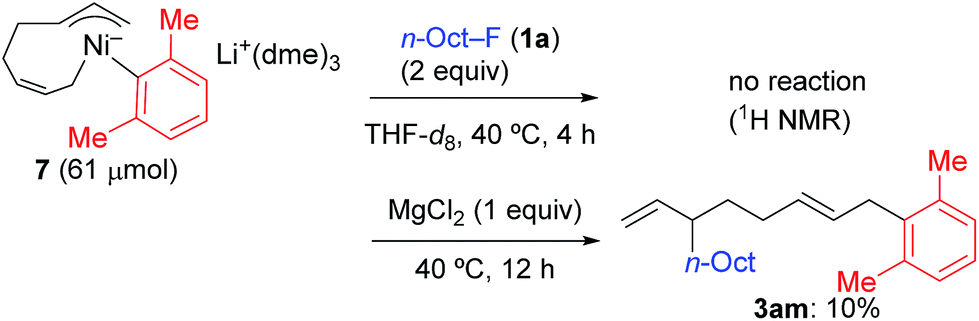 | ||
| Scheme 6 The stoichiometric reaction of nickelate complex 7 with 1a: effect of the Mg ion. | ||
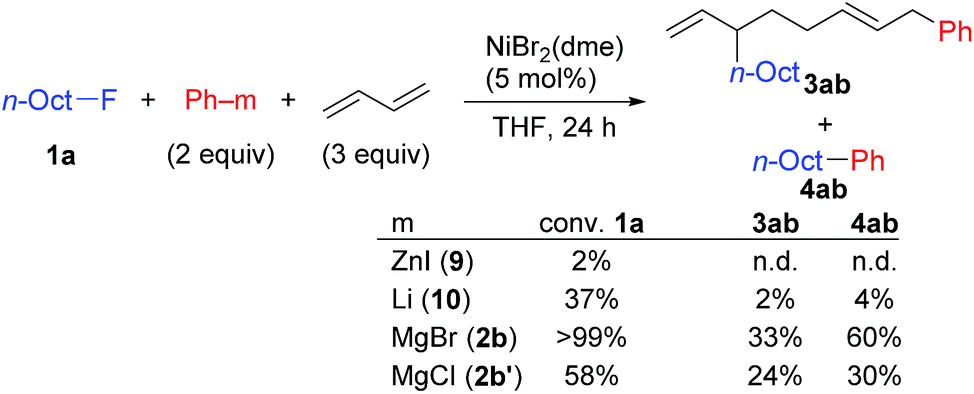 | ||
| Scheme 7 The reaction of arylzinc, aryllithium, and aryl Grignard reagents. Conversion and yields were determined by GC. | ||
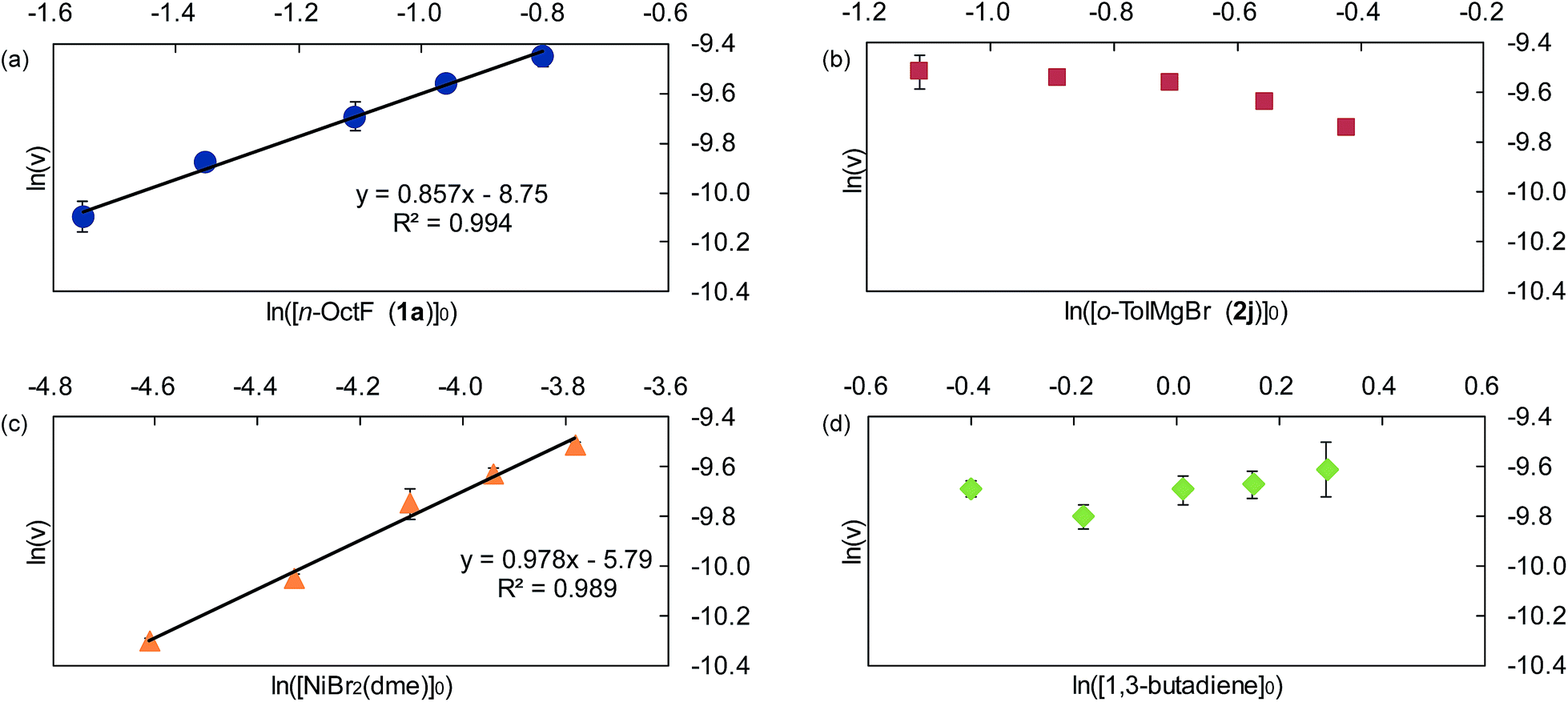 | ||
| Fig. 3 Double logarithm plots of the initial reaction rates against initial concentrations of each substrate. (a) [1a]0 = 0.21 to 0.45 M, (b) [2j]0 = 0.33 to 0.66 M, (c) [NiBr2(dme)]0 = 0.010 to 0.023 M, and (d) [1,3-butadiene]0 = 0.67 to 1.34 M. | ||
Next, we conducted the reaction at different temperatures (30–50 °C) using o-tolyl- and 2,6-dimethylphenylmagnesium bromide (2j and 2m), and the results are plotted in Fig. 4a and b, respectively. Eyring plots of these data employing the rate law d[3a]/dt = kobs[1a] showed good linear relationships (Fig. 4c), and the resulting parameters are summarized in Table 5. A similar analysis was also conducted for the reaction of p-fluorophenylmagnesium bromide (2a).
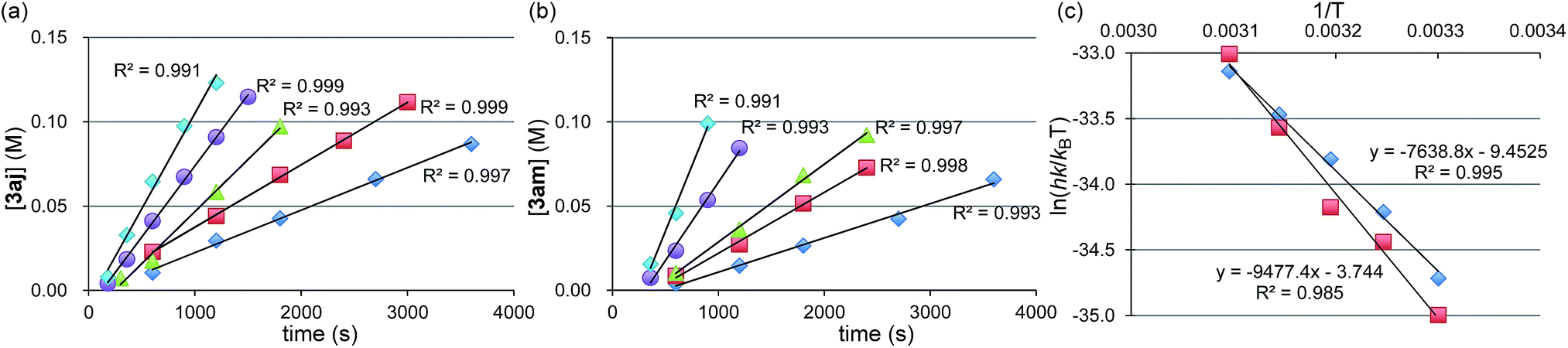 | ||
Fig. 4 (a) Time-course of the reaction of 1a with 2j at 30 °C ( ), 35 °C ( ), 35 °C ( ), 40 °C ( ), 40 °C ( ), 45 °C ( ), 45 °C ( ), and 50 °C ( ), and 50 °C ( ). (b) Time-course of the reaction of 1a with 2m at 30 °C ( ). (b) Time-course of the reaction of 1a with 2m at 30 °C ( ), 35 °C ( ), 35 °C ( ), 40 °C ( ), 40 °C ( ), 45 °C ( ), 45 °C ( ), and 50 °C ( ), and 50 °C ( ). (c) Eyring plot of the reaction using 2j ( ). (c) Eyring plot of the reaction using 2j ( ) and 2m ( ) and 2m ( ). ). | ||
| ArMgBr | ΔG‡273 (kJ mol−1) | ΔH‡ (kJ mol−1) | ΔS‡ (J Kmol−1) |
|---|---|---|---|
| 2a | 87.4 ± 7.6 | 62.9 ± 3.9 | −89.9 ± 12.4 |
| 2j | 85.0 ± 5.1 | 63.5 ± 2.7 | −78.6 ± 8.7 |
| 2m | 87.3 ± 10.6 | 78.8 ± 5.6 | −31.1 ± 18.0 |
These data indicate that the present four-component coupling reaction is mainly controlled by enthalpy factors in all cases employing Grignard reagents (2a, 2j, and 2m). The introduction of one methyl group into the ortho-position (o-TolMgBr (2j) vs. p-FC6H4MgBr (2a)) showed a small effect on the activation parameters, probably because the ortho-methyl group is located far from the reacting γ-allylic carbon in the transition state. In contrast, the activation parameters changed significantly upon the introduction of methyl groups into both ortho-positions (2m), which increased the activation enthalpy.32
Activation parameters of the Ni-catalyzed cross-coupling reaction of alkyl(pseudo)halides with n-BuMgCl in the presence of 1,3-butadiene were also determined: ΔG‡273 = 55.3 ± 6.0 kJ mol−1, ΔH‡ = 51.2 ± 2.6 kJ mol−1, and ΔS‡ = −14.8 ± 12.3 J Kmol−1 for iodide; ΔG‡273 = 61.9 ± 8.6 kJ mol−1, ΔH‡ = 57.3 ± 4.0 kJ mol−1, and ΔS‡ = −16.9 ± 16.7 J Kmol−1 for bromide; and ΔG‡273 = 71.1 ± 9.8 kJ mol−1, ΔH‡ = 52.7 ± 4.9 kJ mol−1, and ΔS‡ = −67.6 ± 18.0 J Kmol−1 for tosylate.25b These data correspond to step E in Scheme 4 and show similar tendency with step C, suggesting similar transition states between the four-component coupling and the cross-coupling reactions.
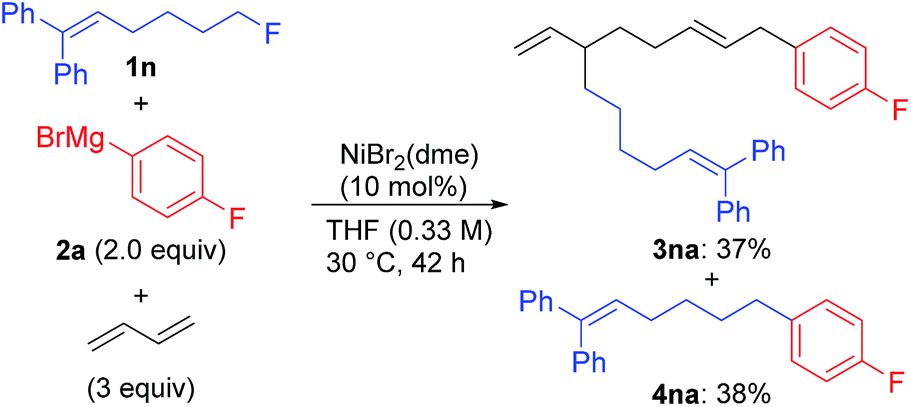 | (5) |
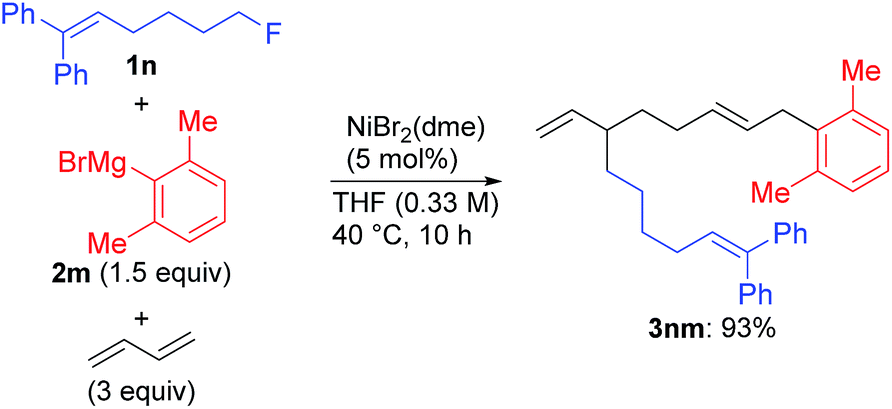 | (6) |
|
|
|||||
|---|---|---|---|---|---|
| R | H (2j) | Cl (2p) | F (2o) | OMe (2q) | Me (2r) |
| k X (10−3 s−1) | 12.7 | 4.80 | 7.47 | 12.8 | 17.8 |

Among Hammett plots of various substituent constants, Yukawa–Tsuno’s σ0, which is determined by the electronic effects of the aromatic substituents on the reaction center connected by methylene tethers, showed a satisfactory linear relationship (R = 0.962) with a negative ρ value of −1.178 (Fig. 5). This negative ρ value is in accord with the SN2 mechanism of the rate-determining step, in which electron-donating substituents accelerate the nucleophilic attack by increasing the electron density of the allylnickel intermediates.
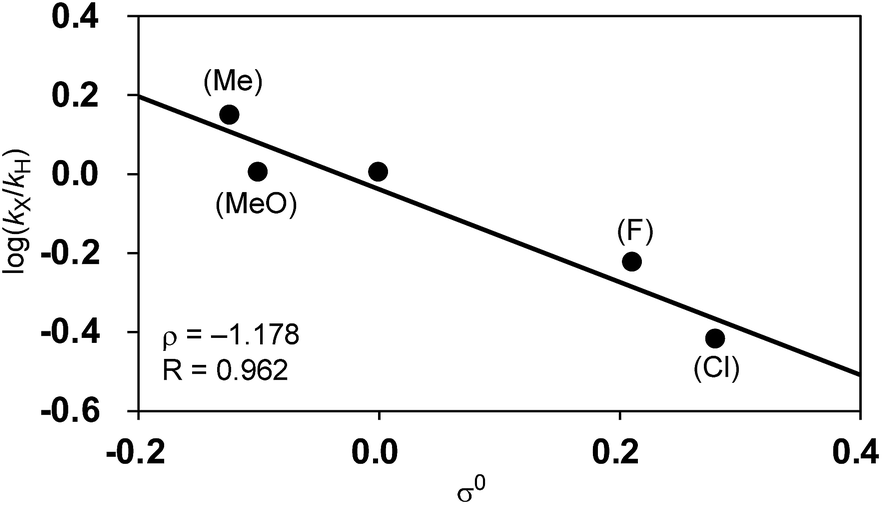 | ||
| Fig. 5 Hammett plot of the relative reaction rates against Yukawa–Tsuno’s σ0. | ||
This result is also in good agreement with the observed electronic effects on the selectivity of the four-component coupling and cross-coupling reactions. As mentioned in Section 2.1 regarding the results in Scheme 2, electron-deficient Grignard reagents showed higher selectivities for four-component coupling over cross-coupling compared to electron-rich ones. For instance, the ratio between the four-component coupling product 3 and cross-coupling product 4 is 1.7:1 for the p-fluorophenyl Grignard reagent (2a), 1:1.9 for the p-tolyl Grignard reagent (2c), and 1:3.3 for the p-dimethylaminophenyl Grignard reagent (2f). This selectivity is related to the relative reaction rates of the nucleophilic attack by the γ-position of the σ-allyl group of nickelate intermediate B (step C) and that by the nickel center (step E). This can be explained by the fact that electron-donating groups on the aryl group increase the electron density on the directly connected Ni more effectively, compared to that on the remote γ-carbon of the σ-allyl group.
Since the selectivity is determined by the relative rates of step C vs. step E in Scheme 4, the product ratio 3/4 can be expressed by the relative rate constants between step C and step E (kC/kE). Therefore, the ratio of selectivity between X and H, (3X/4X)/(3H/4H), is equal to (kCX/kEX)/(kCH/kEH) = (kCX/kCH)/(kEX/kEH) = ρC/ρE. A plot of log[(3X/4X)/(3H/4H)] against the substituent constants (Yukawa–Tsuno’s σ0) shows a good linear relationship (R = 0.945) with a slope of +0.862 (Fig. 6).34 With ρC = −1.178 for step C, ρE for the cross-coupling reaction (step E) is calculated to be −1.366. The relatively large negative value of ρE in comparison to ρC is also ascribable to the direct connection of the Ni reaction site with the Ar group.
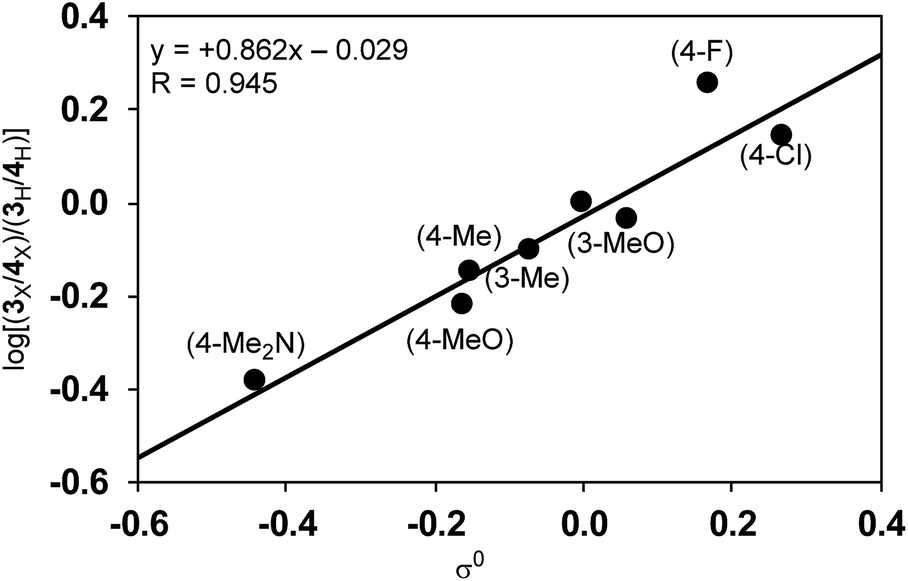 | ||
| Fig. 6 The relationship between the product ratio 3/4 of substituted Grignard reagents against σ0. | ||
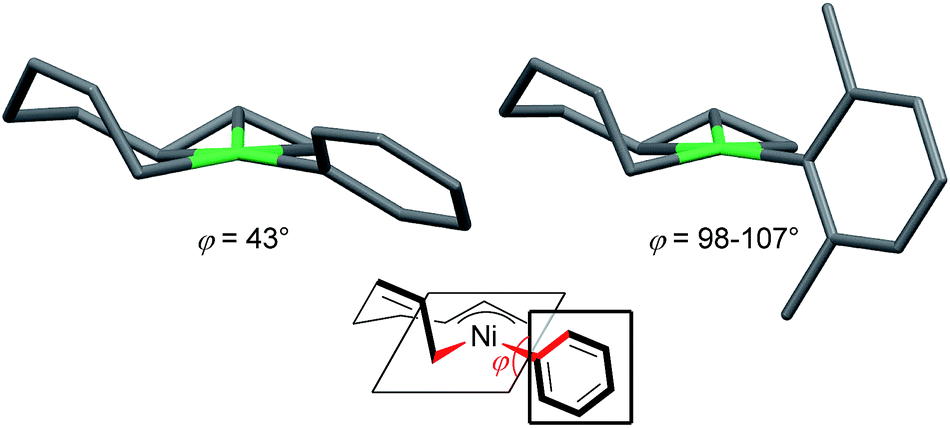 | ||
| Fig. 7 The molecular structures of nickelate complexes 11 (left) and 8 (right). Bottom: the dihedral angle between the square planar Ni plane and the aryl ring. | ||
Fig. 8 shows the space-filling model of complexes 11 and 8. The Ni center (green) of phenyl complex 11 is open to the approach of alkyl fluorides (Fig. 7, left) while such an approach to the Ni of complex 8 is blocked by the orthogonally oriented 2,6-dimethylphenyl group (right). Therefore, alkyl fluorides react with the less hindered γ-carbon of the σ-allyl group (orange), giving rise to the four-component coupling product 3.
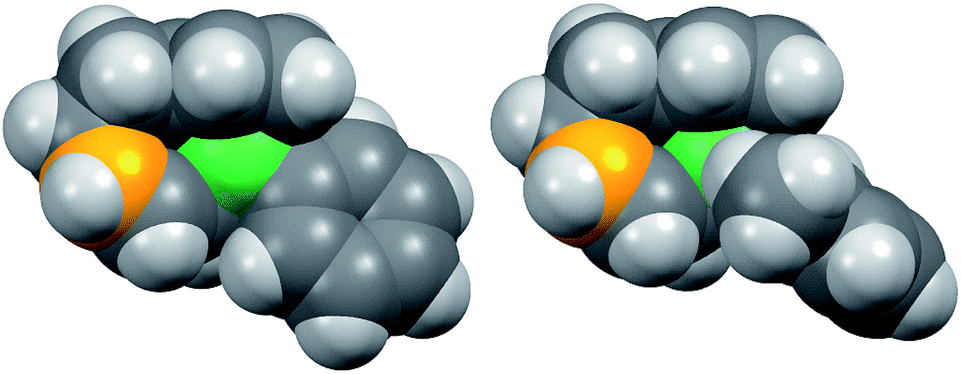 | ||
| Fig. 8 Space-filling models of the nickelate complexes (top view) in Fig. 7. The reacting carbon of the σ-allyl group and Ni atom are shown in orange and green, respectively. | ||
|
|
|||
|---|---|---|---|
| Entry | R | k X/kH | Total yield (%)a |
| a Total yield of the products at 40 min. b Due to the difficulty in analysis, the competitive reaction with 2,4-dimethylphenylmagnesium bromide (2q) instead of 2j was performed to give kF/kMe = 1.12 and calculated with kMe/kH = 0.61. | |||
| 1 | H (2j) | — | 36 |
| 2 | Cl (2p) | 1.12 | 25 |
| 3 | F (2o)b | 0.68 | 34 |
| 4 | Me (2q) | 0.61 | 46 |
| 5 | OMe (2r) | 0.18 | 43 |
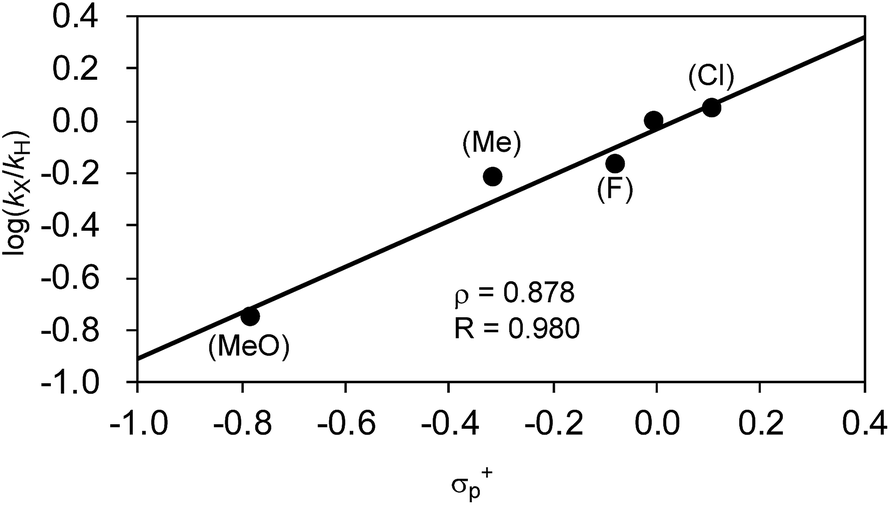 | ||
| Fig. 9 Hammett plot of the relative reaction rates obtained in the competitive reaction against σ+p. | ||
Another important finding from Table 7 is that electron-withdrawing substituents retard the reaction and produce lower total yields. For instance, when the o-tolyl Grignard reagent (2j) was used alone, 36% of 3aj was yielded after 40 min (entry 1). The competitive reaction of 2j and the electron-deficient 2p produced a mixture of 3aj and 3ap with a combined yield of 25% (entry 2). The use of the electron-rich Grignard reagent 2r along with 2j afforded products in 43% total yield (entry 5). These results may indicate that the nickelate complex-forming step (step B) prefers electron-deficient Grignard reagents. However, the formed nickelate with the electron-deficient Ar group reacts with alkyl fluorides at relatively slow rates compared to those formed with electron-rich ones.
2.4. Computational studies
The mechanisms of the four-component coupling and cross-coupling reactions were theoretically investigated using density functional theory calculations. Molecular structures were optimized at the M06 (ref. 36)/6-31G(d,p) level of theory using the Gaussian 09 (ref. 37) program, followed by single-point energy calculations at the M06/6-311+G(d,p) level with the SMD38 model (THF). The energy reported here includes the electronic energy, zero-point energy, thermal correction at 298 K and 1 atm, and solvent effect correction. The molecular structures were drawn with CYLview.39 The experimental findings discussed in Section 2.3 revealed that nickelate complexes are readily formed under the reaction conditions, and the reaction of the nickelate complex with alkyl fluorides is the rate- and selectivity-determining step of the whole catalytic cycle. In order to reveal two competing reaction pathways, we thus focused on the reaction of the nickelate complex with alkyl fluorides to give the four-component coupling and cross-coupling products.One of the most critical reasons to give such inaccurate results is due to the structure and position of the counter cation, which coordinates to not only the leaving group (Br) but also a π-ligand in the anionic part, resulting in the strained structure of the transition states.41,42 Such cation–π interactions are often encountered in theoretical calculations of ate complex-mediated transformations.43,44
As mentioned above, the Mg cation contributes to both the four-component coupling and the cross-coupling reactions (Scheme 7), and the cationic part (Mg+Br) of the nickelate complex would exist as solvent separated ion pair in THF.45 Therefore, we chose models that have four THF molecules on the MgBr cation to fulfill the six coordination sites of Mg in the TSs. The chosen solvent separated ion pair mode led many local minima throughout the theoretical calculations because of not only the many possibilities of the relative position of the anionic Ni moiety and Mg cation46 but also the flexibility of the octadienediyl moiety and alkyl fluorides. However, this model provides more reasonable results compared to a model containing MgBr as a naked counter cation. The following discussions are carried out by typical optimized structures employing Mg+Br·4THF as the counter cation.
The energy profiles and the corresponding key transition states are shown in Fig. 10 and 11, respectively. All the other geometric structures are presented in the ESI.† From INT1, there are two different pathways that lead to four-component coupling and cross-coupling products. On one hand, the methyl fluoride is inserted with the methyl group pointing to the γ-carbon, forming an intermediate INT2 with a relative energy of 42.6 kJ mol−1. Via a transition state TS3 (78.5 kJ mol−1), the methyl group is transferred to the γ-carbon of the σ-allyl group giving rise to INT4. The energy of INT4 is as low as −210.8 kJ mol−1, revealing that the bond formation between the methyl group and γ-carbon is largely exothermic. Then, MgBrF·4THF dissociates from the reaction system, and INT5 is formed with an energy of −227.7 kJ mol−1. The subsequent reductive elimination takes place between the phenyl group and the terminal carbon viaTS6 with a low energy barrier of 60.8 kJ mol−1 to produce INT7. Finally, INT7 exchanges with two 1,3-butadiene molecules to form the product P8.
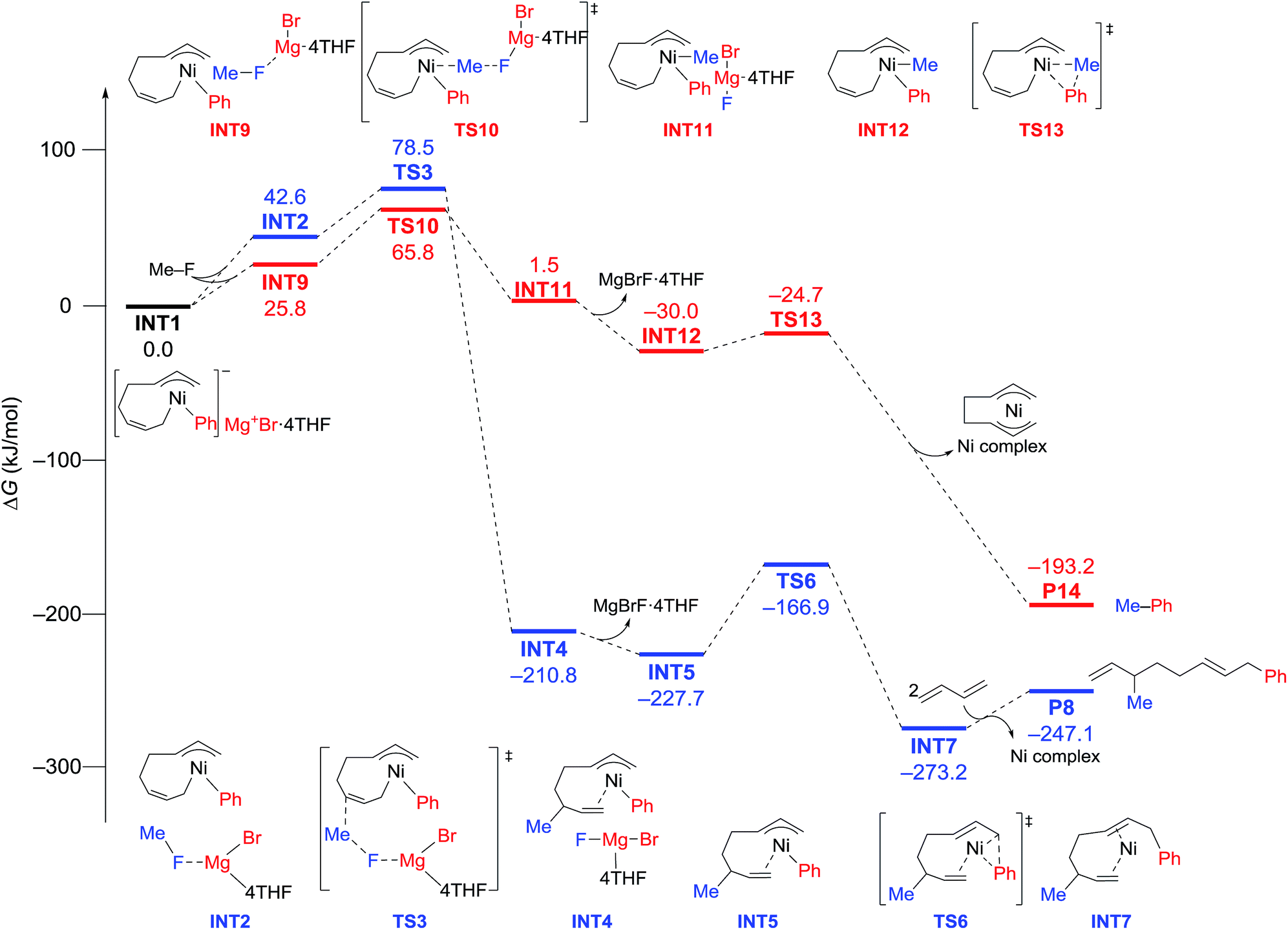 | ||
| Fig. 10 Reaction pathways of nickel-catalyzed four-component coupling (blue) and cross-coupling (red) reactions of methyl fluoride, phenyl Grignard reagent, and 1,3-butadiene. | ||
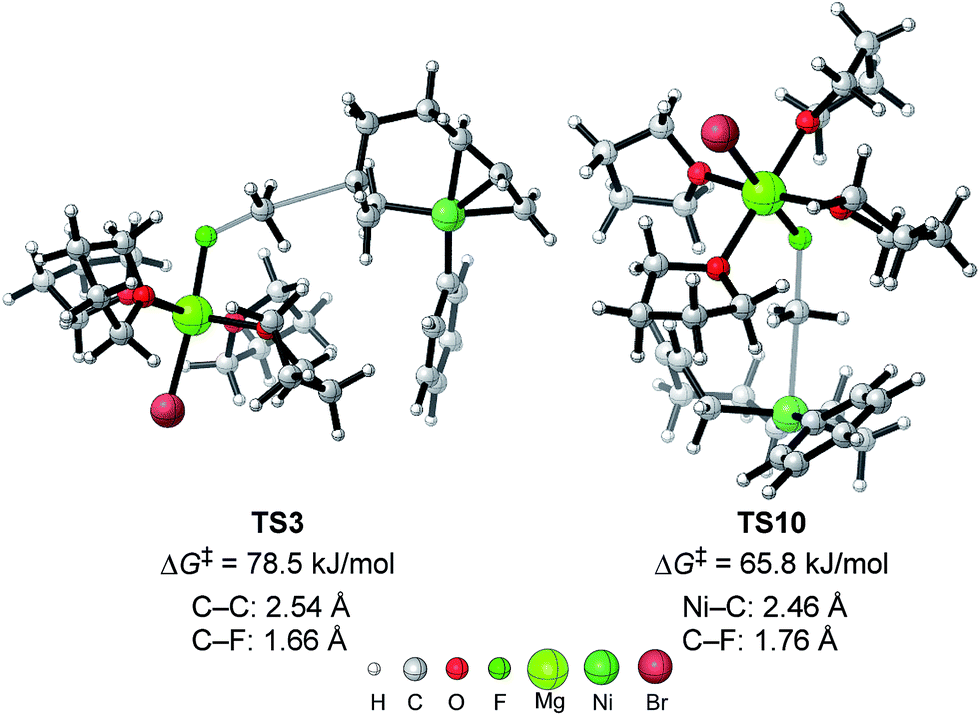 | ||
| Fig. 11 The molecular structures of important transition states in Fig. 10 with selected bond distances. | ||
On the other hand, MeF can be inserted into INT1 to form INT9 (25.8 kJ mol−1), with the methyl group pointing to Ni. Next, the methyl group is transferred to Ni via a transition state TS10 (65.8 kJ mol−1) to form INT11 with an energy of 1.5 kJ mol−1. After elimination of MgBrF·4THF with an energy release of 31.5 kJ mol−1, INT12 is generated, in which both phenyl and methyl groups are bound to Ni. Via a transition state TS13, which has an energy barrier of 5.3 kJ mol−1, reductive elimination occurs between the phenyl and methyl groups, giving rise to the product P14.
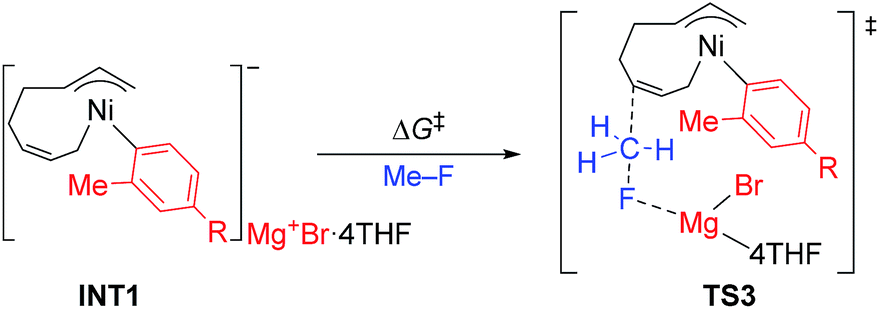
The above results revealed that the rate-determining step is the methyl group transfer, which corresponds to TS3 and TS10 for the two competing pathways, respectively. The overall energy barriers of the four-component coupling and cross-coupling reactions are 78.5 (INT1 to TS3) and 65.8 kJ mol−1 (INT1 to TS10), respectively. This appreciably large difference may be due, in part, to overestimation of the C–H–π interaction between THF on the Mg cation and the Ph group in TS10 that would not be probable, or would exert only a small effect if possible, in the practical conditions surrounded by THF molecules as the solvent (vide infra).
As shown in Fig. 11, in both TS3 and TS10 the Mg cation moiety (Mg+Br·4THF) binds to the fluorine atom to associate the elimination process, where no other short contact between the Mg+Br·4THF moiety and the anionic part is found, and therefore the angle of the participating atoms, C–C–F for TS3 and Ni–C–F for TS10, is 171° and 177°, respectively. These linear structures of the TSs clearly agree with the SN2 mechanism of the C–C bond forming step of both reaction pathways.
Other important findings in the introductory investigation are as follows: (i) a configuration orienting the ortho-methyl group toward the reacting face is energetically favored in TS3 (TS3-2-methyl-up) compared to the opposite orientation (TS3-2-methyl-down) (ΔΔG‡ = 2.5 kJ mol−1), (ii) in TS10, a configuration orienting the ortho-methyl group to the opposite side (TS10-2-methyl-down) is more favorable than TS10-2-methyl-up (ΔΔG‡ = 18.7 kJ mol−1), and (iii) the β-hydrogens of the alkyl fluorides are close to the aryl group in both cases of TS-10-2-methyl-up (H–H distances with the ortho-methyl group are 2.13 and 2.32 Å) and TS-10-2-methyl-down (H–H distances with the ortho-C–H bond are 2.24 and 2.24 Å). These short H–H distances may contribute to the increased energy barrier of TS10, resulting in high four-component coupling selectivity.24
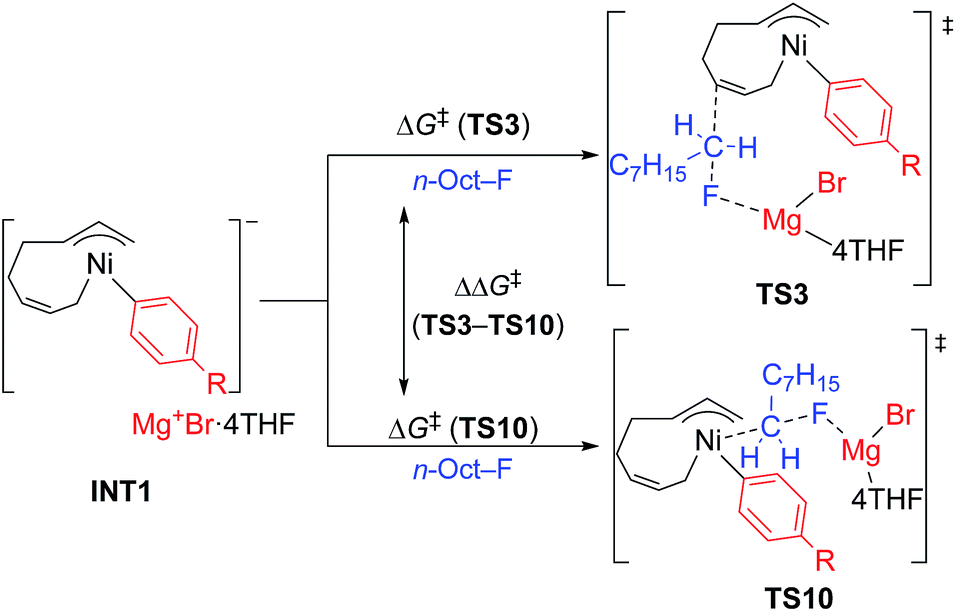
Next, TS3 and TS10 with various aryl Grignard reagents and alkyl fluorides were calculated at the higher level of theory (M06/6-311+G(d,p)), and the results are summarized in Table 8, Fig. 12 (for MeF), and Fig. 13 (for n-OctF). Introducing the first ortho-methyl group significantly increases the energy barriers of both TS3 and TS10. In particular, irrespective to the alkyl fluorides, TS10 has an energy barrier above 106 kJ mol−1, and therefore is clearly disfavored compared with TS3. In the case of the 2,6-dimethylphenyl Grignard reagent, the energy barrier of TS10 increases to 134.2 kJ mol−1. However, in this case, the energy barrier of TS3 is much lower (97.3 kJ mol−1). From these results, it is clear that the ortho-methyl group significantly influences the selectivity between the four-component coupling and cross-coupling reactions by favoring the former.
| R1 | R2 | TS3 | TS10 | ΔΔG‡ (TS3–TS10) |
|---|---|---|---|---|
| Ph | Me | 78.5 | 65.8 | 12.7 |
| Ph | n-Oct | 86.7 | 80.2 | 6.5 |
| 2-Methylphenyl | Me | 97.0 | 116.3 | −19.3 |
| 2-Methylphenyl | n-Oct | 98.3 | 106.9 | −8.6 |
| 2,6-Dimethylphenyl | Me | 97.3 | 134.2 | −36.9 |
| 2,6-Dimethylphenyl | n-Oct | 94.5 | 117.5 | −23.0 |
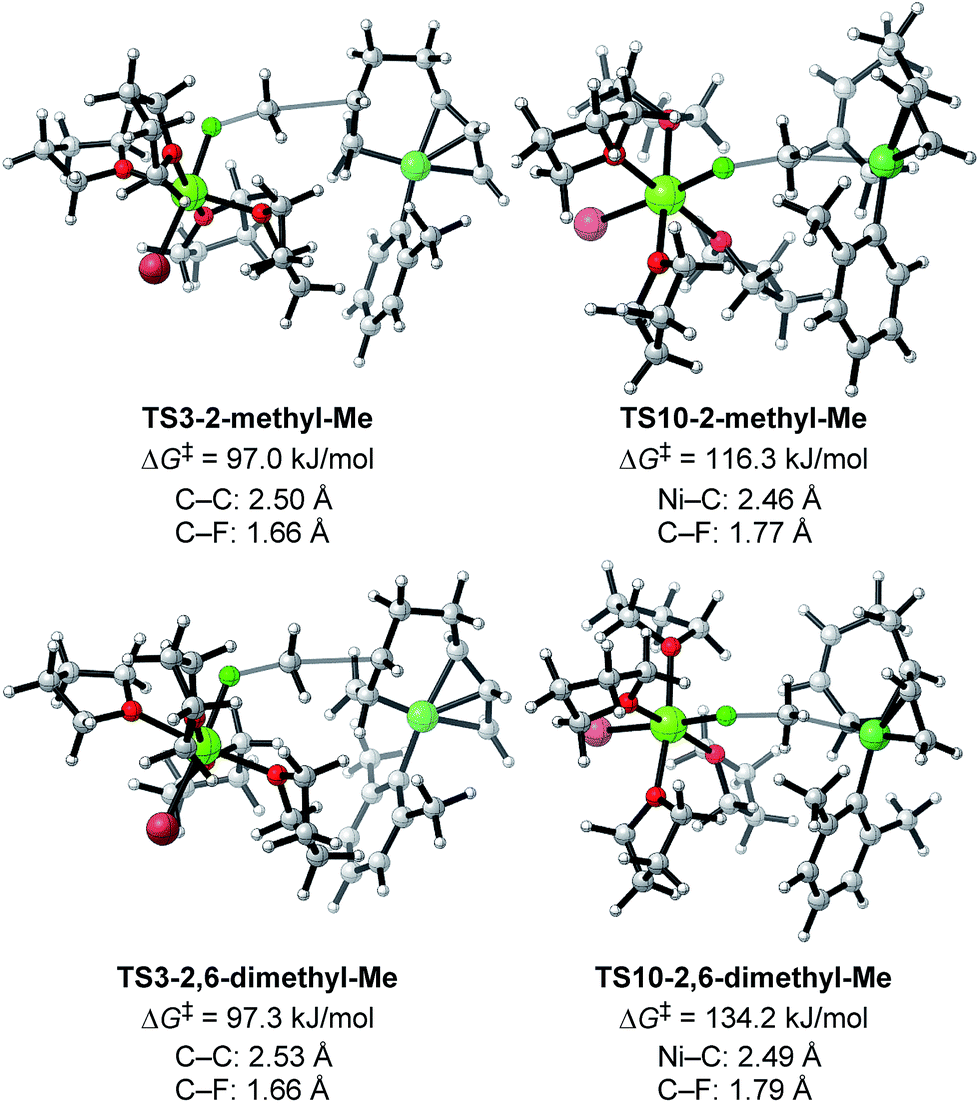 | ||
| Fig. 12 Optimized structures and the corresponding energy barriers for the transition states of γ-carbon attack (TS3) and Ni-attack (TS10) toward MeF for 2-methylphenyl and 2,6-dimethylphenyl Grignard reagents. Bond distances between key atoms are given. | ||
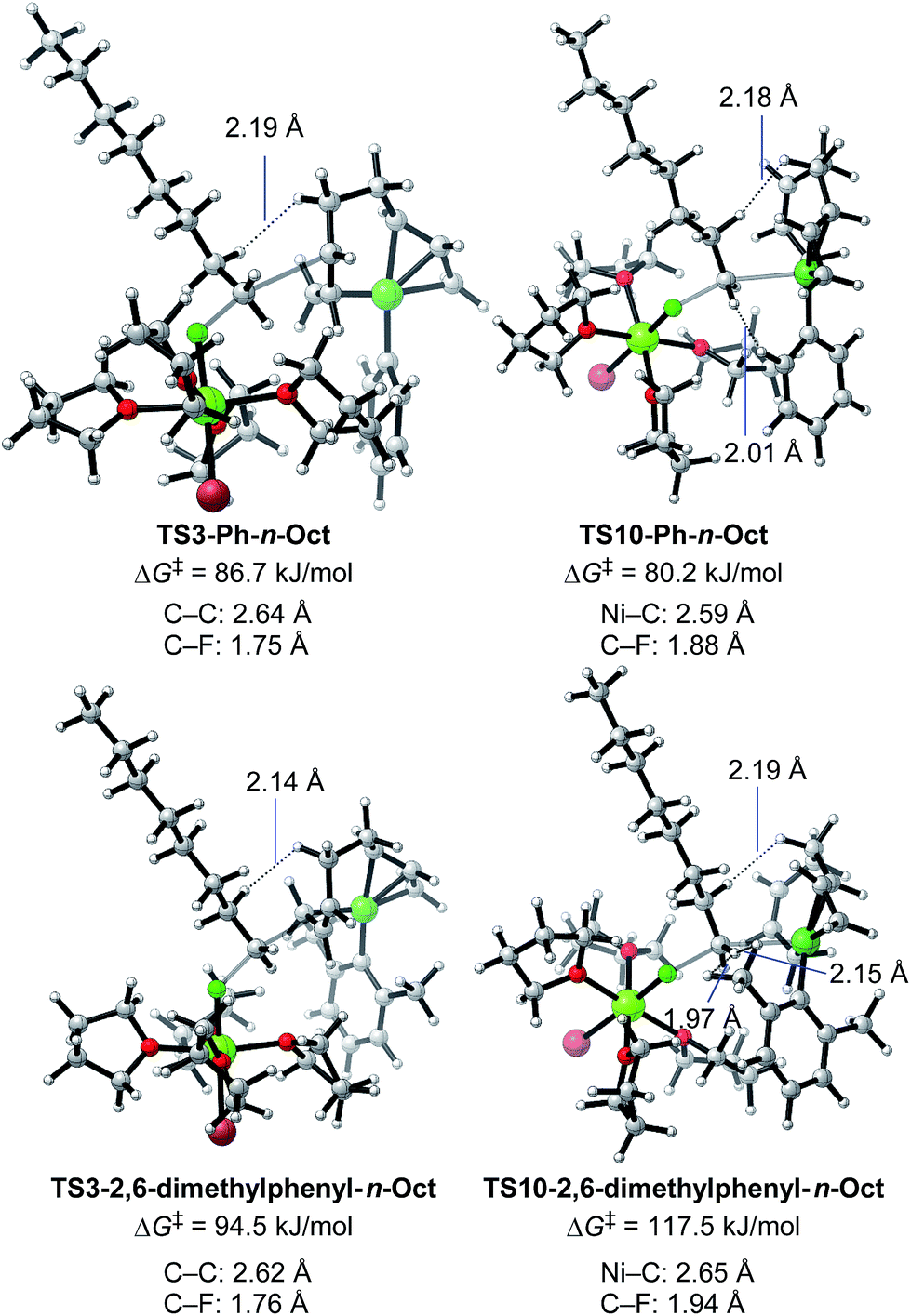 | ||
| Fig. 13 Optimized structures and the corresponding energy barriers for the transition states of γ-carbon attack (TS3) and Ni-attack (TS10) toward n-OctF for phenyl and 2,6-dimethylphenyl Grignard reagents. Bond distances between key atoms and representative short H–H distances are given. | ||
We then calculated TS3 and TS10 using n-OctF instead of MeF (Table 8). The structure of the alkyl fluoride affects the energy barriers at the transition states (especially for TS10), but it has no effect on the selectivity once the ortho-methyl group is present. Those computational results agree well with the experimental observations (Table 5). In the case of TS10, the energy barrier of MeF rises by far more than that of n-OctF by increasing steric hindrance of the aryl group. These results are due to their tight structure, where the sum of the C–F and C–Ni bond lengths of TS10 with MeF is 4.22 Å (R1 = Ph, R2 = Me: Fig. 11) in sharp contrast to the long bond length of TS10 with n-OctF (4.47 Å for R1 = Ph, R2 = n-Oct: Fig. 13).24 These differences in TS10 by alkyl fluorides reflect the reaction mechanism, where TS10 with MeF is a pure SN2 mechanism due to the lower stability of the Me cation, but TS10 with n-OctF has a somewhat SN1 character. The natural bond orbital (NBO)48 analysis of these TSs at the M06/6-311+G(d,p)-SMD(THF) level of theory supports the difference in mechanism, where the atomic charge of the reacting carbon in TS10 with n-OctF is more positive than that of TS10 with MeF.24 Therefore, in the case of TS10 with n-OctF, the bond distances of C–F and C–Ni could be elongated to minimize steric repulsions. The TS10 with n-OctF seems to be more likely and well-describes the effect of the counter cation in the C–F bond cleavage step.
As discussed in Section 2.3.7, the ortho-substitution effect mainly stems from the steric effects in the nickelate complexes. Due to the ortho-substitution(s), the Ni center that acts as the reacting site at transition state TS10 is partly covered, resulting in an increased energy barrier for TS10. To reveal the origin of the steric effect, the structures of TS3 and TS10 based on phenyl and 2,6-dimethylphenyl Grignard reagents are shown in Fig. 13 with representative short contacts. A short H–H distance is found between the β–H of the n-Oct group and the δ–H of the σ-allyl group in both cases of TS3-Ph-n-Oct (2.19 Å) and TS3-2,6-dimethylphenyl-n-Oct (2.14 Å) though a similar short distance is also found in TS10, where the β–H of the n-Oct is close to a methylene proton (2.18 and 2.19 Å). These short H–H distances are not the principal factor on the observed selectivity. On the other hand, compared with the H–H distance of 2.01 Å in TS10-Ph-n-Oct, the H–H distance in TS10-2,6-dimethyl-n-Oct is as short as 1.97 Å, indicating that there is an obvious repulsive interaction between the transferred n-Oct group and ortho-methyl group, which is responsible for the large increment in the energy barrier of TS10.
|
|
||||||
|---|---|---|---|---|---|---|
| R | H | F | Cl | Me | OMe | NMe2 |
| ΔG‡ | 97.0 | 98.3 | 97.6 | 94.9 | 92.2 | 91.2 |
| γ-C | −0.388 | −0.387 | −0.376 | −0.390 | −0.388 | −0.390 |
NBO analysis was performed to further study the charge on the γ-carbon of the σ-allyl group in TS3. As shown in Table 9, the 4-position substitution moderately affects the charge state of the γ-carbon: electron-donating groups here increase the charge while electron-withdrawing groups reduce it.49 This is in good agreement with the reaction mechanism and observed electronic effects on the reaction rate (Section 2.3.6).
To further study the electronic effects on the selectivity between four-component coupling and cross-coupling reactions, the energy barriers of TS3 and TS10 based on para-substituted phenyl Grignard reagents and n-OctF were calculated, and ΔΔG‡ of the two reaction pathways (TS3–TS10) and selectivities based on experimental results are summarized in Table 10. The calculated results generally agree well with the experimental observations. The electron-withdrawing groups such as F and Cl at the 4-position obviously destabilize TS10 by 2.7 and 1.9 kJ mol−1 compared to TS3, respectively, and therefore, favor the four-component coupling product 3. The electron-donating groups (Me and NMe2), on the other side, disfavor TS3 and gives rise to the reverse ratio of 3:4. Those results suggest that the electronic effects play an important role in the reactions with phenyl Grignard reagents (no ortho-substitution) though the impact on selectivity is relatively smaller than that from steric effects (Section 2.4.2).
|
|
||||||
|---|---|---|---|---|---|---|
| R | H | F | Cl | Me | OMe | NMe2 |
| ΔΔG‡ | 6.5 | −2.7 | −1.9 | 5.0 | 5.0 | 8.9 |
| Exp. ratio (4/3) | 1.4 | 0.60 | 1.0 | 1.9 | 2.3 | 3.3 |
3 Conclusions
We examined the reaction mechanism, substituent effects, and origins of the selectivity of the Ni-catalyzed four-component coupling of alkyl fluorides, aryl Grignard reagents, and two molecules of 1,3-butadiene and the competing cross-coupling reaction. The introduction of ortho-substituents into the aryl Grignard reagents efficiently suppresses the cross-coupling reaction, giving four-component coupling products in good yields with perfect regio- and stereoselectivity. The present catalysis selectively provides four-component coupling products and does not form three-component coupling products without the dimerization of 1,3-butadiene, in sharp contrast to previous reports of a similar Ni-catalyzed multicomponent coupling reaction employing 1,3-dienes and carbonyl electrophiles.7 This difference arises from the different mechanism in the present reaction: the anionic Ni intermediates generated by the reaction of Ni(0) with two 1,3-butadiene molecules and aryl Grignard reagents are the key intermediate, and they enable the use of alkyl fluorides as the prospective alkylating reagent through C–F bond cleavage.Mechanistic studies including observation and isolation of the anionic nickel complexes as well as kinetic studies clarified the detail of the reaction mechanism as follows: (i) anionic nickel complexes are the key intermediate at the C–C bond forming step with alkyl fluorides, and the corresponding neutral Ni complex is inert toward alkyl fluorides, (ii) the C–C and C–Ni bond forming steps are the rate-determining step in the four-component coupling and cross-coupling reactions, respectively, (iii) therefore, the selectivity of these two products is determined by the relative reactivity between the γ-carbon of the σ-allyl group and the Ni atom of the anionic nickel complexes, (iv) electron-donating groups on the aryl group accelerate the C–C and C–Ni bond formations with alkyl fluorides due to the SN2 type reaction mechanism in both reactions, where Ni is more effectively activated than the remote γ-carbon, (v) the Mg cation plays crucial roles in the activation of the C–F bond by its coordination, whereas other typical metal cations such as Li and Zn are not effective, and (vi) the bis(π-allyl)nickel complex prefers electron-deficient aryl Grignard reagents to form anionic Ni complexes. On the basis of the above experimental observations and DFT calculations, the catalytic cycles of both the four-component coupling and cross-coupling reactions are accomplished, in which the cation Mg+Br·4THF rather than Mg+Br plays a crucial role to activate the C–F bond. The theoretical calculations with the Mg+Br·4THF model not only explain very well the experimental results such as the activation energy barriers but also unravel the origins of the substituent effect (ortho-substituent and para-substituent) on the selectivity and reaction rates in terms of steric and electronic effects.
Although such anionic species of transition metals have sometimes been proposed as the key intermediates in catalytic bond formation, their structure as well as chemical characteristics are unclear in many cases. Our results demonstrate their synthetic utility as promising intermediates for C–C bond forming reactions. The present study provides useful information in organometallic and synthetic organic chemistry for developing efficient and straightforward multicomponent reactions as well as anionic transition metal complexes as unique and powerful catalytic intermediates.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by JSPS KAKENHI grant numbers JP25248025, JP16H06039, JP16H06511, JP16H04104, and JP25105734 in “Molecular Activation Directed toward Straightforward Synthesis”, JP16H01150 in “Middle Molecular Strategy”, JSPS Japanese-German Graduate Externship from JSPS, and the Nanotechnology Platform Japan program from MEXT. Computations were performed at the Research Center for Computational Science, Okazaki, Japan. T. I. thanks the UBE Foundation, Sumitomo Foundation, Uehara Memorial Foundation, and Iketani Science and Technology Foundation. T. Y. thanks the Alexander von Humboldt Foundation for a postdoctoral fellowship. We are grateful to Prof. Satoshi Minakata and Prof. Masato Ohashi (Osaka University) for numerous fruitful discussions.Notes and references
- For reviews: (a) G. H. Posner, Chem. Rev., 1986, 86, 831 CrossRef CAS; (b) R. W. Armstrong, A. P. Combs, P. A. Tempest, S. D. Brown and T. A. Keating, Acc. Chem. Res., 1996, 29, 123 CrossRef CAS; (c) H. Bienaymé, C. Hulme, G. Oddon and P. Schmitt, Chem.–Eur. J., 2000, 6, 3321 CrossRef.
- For reviews: (a) R. Baker, Chem. Rev., 1973, 73, 487 CrossRef CAS; (b) W. Keim, Angew. Chem., Int. Ed., 1990, 29, 235 CrossRef; (c) S. M. Sieburth and N. T. Cunard, Tetrahedron, 1996, 52, 6251 CrossRef CAS; (d) M. Lautens, W. Klute and W. Tam, Chem. Rev., 1996, 96, 49 CrossRef CAS PubMed.
- (a) J. Tsuji, Acc. Chem. Res., 1973, 6, 8 CrossRef CAS; (b) A. Behr, M. Becker, T. Beckmann, L. Johnen, J. Leschinski and S. Reyer, Angew. Chem., Int. Ed., 2009, 48, 3598 CrossRef CAS PubMed.
- N. D. Clement, L. Routaboul, A. Grotevendt, R. Jackstell and M. Beller, Chem.–Eur. J., 2008, 14, 7408 CrossRef CAS PubMed.
- (a) R. Baker and M. J. Crimmin, J. Chem. Soc., Perkin Trans. 1, 1979, 1264 RSC; (b) S. Akutagawa, Bull. Chem. Soc. Jpn., 1976, 49, 3646 CrossRef CAS; (c) R. Baker, M. S. Nobbs and P. M. Winton, J. Organomet. Chem., 1977, 137, C43 CrossRef CAS; (d) P. Brun, A. Tenaglia and B. Waegell, Tetrahedron, 1985, 41, 5019 CrossRef CAS.
- (a) W. E. Billups, W. E. Walker and T. C. Shields, Chem. Commun., 1971, 1067 RSC; (b) J. Tsuji, Y. Mori and M. Hara, Tetrahedron, 1972, 28, 3721 CrossRef CAS. For a review: (c) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510 CrossRef CAS PubMed.
- Using aldehydes: (a) M. Kimura, S. Matsuo, K. Shibata and Y. Tamaru, Angew. Chem., Int. Ed., 1999, 38, 3386 CrossRef CAS. Using ketones: (b) M. Kimura, A. Ezoe, M. Mori and Y. Tamaru, J. Am. Chem. Soc., 2005, 127, 201 CrossRef CAS PubMed. Using imines: (c) K. Kojima, M. Kimura and Y. Tamaru, Chem. Commun., 2005, 4717 RSC; (d) M. Kimura, Y. Tatsuyama, K. Kojima and Y. Tamaru, Org. Lett., 2007, 9, 1871 CrossRef CAS PubMed.
- For reactions involving alkyne insertion, see: (a) M. Kimura, K. Kojima, Y. Tatsuyama and Y. Tamaru, J. Am. Chem. Soc., 2006, 128, 6332 CrossRef CAS PubMed; (b) M. Kimura, M. Togawa, Y. Tatsuyama and K. Matsufuji, Tetrahedron Lett., 2009, 50, 3982 CrossRef CAS; (c) D. Takushima, M. Fukushima, H. Satomura, G. Onodera and M. Kimura, Heterocycles, 2012, 86, 171 CrossRef CAS. For reactions involving alkene insertion, see: (d) T. Nakamura, T. Mori, M. Togawa and M. Kimura, Heterocycles, 2012, 84, 339 CrossRef CAS.
- For reviews on the reaction of carbonyl compounds, 1,3-butadiene, and organometallic reagents in a 1:1:1 ratio, see:
(a) Y. Tamaru, J. Organomet. Chem., 1999, 576, 215 CrossRef CAS;
(b) S.-i. Ikeda, Angew. Chem., Int. Ed., 2003, 42, 5120 CrossRef CAS PubMed;
(c) J. Montgomery, Angew. Chem., Int. Ed., 2004, 43, 3890 CrossRef CAS PubMed;
(d) M.-Y. Ngai, J.-R. Kong and M. J. Krische, J. Org. Chem., 2007, 72, 1063 CrossRef CAS PubMed.
- S. Ogoshi, K.-i. Tonomori, M.-a. Oka and H. Kurosawa, J. Am. Chem. Soc., 2006, 128, 7077 CrossRef CAS PubMed.
- For multicomponent reactions using chlorosilanes as heteroatom electrophiles, see: (a) J. Terao, A. Oda, A. Ikumi, A. Nakamura, H. Kuniyasu and N. Kambe, Angew. Chem., Int. Ed., 2003, 42, 3412 CrossRef CAS PubMed; (b) J. Terao, A. Oda and N. Kambe, Org. Lett., 2004, 6, 3341 CrossRef CAS PubMed.
- T. Iwasaki, X. Min, A. Fukuoka, H. Kuniyasu and N. Kambe, Angew. Chem., Int. Ed., 2016, 55, 5550 CrossRef CAS PubMed.
- T. Iwasaki, A. Fukuoka, X. Min, W. Yokoyama, H. Kuniyasu and N. Kambe, Org. Lett., 2016, 18, 4868 CrossRef CAS PubMed.
- (a) J. Terao, A. Ikumi, H. Kuniyasu and N. Kambe, J. Am. Chem. Soc., 2003, 125, 5646 CrossRef CAS PubMed; (b) J. Terao, H. Todo, H. Watanabe, A. Ikumi and N. Kambe, Angew. Chem., Int. Ed., 2004, 43, 6180 CrossRef CAS PubMed; (c) J. Terao, H. Todo, S. A. Begum, H. Kuniyasu and N. Kambe, Angew. Chem., Int. Ed., 2007, 46, 2086 CrossRef CAS PubMed; (d) T. Iwasaki, R. Imanishi, R. Shimizu, H. Kuniyasu, J. Terao and N. Kambe, J. Org. Chem., 2014, 79, 8522 CrossRef CAS PubMed; (e) T. Iwasaki, K. Yamashita, H. Kuniyasu and N. Kambe, Org. Lett., 2017, 19, 3691 CrossRef CAS PubMed.
- (a) J. Terao, H. Watanabe, A. Ikumi, H. Kuniyasu and N. Kambe, J. Am. Chem. Soc., 2002, 124, 4222 CrossRef CAS PubMed. For synthetic applications: (b) T. Iwasaki, K. Higashikawa, V. P. Reddy, W. W. S. Ho, Y. Fujimoto, K. Fukase, J. Terao, H. Kuniyasu and N. Kambe, Chem.–Eur. J., 2013, 19, 2956 CrossRef CAS PubMed; (c) A. Ghaderi, T. Iwasaki, A. Fukuoka, J. Terao and N. Kambe, Chem.–Eur. J., 2013, 19, 2951 CrossRef CAS PubMed; (d) T. Aiba, M. Sato, D. Umegaki, T. Iwasaki, N. Kambe, K. Fukase and Y. Fujimoto, Org. Biomol. Chem., 2016, 14, 6672 RSC.
- (a) R. B. King, Coord. Chem. Rev., 2000, 200–202, 813 CrossRef CAS; (b) J. E. Ellis, Organometallics, 2003, 22, 3322 CrossRef CAS; (c) F. Mongin and A. Harrison-Marchand, Chem. Rev., 2013, 113, 7563 CrossRef CAS PubMed; (d) P. K. Sazonov and I. P. Beletskaya, Chem.–Eur. J., 2016, 22, 3644 CrossRef CAS PubMed.
- (a) B. H. Lipshutz and S. Sengupta, Org. React., 1992, 41, 135 CAS; (b) N. Yoshikai and E. Nakamura, Chem. Rev., 2012, 112, 2339 CrossRef CAS PubMed.
- For Zr: (a) E.-i. Negishi and T. Takahashi, Bull. Chem. Soc. Jpn., 1998, 71, 755 CrossRef CAS. For Fe: (b) A. Fürstner, H. Krause and C. W. Lehmann, Angew. Chem., Int. Ed., 2006, 45, 440 CrossRef PubMed; (c) R. B. Bedford, P. B. Brenner, E. Carter, P. M. Cogswell, M. F. Haddow, J. N. Harvey, D. M. Murphy, J. Nunn and C. H. Woodall, Angew. Chem., Int. Ed., 2014, 53, 1804 CrossRef CAS PubMed.
- Selected examples in which anionic intermediates were proposed as key intermediates without clarifying their actual structures. V: (a) S. Yasuda, H. Yorimitsu and K. Oshima, Bull. Chem. Soc. Jpn., 2008, 81, 287 CrossRef CAS. Cr: (b) T. Nishikawa, H. Kakiya, H. Shinokubo and K. Oshima, J. Am. Chem. Soc., 2001, 123, 4629 CrossRef CAS PubMed. Mn: (c) K. Oshima, J. Organomet. Chem., 1999, 575, 1 CrossRef CAS; (d) G. Cahiez, C. Duplais and J. Buendia, Chem. Rev., 2009, 109, 1434 CrossRef CAS PubMed. Fe: (e) Y. Hayashi, H. Shinokubo and K. Oshima, Tetrahedron Lett., 1998, 39, 63 CrossRef CAS; (f) G. Cahiez, V. Habiak, C. Duplais and A. Moyeux, Angew. Chem., Int. Ed., 2007, 46, 4364 CrossRef CAS PubMed; (g) S. K. Ghorai, M. Jin, T. Hatakeyama and M. Nakamura, Org. Lett., 2012, 14, 1066 CrossRef CAS PubMed; (h) Z. Mo, Q. Zhang and L. Deng, Organometallics, 2012, 31, 6518 CrossRef CAS. Co: (i) K. Wakabayashi, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2001, 123, 5374 CrossRef CAS PubMed.
- Our reports on the catalytic cross-coupling reaction involving ate complexes as intermediates. Pd: (a) J. Terao, Y. Naitoh, H. Kuniyasu and N. Kambe, Chem. Lett., 2003, 32, 890 CrossRef CAS. Co: (b) T. Iwasaki, H. Takagawa, S. P. Singh, H. Kuniyasu and N. Kambe, J. Am. Chem. Soc., 2013, 135, 9604 CrossRef CAS PubMed; (c) T. Iwasaki, H. Takagawa, K. Okamoto, S. P. Singh, H. Kuniyasu and N. Kambe, Synthesis, 2014, 46, 1583 CrossRef. Rh: (d) T. Iwasaki, Y. Miyata, R. Akimoto, Y. Fujii, H. Kuniyasu and N. Kambe, J. Am. Chem. Soc., 2014, 136, 9260 CrossRef CAS PubMed. Fe: (e) T. Iwasaki, R. Akimoto, H. Kuniyasu and N. Kambe, Chem.–Asian J., 2016, 11, 2834 CrossRef CAS PubMed.
- For recent reports on nickelate complexes in organic synthesis: (a) C. Zarate, M. Nakajima and R. Martin, J. Am. Chem. Soc., 2017, 139, 1191 CrossRef CAS PubMed; (b) A. Obata, Y. Ano and N. Chatani, Chem. Sci., 2017, 8, 6650 RSC; (c) J. Wei, W.-X. Zhang and Z. Xi, Angew. Chem., Int. Ed., 2015, 54, 5999 CrossRef CAS PubMed; (d) H. Ogawa, H. Minami, T. Ozaki, S. Komagawa, C. Wang and M. Uchiyama, Chem.–Eur. J., 2015, 21, 13904 CrossRef CAS PubMed; (e) M. Tobisu and N. Chatani, Acc. Chem. Res., 2015, 48, 1717 CrossRef CAS PubMed; (f) K. Murakami, Y. Yamamoto, H. Yorimitsu and A. Osuka, Chem.–Eur. J., 2013, 19, 9123 CrossRef CAS PubMed; (g) D.-G. Yu, B.-J. Li, S.-F. Zheng, B.-T. Guan, B.-Q. Wang and Z.-J. Shi, Angew. Chem., Int. Ed., 2010, 49, 4566 CrossRef CAS PubMed; (h) T. Hatakeyama, S. Hashimoto, K. Ishizuka and M. Nakamura, J. Am. Chem. Soc., 2009, 131, 11949 CrossRef CAS PubMed.
- For representative reviews on C–F bond cleavage in organic syntheses, see: (a) J. L. Kiplinger, T. G. Richmond and C. E. Osterberg, Chem. Rev., 1994, 94, 373 CrossRef CAS; (b) T. Braun and R. N. Perutz, Chem. Commun., 2002, 2749 RSC; (c) H. Torrens, Coord. Chem. Rev., 2005, 249, 1957 CrossRef CAS; (d) J. Terao, H. Todo, H. Watabe, A. Ikumi, Y. Shinohara and N. Kambe, Pure Appl. Chem., 2008, 80, 941 CrossRef CAS; (e) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119 CrossRef CAS PubMed.
- For C–C bond formation of alkyl fluorides, see: (a) T. Ooi, D. Uraguchi, N. Kagoshima and K. Maruoka, Tetrahedron Lett., 1997, 38, 5679 CrossRef CAS; (b) K. Hirano, K. Fujita, H. Yorimitsu, H. Shinokubo and K. Oshima, Tetrahedron Lett., 2004, 45, 2555 CrossRef CAS; (c) T. Hatakeyama, S. Ito, M. Nakamura and E. Nakamura, J. Am. Chem. Soc., 2005, 127, 14192 CrossRef CAS PubMed; (d) J. Terao, S. A. Begum, Y. Shinohara, M. Tomita, Y. Naitoh and N. Kambe, Chem. Commun., 2007, 855 RSC; (e) M. Sai, H. Someya, H. Yorimitsu and K. Oshima, Org. Lett., 2008, 10, 2545 CrossRef CAS PubMed; (f) L. W. Erickson, E. L. Lucas, E. J. Tollefson and E. R. Jarvo, J. Am. Chem. Soc., 2016, 138, 14006 CrossRef CAS PubMed . Also see, ref. 19h.
- See the ESI† for details.
- For mechanistic studies on the cross-coupling reaction using a Ni/1,3-butadiene catalytic system, see: (a) J. Terao, Y. Naitoh, H. Kuniyasu and N. Kambe, Chem. Commun., 2007, 825 RSC; (b) T. Iwasaki, A. Tsumura, T. Omori, H. Kuniyasu, J. Terao and N. Kambe, Chem. Lett., 2011, 40, 1024 CrossRef CAS . See also ESI.†.
- (a) G. Wilke, Angew. Chem., Int. Ed., 1963, 2, 105 CrossRef; (b) B. Barnett, B. Büssemeier, P. Heimbach, P. W. Jolly, C. Krüger, I. Tkatchenko and G. Wilke, Tetrahedron Lett., 1972, 1457 CrossRef CAS; (c) R. Benn, B. Büssemeier, S. Holle, P. W. Jolly, R. Mynott, I. Tkatchenko and G. Wilke, J. Organomet. Chem., 1985, 279, 63 CrossRef CAS.
- For a computational study, see: S. Tobisch, Chem.–Eur. J., 2003, 9, 1217 CrossRef CAS PubMed.
- (a) S. Holle, P. W. Jolly, R. Mynott and R. Salz, Z. Naturforsch., 1982, 37b, 675 CAS; (b) W. Kaschube, K.-R. Pörschke, K. Angermund, C. Krüger and G. Wilke, Chem. Ber., 1988, 121, 1921 CrossRef CAS. The formation of ate complexes by oxidative dimerization of 1,3-butadiene on arylnickel(0) is also reported: (c) K. Jonas and C. Krüger, Angew. Chem., Int. Ed., 1980, 19, 520 CrossRef.
- CCDC-1572238 contains the supplementary crystallographic data for this paper.†.
- (a) When n-OctF (1a) was treated with 1 equiv. of MgBr2 in THF at 40 °C, only 7% of the corresponding n-OctBr was observed after 10 h. This result indicates that the halogen exchange reaction with Mg salt is not the major pathway in this reaction; (b) When an alkyl bromide instead of alkyl fluoride was employed in the reaction of isolated nickelate complexes bearing Li as the cation, the cross-coupling reaction took place in the absence of an additional Mg salt in sharp contrast with the case of alkyl fluorides. This result suggests that the reaction of nickelates with alkyl bromides proceeds without the assistance of the Mg cation in the TS due to the higher reactivity of alkyl bromides. See the ESI† for details.
- Under the standard conditions, there is a short (within 5 min) induction period, probably arising from the reduction of the Ni salt to Ni(0). Therefore, the fitting lines do not cross the origin. The starting point of the plots is in the range of 3–5 min, and the initial reaction rates were evaluated from the slope of the plots.
- The enthalpy-entropy compensation plot showed an excellent linear relationship with R2 = 0.999, suggesting the same reaction mechanisms of these Grignard reagents. See: M. S. Searle, M. S. Westwell and D. H. Williams, J. Chem. Soc., Perkin Trans. 2, 1995, 141 RSC.
- The rate constant for the cyclization is known to be k = 4.0 × 107 s–1 at 25 °C. See: M. Newcomb, S.-Y. Choi and J. H. Horner, J. Org. Chem., 1999, 64, 1225 CrossRef CAS.
- According to the Yukawa–Tsuno equation, log(kX/kH) = ρ(σ0 + rΔ
![[small sigma, Greek, macron]](https://www.rsc.org/images/entities/char_e0d2.gif) R+), the value of r was estimated to be 0. This result may indicate that in both reactions the contribution of the resonance effect is very small.
R+), the value of r was estimated to be 0. This result may indicate that in both reactions the contribution of the resonance effect is very small. - The X-ray crystallography data of 11 has been reported earlier, see ref. 13. CCDC-1478800.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, Canada, 2009; http://www.cylview.org Search PubMed.
- L. M. Pratt, S. Voit, F. N. Okeke and N. Kambe, J. Phys. Chem. A, 2011, 115, 2281 CrossRef CAS PubMed.
- G. A. Chass, E. A. B. Kantchev and D.-C. Fang, Chem. Commun., 2010, 46, 2727 RSC.
- Although the reported TS is a Mg assisted SN2-like model, the angle of the participating atoms, Ni, C, and Br, is bended ca. 140°.
- For a recent example, see: H. Ogawa, H. Minami, T. Ozaki, S. Komagawa, C. Wang and M. Uchiyama, Chem.–Eur. J., 2015, 21, 13904 CrossRef CAS PubMed.
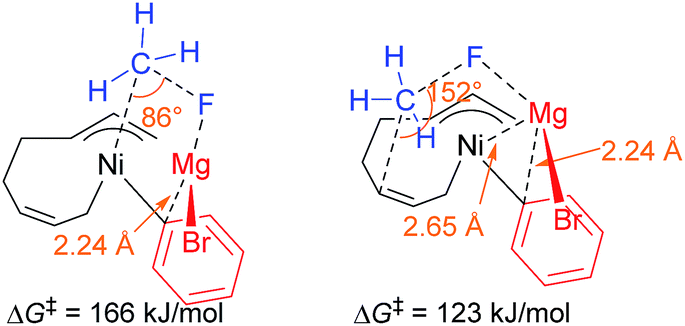
- Our initial attempts to estimate the TSs of both the four-component coupling and cross-coupling reactions employing Mg+Br as a model of the counter cation resulted in unsatisfactory results. As shown in the image below, in the TS of the cross-coupling reaction (left), the MgBr cation has a short contact to the ipso-carbon of the Ph group (2.24 Å), and the Ni atom attacks from the same side of the F atom (angle of Ni–C–F = 86°). Similarly, the MgBr cation bonds to the Ni atom and the ipso-carbon of the Ph group (Mg–Ni = 2.65 Å and Mg–C = 2.24 Å) with a narrow angle of Ni–C–F (152°) in the TS of the four-component coupling reaction (right). Due to their strained structure, the activation energy of these TSs was calculated to be 166 and 123 kJ mol−1, respectively. These preliminary results clearly indicate the necessity of more feasible models
. - It is known that cuprates form solvent separated ion pairs in THF although they exist as contact ion pairs in Et2O. See: (a) M. John, C. Auel, C. Behrens, M. Marsch, K. Harms, F. Bosold, R. M. Gschwind, P. R. Rajamohanan and G. Boche, Chem.–Eur. J., 2000, 6, 3060 CrossRef CAS PubMed; (b) R. M. Gschwind, P. R. Rajamohanan, M. John and G. Boche, Organometallics, 2000, 19, 2868 CrossRef CAS . Also see ref. 17.
- Theoretical studies on Ni-catalyzed cross-coupling reactions using Grignard reagents have been reported, in which the Mg cation coordinates to a ligand on Ni. For examples, see: N. Yoshikai, H. Matsuda and E. Nakamura, J. Am. Chem. Soc., 2009, 131, 9590 CrossRef CAS PubMed . Also see ref. 21h.
- The rotation barrier was estimated to be 55 kJ mol−1 by calculation, suggesting the possibility of both isomers. Indeed, VT NMR of the nickelate complex having an o-Tol group showed two isomers, and the rotation barrier was determined to be 52.7 ± 6.0 kJ mol−1 at 273 K. See the ESI† for details.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO, version 3.1, University of Wisconsin, Madison, WI, 1996 Search PubMed.
- The NBO charge of Ni did not change due to the para-substituent in all cases. This is due to the π-allyl ligand in the trans-position. Indeed, the total NBO charges of the Ni atom and the π-allyl ligand are –0.818 (H), –0.815 (F), –0.815 (Cl), –0.820 (Me), –0.819 (OMe), and –0.820 (NMe2). See the ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental and computational results, procedures, characterization data, copies of NMR charts, and crystallographic data. CCDC 1572238. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04675h |
| This journal is © The Royal Society of Chemistry 2018 |