DOI:
10.1039/C7SC04450J
(Edge Article)
Chem. Sci., 2018,
9, 1534-1539
Aromatization modulates the activity of small organic molecules as promoters for carbon–halogen bond activation†
Received
15th October 2017
, Accepted 13th December 2017
First published on 5th January 2018
Abstract
The combination of small organic molecules and a base serves as a unique system for the activation carbon–halogen bonds in haloarenes by single electron transfer (SET). However, most of the molecules employed as promoters only allow for the activation of aryl iodides, and efficient activation of aryl bromides and chlorides under this mode is still rather challenging. Herein, we report the discovery of a structurally simple yet powerful promoter molecule, indoline, which exhibits unusually high activity in promoting the activation of haloarenes by SET. In the presence of t-BuOK and a trace amount of oxygen, indoline promotes the formation of aryl radicals not only from aryl iodides and bromides, but also from unactivated aryl chlorides (e.g., chlorobenzene) under relatively mild conditions. Mechanistic studies reveal the molecular basis for its high activity, for which the aromatization process plays a key role in modulating the electron transfer process.
Introduction
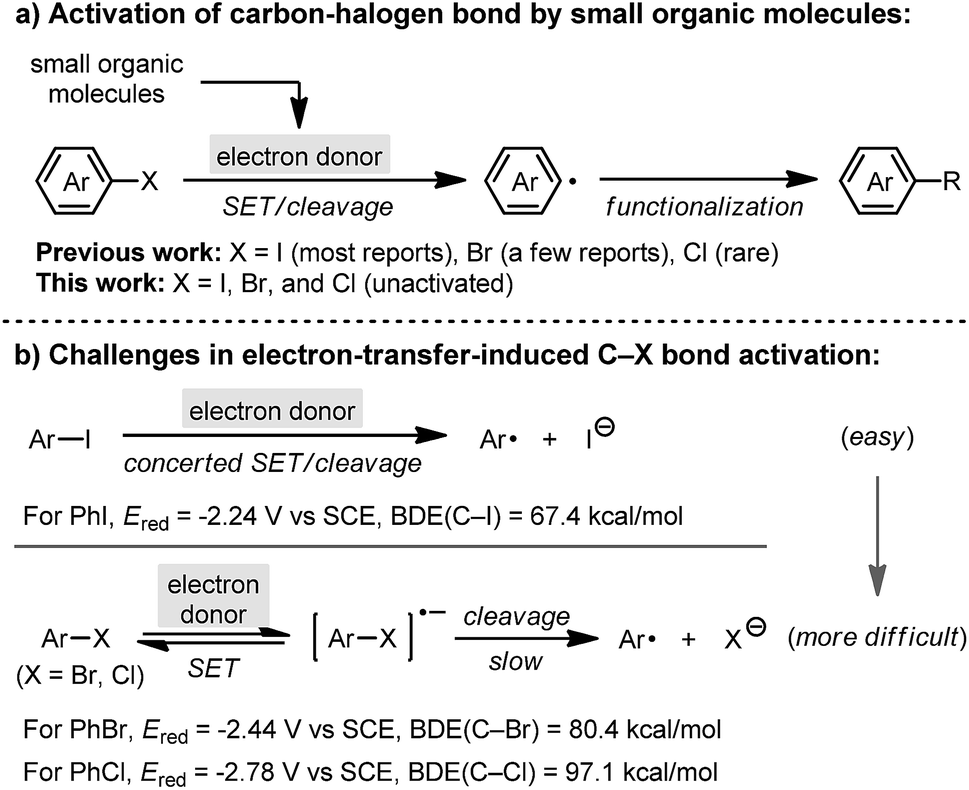
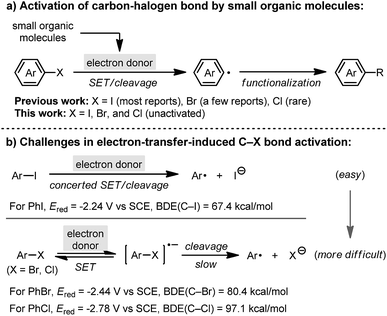
Aryl halides constitute an important category of intermediates in organic chemistry, and the activation of carbon–halogen (C–X) bonds in aryl halides plays a significant role in organic synthesis.1 In addition to stoichiometric metalation2 or transition-metal catalysis3 for C–X bond activation, in recent years the development of small-organic-molecule-promoted C–X bond activation has attracted great attention. A series of small organic molecules, such as 1,10-phenanthrolines,4 1,2-diamines,5 1,2-diols6 and N-methylanilines,7 were found to promote the activation of haloarene in the presence of a base, producing a reactive aryl radical intermediate which undergoes substitution processes (referred to as base-promoted homolytic aromatic substitution, BHAS).8 Careful mechanistic studies revealed that the promoter is transformed into a “super electron donor” under the reaction conditions, which then initiates the cleavage of the C–X bond by single electron transfer (SET) to the haloarene substrate (Scheme 1a).9 This process represents a new mode of carbon–halogen bond activation, which enables a series of synthetic methods utilizing haloarenes as starting materials in a transition-metal-free manner.10
 |
| | Scheme 1 Reaction mode of carbon–halogen bond activation promoted by small organic molecules and challenges therein. | |
Despite these achievements, efficient activation of relatively inert C–X bonds in the BHAS reaction remains a major challenge. To date, most promoters could merely activate iodoarenes,5 and only a few could activate bromoarenes.11 The activation of the C–Cl bond in unactivated chloroarenes is still rather challenging.12 This is due to the stepwise C–Br/C–Cl bond activation process with a slow C–X cleavage step,13 as well as the increasing difficulty for bromo-/chloroarene activation with respect to both reduction potential14 and bond strength15 (Scheme 1b). Herein, we report the discovery of a highly active promoter, indoline, based on the structural modification of existing promoters for carbon–halogen bond activation. It ranked among the most active small molecule promoters to date, which enables the activation of bromo- and chloroarenes, and more attractively, a series of single electron reduction reactions traditionally carried out using alkali metal reductants.
Results and discussion
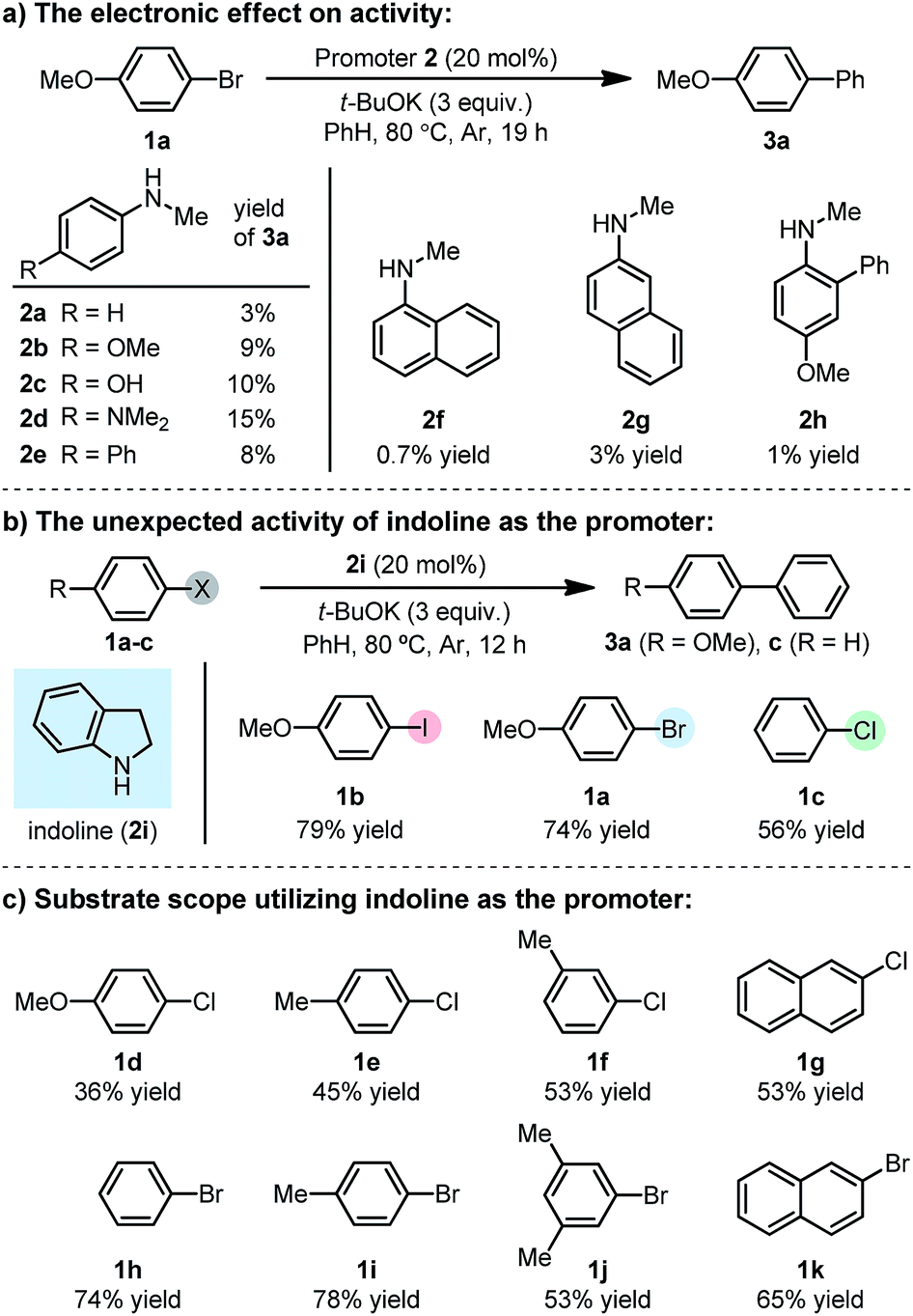
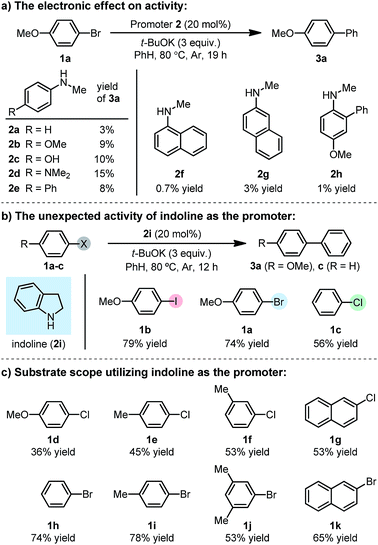
Our previous study showed that, N-methylaniline serves as an efficient promoter for the activation of iodoarenes in the presence of t-BuOK, where N-methylanilide anion acts as an electron donor.7 However, attempts to enable the activation of bromoarene 1a by structural tuning of the N-methylaniline promoter proved unsuccessful (Scheme 2a). Although electron-rich anilines exhibited slightly better activities (2a–e), the results were far from satisfactory, and the attachment of an additional aromatic ring to aniline (either as a substituent or as a fused cycle, 2f–h) with the aim to assist SET by stabilizing the formed N-centered radical, was also useless. With the hypothesis that ring strain might help to increase the activity, we attempted to use a cyclic aniline derivative, indoline (2i), as the promoter. To our surprise, the indoline/t-BuOK system exhibited great activity for activating iodo-, bromo- and even chloroarenes (Scheme 2b). A screen of substrates showed that the indoline/t-BuOK system is able to promote the BHAS reaction of a series of bromo- and chloroarenes, although chloroarenes are generally less reactive than bromoarenes (Scheme 2c). Furthermore, compared with several of the most active organic promoters reported to date, indoline exhibited the highest activity under 80 °C (Fig. 1). The unprecedented high activity of indoline in the BHAS reaction attracted our interest and prompted us to elucidate its molecular basis.
 |
| | Scheme 2 Effect of the promoter structure on carbon–halogen bond activation. | |
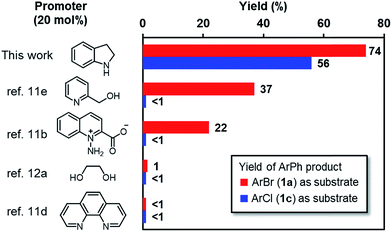 |
| | Fig. 1 Yields of the coupling products from the reaction of ArBr 1a (red) and ArCl 1c (blue) employing various promoters. Reaction conditions: 1a or 1c (0.5 mmol), promoter (20 mol%), t-BuOK (1.5 mmol), 4 mL benzene, argon atmosphere, 80 °C for 24 h. | |
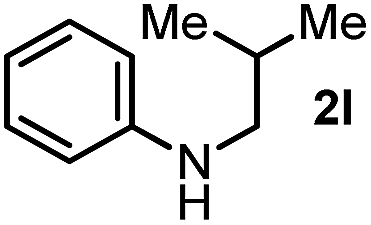
First, we found that indole was generated in a high yield as the end-product in this indoline-promoted BHAS-type reaction. This transformation involves both oxidation and aromatization, which might be responsible for the observed SET-initiated C–X activation. Second, aniline-type promoters that were not able to form indole through aromatization were found to be much less active (Table 1). 3,3-Dimethylindoline (2j), tetrahydroquinoline (2k), and N-isobutylaniline (2l) were found to exhibit activity for iodoarene, but not for bromo- and chloroarenes. These results suggest that the activation of iodoarenes and bromo-/chloroarenes is rather different, and the aromatization process, rather than the ring-strain, plays a key role in the activation of more challenging C–X bonds.
Table 1 Effect of promoter structure on activitya
| Entry |
Promoter |
ArI (1b) |
ArBr (1a) |
ArCl (1c) |
| Conv. |
Yield |
Conv. |
Yield |
Conv. |
Yield |
|
Reaction conditions: haloarene 1 (0.5 mmol), promoter 2 (20 mol%), t-BuOK (1.5 mmol), 4 mL benzene, Ar atmosphere, 80 °C for 12 h. Conversions and yields were determined by gas chromatography.
|
| 1 |
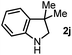
|
95% |
75% |
19% |
6% |
10% |
<1% |
| 2 |
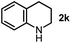
|
99% |
73% |
16% |
7% |
7% |
<1% |
| 3 |
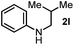
|
76% |
59% |
16% |
1% |
10% |
<1% |
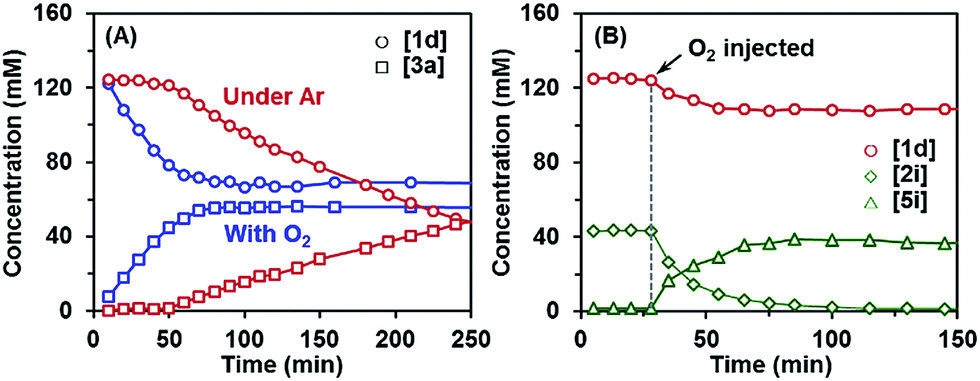
Another observation also provided important clues for the initiation mechanism. The kinetic profile of the reaction revealed that the indoline-promoted BHAS reaction of 4-chloroanisole (1d) had a significant induction period under argon, whereas the reaction exhibited no induction period in the presence of O2 (Fig. 2A). The generation of indole from indoline in these reactions followed the same trend. Injection of pure O2 into the reaction system directly initiated the reaction, but led to a low final conversion (Fig. 2B). The control experiment also showed that under rigorous oxygen-free conditions, the reactions of chloro- and bromoarene were almost completely suppressed, but the reaction of iodoarene could still proceed (Scheme 3). The transformation of indoline to indole proceeded without a haloarene substrate with trace O2, but could not proceed under O2-free conditions (Scheme 3).
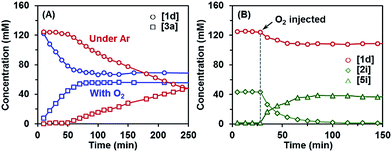 |
| | Fig. 2 Kinetic profiles of the indoline-promoted BHAS reaction of 4-chloroanisole (1d) to produce biaryl 3a under Ar and in the presence of O2 (A) and under Ar with injection of O2 (B). Reaction conditions: 1d (1 mmol), indoline (40 mol%), t-BuOK (3 mmol), 8 mL benzene, 80 °C. | |
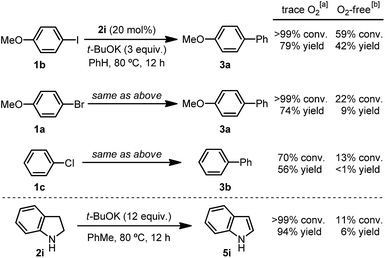 |
| | Scheme 3 Effect of oxygen on the C–X activation activity of indoline. [a] “Trace O2” condition refers to setting up the reaction in a Schlenk tube sealed with a rubber septum under Ar. [b] “O2-free” condition refers to carefully degassing the solvent by three freeze–pump–thaw cycles and then setting up the reaction in a reaction tube sealed with a Teflon screw valve under Ar. | |
The above experimental results indicate that the aromatization of indoline plays a critical role in the activation of chloro- and bromoarene, and trace oxygen existing in the reaction system serves as a key factor to initiate the activation process. Based on this, the mechanism of carbon–halogen bond activation in the indoline-promoted BHAS reaction is proposed in Scheme 4. Deprotonation of indoline by t-BuOK led to anilide anion A, which serves as a weak electron donor. For iodoarenes, anion A could directly activate the C–I bond by SET, and radical intermediate B is formed simultaneously. Subsequent proton abstraction by t-BuOK affords intermediate C (equivalent to a radical anion), which acts as a better electron donor and is able to activate another iodoarene molecule. This process is similar to the initiation mechanism of the BHAS reaction promoted by N-methylaniline. On the other hand, for bromo-/chloroarenes, anion A is not able to act as a direct electron donor. Alternatively, in the presence of a trace amount of O2, single electron oxidation of A takes place to generate B, which then affords C as a more efficient electron donor. Due to the slow C–X bond cleavage step, the electron transfer between C and the ArBr/ArCl substrate to form ArX˙− and isoindole D is reversible. Under this circumstance, the isomerization of D to indole is rather important because the irreversible aromatization favors the electron transfer equilibrium and thus enables the activation of more challenging bromo-/chloroarenes. Thus, the proposed mechanism well rationalizes the superior activity of indoline over many aniline-type promoters, and clarifies the key role of trace O2 in the initiation process.
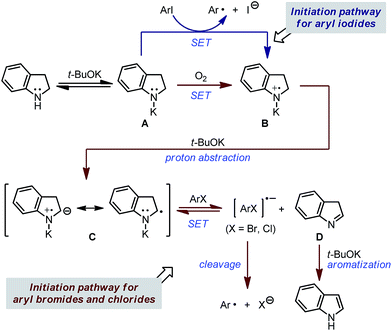 |
| | Scheme 4 Proposed initiation mechanism in the indoline-promoted BHAS reaction. | |
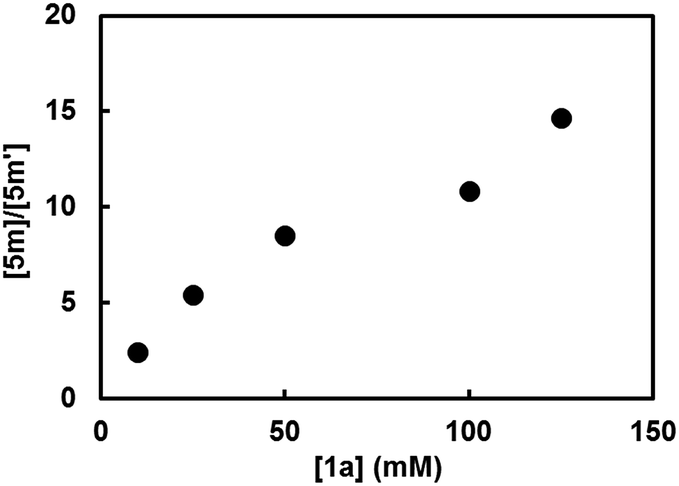
This mechanism was further supported by the reaction employing 2-cyclopropylindoline (2m) as a probe16 (Scheme 5a). In the reactions of bromo- and chloroarenes 1a and 1c promoted by 2m, both 2-cyclopropylindole (5m) and 2-propylindole (5m′) were observed as the end-product of indoline 2m. Under O2-free conditions no reaction occurred and none of the end-products were formed. The formation of the ring-opening product 5m′ serves as key evidence for the existence of radical anion-type intermediate E in the reaction system (Scheme 5b). Once formed, radical anion E could undergo either SET to ArX to afford indole 5m, or cyclopropane ring-opening to produce the ring-opened radical F, which is then transformed to 5m′ by hydrogen atom transfer or reduction. In this kinetic competition scenario, the ratio of the two end-products is expected to be dependent on [ArX]. This was proven to be true by experiments performed with different [1a] (Fig. 3), which clearly indicate that intermediate E acts as the key electron donor in the SET-induced carbon–halogen activation.
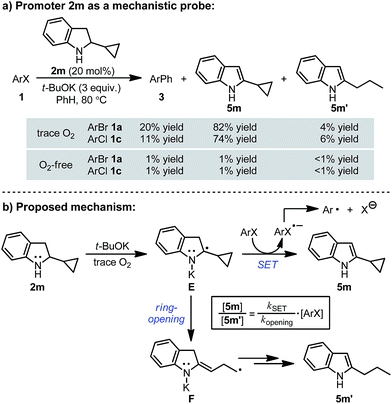 |
| | Scheme 5 Cyclopropyl substituted indoline as the mechanistic probe. | |
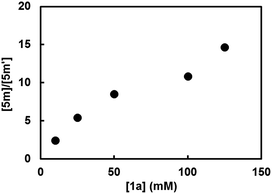 |
| | Fig. 3 Ratio between the indole products 5m and 5m′ in the BHAS reaction of 1a employing indoline 2m as a mechanistic probe. | |
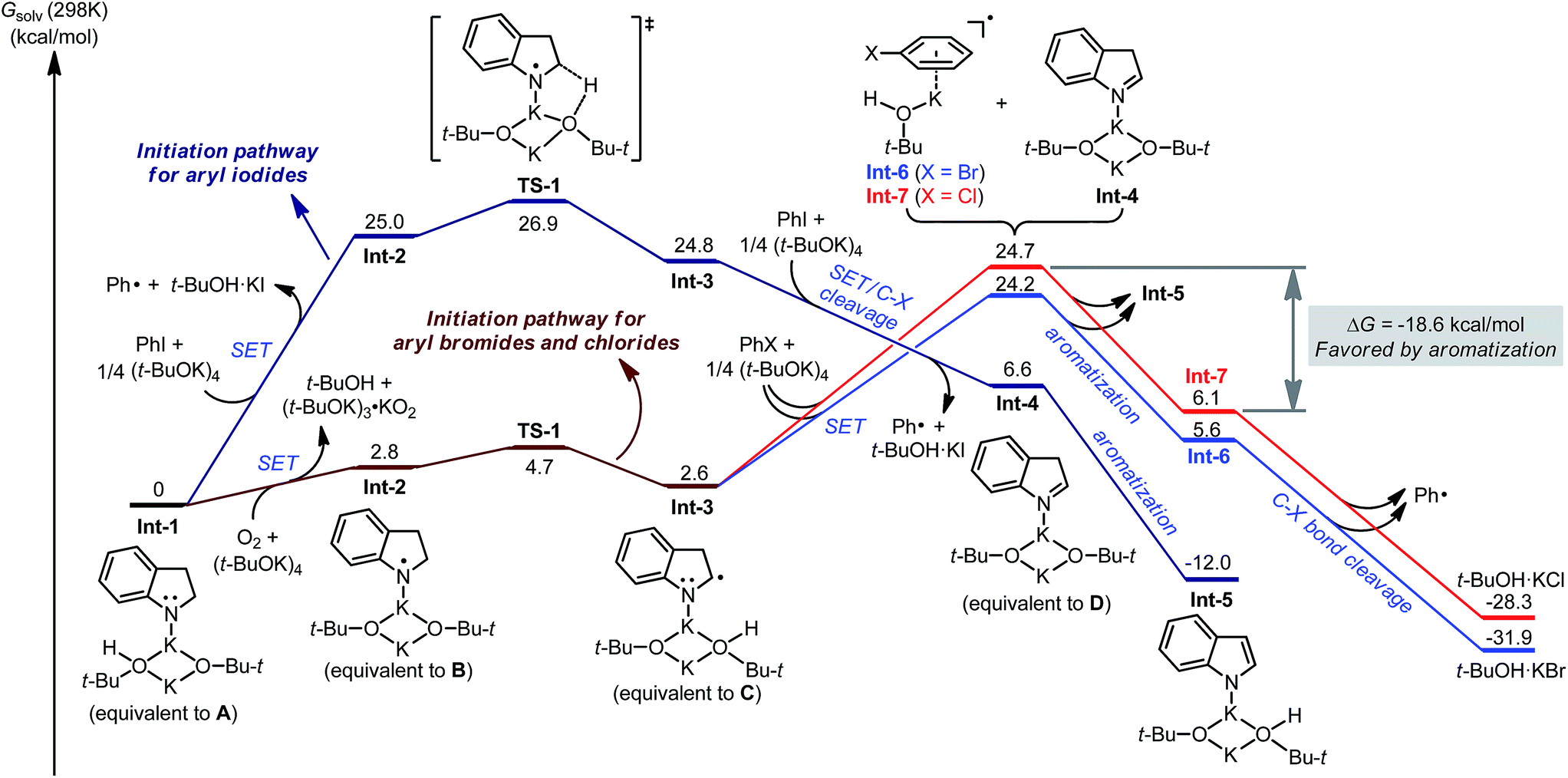
DFT calculation was conducted to provide a quantitative understanding of the indoline-promoted C–X bond activation (Fig. 4). Starting from the deprotonated indoline Int-1 (equivalent to intermediate A), direct electron transfer to PhI is possible, though endergonic by 25.0 kcal mol−1 in terms of Gibbs free energy, to produce phenyl radical and radical complex Int-2 (equivalent to intermediate B). Deprotonation of Int-2 at the C–H bond neighboring the N-centered radical viaTS-1 is rather facile, affording the radical anion complex Int-3 (equivalent to intermediate C). Though endergonic by 24.8 kcal mol−1, the direct activation of PhI by anilide Int-1 is feasible. In contrast, for PhBr and PhCl direct electron transfer is not possible since the SET step is rather endergonic (>60 kcal mol−1, see the ESI†). In such cases, trace oxygen reacts with Int-1 to afford radical complex Int-2 with a much more favorable free energy barrier,17 and Int-3 is subsequently generated by deprotonation.
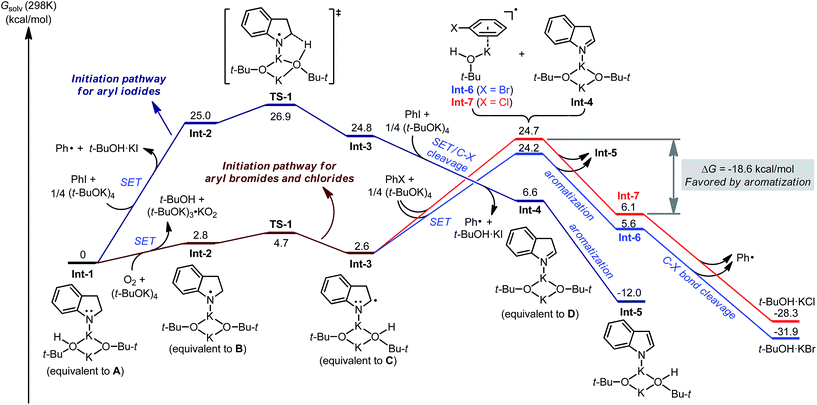 |
| | Fig. 4 Partial potential energy surface of different initiation pathways calculated at the M06-2X/6-311+G(d,p)//B3LYP/6-31G(d) level of theory. | |
As expected, complex Int-3 is a more efficient electron donor, enabling exergonic SET to PhI to form the phenyl radical and isoindole complex Int-4 (equivalent to intermediate D). The following aromatization process from Int-4 to Int-5 is also exergonic (by 18.6 kcal mol−1). For the reaction of bromo-/chloroarenes, this aromatization process is of great significance. SET from Int-3 to PhBr/PhCl is endergonic by >20 kcal mol−1, which hampers efficient activation of these substrates given that the following rate-limiting C–X bond cleavage requires even greater energy barriers. In combination with the aromatization process, the SET process is endergonic by ca. 3 kcal mol−1, leading to a reasonable overall energy barrier for C–Br/C–Cl bond activation. Therefore, it is clear that the aromatization process helps to overcome the SET energy barrier of the challenging haloarene substrates.
Since the mechanistic study revealed that the indoline/t-BuOK/O2 reaction system could generate a potent electron donor (Int-3) in situ, we sought to exploit its potential use in single electron reduction. A preliminary study showed that this system is able to promote a series of reduction and reductive cleavage reactions (Scheme 6), such as the reduction of benzophenone (6)18 and diphenylacetylene (8),19 as well as the reductive cleavage of diphenylsulfone (10)20 and benzyl trityl ether (11).21 These reactions traditionally required the use of potent single electron reductants (e.g., alkali metals), thus the unprecedented reactivity of the present system highlights its synthetic potential. Interestingly, for the reduction of benzophenone, the formation of the benzophenone radical anion was observed by its characteristic blue color and confirmed by EPR spectroscopy,22 which further evidence the superior electron reduction ability of this system.
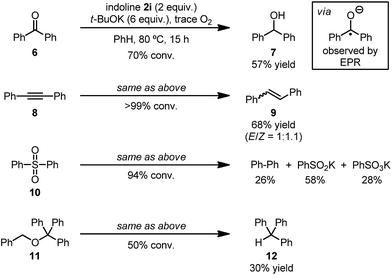 |
| | Scheme 6 Reduction and reductive cleavage reactions using the indoline/t-BuOK/O2 system. | |
Conclusions
In this work, we have shown the unusual high activity of the indoline/t-BuOK/O2 system for promoting carbon–halogen bond activation in the BHAS reaction, as well as its molecular basis. A trace amount of oxygen is essential for initiating the reaction, while the aromatization process of the generated isoindole intermediate plays a crucial role in modulating the electron transfer reaction. The present system enabled efficient activation of more challenging carbon–halogen bonds, and demonstrated for the first time that the aromatization energy could be utilized to boost the activity of a small molecule promoter in the BHAS reaction. This concept may be utilized in other reactions to modulate the electron transfer processes therein.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
The National Natural Science Foundation of China (Grant No. 21390403) and the Thousand Talents Plan for Young Professionals are acknowledged for financial support. The technology platform of CBMS and the Tsinghua Xuetang Talents Program are acknowledged for providing instrumentation and computational resources.
Notes and references
-
(a)
G. W. Gribble, Structure and Biosynthesis of Halogenated Alkaloids, in Modern Alkaloids: Structure, Isolation, Synthesis, and Biology, ed. E. Fattorusso and O. Tagilialatela-Scafati, Wiley-VCH, Weinheim, 2008 Search PubMed;
(b)
T. Kosjek and E. Heath, Halogenated Heterocycles as Pharmaceuticals, in Halogenated Heterocycles: Synthesis, Application and Environment, ed. J. Iskra, Springer, New York, 2012 Search PubMed;
(c) D. A. Petrone, J. Ye and M. Lautens, Chem. Rev., 2016, 116, 8003–8104 CrossRef CAS PubMed.
-
(a) W. F. Bailey and J. J. Patricia, J. Organomet. Chem., 1988, 352, 1–46 CrossRef CAS;
(b) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2002, 102, 4009–4091 CrossRef CAS PubMed.
-
(a) D. Baranano, G. Mann and J. F. Hartwig, Curr. Org. Chem., 1997, 1, 287–305 CrossRef CAS;
(b) J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046–2067 CrossRef CAS;
(c) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176–4211 CrossRef CAS.
-
(a) C.-L. Sun, H. Li, D.-G. Yu, M. Yu, X. Zhou, X.-Y. Lu, K. Huang, S.-F. Zheng, B.-J. Li and Z.-J. Shi, Nat. Chem., 2010, 2, 1044–1049 CrossRef CAS PubMed;
(b) C.-L. Sun, Y.-F. Gu, W.-P. Huang and Z.-J. Shi, Chem. Commun., 2011, 47, 9813–9815 RSC.
-
(a) W. Liu, H. Cao, H. Zhang, H. Zhang, K. H. Chung, C. He, H. Wang, F. Y. Kwong and A. Lei, J. Am. Chem. Soc., 2010, 132, 16737–16740 CrossRef CAS PubMed;
(b) S. De, S. Ghosh, S. Bhunia, J. A. Sheikh and A. S. Bisai, Org. Lett., 2012, 14, 4466–4469 CrossRef CAS PubMed.
-
(a) E. Shirakawa, X. Zhang and T. Hayashi, Angew. Chem., Int. Ed., 2011, 50, 11125–11128 CrossRef PubMed;
(b) S. Sharma, M. Kumar, V. Kumar and N. Kumar, Tetrahedron Lett., 2013, 54, 4868–4871 CrossRef CAS.
- H. Yang, L. Zhang and L. Jiao, Chem.–Eur. J., 2017, 23, 65–69 CrossRef CAS PubMed.
-
(a) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2011, 50, 5018–5022 CrossRef CAS PubMed;
(b) T. L. Chan, Y. Wu, P. Y. Choy and F. Y. Kwong, Chem.–Eur. J., 2013, 19, 15802–15814 CrossRef CAS PubMed;
(c) A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765–773 CrossRef CAS PubMed.
-
(a) S. Zhou, E. Doni, G. M. Anderson, R. G. Kane, S. W. MacDougall, V. M. Ironmonger, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2014, 136, 17818–17826 CrossRef CAS PubMed;
(b) H. Yi, A. Jutand and A. Lei, Chem. Commun., 2015, 51, 545–548 RSC;
(c) L. Zhang, H. Yang and L. Jiao, J. Am. Chem. Soc., 2016, 138, 7151–7160 CrossRef CAS PubMed;
(d) J. P. Barham, G. Coulthard, R. G. Kane, N. Delgado, M. P. John and J. A. Murphy, Angew. Chem., Int. Ed., 2016, 55, 4492–4496 CrossRef CAS PubMed.
- For selected recent examples, see:
(a) Y. Gao, P. Tang, H. Zhou, W. Zhang, H. Yang, N. Yan, G. Hu, D. Mei, J. Wang and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 3124–3128 CrossRef CAS PubMed;
(b) J. P. Barham, G. Coulthard, R. G. Kane, N. Delgado, M. P. John and J. A. Murphy, Angew. Chem., 2016, 128, 4568–4572 (
Angew. Chem. Int. Ed.
, 2016
, 55
, 4492–4496
) CrossRef.
-
(a) E. Shirakawa, K. Itoh, T. Higashino and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 15537–15539 CrossRef CAS PubMed;
(b) Y. Qiu, Y. Liu, K. Yang, W. Hong, Z. Li, Z. Wang, Z. Yao and S. Jiang, Org. Lett., 2011, 13, 3556–3559 CrossRef CAS PubMed;
(c) B. Li, X. Qin, J. You, X. Cong and J. Lan, Org. Biomol. Chem., 2013, 11, 1290–1293 RSC;
(d) Y. Wu, P. Y. Choy and F. Y. Kwong, Org. Biomol. Chem., 2014, 12, 6820–6823 RSC.
- The activation of aryl chlorides by SET required activated chloroarene substrates, high reaction temperature or photoactivation. For examples, see:
(a) Y. Wu, S. M. Wong, F. Mao, T. L. Chan and F. Y. Kwong, Org. Lett., 2012, 14, 5306–5309 CrossRef CAS PubMed;
(b) I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed;
(c) D. Ghosh, J.-Y. Lee, C.-Y. Liu, Y.-H. Chiang and H. M. Lee, Adv. Synth. Catal., 2014, 356, 406–410 CrossRef CAS;
(d) M. Jiang, H. Li, H. Yang and H. Fu, Angew. Chem., Int. Ed., 2017, 56, 874–879 CrossRef CAS PubMed;
(e) T. Hokamp, A. Dewanji, M. Lübbesmeyer, C. Mück-Lichtenfeld, E.-U. Würthwein and A. Studer, Angew. Chem., Int. Ed., 2017, 56, 13275–13278 CrossRef CAS PubMed.
-
(a) N. Takeda, P. V. Poliakovr, A. R. Cook and J. R. Miller, J. Am. Chem. Soc., 2004, 126, 4301–4309 CrossRef CAS PubMed;
(b) C. Coostentin, M. Robert and J.-M. Saveant, J. Am. Chem. Soc., 2004, 126, 16051–16057 CrossRef PubMed;
(c) J. J. Devery, J. D. Nguyen, C. Dai and C. R. J. Stephenson, ACS Catal., 2016, 6, 5962–5967 CrossRef CAS.
- L. Pause, M. Robert and J.-M. Saveant, J. Am. Chem. Soc., 1999, 121, 7158–7159 CrossRef CAS.
- The bond dissociation energy (BDE) data were taken from the Internet Bond-energy Databank (iBond), see: http://ibond.chem.tsinghua.edu.cnor http://ibond.naikai.edu.cn.
-
(a) D. Griller and K. U. Ingold, Acc. Chem. Res., 1980, 13, 317–323 CrossRef CAS;
(b) A. L. J. Beckwith and V. W. Bowry, J. Am. Chem. Soc., 1994, 116, 2710–2716 CrossRef CAS.
- W. B. Liu, D. P. Schuman, Y. F. Yang, A. A. Toutov, Y. Liang, H. F. T. Klare, N. Nesnas, M. Oestreich, D. G. Blackmond, S. C. Virgil, S. Banerjee, R. N. Zare, R. H. Grubbs, K. N. Houk and B. M. Stoltz, J. Am. Chem. Soc., 2017, 139, 6867–6879 CrossRef CAS PubMed.
-
(a) M. Gomberg and W. E. Bachmann, J. Am. Chem. Soc., 1927, 49, 236–257 CrossRef CAS;
(b) R. Sato, T. Nagaoka, T. Goto and M. Sarro, Bull. Chem. Soc. Jpn., 1990, 63, 290–292 CrossRef CAS;
(c) G. Kleiner, A. Tarnopolsky and S. Hoz, Org. Lett., 2005, 7, 4197–4200 CrossRef CAS PubMed.
-
(a) A. Maercker, M. Kemmer, H. C. Wang, D.-H. Dong and M. Szwarc, Angew. Chem., Int. Ed., 1998, 37, 2136–2138 CrossRef CAS;
(b) L. H. Li, L. C. Li and C. H. Xu, Chin. Chem. Lett., 2012, 23, 69–72 CrossRef;
(c) U. Rayhan, Z. Kowser, C. Redshaw and T. Yamato, Tetrahedron, 2016, 72, 6943–6947 CrossRef CAS.
-
(a) E. Constantinescu, M. Hillebrand, E. Volanschi, M. Andrei, G. Ivanescu and O. Maior, J. Electroanal. Chem., 1995, 395, 211–220 CrossRef;
(b) D. P. Morales, A. S. Taylor and S. C. Farmer, Molecules, 2010, 15, 1265–1269 CrossRef CAS PubMed;
(c) F. Schoenebeck, J. A. Murphy, S.-Z. Zhou, Y. Uenoyama, Y. Miclo and T. Tuttle, J. Am. Chem. Soc., 2007, 129, 13368–13369 CrossRef CAS PubMed.
- M. Yus, C. Behloul and D. Guijarro, Synthesis, 2003, 14, 2179–2184 CrossRef.
- J. Säuberlich, O. Brede and D. Beckert, J. Phys. Chem., 1996, 100, 18101–18107 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04450j |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*