
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Detection of antimicrobial resistance-associated proteins by titanium dioxide-facilitated intact bacteria mass spectrometry†
Yingdi
Zhu
 a,
Natalia
Gasilova
ab,
Milica
Jović
a,
Liang
Qiao
c,
Baohong
Liu
c,
Lysiane Tissières
Lovey
d,
Horst
Pick
e and
Hubert H.
Girault
*a
a,
Natalia
Gasilova
ab,
Milica
Jović
a,
Liang
Qiao
c,
Baohong
Liu
c,
Lysiane Tissières
Lovey
d,
Horst
Pick
e and
Hubert H.
Girault
*a
aLaboratoire d’Electrochimie Physique et Analytique, École Polytechnique Fédérale de Lausanne, Rue de l’industrie 17, CH-1951 Sion, Switzerland. E-mail: hubert.girault@epfl.ch
bISIC-GE-VS, École Polytechnique Fédérale de Lausanne, Rue de l’industrie 17, CH-1951 Sion, Switzerland
cDepartment of Chemistry, Fudan University, Handan Road 220, 200433 Shanghai, China
dICH, Hôpital du Valais, Avenue du Grand Champsec 86, CH-1951 Sion, Switzerland
eLaboratoire de Chimie Biophysique des Macromolécules, École Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland
First published on 18th January 2018
Abstract
Titanium dioxide-modified target plates were developed to enhance intact bacteria analysis by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. The plates were designed to photocatalytically destroy the bacterial envelope structure and improve the ionization efficiency of intracellular components, thereby promoting the measurable mass range and the achievable detection sensitivity. Accordingly, a method for rapid detection of antimicrobial resistance-associated proteins, conferring bacterial resistance against antimicrobial drugs, was established by mass spectrometric fingerprinting of intact bacteria without the need for any sample pre-treatment. With this method, the variations in resistance proteins’ expression levels within bacteria were quickly measured from the relative peak intensities. This approach of resistance protein detection directly from intact bacteria by mass spectrometry is useful for fast discrimination of antimicrobial-resistant bacteria from their non-resistant counterparts whilst performing species identification. Also, it could be used as a rapid and convenient way for initial determination of the underlying resistance mechanisms.
Introduction
Infectious diseases caused by pathogenic bacteria are serious threats to human health. Misuse and overuse of antimicrobial drugs over many years have led to the emergence of antimicrobial resistance among microbes worldwide.1 For fast diagnosis and efficient treatment, it is crucial to perform pathogen identification and a rapid analysis of their antimicrobial resistance phenotypes. With the ability to generate characteristic mass spectral fingerprints directly from intact bacteria, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI–TOF MS) provides a rapid method for bacteria identification (e.g. ∼30 min for 48 samples) and has received clearance from the US Food and Drug Administration (FDA).2,3 Commercial systems, including Vitek MS (bioMérieux) and MALDI Biotyper (Bruker Daltonics), have been installed and constantly used in many hospitals. Meanwhile, antimicrobial resistance detection in hospitals still mainly relies on antimicrobial drug culture approaches like the broth (or agar) microdilution method and disk diffusion method, which need several hours or even several days.4 Therefore, performing a complete clinical diagnosis remains a lengthy process.In addition to the classical culture-based methods, several new strategies have been proposed for antimicrobial resistance detection. Examples include nucleic acid-based resistance gene detection,5 single-cell morphological analysis,6 surface-enhanced Raman spectroscopic biomarker detection,7 atomic force microscope cantilever-based nanomechanical sensors,8etc.
Recently, continuous efforts have been made to explore the potential of MALDI–TOF MS for rapid antimicrobial resistance analysis. Related studies were mainly carried out with three approaches. The first one is an indirect evaluation by detection of resistance enzyme activity, such as the degradation of β-lactam antibiotics through hydrolysis (mass increased by 18 Da) by β-lactamases9 and the alternation of rRNA through methylation (mass increased by 14 Da) by rRNA methyltransferase.10 This is a fast method for resistance mechanism investigation, but is limited to certain enzyme-related resistance types. The second approach is an isotope labelling-bacteria culture method, with the appearance of peak mass shifts in bacterial fingerprinting patterns if a resistant strain is incubated with culture medium supplemented with stable (non-radioactive) isotope labelled amino acids and corresponding antibiotics.11,12 Based on the machinery of protein biosynthesis, this approach is applicable to determining bacterial resistance or susceptibility to a wide range of antibiotics, but limited by the need for special isotope labelled culture medium. The third one is also a culture-based method, in which semi-quantitative MALDI–TOF MS using an internal standard is employed to investigate bacterial growth status with the presence or absence of antibiotics by measuring the quantity of biomass within a spectrum.13,14 This method has been demonstrated to be feasible for different antibiotic classes/bacterial species combinations. In addition to the above three approaches, bacteria subtyping assays have also been conducted to study the correlation between antibiotic-susceptible and resistant strains by comparing their fingerprint patterns. For example, it has been used to discriminate major methicillin-resistant Staphylococcus aureus lineages15 and to identify vancomycin-resistant Enterococcus spp.16
Proteins encoded by antimicrobial resistance genes are directly involved in bacterial resistance process against antimicrobial drugs.17 Antimicrobial resistance can be analysed by tracing these resistance-associated proteins within bacterial cells. Ideally, they should be read out directly from MALDI–TOF MS fingerprint patterns of intact bacteria without any sample pre-treatment, a useful procedure that would be comparable to fast bacteria identification. But many of those proteins are large ones (>15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) expressed in low abundance, and are difficult to detect directly from intact cells by classic MALDI–TOF MS measurements, which typically focus on smaller proteins (<15000 Da) expressed in high abundance.18 Until now, to the best of our knowledge no studies have reported the success of this procedure, as pointed out in a review by Walkova et al.19 In order to detect these resistance-associated proteins, preparatory extraction and enrichment processes are required prior to their identification by MS, which is labour-intensive and time-consuming.20–22 Very recently, a surrogate marker around 11 kDa was detected from carbapenem-resistant bacteria strains containing blaKPC-harboring plasmids by a MALDI–TOF MS fingerprinting approach. But it is a particularly small protein and an additional step of protein extraction was required prior to MS analysis.23
000 Da) expressed in low abundance, and are difficult to detect directly from intact cells by classic MALDI–TOF MS measurements, which typically focus on smaller proteins (<15000 Da) expressed in high abundance.18 Until now, to the best of our knowledge no studies have reported the success of this procedure, as pointed out in a review by Walkova et al.19 In order to detect these resistance-associated proteins, preparatory extraction and enrichment processes are required prior to their identification by MS, which is labour-intensive and time-consuming.20–22 Very recently, a surrogate marker around 11 kDa was detected from carbapenem-resistant bacteria strains containing blaKPC-harboring plasmids by a MALDI–TOF MS fingerprinting approach. But it is a particularly small protein and an additional step of protein extraction was required prior to MS analysis.23
Herein, we have developed a MALDI–TOF MS fingerprinting approach for intact bacteria analysis using photo-reactive titanium dioxide (TiO2)-modified target plates, providing access to a high mass range with enhanced detection sensitivity. TiO2-modified target plates or more generally metal oxide-modified plates have been used for many different MALDI–TOF MS applications.24,25 In the current work, the rationale of the proposed approach is to take advantage of the photo-reactivity of TiO2 to destroy bacterial cell membranes and to facilitate inner component desorption/ionization. Such experimental improvement makes feasible a direct fast read out of resistance-associated proteins from intact bacteria cells without any sample pre-treatment.
Results and discussion
TiO2-facilitated intact bacteria MALDI–TOF MS fingerprinting
One important factor affecting MALDI–TOF MS measurements is the choice of matrix. Sinapinic acid was utilized as a matrix throughout this work, as it provides satisfying reproducibility and facilitates the detection of large proteins (Part S1, ESI†). Based on our experience in designing photo-reactive TiO2-modified target plates for inducing in-source electrochemical reactions,26,27 we have developed here a plate able not only to absorb bacteria on a porous structure but also to lyse them by photocatalytic oxidation, improving intact bacteria fingerprinting in a broad mass range as demonstrated below.This target plate was prepared by depositing an aqueous suspension of TiO2 nanoparticles (NPs) on the spots (3 mm diameter) of a classic bare stainless steel target plate, or by dropping TiO2 suspension as an array of spots on a stainless steel foil (20 μm thick), which was afterwards affixed onto a bare target plate by an adhesive tape (Fig. 1a). The TiO2 NPs were subsequently thermally or photonically sintered. The sintered NPs exhibited strong adherence to the steel substrate and provided a stable support layer (∼3 μm thick) for the bacteria and matrix, with small particles (of 20–25 nm size) densely covering the bottom and large particles (of 0.5–3 μm size) observed on the surface (Fig. 1b). The TiO2 used is a commercial P25 nanopowder, a mixture of anatase (80%) and rutile (20%) crystalline phases. The anatase is more photo-reactive than the rutile, but the latter is more thermodynamically stable. The crystalline phases of TiO2 were not changed after sintering, and the corresponding X-ray powder diffraction patterns are shown in Fig. 1c. Compared to bare steel target spots, the spots with TiO2 NPs had rough and mesoporous surfaces (see the surface roughness profiles in Part S2, ESI†), with a larger surface area and lower water contact angle (decreased from 70° to 38°, Part S3, ESI†). As the surface of the TiO2 spots are more hydrophilic than the steel substrate, this kind of TiO2-modified plate can be used as “AnchorChip” targets. Upon deposition, bacteria cells (mostly of 0.2–2 μm size) entered into the porous TiO2 NPs structure. Due to the high affinity between the bacterial membrane and TiO2,28 the cells tended to be absorbed on the surface of TiO2. Matrix drop casting consequently led to the formation of fine and well dispersed bacteria/matrix crystals, highly favourable for an efficient desorption/ionization process (Fig. 1d). MALDI–TOF MS analysis of intact Escherichia coli (E. coli, strain DH5α) yielded much higher quality fingerprint patterns using a TiO2-modified target plate in comparison with a bare steel target plate (Fig. 1e). Such significant improvement, especially in the mass range m/z = 15000–60000, could not be solely caused by the high quality of bacteria/matrix co-crystals resulting from the mesoporous spots’ surface. It could also be explained by the ability of TiO2 to destroy the bacterial cell membrane and to improve analyte desorption/ionization due to its well-known photo-reactivity.28,29
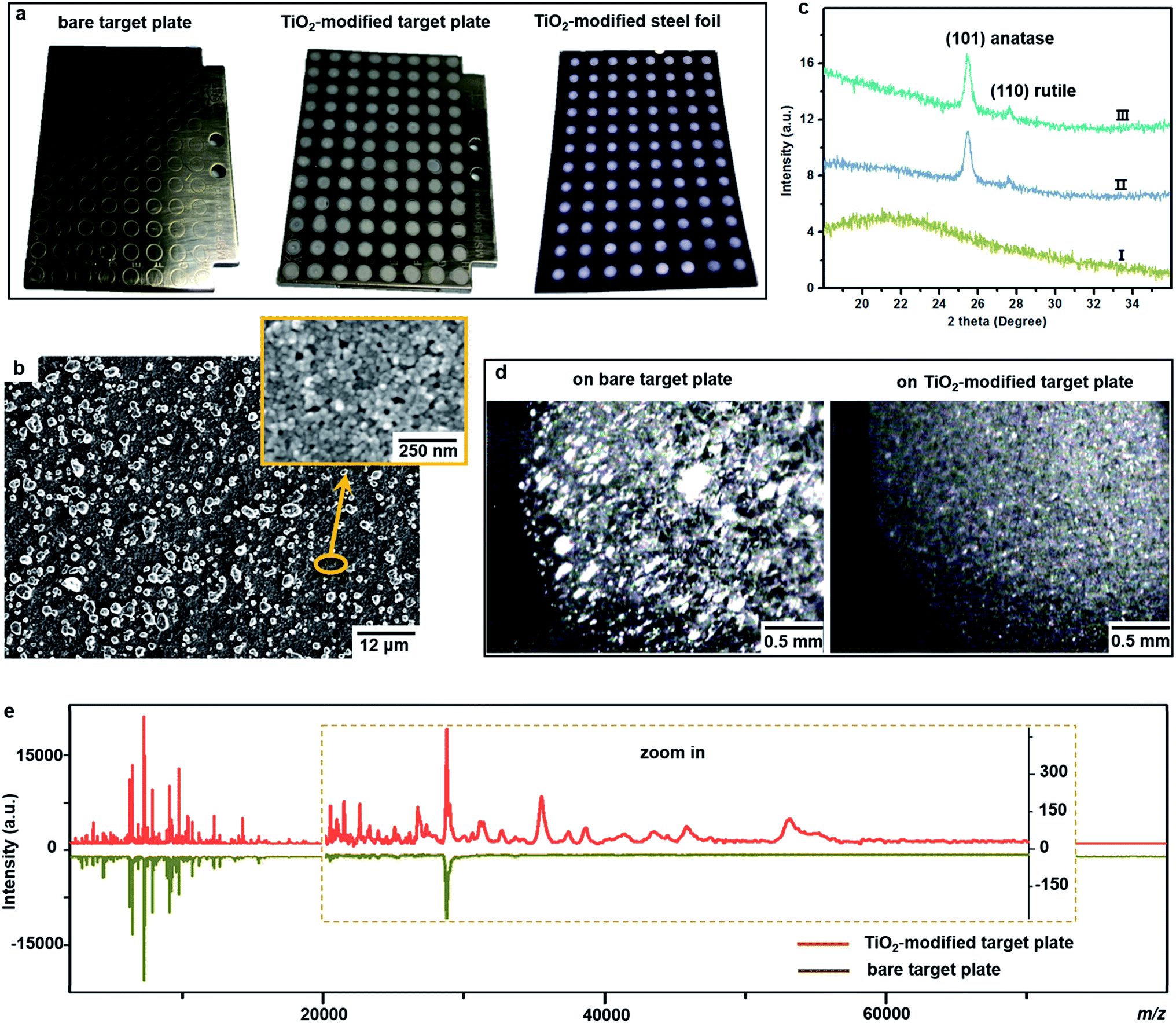 | ||
| Fig. 1 (a) Photos of a classic bare stainless steel target plate (MSP 96 ground steel MALDI target, Bruker Daltonics), a TiO2 NP-modified target plate and a piece of TiO2 NP-modified stainless steel foil, which was affixed onto a bare target plate before MALDI–TOF MS measurements; (b) scanning electron microscope image of a TiO2 NP layer after sintering at 400 °C; (c) X-ray powder diffraction patterns of (I) the steel substrate, (II) TiO2 NPs on the steel surface before sintering at 400 °C and (III) TiO2 NPs on the steel surface after sintering at 400 °C; (d) microscope image of intact bacteria (E. coli)/sinapinic acid matrix crystals (the shining clusters) on a spot of a classic bare target plate and a TiO2 NP-modified target plate; (e) MALDI–TOF MS fingerprint patterns (each was averaged from three replicates) of intact E. coli in a mass range of m/z = 2000–80000 obtained using a classic bare and a TiO2 NP-modified target plate. | ||
As proof of cell membrane disruption, the morphological changes of E. coli were visualized by scanning electron microscopy (Fig. 2). E. coli cells showed a straight, rod-like shape when they were deposited on the spots of a bare and a TiO2-modified target plate with no matrix covering and no MALDI laser irradiation (Fig. 2a and b). The spots with E. coli were then covered with matrix and underwent the running of a typical MS measurement (500 nitrogen laser shots on each sample spot, 20 Hz laser frequency). On the spots of the bare target plate, most E. coli cells (>95% according to microscopic observation) generally maintained their rod-like shape (Fig. 2c). The diameter of the laser beam used in the MALDI–TOF MS instrument (Bruker Microflex) is about 100 μm, 30 times smaller than the sample spot size (3 mm diameter). Thus, a typical MS measurement is accomplished with many “blind shots”, and only the cells exactly shot by the laser could be lysed. However, the situation was different on the spots with TiO2: most cells were seriously damaged with apparent deformation and membrane rupture, and the “melted” cells were embedded into the mesoporous spot surface (Fig. 2d). Interestingly, it was found that the crystal shape of the matrix on the spots with TiO2 was quite different from that on the bare spots (Fig. 2c and d). These microscopic observations show that the presence of TiO2 can indeed cause the disruption of more bacteria cells during MALDI–TOF MS measurements. With a band gap of 3.0–3.2 eV,30 TiO2 has strong light absorption in the UV range (Part S4, ESI†). Accordingly, during MS measurements, TiO2 absorbed energy from the nitrogen laser source (337.1 nm), generating electron–hole pairs, and triggering electron-transfer and radical reactions (see equations in Part S5, ESI†). The generated reactive oxygen species, like positive hole h+, hydroxyl radical ˙OH and peroxide H2O2, on the TiO2 surface caused oxidative disruption of the bacterial envelope, as previously reported.31,32 The disruption of more bacteria cells facilitated the detection of barely accessible inner cell components. The importance of oxidative cell disruption for enhancing bacteria MS analysis was confirmed by sample treatment with scavengers of these reactive oxygen species. Bacteria aqueous solutions containing different scavengers, i.e., sodium oxalate, isopropanol and ferrocenemethanol, were deposited onto TiO2-modified target plates for MALDI–TOF MS measurements. The concentration of each scavenger was set to an appropriate value to eliminate its possible influence on bacterial cells (initial pH 7.44): 2 mM sodium oxalate (pH 7.87), 2 mM isopropanol (pH 7.11) and 0.4 mM ferrocenemethanol (pH 7.90).32,33 The MALDI–TOF MS analysis in the presence of these scavengers showed low quality bacterial fingerprint patterns throughout the mass range m/z = 2000–80000 (Part S6, ESI†). In addition to disrupting bacteria cells, the high photo-reactivity of TiO2 favours efficient energy absorption from the laser source and transfer of this energy to matrix/analyte.34 This process occurs in addition to laser energy absorption directly by the matrix, and thus can facilitate analyte desorption/ionization even further. This was demonstrated by the analysis of bacterial protein extracts and standard protein mixtures. For the bacterial protein extracts, prepared according to the often-used ethanol/formic acid/acetonitrile extraction protocol, much higher quality MS patterns were observed using a TiO2-modified target plate than when using a classic bare one (Part S7, ESI†). For the standard protein mixtures, containing cytochrome c (∼12 kDa), myoglobin (∼17 kDa), bovine serum albumin (BSA, ∼66 kDa) and lactoferrin (∼82 kDa), the MS peak intensity of each protein was increased by the presence of TiO2. Consequently, the detection sensitivity was also improved, especially for the two hardly-ionized large proteins BSA and lactoferrin (Part S8, ESI†).
 | ||
| Fig. 2 Scanning electron microscope images of E. coli: (a) deposited on a spot of the bare target plate with no matrix covering and no MALDI laser irradiation (no TiO2/no matrix/no laser irradiation), (b) deposited on a spot of the TiO2 NP-modified target plate with no matrix covering and no MALDI laser irradiation (with TiO2/no matrix/no laser irradiation), (c) deposited on a spot of the bare target plate with matrix covering and with MALDI–TOF MS measurement (no TiO2/with matrix/with laser irradiation), (d) deposited on a spot of the TiO2 NP-modified target plate with matrix covering and with MALDI–TOF MS measurement (with TiO2/with matrix/with laser irradiation); the yellow arrows point to some of the E. coli cells. The matrix used is 15 μg mL−1 of sinapinic acid in 50/49.9/0.1 (v/v/v) acetonitrile/water/trifluoroacetic acid. | ||
For comparison, we have tested the performance of non-photo-reactive nanomaterials like Al2O3 NPs (<50 nm in particle size) and SiO2 NPs (200 nm in particle size). They were shown to have detrimental effects on MS results for both standard protein mixtures and intact bacteria (Part S9 and S10, ESI†).
TiO2-facilitated intact bacteria MALDI–TOF MS fingerprinting was further tested on different bacteria species. In addition to E. coli (strain DH5α), two more species, i.e., Pseudomonas aeruginosa (P. aeruginosa, strain ATCC 27853) and Bacillus subtilis (B. subtilis, strain 168), were chosen as model analytes. All bacteria were measured in their intact whole state, without any preparatory protein extraction. Corresponding fingerprint patterns generated with a classic bare target plate and a TiO2-modified one with exactly the same measurement parameters are compared in Fig. 3a–c. Notably, each MALDI–TOF MS test in the present work was repeated three times. In each replicate, a freshly cultured bacteria strain was measured. Collected fingerprint patterns demonstrated high reproducibility, and each displayed pattern is an average of the three replicates (see examples in Part S11, ESI†). To facilitate data interpretation, the patterns were compared in three separate sections, i.e., m/z = 2000–15000 (Fig. 3a/b/c-I), m/z = 15000–29000 (Fig. 3a/b/c-II) and m/z = 29000–80000 (Fig. 3a/b/c-III). Substantial improvements in pattern quality, both in terms of peak number and peak intensities, especially visible in the high mass range (m/z >15000), were observed for all three bacteria samples when a photo-reactive TiO2-modified target plate was utilized. The total peak numbers (S/N > 3, counted with an open source mass spectrometry tool mMass, http://www.mmass.org/) were increased by 50–70%, i.e., from 103 ± 1 to 174 ± 1 for E. coli, 87 ± 1 to 139 ± 2 for P. aeruginosa and 52 ± 1 to 88 ± 1 for B. subtilis (Fig. 3a/b/c-IV). The newly detected peaks, completely absent in the case of bare steel target plates, are mostly low intensity ones, corresponding to low-abundance or hardly-ionized bacterial components.
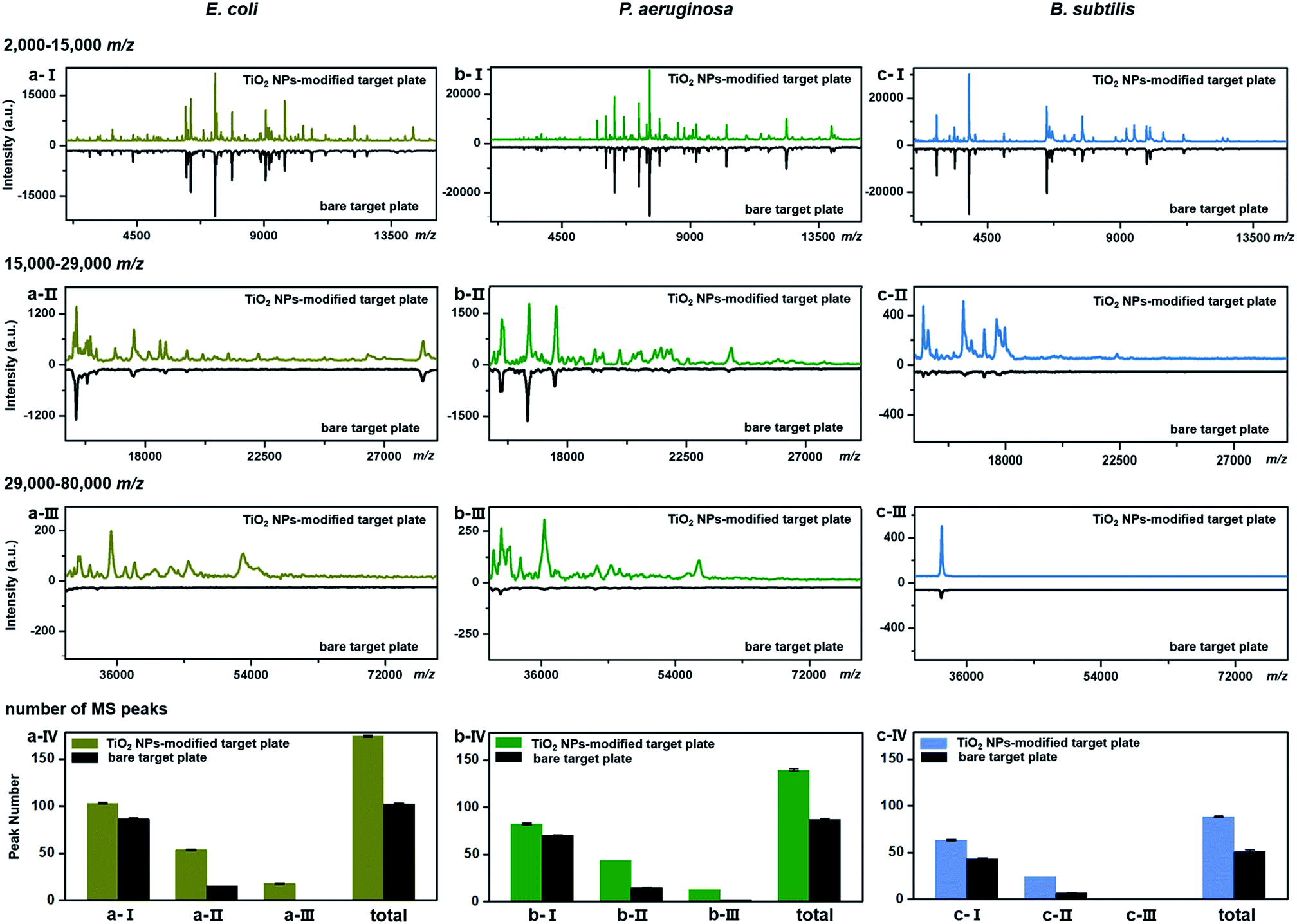 | ||
| Fig. 3 Comparison of a classic bare and a TiO2 NP-modified target plate for intact bacteria MALDI–TOF MS fingerprinting. The fingerprint patterns (each was averaged from three replicates) were generated from intact (a) E. coli, (b) P. aeruginosa and (c) B. subtilis in the mass range of (a/b/c-I) m/z = 2000–15000, (a/b/c-II) m/z = 15000–29000 and (a/b/c-III) m/z = 29000–80000, with peak numbers counted in a/b/c-IV. During the peak number counting process, the threshold value of signal-to-noise ratio (S/N) of counted peaks was set as 3, and the threshold value of relative peak intensity was set as 2.0% for the mass range m/z = 2000–15000 and 0.1% for the mass range m/z = 15000–80000. The number of bacterial cells on each sample spot was around 5 × 105. All measurements were conducted under the exact same instrumental parameters. | ||
As the genomes of B. subtilis 168 have been completely sequenced, the 36 fingerprint peaks additionally detected using the TiO2-modified target plate (Fig. 3c) were analysed by a proteome database search. According to the search method provided by Fenselau et al.,35,36 33 of the 36 peaks were tentatively assigned to proteins in the B. subtilis 168 proteome database (UniProtKB, proteome ID UP000001570) based on their m/z values. The list of assigned proteins is given in Part S12, ESI.† Each possibly matching protein was characterized by its subcellular location, isoelectric point (pI) and grand average value of hydropathicity (GRAVY). Results showed that these proteins mainly came from the cytoplasmic membrane and interior region (cytosol) of the bacteria cells (Part S13a, ESI†). The distribution of their pI values, varying between 3.72 and 10.73, indicated that TiO2 had no special preference in improving MALDI–TOF MS-based detection of basic (pI > 7) or acidic (pI < 7) proteins (Part S13b, ESI†). The distribution of the GRAVY index, normally used to evaluate the average protein hydrophobicity (GRAVY > 0) and hydrophilicity (GRAVY < 0), showed that most of these proteins were moderately hydrophilic (Part S13c, ESI†).
Overall, MALDI–TOF MS analysis of bacteria was promoted by TiO2, with significant improvement in both peak numbers and peak intensities of bacterial fingerprint patterns within a broad mass range. Attributed to its high photo-reactivity and photocatalytic bacteria disruption ability, TiO2 helped not only to break the cellular envelope structure, but also to enhance the desorption/ionization efficiency of intracellular components. By generating high quality bacterial fingerprint patterns, the TiO2-modified target plate could greatly boost the reliability of bacteria identification, which is based on fingerprint pattern matching. More importantly, it facilitates the extraction of more bacterial cellular information, and enables the detection of large molecular weight and low abundance bacterial components, especially those related to antimicrobial drug resistance, as discussed further.
Detection of antimicrobial resistance-associated proteins from intact bacteria
The possibility of antimicrobial resistance-associated protein detection by an intact bacteria MS fingerprinting approach was investigated with the TiO2-modified target plate. The detection was firstly conducted with bacteria samples that were modified by gene transfer. Corresponding plasmid DNAs, carrying specific resistance genes, were artificially transformed into recipient bacteria using recombinant techniques.37 Following this strategy, defined non-resistant (or antibiotic-susceptible) E. coli strains were equipped with the desired antimicrobial resistance, i.e., resistance against ampicillin, kanamycin, gentamicin and chloramphenicol, respectively. MALDI–TOF MS fingerprint patterns of the resistant strains were measured within the mass range of m/z = 2000–80000 and compared with those of non-resistant strains. To ensure result reliability, each type of resistance was repeatedly developed within two E. coli strains, i.e., two DH5α, XL1-Blue or BL21. The MS results showed that resistance-associated proteins were successfully detected from all of these resistant strains (Fig. 4), as explained in further detail below.
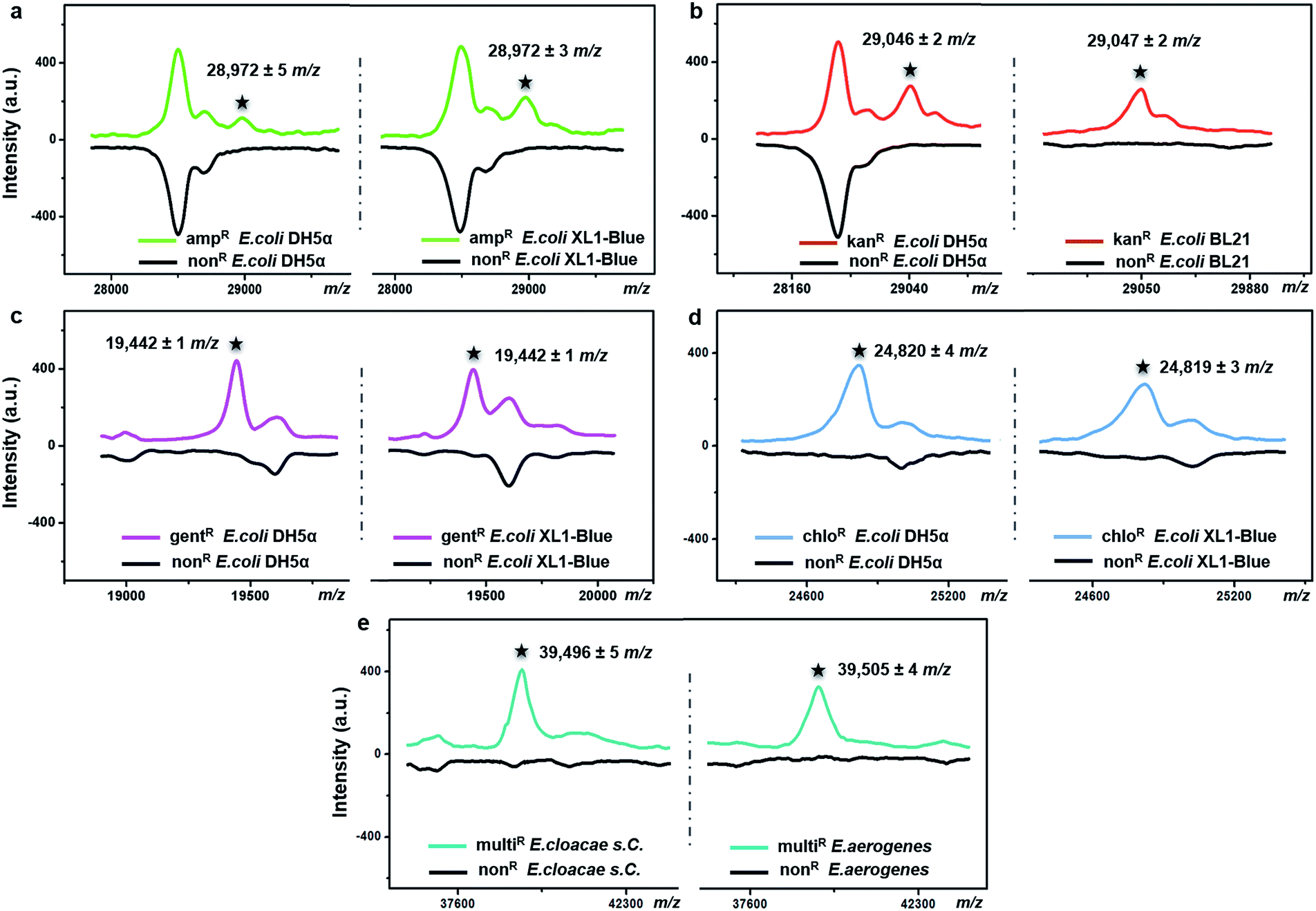 | ||
| Fig. 4 Detection of antimicrobial resistance-associated proteins from (a) ampicillin-resistant (ampR), (b) kanamycin-resistant (kanR), (c) gentamicin-resistant (gentR), and (d) chloramphenicol-resistant (chloR) E. coli strains (two strains in each case) and (e) multidrug-resistant (multiR) E. cloacae s. C. and E. aerogenes by intact bacteria MALDI–TOF MS fingerprinting with TiO2 NP-modified target plates. Each pattern was averaged from three replicates. The number of bacterial cells on each sample spot was around 5 × 105. | ||
Gene blaTEM-1, encoding a TEM-1 β-lactamase, conferred resistance against ampicillin. With a molecular weight around 29 kDa, TEM-1 β-lactamase inactivates ampicillin by hydrolysis of the β-lactam ring in the ampicillin molecule.38 Compared to the ampicillin-susceptible E. coli, the ampicillin-resistant ones exhibited almost the same MALDI–TOF MS fingerprint patterns except for an additional peak at m/z = 28972 ± 5 for strain DH5α and m/z = 28972 ± 3 for strain XL1-Blue (Fig. 4a). This result coincides with a previous study, in which a special preparatory protein extraction was conducted before MS measurement.38 The resistance against kanamycin resulted from the expression of neomycin-kanamycin phosphotransferase type II (29048 Da, UniProtKB-P00552), which inactivates kanamycin by phosphoryl transfer at its 3′-hydroxyl group.39 Using TiO2-modified target plates, this phosphotransferase was successfully detected in two kanamycin-resistant E. coli strains (at m/z = 29046 ± 2 for strain DH5α, and m/z = 29047 ± 2 for strain BL21), but not in their non-resistant counterparts (Fig. 4b). The resistance against gentamicin was conferred by gene aacC1, encoding gentamicin acetyltransferase I (19442 Da, UniProtKB-P23181), which inactivates gentamicin by acetylating its 3-amino deoxystreptamine moiety.40 This protein was detected exclusively in the gentamicin-resistant E. coli at m/z = 19442 ± 1 for both strains DH5α and XL1-Blue (Fig. 4c). The resistance against chloramphenicol was caused by the synthesis of chloramphenicol acetyltransferase (CAT, 24–26 kDa),41 which catalyses the transfer of an acetyl moiety from bacterial coenzyme A to the chloramphenicol molecules, and, therefore, results in antibiotic inactivation. Here, in contrast with non-resistant E. coli, a peak around m/z = 24820 was clearly detected for the chloramphenicol-resistant E. coli (24820 ± 4 for strain DH5α, 24819 ± 3 for strain XL1-Blue), confirming the expression of CAT (Fig. 4d). For all of the above measurements, detection of each resistance protein showed high reproducibility for both tested E. coli strains. The allowed tolerance between the measured and the theoretical masses was 300 ppm, due to the limited resolving power of the MALDI–TOF MS instrument used.
To investigate the expression of the same resistance gene within different bacteria species, Enterobacter cloacae ssp. cloacae (E. cloacae s. C.) and Enterobacter aerogenes (E. aerogenes) were artificially transformed with an ampC gene encoding AmpC type β-lactamase (∼39.5 kDa)42,43 and measured with MALDI–TOF MS. After the gene transfer, both E. cloacae s. C. and E. aerogenes acquired resistance against 10 different β-lactam antibiotics, becoming multidrug resistant. Their detailed antimicrobial susceptibility profiles (measured with a bioMérieux VITEK 2 automated AST system based on an antimicrobial drug culture method) before and after the gene transfer are shown in Part S16, Tables S1–S4, ESI.† For the multidrug-resistant E. cloacae s. C. and E. aerogenes, the minimum inhibitory concentrations (MICs) of the 10 antibiotics varied from 16 to 128 μg mL−1. Compared to their non-resistant counterparts, the two resistant strains both exhibited an additional peak around m/z = 39500 (m/z = 39496 ± 5 and 39505 ± 4, respectively) (Fig. 4e), confirming the expression of the AmpC type β-lactamase.
It should be mentioned that none of the above resistance-associated proteins were detectable when classic bare stainless steel plates were used (Part S14, ESI†), showing the importance of TiO2-modified target plates in bacteria analysis.
To further confirm the identity of the detected resistance-associated proteins, the antibiotic-resistant or non-resistant strains were analysed with a widely used proteomic approach. Bacteria cells were lysed in sodium dodecyl sulphate loading buffer, and the extracted proteins were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently identified by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Taking gentamicin-resistant E. coli DH5α and kanamycin-resistant E. coli BL21 as examples, a protein band around 19 kDa or 29 kDa was clearly observed on their corresponding SDS-PAGE gel running lanes, but not observed for their non-resistant counterparts (Part S15, ESI†). Excision of the ∼19 kDa band from both gentamicin-resistant and non-resistant E. coli DH5α (as control) lanes, followed by digestion in trypsin, revealed the presence of 60 gentamicin acetyltransferase I exclusive unique peptides in the gentamicin-resistant strain, with 100% protein identification probability and 93% (164/177) amino acid coverage (Table S1 in Part S15, ESI†). The ∼29 kDa bands from kanamycin-resistant and non-resistant E. coli BL21 (as control) lanes were analysed in the same way, revealing the presence of 72 neomycin-kanamycin phosphotransferase type II exclusive unique peptides in the kanamycin-resistant strain, with 100% protein identification probability and 93% (246/264) amino acid coverage (Table S2 in Part S15, ESI†). The above results coincide with the MALDI–TOF MS intact bacteria fingerprinting results in Fig. 4, confirming the expression and identity of antibiotic resistance-associated proteins in corresponding resistant strains.
The described TiO2-facilitated MALDI–TOF MS approach can also quickly sense the variations in resistance genes’ expression levels within bacteria. To demonstrate this, antibiotic-resistant bacteria were cultured in Luria-Bertani (LB) medium that contained different concentrations of corresponding antibiotics. A gradual increase of a given antibiotic concentration brings a proportionally higher selection pressure to the bacterial cells. As a response, bacterial cells modulate the resistance genes’ expression level to increase the synthesis of resistance proteins for survival.44 Such kinds of change were measured for ampicillin-, kanamycin- and chloramphenicol-resistant E. coli DH5α by comparing the relative peak intensities (r.t.) of the corresponding resistance proteins in the MALDI–TOF MS fingerprint patterns (Fig. 5a–c). The r.t. of the resistance proteins (i.e., TEM-1 β-lactamase at m/z = 28972 ± 5, neomycin-kanamycin phosphotransferase type II at m/z = 29046 ± 2 and CAT at m/z = 24820 ± 4) were calculated using signals from E. coli DH5α D-ribose-binding periplasmic protein (RbsB, ∼28.5 kDa)45 as an internal intensity standard (r.t.RbsB = 1). For all three proteins, their r.t. increased with an increase in corresponding antibiotic concentration. These data confirm that higher levels of antibiotic resistance would accompany higher expression levels of resistance proteins and consequently higher r.t. values of the corresponding MS peaks. For the chloramphenicol-resistant strain, however, the r.t. of CAT decreased when the chloramphenicol concentration reached 120 μg mL−1 (Fig. 5c). Probably, this concentration was already too high and started to negatively affect the bacterial physiological state. In addition to the antibiotics present, the type of culture medium can also affect the expression level of resistance proteins. Synthesis of resistance proteins to fight against antibiotics is an energy-consuming process, which can be positively influenced by the use of nutritionally rich growth medium.46 To observe this effect, ampicillin-resistant E. coli DH5α was cultured in different growth media containing a fixed concentration (60 μg mL−1) of ampicillin. The corresponding MALDI–TOF MS fingerprint patterns indicated that 2xYT medium, specifically rich in amino acids and peptides, favoured the up-regulation of gene blaTEM-1 expression. In particular, when the growth medium was changed from LB to 2xYT, the averaged r.t. of TEM-1 β-lactamase increased from 0.60 to 6.66 (r.t.RbsB = 1) (Fig. 5d).
 | ||
| Fig. 5 Measurement of variations in resistance protein expression levels within bacteria. Relative MS peak intensities of (a) TEM-1 β-lactamase, (b) neomycin-kanamycin phosphotransferase type II or (c) CAT detected from ampicillin-resistant, kanamycin-resistant or chloramphenicol-resistant E. coli DH5α that were cultured with LB medium containing ampicillin, kanamycin or chloramphenicol, respectively. The concentration of each antibiotic was set as 0, 15, 30, 60 and 120 μg mL−1. Corresponding MALDI–TOF MS patterns of related peaks (with overlapping of three replicates for each test) are shown in the insert graphs. (d) MALDI–TOF MS patterns in the mass range of m/z = 28000–30000 (with overlapping of three replicates) of ampicillin-resistant E. coli DH5α that was cultured with LB or 2xYT medium containing 60 μg mL−1 of ampicillin. Relative intensities (r.t.) of all peaks were calculated using a signal from protein RbsB as an internal intensity standard (r.t.RbsB = 1). The data were obtained by intact bacteria MALDI–TOF MS fingerprinting using TiO2 NP-modified target plates. The number of bacterial cells on each sample spot was around 5 × 105. | ||
Simultaneous bacteria identification and antimicrobial resistance-associated protein detection with clinical pathogens
The proposed method can be used for antimicrobial resistance protein detection whilst performing bacteria species identification. The feasibility was explored with three clinical pathogens: extended-spectrum β-lactamase-producing E. coli (ESBL-E. coli), multidrug-resistant Pseudomonas aeruginosa (MDR-P. aeruginosa) and methicillin-resistant Staphylococcus aureus (MRSA).ESBL, first reported in Germany in 1983, confers resistance to a broad spectrum of β-lactam antibiotics.47 Worldwide emergence of ESBL-E. coli raises serious therapeutic problems. Resistance in the ESBL-E. coli tested here was conferred by the expression of CTX-M type β-lactamase (∼28 kDa).48E. coli ATCC25922, a strain without such kind of resistance, was used as the reference for species identification and resistance protein detection of the testing strain (ESBL-E. coli). Detailed antimicrobial susceptibility profiles of the two strains, measured with a bioMérieux VITEK 2 automated AST system, are shown in Part S16, Tables S5 and S6, ESI.† For the testing strain, the MICs of the corresponding antibiotics were 4–320 μg mL−1 (Table S6†). The averaged MALDI–TOF MS fingerprint patterns of the two strains are displayed in Fig. 6a. The similarity score between the two patterns was calculated using a public bacteria identification platform, BacteriaMS, with a cosine correlation algorithm (http://bacteriams.fudan.edu.cn/#/). This algorithm gives the maximum score as 1.0. Here, with the pattern similarity as high as 0.9427, the testing strain was identified to be the same species as the reference one, i.e.,E. coli. The two strains shared almost all MS peaks (S/N > 3, r.i. > 0.1%) in the mass range of m/z = 10000–80000, except for a peak at m/z = 28074 ± 4 only detected for the testing strain (Fig. 6a, zoom-in). The appearance of this peak most probably results from the expression of CTX-M type β-lactamase. Therefore, together with the species identification, the CTX-M type ESBL resistance was recognized in the testing strain.
 | ||
| Fig. 6 Simultaneous species identification and antimicrobial resistance-associated protein detection of clinical pathogens, i.e., (a) ESBL-E. coli, (b) MDR-P. aeruginosa and (c) MRSA, by comparing their MS fingerprint patterns with those of reference strains (i.e., E. coli ATCC25922, P. aeruginosa ATCC 27853 and S. aureus ATCC29213). The data were obtained by intact bacteria MALDI–TOF MS fingerprinting using TiO2 NP-modified target plates. Each pattern was averaged from three replicates. The number of bacterial cells on each sample spot was around 5 × 105. | ||
Similarly, the MDR-P. aeruginosa and MRSA were also identified at the species level by comparison of their fingerprint patterns with those of corresponding reference strains (i.e., P. aeruginosa ATCC 27853 and S. aureus ATCC 29213), resulting in pattern similarity scores of 0.8546 and 0.9578, respectively (Fig. 6b and c). Simultaneously, two mass spectral peaks, at m/z = 38080 ± 6 and m/z = 40900 ± 8, were exclusively observed from the MDR-P. aeruginosa (Fig. 6b, zoom-in). They most likely come from efflux pump proteins MexA (∼38 kDa)49,50 and MexX (40.9 kDa, UniProtKB-Q9ZNG9), which confer the multidrug resistance of the MDR-P. aeruginosa. These two proteins are involved in the extrusion of β-lactam antibiotics (e.g., tazobactam, ceftazidime, and cefepime) and aminoglycosides (e.g., amikacin, gentamicin, netilmicin, and tobramycin) from within bacteria cells into the external environment.51 Meanwhile, antimicrobial resistance in MRSA, one of the most common multidrug resistant pathogens, arises from the expression of gene mecA, which causes the alteration of penicillin binding protein (PBP) and triggers the expression of its alternative, i.e., PBP 2a (∼78 kDa). PBP 2a has a low affinity for most β-lactam antibiotics including methicillin, thereby making bacteria resistant against them.52 According to previous studies, a characteristic fragment of PBP 2a (∼13 kDa) can be detected for MRSA by a proteomics-based method.53–55 In the present work, a peak at m/z = 13080 ± 2 was exclusively detected for the MRSA strain (Fig. 6c, zoom-in), which could come from the PBP 2a fragment. To confirm this assumption, two more MRSA strains were tested and the peaks around 13 kDa (at m/z = 13083 ± 3 and m/z = 13081 ± 4, respectively) were repeatedly detected, as shown in Part S17, ESI.† The detailed antimicrobial susceptibility profiles of MDR-P. aeruginosa, MRSA and their reference strains are shown in Part S16, Tables S7–S12, ESI.† As shown in these profiles, antibiotic MICs for the resistant strains were measured as 0.5–8 μg mL−1.
Herein, antimicrobial resistance-associated proteins were successfully detected directly in intact bacteria without any sample pre-treatment, by TiO2-facilitated MALDI–TOF MS. The developed approach showed feasibility for both Gram-negative and Gram-positive bacteria species bearing different types of antimicrobial resistance. Each of the resistance proteins were specifically detected from the corresponding antibiotic-resistant strains, not from the non-resistant reference strains or the strains resistant to other antibiotics. For certain resistance types tested in this work, it was also shown that higher levels of antibiotic resistance could accompany higher expression levels of the resistance proteins. Sensing of the expression level variations was proven possible through direct readout of the relative intensities of the corresponding MS peaks.
The described method performed resistance protein recognition according to their m/z values. Due to the limited resolving power of current MALDI–TOF MS instruments, it would be difficult to distinguish closely-related protein isoforms with quite similar molecular weights like TEM-1, TEM-2 and TEM-3 β-lactamases that differ only in a few amino acid substitutions. This is a drawback for MALDI–TOF MS-based analysis of proteins in comparison with nucleic acid-based molecular detection of the related genes or proteomics-based approaches. In this work, it has been confirmed that the expression levels of resistance proteins directly determine their MS peak appearances in bacterial fingerprint patterns. As intact bacteria are analysed directly without preparatory protein extraction, enrichment or selective separation, the proposed method could lack some sensitivity when the resistance proteins are expressed at a very low level. Here, the method was shown to be sensitive enough for resistant strains with antibiotic MICs as low as a few microgram per millilitre when 1 uL of the bacteria sample (∼5 × 105 cells) was measured.
Nonetheless, compared to existing methods for resistance gene or protein detection such as nucleic acid-based molecular techniques or proteomics-based approaches, the proposed MALDI–TOF MS-based method has clear advantages of simplicity and rapidity of sample preparation, measurement protocol and data analysis. It is a useful procedure for quick discrimination of antimicrobial-resistant bacteria strains from their non-resistant counterparts, as well as a fast method for the initial determination of resistance mechanisms and prediction of antibiotic types or classes that the strains could be resistant to.
Conclusions
In this work, intact bacteria MALDI–TOF MS analysis was improved by TiO2 due to its ability to photo-catalytically destroy bacterial envelopes and to facilitate analyte desorption/ionization. Impressive improvement in detection sensitivity and working mass range was achieved, pushing the current limits of the bacteria MALDI–TOF MS fingerprinting approach. Accordingly, antimicrobial resistance-associated proteins, especially those larger than 15 kDa, were successfully detected from intact bacteria by the direct readout of the corresponding MS peaks from the fingerprint patterns, together with a rapid sensing of their expression level variations. With the potential of simultaneous species identification and antimicrobial resistance analysis, the TiO2-facilitated MALDI–TOF MS opens new avenues for bacteria analysis.Conflicts of interest
There are no conflicts to declare.References
- M. Sharland, P. Saroey and E. N. Berezin, J. Pediatr., 2015, 91, 410–412 CrossRef PubMed.
- P. Seng, M. Drancourt, F. Gouriet, B. La Scola, P. E. Fournier, J. M. Rolain and D. Raoult, Clin. Infect. Dis., 2009, 49, 543–551 CrossRef PubMed.
- N. Matsuda, M. Matsuda, S. Notake, H. Yokokawa, Y. Kawamura, K. Hiramatsu and K. Kikuchi, J. Clin. Microbiol., 2012, 50, 3862–3866 CrossRef PubMed.
- J. H. Jorgensen and M. J. Ferraro, Clin. Infect. Dis., 2009, 49, 1749–1755 CrossRef PubMed.
- A. C. Fluit, M. R. Visser and F. J. Schmitz, Clin. Microbiol. Rev., 2001, 14, 836–871 CrossRef PubMed.
- J. Choi, J. Yoo, M. Lee, E. G. Kim, J. S. Lee, S. Lee, S. Joo, S. H. Song, E. C. Kim, J. C. Lee, H. C. Kim, Y. G. Jung and S. Kwon, Sci. Transl. Med., 2014, 6, 267ra174 CrossRef PubMed.
- C. Y. Liu, Y. Y. Han, P. H. Shih, W. N. Lian, H. H. Wang, C. H. Lin, P. R. Hsueh, J. K. Wang and Y. L. Wang, Sci. Rep., 2016, 6, 23375 CrossRef PubMed.
- G. Longo, L. Alonso-Sarduy, L. M. Rio, A. Bizzini, A. Trampuz, J. Notz, G. Dietler and S. Kasas, Nat. Nanotechnol., 2013, 8, 522–526 CrossRef PubMed.
- K. Sparbier, S. Schubert, U. Weller, C. Boogen and M. Kostrzewa, J. Clin. Microbiol., 2012, 50, 927–937 CrossRef PubMed.
- M. Savic, J. Lovric, T. I. Tomic, B. Vasiljevic and G. L. Conn, Nucleic Acids Res., 2009, 37, 5420–5431 CrossRef PubMed.
- K. Sparbier, C. Lange, J. Jung, A. Wieser, S. Schubert and M. Kostrzewa, J. Clin. Microbiol., 2013, 51, 3741–3748 CrossRef PubMed.
- J. S. Jung, T. Eberl, K. Sparbier, C. Lange, M. Kostrzewa, S. Schubert and A. Wieser, Eur. J. Clin. Microbiol., 2014, 33, 949–955 CrossRef PubMed.
- C. Lange, S. Schubert, J. Jung, M. Kostrzewa and K. Sparbier, J. Clin. Microbiol., 2014, 52, 4155–4162 CrossRef PubMed.
- K. Sparbier, S. Schubert and M. Kostrzewa, Methods, 2016, 104, 48–54 CrossRef CAS PubMed.
- M. Wolters, H. Rohde, T. Maier, C. Belmar-Campos, G. Franke, S. Scherpe, M. Aepfelbacher and M. Christner, Int. J. Med. Microbiol., 2011, 301, 64–68 CrossRef CAS PubMed.
- P. M. Griffin, G. R. Price, J. M. Schooneveldt, S. Schlebusch, M. H. Tilse, T. Urbanski, B. Hamilton and D. Venter, J. Clin. Microbiol., 2012, 50, 2918–2931 CrossRef CAS PubMed.
- J. M. A. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. V. Piddock, Nat. Rev. Microbiol., 2015, 13, 42–51 CrossRef CAS PubMed.
- S. Sauer and M. Kliem, Nat. Rev. Microbiol., 2010, 8, 74–82 CrossRef PubMed.
- J. Hrabak, E. Chudackova and R. Walkova, Clin. Microbiol. Rev., 2013, 26, 103–114 CrossRef PubMed.
- L. Pinto, P. Poeta, S. Vieira, C. Caleja, H. Radhouani, C. Carvalho, M. Vieira-Pinto, P. Themudo, C. Torres, R. Vitorino, P. Domingues and G. Igrejas, J. Proteomics, 2010, 73, 1535–1541 CrossRef PubMed.
- K. V. dos Santos, C. G. Diniz, L. D. Veloso, H. M. de Andrade, M. D. Giusta, S. D. Pires, A. V. Santos, A. C. M. Apolonio, M. A. R. de Carvalho and L. D. Farias, Res. Microbiol., 2010, 161, 268–275 CrossRef PubMed.
- F. Boulund, R. Karlsson, L. Gonzales-Siles, A. Johnning, N. Karami, O. Al-Bayati, C. Ahren, E. R. B. Moore and E. Kristiansson, Mol. Cell. Proteomics, 2017, 16, 1052–1063 Search PubMed.
- J. H. Youn, S. K. Drake, R. A. Weingarten, K. M. Frank, J. P. Dekker and A. F. Lau, J. Clin. Microbiol., 2016, 54, 35–42 CrossRef PubMed.
- H. Y. Bi, L. Qiao, J. M. Busnel, V. Devaud, B. H. Liu and H. H. Girault, Anal. Chem., 2009, 81, 1177–1183 CrossRef PubMed.
- C. R. Cox, K. R. Jensen, N. R. Saichek and K. J. Voorhees, Sci. Rep., 2015, 5, 10470 CrossRef PubMed.
- L. Qiao, C. Roussel, J. J. Wan, J. Kong, P. Y. Yang, H. H. Girault and B. H. Liu, Angew. Chem., Int. Ed., 2008, 47, 2646–2648 CrossRef PubMed.
- L. Qiao, H. Y. Bi, J. M. Busnel, J. Waser, P. Y. Yang, H. H. Girault and B. H. Liu, Chem.–Eur. J., 2009, 15, 6711–6717 CrossRef PubMed.
- G. Gogniat, M. Thyssen, M. Denis, C. Pulgarin and S. Dukan, FEMS Microbiol. Lett., 2006, 258, 18–24 CrossRef PubMed.
- R. Nakano, M. Hara, H. Ishiguro, Y. Y. Yao, T. Ochiai, K. Nakata, T. Murakami, J. Kajioka, K. Sunada, K. Hashimoto, A. Fujishima and Y. Kubota, Catalysts, 2013, 3, 310–323 CrossRef CAS.
- H. J. Zhai and L. S. Wang, J. Am. Chem. Soc., 2007, 129, 3022–3026 CrossRef CAS PubMed.
- A. Kubacka, M. S. Diez, D. Rojo, R. Bargiela, S. Ciordia, I. Zapico, J. P. Albar, C. Barbas, V. A. P. Martins dos Santos, M. Fernandez-Garcia and M. Ferrer, Sci. Rep., 2014, 4, 4134 CrossRef PubMed.
- S. L. Ma, S. H. Zhan, Y. N. Jia and Q. X. Zhou, ACS Appl. Mater. Interfaces, 2015, 7, 21875–21883 CAS.
- W. J. Wang, L. Z. Zhang, T. C. An, G. Y. Li, H. Y. Yip and P. K. Wong, Appl. Catal., B, 2011, 108, 108–116 CrossRef.
- S. A. Ansari and M. H. Cho, Sci. Rep., 2016, 6, 25405 CrossRef CAS PubMed.
- V. Ryzhov and C. Fenselau, Anal. Chem., 2001, 73, 746–750 CrossRef CAS PubMed.
- P. A. Demirev, Y. P. Ho, V. Ryzhov and C. Fenselau, Anal. Chem., 1999, 71, 2732–2738 CrossRef CAS PubMed.
- P. M. Bennett, Br. J. Pharmacol., 2008, 153, S347–S357 CrossRef CAS PubMed.
- J. E. Camara and F. A. Hays, Anal. Bioanal. Chem., 2007, 389, 1633–1638 CrossRef CAS PubMed.
- D. M. Daigle, G. A. McKay, P. R. Thompson and G. D. Wright, Chem. Biol., 1999, 6, 11–18 CrossRef CAS PubMed.
- L. R. Schwocho, C. P. Schaffner, G. H. Miller, R. S. Hare and K. J. Shaw, Antimicrob. Agents Chemother., 1995, 39, 1790–1796 CrossRef CAS PubMed.
- I. A. Murray and W. V. Shaw, Antimicrob. Agents Chemother., 1997, 41, 1–6 CAS.
- K. E. Preston, C. C. A. Radomski and R. A. Venezia, Antimicrob. Agents Chemother., 2000, 44, 3158–3162 CrossRef CAS PubMed.
- G. A. Jacoby, Clin. Microbiol. Rev., 2009, 22, 161–182 CrossRef CAS PubMed.
- F. Depardieu, I. Podglajen, R. Leclercq, E. Collatz and P. Courvalin, Clin. Microbiol. Rev., 2007, 20, 79–114 CrossRef CAS PubMed.
- X. X. Xia, Z. G. Qian and S. Y. Lee, FEMS Microbiol. Lett., 2011, 314, 119–124 CrossRef CAS PubMed.
- Y. Wang, M. Ran, J. Wang, Q. Ouyang and C. X. Luo, PLoS One, 2015, 10, e0127115 Search PubMed.
- H. Knothe, P. Shah, V. Krcmery, M. Antal and S. Mitsuhashi, Infection, 1983, 11, 315–317 CrossRef CAS PubMed.
- R. Bonnet, Antimicrob. Agents Chemother., 2004, 48, 1–14 CrossRef CAS PubMed.
- Y. Ferrandez, L. Monlezun, G. Phan, H. Benabdelhak, P. Benas, N. Ulryck, P. Falson, A. Ducruix, M. Picard and I. Broutin, Electrophoresis, 2012, 33, 1282–1287 CrossRef CAS PubMed.
- J. Mikolosko, K. Bobyk, H. I. Zgurskaya and P. Ghosh, Structure, 2006, 14, 577–587 CrossRef CAS PubMed.
- C. Llanes, D. Hocquet, C. Vogne, D. Benali-Baitich, C. Neuwirth and P. Plesiat, Antimicrob. Agents Chemother., 2004, 48, 1797–1802 CrossRef CAS PubMed.
- C. Fuda, M. Suvorov, S. B. Vakulenko and S. Mobashery, J. Biol. Chem., 2004, 279, 40802–40806 CrossRef CAS PubMed.
- S. Haghighat, S. D. Siadat, S. M. Sorkhabadi, A. A. Sepahi and M. Mahdavi, Avicenna J. Med. Biotechnol., 2013, 5, 204–211 CAS.
- J. S. Keseru, Z. Gal, G. Barabas, I. Benko and I. Szabo, Chemotherapy, 2005, 51, 300–304 CrossRef CAS PubMed.
- A. M. Di Guilmi, A. Dessen, O. Dideberg and T. Vernet, J. Bacteriol., 2003, 185, 1650–1658 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04089j |
| This journal is © The Royal Society of Chemistry 2018 |