
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Traceless β-mercaptan-assisted activation of valinyl benzimidazolinones in peptide ligations†
Yinglu
Wang
a,
Lin
Han
a,
Ning
Yuan
a,
Hanxuan
Wang
a,
Hongxing
Li
a,
Jinrong
Liu
a,
Huan
Chen
b,
Qiang
Zhang
 *b and
Suwei
Dong
*a
*b and
Suwei
Dong
*a
aState Key Laboratory of Natural and Biomimetic Drugs, Department of Chemical Biology, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China. E-mail: dongs@hsc.pku.edu.cn
bDepartment of Chemistry, University at Albany, Albany, New York 12222, USA. E-mail: qzhang5@albany.edu
First published on 5th January 2018
Abstract
Peptidyl thioesters or their surrogates with C-terminal β-branched hydrophobic amino acid residues usually exhibit poor reactivities in ligation reactions. Thus, activation using exogenous additives is required to ensure an acceptable reaction efficiency. Herein, we report a traceless ligation at Val-Xaa sites under mild thiol additive-free reaction conditions, whereby the introduction of β-mercaptan on the C-terminal valine residue effectively activates the otherwise unreactive N-acyl-benzimidazolinone (Nbz), and enables the use of a one-pot ligation–desulfurization strategy to generate the desired peptide products. The orthogonality between β-thiovaline-Nbz and a conventional alkyl thioester, as well as the convenient access to the former from readily available penicillamine, also allowed expedited assembly of the peptidic hormone β-LPH and hPTH analogues, based on a kinetically controlled one-pot three-segment ligation and desulfurization strategy.
Introduction
Native chemical ligation (NCL) is one of the most powerful and frequently used methods employed in the fields of chemistry and chemical biology1 for the construction of polypeptides and proteins.2 The versatility of the approach has encouraged continued development of surrogates, or “masked forms” of the required peptidyl thioesters,3 such as peptidyl hydrazide,4 peptide N-acyl-benzimidazolinone (peptide-Nbz),5 peptidyl selenoesters,6 and other amide-based precursors.7 Moreover, the logic of NCL has also inspired the desulfurization-enabled methodological expansion of optional peptide N-terminal residues in addition to the cysteines,8 along with other biocompatible ligation methods, including Staudinger ligation,9 α-ketoacid-hydroxylamine (KAHA) ligation,10 seleno-amino acid-based ligation,11 serine/threonine ligation (STL),12 and so on. While the ligation–desulfurization strategy has been widely applied in chemical protein synthesis, overcoming the detrimental steric effects involved in ligation has remained a challenge even with the assistance of exogenous thiol additives.13 Thus, innovative approaches to accelerate the reaction rate at bulky amino acid sites (e.g. Pro, Val, Ile, etc.) are desirable. Mild reaction conditions that are compatible with the broadly applied metal-free desulfurization (MFD) protocol,8b rather than those that utilize radical-quenching thiol additives such as 4-mercaptophenylacetic acid (MPAA),14would also be of benefit. Besides the search for more effective and practical additives,15 studies have also been conducted exploiting the presence of –SH functionalities within the peptide segments to activate the C-termini under thiol additive-free conditions,16 including a recent report from our group on a 4-thioproline-based ligation at the Pro-Xaa sites.17 In contrast to the internal activation originating from the cysteine residues within the sequence, a constrained thiolactone structure was suggested to be crucial in accounting for the drastically increased reactivity in comparison to the regular prolyl thioesters.18 Despite these improvements, methods that can improve efficiencies at other sterically demanding sites under mild reaction conditions are still of great importance, and call for further investigations.Despite being one of the most abundant amino acids in nature, connection sites in which valine is present are usually avoided in chemical ligations because of the sluggish reaction rate. Overcoming this constraint would afford more flexibility in choosing ligation sites for synthesizing intriguing peptides and proteins.19 Along this line, Seitz and co-workers have demonstrated that N-terminal penicillamine (Pen) could effectively mediate peptide ligation that eventually leads to an Xaa-Val linkage.20 While most of the developed methods accomplishing ligation at valine sites rely on a large excess of activating agents (Fig. 1a), we envisaged that the introduction of a C-terminal β-thiolactone moiety, possibly generated from a penicillamine-derived precursor in situ, might effect an increased reaction rate driven by the strain-releasing power of the reaction (Fig. 1b). To ensure the incorporation of the β-thio-valine structure in the peptides, the epimerization-free peptidyl-Nbz protocol developed by Dawson et al. could be applied, which would avoid extra modifications after SPPS.5
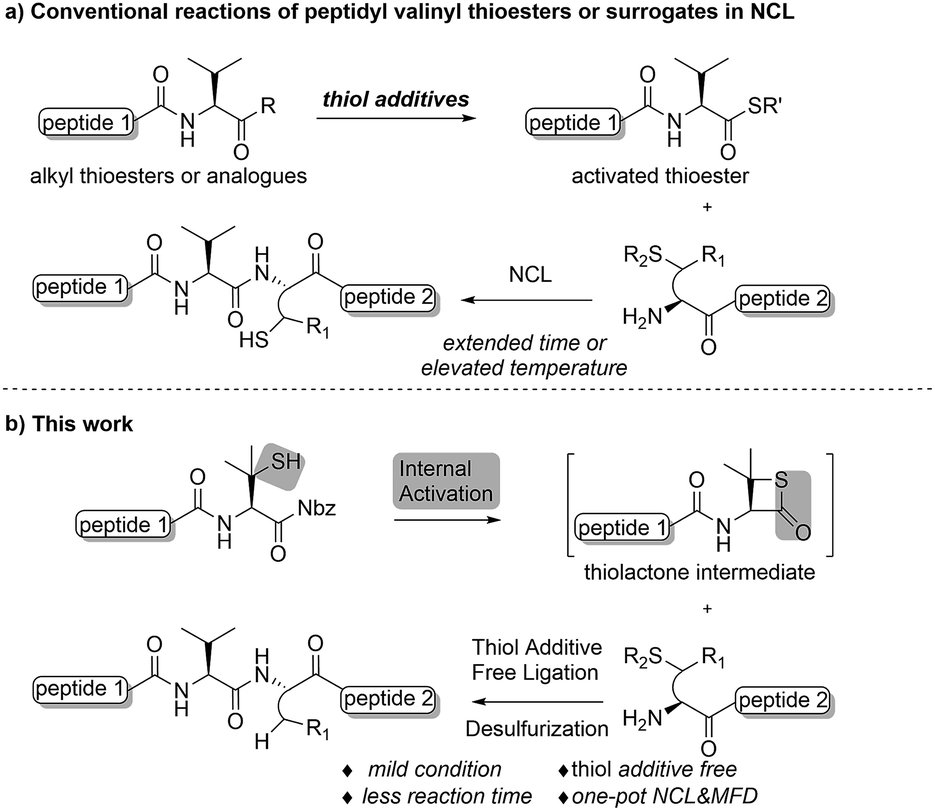 | ||
| Fig. 1 (a) Conventional reactions of peptidyl valinyl thioesters or surrogates versus (b) mercaptan-assisted internal activation of peptide valinyl-Nbz in NCL. | ||
Results and discussion
We commenced our investigation by synthesizing the Pen-containing peptide-Nbz (Scheme 1). From readily available penicillamine (1), N-Fmoc protected amino acid derivative 2 was synthesized in two steps. It was subsequently installed on the 3,4-diaminobenzoic acid (Dbz)-preloaded Rink-amide resin 3 under DIC/oxyma pure conditions. The resulting resin was further employed in Fmoc-based SPPS, followed by the conversion of Dbz to Nbz based on literature-reported procedures.5 After the standard resin cleavage and global deprotections using a TFA cocktail, purification of the crude peptide using preparative reverse-phase HPLC provided the desired peptidyl-Nbz. Separately, the right-side peptides containing cysteines or other thio-amino acid derivatives at the N-termini were synthesized after the Fmoc-based SPPS and deprotection procedures.21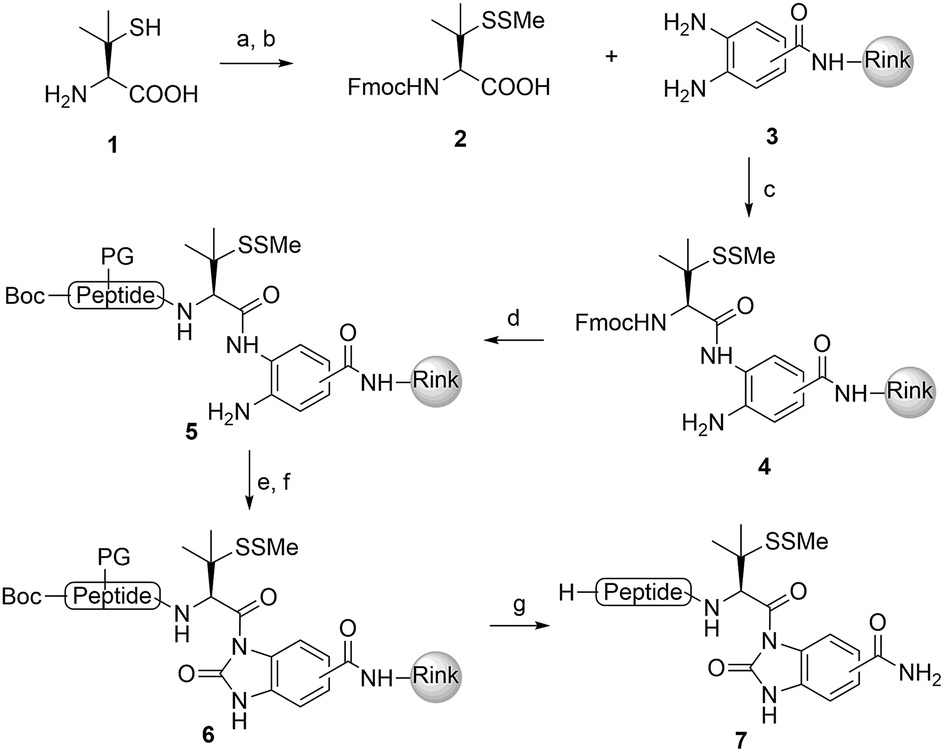 | ||
Scheme 1 Synthesis of peptidyl-Nbz. Reagents and conditions: (a) MMTS, EtOH, rt, 4 h; (b) Fmoc-Cl, TEA, CH2Cl2, rt, overnight; (c) 3, DIC, oxyma pure, DMF, 8 h; (d) Fmoc-SPPS; (e) p-nitrophenylchoroformate, DCM, 40 min to 1 h; (f) DIEA, DMF, 15 min; (g) TFA/TIS/H2O (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:2.5). DMF = N,N-dimethylformamide, DIEA = N,N-diisopropylethylamine, HATU = 1-[bis (dimethylamino)-methyl-ene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexa-fluorophosphate, DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene, DIC = N,N-diisopropylcarbodiimide, oxyma pure = ethyl cyanoglyoxylate-2-oxime, TFA = trifluoroacetic acid, TIS = triisopropylsilane, and PG = protecting groups. :2.5:2.5). DMF = N,N-dimethylformamide, DIEA = N,N-diisopropylethylamine, HATU = 1-[bis (dimethylamino)-methyl-ene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexa-fluorophosphate, DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene, DIC = N,N-diisopropylcarbodiimide, oxyma pure = ethyl cyanoglyoxylate-2-oxime, TFA = trifluoroacetic acid, TIS = triisopropylsilane, and PG = protecting groups. | ||
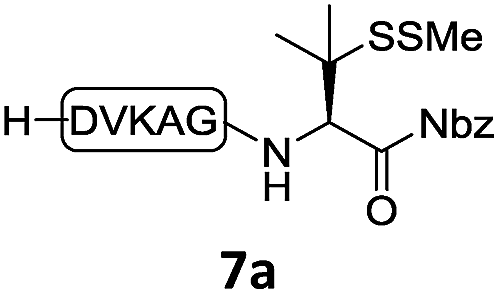
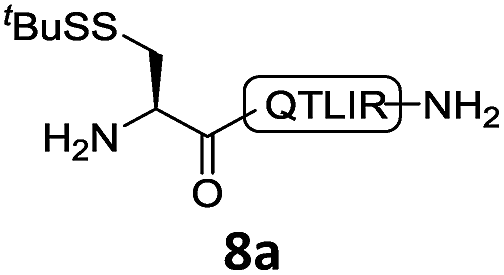
With the requisite peptide segments in hand, the ligation efficiency was evaluated using peptides H-DVKAGPen(SMe)-Nbz (7a, 1.0 equiv., Fig. 2) and H–C(StBu)QTLIR-NH2 (8a, 1.2 equiv.) under typical NCL reaction conditions at room temperature (6 M Gn·HCl, 200 mM Na2HPO4, 20 mM TCEP·HCl, pH 7.0). The reaction progress was monitored using HPLC-MS, and, as shown in Fig. 2a, peptidyl Pen-Nbz was efficiently transformed to product 9a with high conversion after two hours, along with a trace amount of desulfurized products 8a and 9a (Fig. 2c, curve A). These desulfurized products may be derived from a TCEP-mediated reaction, where a phosphoranyl radical-based mechanism is possibly involved.8b,22 Similar observations have also been reported previously in the cases of ligation reactions in TCEP-containing buffer without any radical scavengers.23 Such side reactions could be suppressed by the addition of sodium ascorbate, which requires an extra HPLC purification before the metal-free desulfurization step,16,23 thus leading to decreased overall efficiency in comparison to the additive-free one-pot ligation–desulfurization protocol. In contrast, when peptide segment H-DVKAGV-Nbz (10), lacking β-thiol substitution at the C-terminal valine, was subjected to the same reaction conditions, a significantly lower reactivity was observed (Fig. 2c, curve C).21 The addition of MPAA accelerated the ligation of peptidyl Val-Nbz (Fig. 2b and c), which is in accordance with a previous report,5 but required a longer time than that of the β-thio Val-Nbz derived peptide to achieve over 90% conversion (i.e. 8 h). These results indicate that the β-mercaptan group on the terminal valine residue drastically increases the reaction rate.
 | ||
| Fig. 2 Evaluation of ligation efficiency using Pen or Val-containing peptide-Nbz in thiol additive-free buffer or MPAA-containing buffer. (a) UV trace of ligation between 7a and 8a in buffer I (6 M Gn·HCl, 200 mM Na2HPO4, 20 mM TCEP·HCl, pH 7.0); (b) UV trace of ligation between 10 and 8a in buffer II (6 M Gn·HCl, 200 mM Na2HPO4, 200 mM MPAA, 20 mM TCEP·HCl, pH 7.0); (c) the reaction conversion as a function of time for the reactions between 7a/10 and 8a in different buffers. Gn = guanidine and TCEP = tris(2-carboxyethyl) phosphine. | ||
Since the proposed peptidyl thiolactone intermediate was not observed in the ligation between 7a and 8a, a series of control experiments were conducted to probe the penicillamine-Nbz-based reactions. When 7a alone was dissolved in ligation buffer and monitored using HPLC-MS (Fig. 3), a new peak was observed after 30 min showing a mass corresponding to the β-thiolactone structure 7a′ (Fig. 3b), which could be either converted to alkyl thioester 7a′′ using excess MESNa (Fig. 3c), or intercepted by cysteinyl peptide 8a to generate the ligated product 9a (Fig. 3d). These results suggest the likely existence of the β-thiolactone structure, and rule out other possible intermediates such as lactams derived from intramolecular cyclizations.
 | ||
| Fig. 3 Mechanistic investigations. (a) Mass trace from the HPLC-MS analysis of 7a dissolved in buffer I for 1 min; (b) mass trace of 7a dissolved in buffer I for 30 min; (c) mass trace of the reaction between the mixture generated from stirring 7a in buffer I for 30 min and 10 equiv. MESNa for 30 min; (d) mass trace of the reaction between the mixture generated from stirring 7a in buffer I for 30 min and peptide 8a for 2 h. | ||
Moreover, when two analogous peptides with either a terminal Cys-Nbz instead of Pen-Nbz (H-DVKAGC(StBu)-Nbz) or an internal Cys residue [H-DVKC(StBu)GV-Nbz] were subjected to the ligation reactions,16,21 an extremely low reactivity accompanied by extreme decomposition was observed (Fig. S24 and S25†). These findings further underscore the activating role played by the β-thiol at the C-terminal Val when compared to other internal activation methods using cysteines. Although our attempts to isolate the peptidyl thiolactone intermediate failed, due to severe decomposition during the HPLC purification and lyophilization of the sample, the experimental results suggest that the gem-dimethyl is crucial for the success of Pen-Nbz-based ligation under the reaction conditions, where the Thorpe-Ingold effect24 can stabilize the in situ-formed reactive four-membered-ring intermediate. Furthermore, the strain releasing power may accelerate the transthioesterification that is usually the rate-limiting step in cases of ligations at sterically hindered amino acid sites, accounting for the high reactivity observed. In another experiment, when N-terminal alanyl peptide H-AQTLIR-NH2 was subjected to ligation with 8a,21 only a trace amount of the ligated product was generated (Fig. S26†), indicating that the reaction of Pen-Nbz likely proceeds through an NCL-like process instead of direct aminolysis.
In the absence of thiol additives, the ligation–desulfurization reaction of C-terminal Pen-Nbz-derived peptides can be conducted in a one-pot manner.15a,25 Several representative peptide-Nbz sequences containing different natural amino acids were subjected to the optimized conditions (Table 1), where the reactions with peptides containing N-cysteinyl peptides proceeded smoothly, affording the desired products in decent isolated yields over two steps (entries 1–3). Glycopeptide 8d bearing an N-linked chitobiose was also found to be compatible under these conditions (entry 4). While the peptide-containing N-terminal β-thiol-Asp provided satisfactory reaction efficiency (entry 5),26 the ones with N-terminal thiol-valine derivatives displayed a much decreased reaction rate (entries 6 and 7),20,27 presumably due to the sterical constraints resulting from the consecutive Val–Val residues. It was notable that during our attempted desulfurization of thio-Asp-containing peptides using VA-044 as the radical initiator, significant amounts of side products were generated,25b which could be eliminated by using ACVA instead.28 Further attempts to react 7a with N-terminal thio-proline-containing peptide failed to provide isolable ligation products,21 indicating that the activated penicillamine derivative was still unable to overcome the extreme steric effects between valine and proline.29 Moreover, a longer sequence, the 56 amino acid peptide γ-lipotropin, could also be efficiently prepared using this two-step one-pot protocol (entry 8).
|
|
|||
|---|---|---|---|
| Entry | Left-side peptide | Right-side peptide | Isolated yield (%) |
| a Reaction conditions: 7 (3 mM), 1.2 eq. 8, NCL buffer (6 M Gn·HCl, 200 mM Na2HPO4, 20 mM TCEP·HCl, pH 7.0), 25 °C, 2 h, then bond-breaker (0.5 M TCEP), 20 μL tBuSH, 100 μL VA-044 (0.1 M in H2O), 37 °C, 3 h. b ACVA used in the desulfurization reaction instead of VA-044, 5 h. c Reaction conditions: 7h (5 mM), 1.2 eq. 8h, 200 μL NCL buffer (6 M Gn·HCl, 200 mM Na2HPO4, 50 mM TCEP·HCl, pH 6.9), 25 °C, 8 h, then 200 μL bond-breaker (0.5 M TCEP), 20 μL tBuSH, 100 μL VA-044 (0.1 M in H2O), 37 °C, 8 h. ACVA = 4,4′-Azobis(4-cyanovaleric acid) and VA-044 = 2,2′-Azobis[2-(2-imidazolin-2-yl) propane] dihydrochloride. | |||
| 1 |
|
|
12a, 65% |
| 2 |

|

|
12b, 62% |
| 3 |
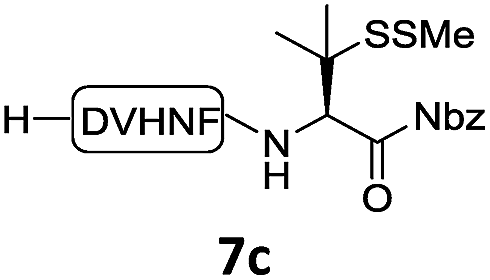
|
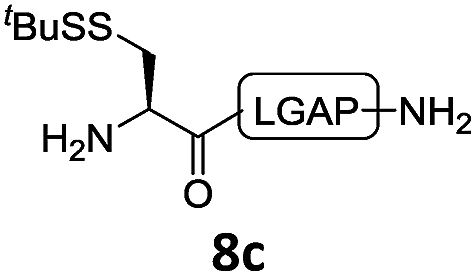
|
12c, 50% |
| 4 |

|

|
12d, 60% |
| 5b |

|
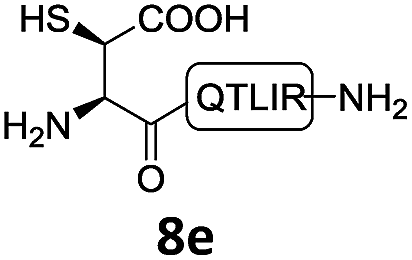
|
12e, 50% |
| 6 |
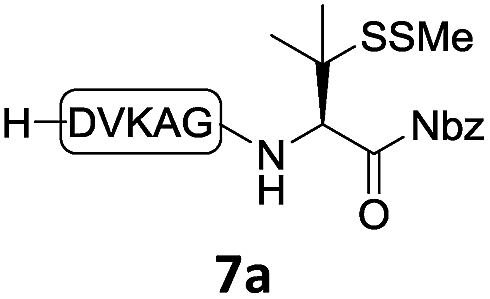
|

|
12f, 34% |
| 7 |

|

|
12f, 18% |
| 8c |
|

|
12h, 55% |

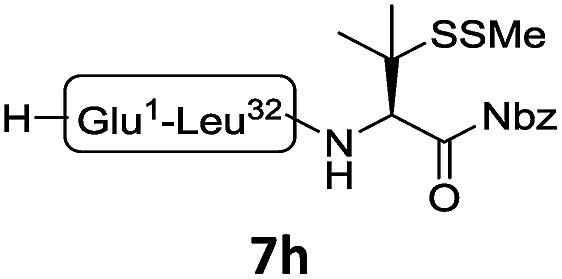
The internal thiol-assisted activation of Val-Nbz-derived peptides has also enabled the utilization of a kinetically controlled ligation (KCL) strategy in the synthesis of longer sequences,15a,30 where a peptidyl alkyl thioester would be unaffected in the thiol additive-free buffer, but could be activated later by an MPAA additive when needed. To validate the applicability of this strategy, we embarked upon the synthesis of β-lipotropin (β-LPH), a human peptidic hormone released from proopiomelanocortin (aka. POMC). β-LPH contains 89 amino acids (Fig. 4a), and can be endogenously cleaved to smaller peptidic hormones, including γ-LPH (Table 1, entry 8), β-MSH, and β-endorphin.31 Examination of the sequence revealed the absence of cysteines and the existence of two valine and two methionine residues, such that the full sequence could be assembled from three segments in a one-pot manner. The forward synthesis was first attempted by choosing Val33-Ala34 and Lys54-Asp55 as the ligation sites. To avoid possible oxidative degradation and enhance peptide stability, the methionines (Met45 and Met63) were replaced with norleucine (Nle) residues, an approach which has been suggested to mitigate oxidative degradation of peptides while maintaining their biological activity.32 Unfortunately, the product of the first ligation degraded rapidly in the MPAA buffer, and failed to connect with the third segment Lys54-Asp55. As we did not observe any lactamization or other obvious side products derived from the activated thioesters, the experimental result underscores the possible instability of peptide segments in the presence of a thiol additive under denaturing conditions, where more optional ligation methods and connection sites would be beneficial for identifying suitable synthetic routes leading to the desired target peptide.33 Alternatively, when the second ligation segment was extended to Leu72 (Fig. 4b), the corresponding ligation product 14 showed significantly improved stability, and was successfully ligated with peptide 15b containing a γ-thiovaline derivative prepared following a palladium-catalyzed γ-C(sp3)–H acetoxylation protocol.34 It is notable that the N-terminal penicillamine-derived peptide 15a was unreactive even at the elevated temperature of 40 °C. After the purification of full length β-LPH bearing three extraneous thiol groups using preparative HPLC to remove both sodium ascorbate and MPAA simultaneously, the subsequent desulfurization proceeded smoothly to generate [Nle45,63]β-LPH (17) in 31% yield over three steps and two HPLC purifications. In a similar manner, [Nle8,18]hPTH (23), an 84-mer protein analogue of human parathyroid hormone (hPTH, Fig. 5a), was successfully assembled with good efficiency and with fewer operational steps in comparison to the previous report,35 featuring a C-terminal β-thiovaline-Nbz ligation followed by an N-terminal γ-thiovaline ligation in the same flask (Fig. 5b). This further demonstrates the applicability of the KCL strategy enabled by the penicillamine-Nbz-derived peptides.
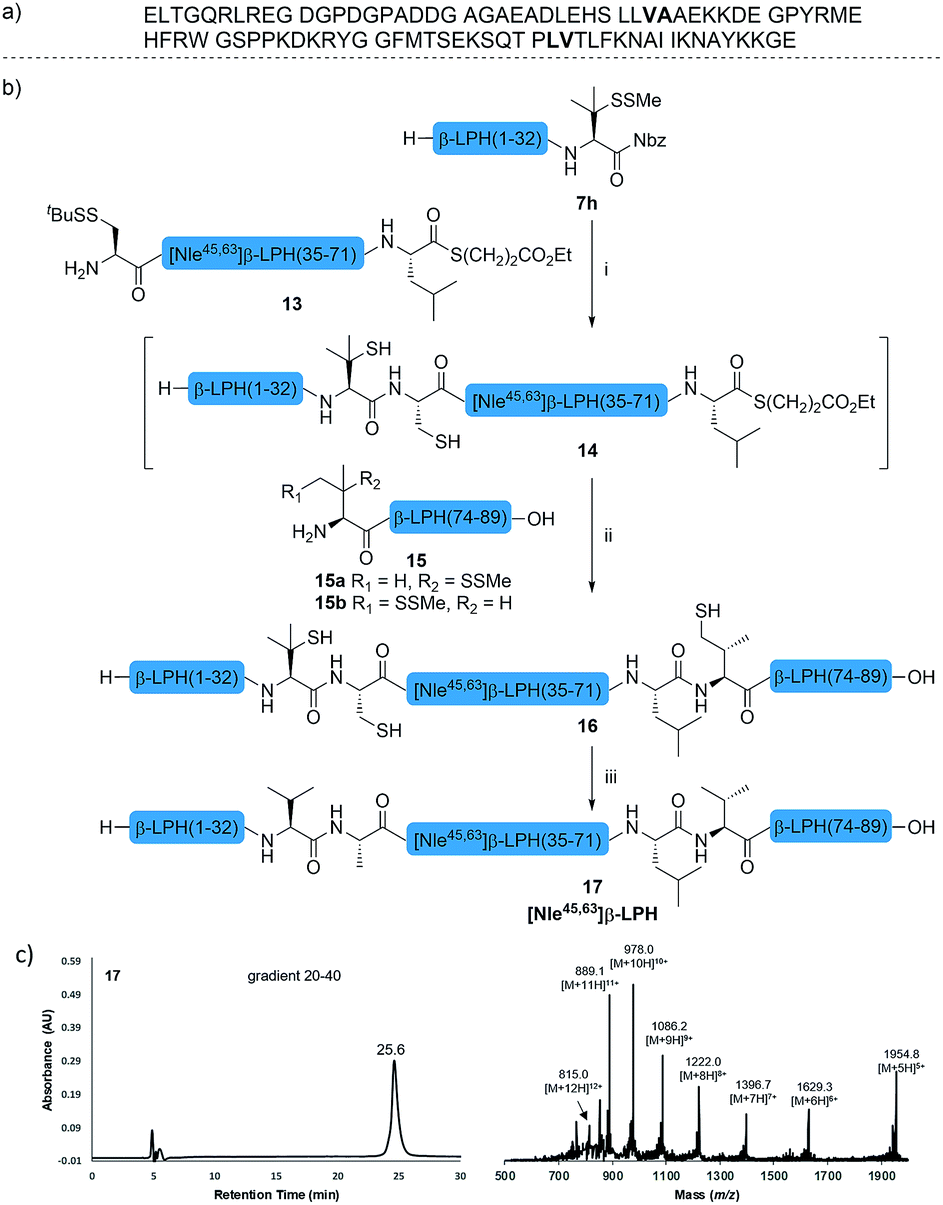 | ||
| Fig. 4 (a) Native sequence of β-LPH; (b) chemical synthesis of [Nle45,63] β-LPH. Reagents and conditions: (i) 6 M Gn·HCl, 100 mM Na2HPO4, 100 mM sodium ascorbate, 50 mM TCEP·HCl, pH 7.2, rt, 10 h; (ii) 6 M Gn·HCl, 200 mM Na2HPO4, 200 mM MPAA, 20 mM TCEP·HCl, rt, pH 7.0, 4 h. (iii) 6 M Gn·HCl, 200 mM Na2HPO4, pH 7.0, TCEP, VA-044, tBuSH, 37 °C, 8 h, 31% yield over 3 steps. (c) HPLC trace and MS data of [Nle45,63] β-LPH. | ||
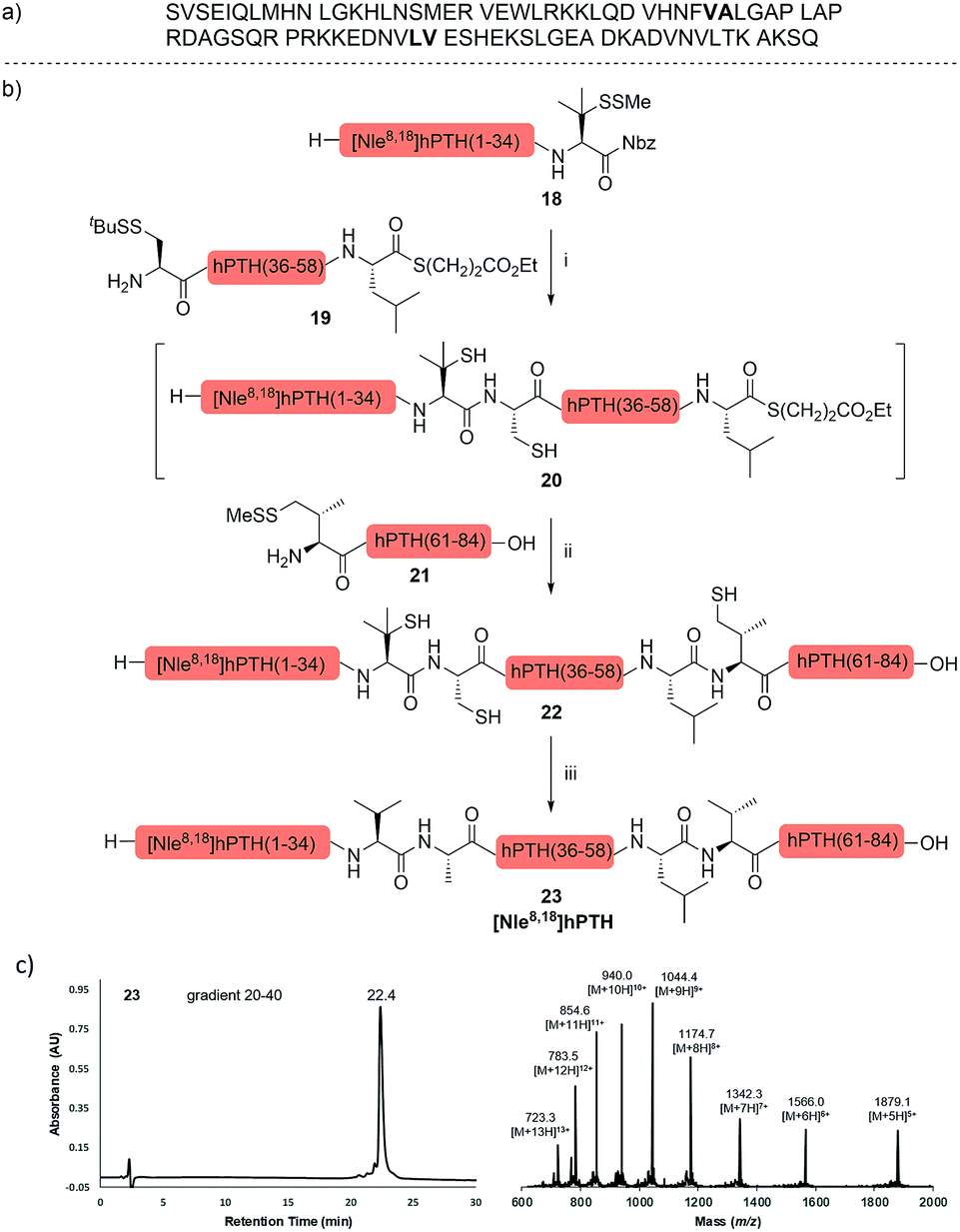 | ||
| Fig. 5 (a) Native sequence of hPTH; (b) chemical synthesis of [Nle8,18] hPTH. Reagents and conditions: (i) 6 M Gn·HCl, 100 mM Na2HPO4, 100 mM sodium ascorbate, 50 mM TCEP·HCl, pH 7.5, rt, 10 h; (ii) 6 M Gn·HCl, 200 mM Na2HPO4, 200 mM MPAA, 20 mM TCEP·HCl, rt, pH 7.2, 10 h. (iii) 6 M Gn·HCl, 200 mM Na2HPO4, pH 7.0, TCEP, VA-044, tBuSH, 37 °C, 5 h, 35% yield over 3 steps. (c) HPLC trace and MS data of [Nle8,18] hPTH. | ||
Conclusions
In summary, we have developed a traceless ligation protocol at sterically demanding C-terminal valine sites, featuring an appropriately designed internal activation step mediated by a β-mercaptan substitution on the corresponding valine residue. In conjunction with the straightforward preparation of peptidyl penicillamine-Nbz segments, this effective thiol additive-free reaction ensured that the tandem desulfurization occurred in the same flask. This illustrates the compatibility of internal activation with alkyl thioesters, which is of practical use in the three-segment one-pot assembly of large polypeptides and proteins. Further studies on the mechanistic aspects of C-terminal penicillamine-based ligation, as well as the extension of the strain-releasing activation strategy in peptide ligation at other difficult sites, are currently in progress and will be reported in due course.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors are grateful for financial support from the National Natural Science Foundation of China (21502005 to S. D.), the National Recruitment Program of Global Youth Experts (1000 Plan to S. D.), the State Key Laboratory of Natural and Biomimetic Drugs, and Peking University Health Science Center (BMU20130354 to S. D.). Q. Zhang thanks the National Science Foundation (CHE 1710174), the University at Albany-SUNY, and the Research Foundation of SUNY for financial support. Thanks are also extended to Prof. Rabi Musah (SUNY Albany) for helpful suggestions, and to Dr Yuan Wang, Weiqing Zhang, and Xulin Sun (Peking University) for assistance with spectroscopic measurements.Notes and references
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CAS.
- For a recent review, see: (a) S. Bondalapati, M. Jbara and A. Brik, Nat. Chem., 2016, 8, 407–418 CrossRef CAS PubMed; For representative examples, see: (b) P. Wang, S. Dong, J. H. Shieh, E. Peguero, R. Hendrickson, M. A. S. Moore and S. J. Danishefsky, Science, 2013, 342, 1357–1360 CrossRef CAS PubMed; (c) A. Reif, S. Siebenhaar, A. Troster, M. Schmalzlein, C. Lechner, P. Velisetty, K. Gottwald, C. Pohner, I. Boos, V. Schubert, S. Rose-John and C. Unverzagt, Angew. Chem., Int. Ed., 2014, 53, 12125–12131 CrossRef CAS PubMed; (d) R. D. Bunker, K. Mandal, G. Bashiri, J. J. Chaston, B. L. Pentelute, J. S. Lott, S. B. Kent and E. N. Baker, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4310–4315 CrossRef CAS PubMed; (e) Z. Wang, W. Xu, L. Liu and T. F. Zhu, Nat. Chem., 2016, 8, 698–704 CrossRef CAS PubMed; (f) M. Murakami, T. Kiuchi, M. Nishihara, K. Tezuka, R. Okamoto, M. Izumi and Y. Kajihara, Sci. Adv., 2016, 2, e1500678 Search PubMed.
- For representative examples, see: (a) J.-S. Zheng, S. Tang, Y.-C. Huang and L. Liu, Acc. Chem. Res., 2013, 46, 2475–2484 CrossRef CAS PubMed; (b) T. Kawakami, in Protein Ligation and Total Synthesis I, ed. L. Liu, Springer International Publishing, Cham, 2015, pp. 107–135, DOI:10.1007/128_2014_575; (c) H. Li and S. Dong, Sci. China: Chem., 2016, 60, 201–213 CrossRef.
- (a) G.-M. Fang, Y.-M. Li, F. Shen, Y.-C. Huang, J.-B. Li, Y. Lin, H.-K. Cui and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 7645–7649 CrossRef CAS PubMed; (b) G.-M. Fang, J.-X. Wang and L. Liu, Angew. Chem., Int. Ed., 2012, 51, 10347–10350 CrossRef CAS PubMed.
- J. B. Blanco-Canosa and P. E. Dawson, Angew. Chem., Int. Ed., 2008, 47, 6851–6855 CrossRef CAS PubMed.
- (a) T. Durek and P. F. Alewood, Angew. Chem., Int. Ed., 2011, 50, 12042–12045 CrossRef CAS PubMed; (b) N. J. Mitchell, L. R. Malins, X. Liu, R. E. Thompson, B. Chan, L. Radom and R. J. Payne, J. Am. Chem. Soc., 2015, 137, 14011–14014 CrossRef CAS PubMed; (c) M. Raj, H. Wu, S. L. Blosser, M. A. Vittoria and P. S. Arora, J. Am. Chem. Soc., 2015, 137, 6932–6940 CrossRef CAS PubMed.
- For representative approaches, see: (a) J. S. Zheng, H. N. Chang, F. L. Wang and L. Liu, J. Am. Chem. Soc., 2011, 133, 11080–11083 CrossRef CAS PubMed; (b) W. Hou, X. Zhang, F. Li and C. F. Liu, Org. Lett., 2011, 13, 386–389 CrossRef CAS PubMed; (c) N. Ollivier, J. Vicogne, A. Vallin, H. Drobecq, R. Desmet, O. El Mahdi, B. Leclercq, G. Goormachtigh, V. Fafeur and O. Melnyk, Angew. Chem., Int. Ed., 2012, 51, 209–213 CrossRef CAS PubMed; (d) R. Okamoto, K. Morooka and Y. Kajihara, Angew. Chem., Int. Ed., 2012, 51, 191–196 CrossRef CAS PubMed; (e) F. Burlina, G. Papageorgiou, C. Morris, P. D. White and J. Offer, Chem. Sci., 2014, 5, 766–770 RSC; (f) S. Tsuda, M. Mochizuki, K. Sakamoto, M. Denda, H. Nishio, A. Otaka and T. Yoshiya, Org. Lett., 2016, 18, 5940–5943 CrossRef CAS PubMed; (g) V. P. Terrier, H. Adihou, M. Arnould, A. F. Delmas and V. Aucagne, Chem. Sci., 2016, 7, 339–345 RSC; (h) L. Raibaut, M. Cargoet, N. Ollivier, Y. M. Chang, H. Drobecq, E. Boll, R. Desmet, J. C. M. Monbaliu and O. Melnyk, Chem. Sci., 2016, 7, 2657–2665 RSC; (i) M. Eto, N. Naruse, K. Morimoto, K. Yamaoka, K. Sato, K. Tsuji, T. Inokuma, A. Shigenaga and A. Otaka, Org. Lett., 2016, 18, 4416–4419 CrossRef CAS PubMed; (j) P. M. M. Shelton, C. E. Weller and C. Chatterjee, J. Am. Chem. Soc., 2017, 139, 3946–3949 CrossRef CAS PubMed; (k) C. Rao and C.-F. Liu, Org. Biomol. Chem., 2017, 15, 2491–2496 RSC; (l) Y. Asahina, T. Kawakami and H. Hojo, Chem. Commun., 2017, 53, 2114–2117 RSC.
- (a) L. Z. Yan and P. E. Dawson, J. Am. Chem. Soc., 2001, 123, 526–533 CrossRef CAS PubMed; (b) Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef CAS PubMed; (c) D. Crich and A. Banerjee, J. Am. Chem. Soc., 2007, 129, 10064–10065 CrossRef CAS PubMed.
- B. L. Nilsson, L. L. Kiessling and R. T. Raines, Org. Lett., 2000, 2, 1939–1941 CrossRef CAS PubMed.
- J. W. Bode, R. M. Fox and K. D. Baucom, Angew. Chem., Int. Ed., 2006, 45, 1248–1252 CrossRef CAS PubMed.
- (a) R. J. Hondal, B. L. Nilsson and R. T. Raines, J. Am. Chem. Soc., 2001, 123, 5140–5141 CrossRef CAS PubMed; (b) M. D. Gieselman, L. Xie and W. A. van der Donk, Org. Lett., 2001, 3, 1331–1334 CrossRef CAS PubMed; (c) N. Metanis, E. Keinan and P. E. Dawson, Angew. Chem., Int. Ed., 2010, 49, 7049–7053 CrossRef CAS PubMed; (d) S. D. Townsend, Z. Tan, S. Dong, S. Shang, J. A. Brailsford and S. J. Danishefsky, J. Am. Chem. Soc., 2012, 134, 3912–3916 CrossRef CAS PubMed; (e) L. R. Malins, N. J. Mitchell, S. McGowan and R. J. Payne, Angew. Chem., Int. Ed., 2015, 54, 12716–12721 CrossRef CAS PubMed; (f) X. Wang, J. Sanchez, M. J. Stone and R. J. Payne, Angew. Chem., Int. Ed., 2017, 56, 8490–8494 CrossRef CAS PubMed.
- (a) X. Li, H. Y. Lam, Y. Zhang and C. K. Chan, Org. Lett., 2010, 12, 1724–1727 CrossRef CAS PubMed; (b) C. L. Lee and X. Li, Curr. Opin. Chem. Biol., 2014, 22, 108–114 CrossRef CAS PubMed.
- T. M. Hackeng, J. H. Griffin and P. D. Dawson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10068–10073 CrossRef CAS.
- E. C. Johnson and S. B. Kent, J. Am. Chem. Soc., 2006, 128, 6640–6646 CrossRef CAS PubMed.
- (a) R. E. Thompson, X. Liu, N. Alonso-García, P. J. B. Pereira, K. A. Jolliffe and R. J. Payne, J. Am. Chem. Soc., 2014, 136, 8161–8164 CrossRef CAS PubMed; (b) Y. C. Huang, C. C. Chen, S. Gao, Y. H. Wang, H. Xiao, F. Wang, C. L. Tian and Y. M. Li, Chem.–Eur. J., 2016, 22, 7623–7628 CrossRef CAS PubMed; (c) K. Sakamoto, S. Tsuda, M. Mochizuki, Y. Nohara, H. Nishio and T. Yoshiya, Chem.–Eur. J., 2016, 22, 17940–17944 CrossRef CAS PubMed.
- (a) S. Tsuda, T. Yoshiya, M. Mochizuki and Y. Nishiuchi, Org. Lett., 2015, 17, 1806–1809 CrossRef CAS PubMed; (b) S. Tsuda, M. Mochizuki, H. Nishio and T. Yoshiya, ChemBioChem, 2016, 17, 2133–2136 CrossRef CAS PubMed.
- Y. Gui, L. Qiu, Y. Li, H. Li and S. Dong, J. Am. Chem. Soc., 2016, 138, 4890–4899 CrossRef CAS PubMed.
- M. I. Ali Shah, Z.-Y. Xu, L. Liu, Y.-Y. Jiang and J. Shi, RSC Adv., 2016, 6, 68312–68321 RSC.
- (a) L. A. Marcaurelle, L. S. Mizoue, J. Wilken, L. Oldham, S. B. Kent, T. M. Handel and C. R. Bertozzi, Chem.–Eur. J., 2001, 7, 1129–1132 CrossRef CAS PubMed; (b) A. A. Vinogradov, E. D. Evans and B. L. Pentelute, Chem. Sci., 2015, 6, 2997–3002 RSC.
- C. Haase, H. Rohde and O. Seitz, Angew. Chem., Int. Ed., 2008, 47, 6807–6810 CrossRef CAS PubMed.
- See the ESI† for complete procedures and details.
- (a) F. W. Hoffmann, T. R. Moore and B. Kagan, J. Am. Chem. Soc., 1956, 78, 6414 CrossRef CAS; (b) C. Walling and R. Rabinowitz, J. Am. Chem. Soc., 1957, 79, 5326 CrossRef CAS; (c) C. Walling, O. H. Basedow and E. S. Savas, J. Am. Chem. Soc., 1960, 82, 2181–2184 CrossRef CAS; (d) Z. Wang, T. Rejtar, Z. S. Zhou and B. L. Karger, Rapid Commun. Mass Spectrom., 2010, 24, 267–275 CrossRef CAS PubMed.
- H. Rohde, J. Schmalisch, Z. Harpaz, F. Diezmann and O. Seitz, ChemBioChem, 2011, 12, 1396–1400 CrossRef CAS PubMed.
- (a) R. M. Beesley, C. K. Ingold and J. F. Thorpe, J. Chem. Soc., Trans., 1915, 107, 1080–1106 RSC; (b) A. Noel, B. Delpech and D. Crich, J. Sulfur Chem., 2013, 34, 104–141 CrossRef CAS.
- (a) T. Moyal, H. P. Hemantha, P. Siman, M. Refua and A. Brik, Chem. Sci., 2013, 4, 2496–2501 RSC; (b) K. M. Cergol, R. E. Thompson, L. R. Malins, P. Turner and R. J. Payne, Org. Lett., 2014, 16, 290–293 CrossRef CAS PubMed.
- (a) X. Guan, M. R. Drake and Z. Tan, Org. Lett., 2013, 15, 6128–6131 CrossRef CAS PubMed; (b) R. E. Thompson, B. Chan, L. Radom, K. A. Jolliffe and R. J. Payne, Angew. Chem., Int. Ed., 2013, 52, 9723–9727 CrossRef CAS PubMed.
- J. Chen, Q. Wan, Y. Yuan, J. Zhu and S. J. Danishefsky, Angew. Chem., Int. Ed., 2008, 47, 8521–8524 CrossRef CAS PubMed.
- L. Markey, S. Giordani and E. M. Scanlan, J. Org. Chem., 2013, 78, 4270–4277 CrossRef CAS PubMed.
- S. Shang, Z. Tan, S. Dong and S. J. Danishefsky, J. Am. Chem. Soc., 2011, 133, 10784–10786 CrossRef CAS PubMed.
- For representative examples using the KCL strategy, see: (a) D. Bang, B. L. Pentelute and S. B. H. Kent, Angew. Chem., Int. Ed., 2006, 45, 3985–3988 CrossRef CAS PubMed; (b) Z. Tan, S. Shang and S. J. Danishefsky, Angew. Chem., Int. Ed., 2010, 49, 9500–9503 CrossRef CAS PubMed; (c) J.-S. Zheng, H.-K. Cui, G.-M. Fang, W.-X. Xi and L. Liu, ChemBioChem, 2010, 11, 511–515 CrossRef CAS PubMed; (d) T. Nakamura, A. Shigenaga, K. Sato, Y. Tsuda, K. Sakamoto and A. Otaka, Chem. Commun., 2014, 50, 58–60 RSC; (e) S. Tsuda, M. Mochizuki, H. Nishio, T. Yoshiya and Y. Nishiuchi, Biopolymers, 2016, 106, 503–511 CrossRef CAS PubMed; (f) E. Boll, H. Drobecq, E. Lissy, F.-X. Cantrelle and O. Melnyk, Org. Lett., 2016, 18, 3842–3845 CrossRef CAS PubMed.
- (a) J. Spiess, C. D. Mount, W. E. Nicholson and D. N. Orth, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 5071–5075 CrossRef CAS PubMed; (b) N. X. Cawley, Z. Li and Y. P. Loh, J. Mol. Endocrinol., 2016, 56, T77–T97 CrossRef CAS PubMed.
- M. Rosenblatt, D. Goltzman, H. T. Keutmann, G. W. Tregear and J. T. Potts, J. Biol. Chem., 1976, 251, 159–164 CAS.
- (a) H. M. Burke, L. McSweeney and E. M. Scanlan, Nat. Commun., 2017, 8, 15655 CrossRef PubMed; (b) L. Liu, Protein Ligation and Total Synthesis I, Springer International, Switzerland, 2015 Search PubMed.
- K. K. Pasunooti, R. Yang, B. Banerjee, T. Yap and C.-F. Liu, Org. Lett., 2016, 18, 2696–2699 CrossRef CAS PubMed.
- S. Dong, S. Shang, J. Li, Z. Tan, T. Dean, A. Maeda, T. J. Gardella and S. J. Danishefsky, J. Am. Chem. Soc., 2012, 134, 15122–15129 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04148a |
| This journal is © The Royal Society of Chemistry 2018 |