
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diazomethane umpolung atop anthracene: an electrophilic methylene transfer reagent†
Maximilian
Joost‡
,
Wesley J.
Transue‡
 and
Christopher C.
Cummins
*
and
Christopher C.
Cummins
*
Department of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: ccummins@mit.edu
First published on 21st December 2017
Abstract
Formal addition of diazomethane's terminal nitrogen atom to the 9,10-positions of anthracene yields H2CN2A (1, A = C14H10 or anthracene). The synthesis of this hydrazone is reported from Carpino's hydrazine H2N2A through treatment with paraformaldehyde. Compound 1 has been found to be an easy-to-handle solid that does not exhibit dangerous heat or shock sensitivity. Effective umpolung of the diazomethane unit imbues 1 with electrophilicity at the methylene carbon center. Its reactivity with nucleophiles such as H2CPPh3 and N-heterocyclic carbenes is exploited for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond formation with elimination of dinitrogen and anthracene. Similarly, 1 is demonstrated to deliver methylene to a nucleophilic singlet d2 transition metal center, W(ODipp)4 (2), to generate the robust methylidene complex [2CH2]. This behavior is contrasted with that of the Wittig reagent H2CPPh3, a more traditional and Brønsted basic methylene source that upon exposure to 2 contrastingly forms the methylidyne salt [MePPh3][2
C bond formation with elimination of dinitrogen and anthracene. Similarly, 1 is demonstrated to deliver methylene to a nucleophilic singlet d2 transition metal center, W(ODipp)4 (2), to generate the robust methylidene complex [2CH2]. This behavior is contrasted with that of the Wittig reagent H2CPPh3, a more traditional and Brønsted basic methylene source that upon exposure to 2 contrastingly forms the methylidyne salt [MePPh3][2![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH].
CH].
Diazomethane is infamous for the dangers associated with its use.1 Despite its synthetic versatility, diazomethane's high toxicity and propensity to explode should give a chemist pause before committing to its use. In an effort to offer an alternate methylene source using an anthracene-based strategy,2–8 we report herein the synthesis and some initial reactivity studies of H2CN2A (1, A = C14H10 or anthracene), a molecule conceived as a formal adduct between diazomethane and anthracene. An initial survey of the reactivity patterns of 1 has revealed it not to be a simple substitute for diazomethane, instead characterizing it as a unique electrophilic methylene source. Its electrophilicity differentiates 1 from common metal-free methylene transfer reagents such as diazomethane and methylene triphenylphosphorane.
Synthesis of hydrazone 1 proceeded from Carpino's hydrazine H2N2A upon paraformaldehyde treatment in a biphasic diethyl ether–water mixture,9,10 providing the target molecule in 74% isolated yield (Scheme 1). An X-ray diffraction study of its structure revealed expected metrical data.11
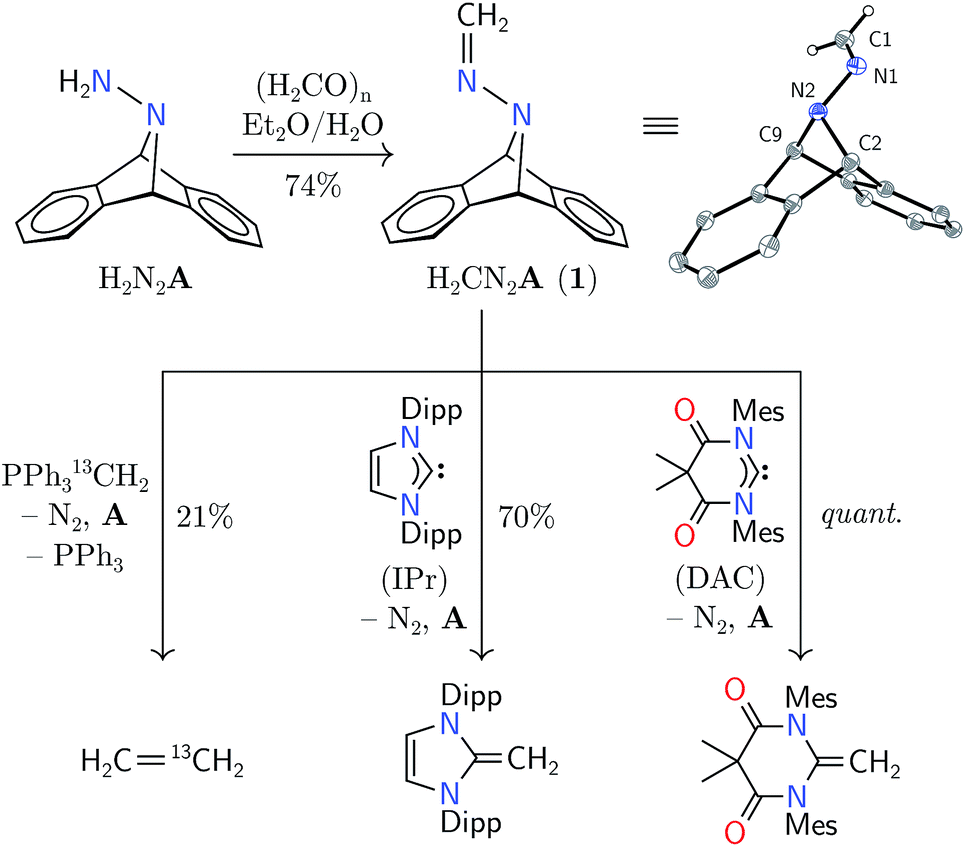 | ||
| Scheme 1 Synthesis of methylene hydrazone 1 and initial studies of methylene transfer (Mes = mesityl, Dipp = 2,6-diisopropylphenyl), shown alongside its structure from an X-ray diffraction study. Thermal ellipsoids are shown at the 50% probability level. Selected distances [Å] and angles [°]: N1–C1 1.275(2), N1–N2 1.389(1), N2–C2 1.508(1), N2–C9 1.521(2); N2–N1–C1 118.3(1). | ||
Hydrazone 1 was found to be an air-stable and crystalline solid, easily manipulable and displaying no propensity for detonation upon heating or shock. The solid was found to be volatile by thermogravimetric analysis (TGA), which showed gradual sample evaporation up to 120 °C without any discrete mass-loss events that would be expected from its fragmentation into diazomethane and anthracene. Within a sealed capillary, 1 melted without explosion (116–119 °C). After heating the melt to 140 °C, NMR spectroscopic analysis of the resolidified solid showed 74% recovery of 1 with 26% anthracene production. Its behavior in solution was similar, evincing only slow fragmentation into anthracene at temperatures greater than 120 °C. The volatility of this compound foiled attempts at analysis of its thermal behavior by molecular beam mass spectrometry (MBMS), limiting our ability to comment on the fragments directly produced by its thermal fragmentation.2–5
Having established 1 to pose a low explosion risk, we were encouraged to proceed to test its reactivity as a methylene synthon. Our initial investigations rapidly uncovered contrasting reactivity patterns vis-à-vis those characteristic of diazomethane. For example, methylation of carboxylic acids, a hallmark of diazomethane reactivity,12 did not proceed upon treatment with excess pivalic acid, acetic acid, or trifluoroacetic acid. These experiments were informative, and led us to consider more closely the electronic structure of 1.
Hydrazones are known to be carbon ambiphiles;13 however, 1 did not demonstrate nucleophilicity. Such behavior is not unexpected, as the πCN is known to be polarized away from the carbon center, although less so than an imine πCN or a ketone πCO bond.14 The polarization of this bond suggests that 1 should be expected to exhibit moderate electrophilicity at its methylene carbon. This would effectively induce umpolung of the diazomethane unit as diazomethane generally reacts as a carbon nucleophile.15
The predicted reversal of philicity was initially confirmed by successful methylene transfer in the reaction between 1 and H2CPPh3. Combination of these two reagents in benzene-d6 yielded ethylene in 21% yield over 12 h in concert with anthracene, triphenylphosphine, and, presumably, dinitrogen. The reaction was found to produce several unidentified byproducts by NMR spectroscopy, explaining the low yield of ethylene; however, isotopic labelling of the ylide led to H2C13CH2 from 1 and H213CPPh3, and H2CCD2 from 1 and D2CPPh3, confirming ethylene formation through the unification of the electro- and nucleophilic methylene units. Although the yield was low, this mode of reactivity was instructive for our further studies.
The electrophilicity of 1 lent itself well to the synthesis of N-heterocyclic olefins from N-heterocyclic carbenes (NHCs).16 In benzene-d6 solution, 1 reacted with nucleophilic IPr (1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) to yield the corresponding olefin in 70% yield after 13 h at 80 °C.16 As a nucleophile with increased electrophilicity, the Bielawski N,N′-diamidocarbene (“DAC”) was found to react in essentially quantitative yield to form a new CC bond over 24 h at 22 °C.17 This mode of reactivity differs markedly from that of diazoalkanes, which have been documented to react with NHCs at their electrophilic N-terminus to produce azines with a new CN–NC moiety.18 Heating 1 with triphenylphosphine or tricyclohexylphosphine has not yielded the analogous yieldes, suggesting a modest Lewis acidity at the carbon center of 1.
It is rare for diazomethane to be used in transition metal chemistry for the synthesis of a stable methylidene complex.19 In fact, the use of diazoalkanes in d-block chemistry is often complicated by their propensity for side reactions other than alkylidene delivery.20,21 The reactivity differences between 1 and diazomethane thus encouraged us to attempt the use of 1 in methylidene complex synthesis to see if engagement of the terminal nitrogen in bonding to anthracene subdues deleterious alternate reaction pathways.
We identified [W(ODipp)4] (2, ODipp = 2,6-diisopropylphenoxide)22,23 as a d2 transition metal complex well poised to behave as a methylene acceptor.24 Complex 2 is synthetically easy to access, and its square-planar geometry features a nucleophilic lone pair of electrons housed in a metal-centered dz2-like orbital, analogously to related tantalum and molybdenum singlet d2 species.8,25 Treatment of 2 with excess 1 gave facile formation of the anticipated methylidene complex [2CH2] after mild heating in benzene to 55 °C for 35 h (Scheme 2). Characteristically deshielded proton and carbon resonances of the CH2 unit were found by NMR spectroscopy: 1H δ 8.95 ppm and 13C δ 232.9 ppm with scalar coupling constants of 2JWH = 156.0 Hz, 1JWC = 185.0 Hz, and 1JCH = 155.6 Hz. The 1JCH coupling constant was typical of metal alkylidenes lacking significant agostic character.26–29 The success of 1 in this capacity was exciting, as the rarity of terminal, isolable methylidene complexes30 makes new methods for their generation welcome developments.
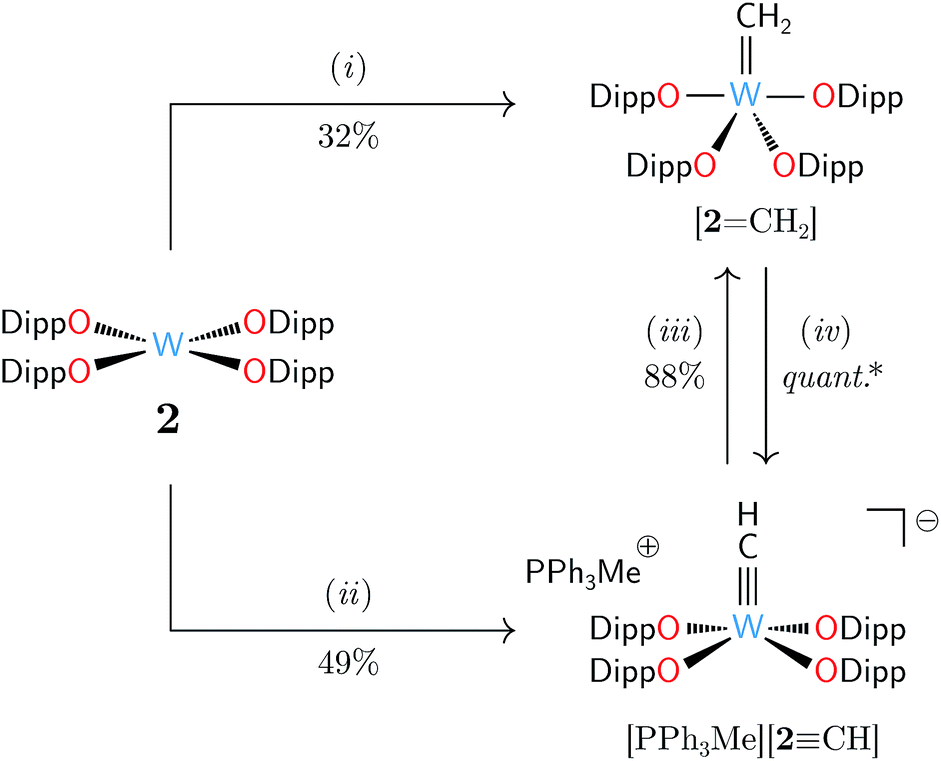 | ||
| Scheme 2 Comparative reactivity of W(ODipp)4 (2): (i) H2CN2A (1, 10 equiv.), benzene, 55 °C, 35 h; (ii) H2CPPh3 (2.0 equiv.), THF, 25 °C, 30 min; (iii) lutidinium triflate (1.0 equiv.), THF, 25 °C, 5 min; (iv) PPh3CH2 (1.0 equiv.), THF, 25 °C, 30 min. (*) NMR spectroscopic analysis showed (iv) to be quantitative. | ||
Crystallization from pentane at −35 °C overnight enabled an X-ray diffraction study of [2CH2] (Fig. 1, left) that confirmed the molecular structure. Although the data were not of high quality, the coordination geometry about the tungsten center was unambiguously identified to be intermediate between square pyramidal and trigonal bipyramidal (τ = 0.48),31 and the alkylidene bond was identified with a W⋯C interatomic distance of 1.864(4) Å. This bond length is typical of a WC double bond32 and similar to values reported for other tungsten(VI) methylidenes.29,33–35 Compound [2CH2] was not found to react productively with ethylene or 1-hexene upon heating to 70 °C in benzene-d6 for 18 h, confirmed by a lack of isotopic migration from [213CH2] to the olefins.36 Under these conditions, [2CH2] also did not react with mesitaldehyde or 4,4′-dimethylbenzophenone to form [2O] and the corresponding olefins. Despite this, [2CH2] is notable as an example of a methylidene complex with aryloxides as the exclusive supporting ligands. As such, it is an interesting structural model for methylidene complexes supported by silica or alumina surfaces implicated in alkane or olefin metathesis.37–39
 | ||
| Fig. 1 Molecular structures of (left) [2CH2] and (right) [MePPh3][2CH] from single-crystal X-ray diffraction studies shown with thermal ellipsoids at the 50% probability level. All hydrogen atoms except for the methylidene and methylidyne hydrogens are omitted for clarity, as is the [MePPh3] cation. Interatomic distances for tungsten-carbon multiple bonds: (left) W1C1 1.864(4) Å, (right) W1C1 1.749(1) Å. | ||
The reactivity of 1 was particularly satisfying after discovery of the contrasting behavior of H2CPPh3, a known reagent for CH2 delivery to transition metal centers.40–42 Treating a solution of 2 with H2CPPh3 (1 equiv.) in THF at 25 °C rapidly consumed 50% of 2 and formed the methylidyne salt [MePPh3][2CH]. Doubling the amount of H2CPPh3 gave total consumption of 2 and provided [MePPh3][2CH] in 49% isolated yield (Scheme 2). Variation of the stoichiometry and temperature of this reaction did not lead to conditions for [2CH2] formation, indicating competitive deprotonation of intermediate [2CH2] by Brønsted basic H2CPPh3. Such acid-base chemistry is postulated to play a critical role in the formation of surface-bound alkylidenes and alkylidynes for alkane and olefin metathesis,37,38 meaning [2CH2] serves also as an interesting reactivity model for alkylidyne synthesis mediated through proton transfer. This was corroborated by independent deprotonation of [2CH2] with H2CPPh3, and highlights the utility of 1 as a weakly Brønsted basic source of methylene. Protonation of [MePPh3][2CH] using lutidinium triflate presents a complementary route to [2CH2].
An X-ray crystallographic study of [MePPh3][2CH] revealed a W⋯C interatomic distance of 1.749(1) Å and a square pyramidal (τ = 0.21) coordination geometry about tungsten. A search of the CSD revealed this to be the first catalogued example of a structurally characterized metal methylidyne in an all-oxygen ligand environment, and the first catalogued example of a tungsten(VI) methylidyne complex.
As interest in metal methylidene species is rapidly growing both in homogeneous and heterogeneous catalysis,37–39,43–45 we hope 1 can be further exploited in their syntheses. Compound 1 has also shown promise in formation of new CC bonds with H2CPPh3 and NHCs, and may find use in construction of terminal olefins.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based on research supported by the National Science Foundation under CHE-1664799. M. J. thanks the Alexander von Humboldt foundation for a Feodor Lynen postdoctoral fellowship. Prof. Richard R. Schrock is gratefully acknowledged for helpful discussions, and Dr Matthew Nava is acknowledged for his assistance in MBMS studies.Notes and references
- J. A. Moore and D. E. Reed, Org. Synth. Coll., 1973, 5, 351 ( Org. Synth. , 1961 , 41 , 16 ) Search PubMed.
- A. Velian and C. C. Cummins, J. Am. Chem. Soc., 2012, 134, 13978–13981 CrossRef CAS PubMed.
- A. Velian, M. Nava, M. Temprado, Y. Zhou, R. W. Field and C. C. Cummins, J. Am. Chem. Soc., 2014, 136, 13586–13589 CrossRef CAS PubMed.
- W. J. Transue, A. Velian, M. Nava, M.-A. Martin-Drumel, C. C. Womack, J. Jiang, G.-L. Hou, X.-B. Wang, M. C. McCarthy, R. W. Field and C. C. Cummins, J. Am. Chem. Soc., 2016, 138, 6731–6734 CrossRef CAS PubMed.
- W. J. Transue, A. Velian, M. Nava, C. García-Iriepa, M. Temprado and C. C. Cummins, J. Am. Chem. Soc., 2017, 139, 10822–10831 CrossRef CAS PubMed.
- D. J. Mindiola and C. C. Cummins, Angew. Chem., Int. Ed., 1998, 37, 945–947 CrossRef CAS.
- D. J. Mindiola, Y.-C. Tsai, R. Hara, Q. Chen, K. Meyer and C. C. Cummins, Chem. Commun., 2001, 125–126 RSC.
- H. S. Soo, J. S. Figueroa and C. C. Cummins, J. Am. Chem. Soc., 2004, 126, 11370–11376 CrossRef CAS PubMed.
- L. A. Carpino, R. E. Padykula, D. E. Barr, F. H. Hall, J. G. Krause, R. F. Dufresne and C. J. Thoman, J. Org. Chem., 1988, 53, 2565–2572 CrossRef CAS.
- C. Pareja, E. Martín-Zamora, R. Fernández and J. M. Lassaletta, J. Org. Chem., 1999, 64, 8846–8854 CrossRef CAS PubMed.
- A. A. Tameem, A. Salhin, B. Saad, I. A. Rahman, M. I. Saleh, S.-L. Ng and H.-K. Fun, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o57–o58 CAS.
- T. Sammakia, Diazomethane, in e-EROS Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd., Chichester, UK, 2001 Search PubMed.
- S. Kim and J. Yoon, Science of Synthesis, Category 4. Compounds with Two Carbon Heteroatom Bonds, Georg Thieme Verlag, Stuttgart, 2005, vol. 27, pp. 671–722 Search PubMed.
- D. Armstrong and G. Walker, J. Mol. Struct.: THEOCHEM, 1987, 149, 369–389 CrossRef.
- R. Huisgen, Angew. Chem., 1955, 67, 439–463 CrossRef CAS.
- K. Powers, C. Hering-Junghans, R. McDonald, M. J. Ferguson and E. Rivard, Polyhedron, 2016, 108, 8–14 CrossRef CAS.
- D. T. Chase, J. P. Moerdyk and C. W. Bielawski, Org. Lett., 2014, 16, 812–815 CrossRef CAS PubMed.
- J. M. Hopkins, M. Bowdridge, K. N. Robertson, T. S. Cameron, H. A. Jenkins and J. A. C. Clyburne, J. Org. Chem., 2001, 66, 5713–5716 CrossRef CAS PubMed.
- W. R. Roper, J. Organomet. Chem., 1986, 300, 167–190 CrossRef CAS.
- M. Dartiguenave, M. Joëlle Menu, E. Deydier, Y. Dartiguenave and H. Siebald, Coord. Chem. Rev., 1998, 178–180, 623–663 CrossRef.
- U. Radius and J. Attner, Inorg. Chem., 2004, 43, 8587–8599 CrossRef CAS PubMed.
- M. L. Listemann, J. C. Dewan and R. R. Schrock, J. Am. Chem. Soc., 1985, 107, 7207–7208 CrossRef CAS.
- M. L. Listemann, R. R. Schrock, J. C. Dewan and R. M. Kolodziej, Inorg. Chem., 1988, 27, 264–271 CrossRef CAS.
- M. Joost, W. J. Transue and C. C. Cummins, Chem. Commun., 2017, 53, 10731–10733 RSC.
- D. R. Neithamer, R. E. LaPointe, R. A. Wheeler, D. S. Richeson, G. D. Van Duyne and P. T. Wolczanski, J. Am. Chem. Soc., 1989, 111, 9056–9072 CrossRef CAS.
- W. A. Nugent and J. M. Mayer, Metal-Ligand Multiple Bonds, Wiley, New York, 1988, pp. 132–136 Search PubMed.
- A. Poater, X. Solans-Monfort, E. Clot, C. Copéret and O. Eisenstein, Dalton Trans., 2006, 3077–3087 RSC.
- X. Solans-Monfort and O. Eisenstein, Polyhedron, 2006, 25, 339–348 CrossRef CAS.
- D. V. Peryshkov and R. R. Schrock, Organometallics, 2012, 31, 7278–7286 CrossRef CAS.
- R. R. Schrock, Chem. Rev., 2002, 102, 145–180 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- A. J. Jiang, J. H. Simpson, P. Müller and R. R. Schrock, J. Am. Chem. Soc., 2009, 131, 7770–7780 CrossRef CAS PubMed.
- R. R. Schrock, A. J. Jiang, S. C. Marinescu, J. H. Simpson and P. Müller, Organometallics, 2010, 29, 5241–5251 CrossRef CAS.
- L. C. H. Gerber and R. R. Schrock, Organometallics, 2013, 32, 5573–5580 CrossRef CAS.
- It has been demonstrated that an empty orbital is required in the respective metallocyclobutane species for productive metathesis to occur: C. P. Gordon, K. Yamamato, W.-C. Liao, F. Allouche, R. A. Anderson, C. Copéret, C. Raynaud and O. Eisenstein, ACS Cent. Sci., 2017, 3, 759 CrossRef CAS PubMed.
- J. M. Basset, C. Copéret, D. Soulivong, M. Taoufik and J. Thivolle Cazat, Acc. Chem. Res., 2010, 43, 323–334 CrossRef CAS PubMed.
- S. Lwin and I. E. Wachs, ACS Catal., 2014, 4, 2505–2520 CrossRef CAS.
- Y. Chen, E. Abou-hamad, A. Hamieh, B. Hamzaoui, L. Emsley and J.-M. Basset, J. Am. Chem. Soc., 2015, 137, 588–591 CrossRef CAS PubMed.
- P. de Frémont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2009, 253, 862–892 CrossRef.
- E. B. Hulley, J. B. Bonanno, P. T. Wolczanski, T. R. Cundari and E. B. Lobkovsky, Inorg. Chem., 2010, 49, 8524–8544 CrossRef CAS PubMed.
- J. Schwartz and K. I. Gell, J. Organomet. Chem., 1980, 184, C1–C2 CrossRef CAS.
- L. N. Grant, S. Ahn, B. C. Manor, M.-H. Baik and D. J. Mindiola, Chem. Commun., 2017, 53, 3415–3417 RSC.
- T. Kurogi, P. J. Carroll and D. J. Mindiola, Chem. Commun., 2017, 53, 3412–3414 RSC.
- T. Kurogi, M. Kamitani, B. C. Manor, P. J. Carroll and D. J. Mindiola, Organometallics, 2017, 36, 74–79 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data. CCDC 1580349–1580351. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04506a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |