
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A three-in-one-bullet for oesophageal cancer: replication fork collapse, spindle attachment failure and enhanced radiosensitivity generated by a ruthenium(II) metallo-intercalator†
Martin R.
Gill
 *a,
Paul J.
Jarman
bc,
Swagata
Halder
a,
Michael G.
Walker
b,
Hiwa K.
Saeed
b,
Jim A.
Thomas
b,
Carl
Smythe
c,
Kristijan
Ramadan
a and
Katherine A.
Vallis
*a
*a,
Paul J.
Jarman
bc,
Swagata
Halder
a,
Michael G.
Walker
b,
Hiwa K.
Saeed
b,
Jim A.
Thomas
b,
Carl
Smythe
c,
Kristijan
Ramadan
a and
Katherine A.
Vallis
*a
aCRUK/MRC Oxford Institute for Radiation Oncology, Department of Oncology, University of Oxford, Oxford, UK. E-mail: martin.gill@oncology.ox.ac.uk; katherine.vallis@oncology.ox.ac.uk
bDepartment of Chemistry, University of Sheffield, Sheffield, UK
cDepartment of Biomedical Science, University of Sheffield, Sheffield, UK
First published on 16th November 2017
Abstract
Substitutionally inert ruthenium(II) polypyridyl complexes have been developed as DNA intercalating agents yet cellular DNA damage responses to this binding modality are largely unexplored. Here, we show the nuclear-targeting complex [Ru(phen)2(tpphz)]2+ (phen = 1,10-phenanthroline, tpphz = tetrapyridophenazine) generates rapid and pronounced stalling of replication fork progression in p53-deficient human oesophageal cancer cells. In response, replication stress and double-strand break (DSB) DNA damage response (DDR) pathways are activated and cell proliferation is inhibited by growth arrest. Moreover, mitotic progression is compromised by [Ru(phen)2(tpphz)]2+, where the generation of metaphase chromosome spindle attachment failure results in spindle assembly checkpoint (SAC) activation. This dual mechanism of action results in preferential growth inhibition of rapidly-proliferating oesophageal cancer cells with elevated mitotic indices. In addition to these single-agent effects, [Ru(phen)2(tpphz)]2+ functions as a radiosensitizer with efficiency comparable to cisplatin, which occurs through a synergistic enhancement of DNA damage. These results establish that DNA replication is the target for [Ru(phen)2(tpphz)]2+ and provide the first experimental evidence that ruthenium-based intercalation targets multiple genome integrity pathways in cancer cells, thereby achieving enhanced selectivity compared to existing DNA-damaging agents such as cisplatin.
Introduction
Small molecules that interfere with DNA replication are widely-used anti-cancer drugs and are often employed in combination therapy alongside ionising radiation (IR) to treat cancer.1,2 One example of a clinical radiosensitizer is the platinum drug cisplatin, which generates both inter and intra-strand Pt–DNA cross-links and double-strand breaks (DSBs) that slow cell-cycle progression through S-phase, exacerbating IR-induced DNA damage.3,4 Although cisplatin-based chemoradiotherapy is highly effective in many cases, oesophageal cancers are marked by poor 5-year survival rates, typically <20%.5 Cisplatin is associated with nephrotoxicity which limits dose escalation and attempts to improve the outcome of patients with oesophageal cancer using alternative DNA-damaging chemotherapy such as doxorubicin have been unsuccessful.6 Application of these potent cytotoxic agents has also been hampered by the fact that the majority (77%) of oesophageal cancers lack p53 function7 and therefore possess a reduced capacity to activate apoptotic pathways in response to significant DNA damage.8 Thus, less toxic compounds that operate by alternative mechanisms of action and can also function as radiosensitizers are required.As typified by [Ru(bpy)2(dppz)]2+ (bpy = 2,2-bipyridine, dppz = dipyridophenazine),9 substitutionally inert ruthenium(II) polypyridyl complexes (RPCs) that interact with DNA solely via intercalation have been developed as site- and structure-specific luminescent DNA binding agents.9,10 Recent X-ray crystal structures have provided molecular insight into RPC metallo-intercalation in unprecedented detail.11–16 This includes evidence that [Ru(phen)2(dppz)]2+ (phen = 1,10-phenanthroline) adopts multiple intercalative geometries12 and Ru(dppz) semi-intercalation induces marked kinking of duplex DNA;11 a structural distortion similar to that observed following platination.17 Based on these studies with isolated DNA, the cellular uptake and anti-cancer properties of RPCs have become of increasing interest.18–20 However, despite the large number of DNA-interactive RPCs that have now been screened for anti-cancer activity,21 few have unequivocally been shown to have genomic DNA as their pharmacological target.22–24 As a result, cellular DNA damage responses to lesions generated by RPC mono-intercalation are completely unknown; a considerable barrier to therapeutic development of this class of DNA-binding agent. Moreover, while RPCs have generated much interest as photosensitizers for photodynamic therapy (PDT),25–27 studies of RPCs in combination with IR have been rare.23,28,29 This is surprising as radiotherapy is a mainstay of cancer medicine: high-energy X-rays or targeted radionuclide therapeutics have a far greater depth of penetration in tissue than visible light and radiotherapy is therefore capable of targeting a wider range of cancers.2,30
[Ru(phen)2(tpphz)]2+ (tpphz = tetrapyrido[3,2-a:2′,3′-c:3′′,2′′-h:2′′′,3′′′-j]phenazine), Ru1 (Fig. 1a), is a water-soluble hydrophilic mono-intercalator (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P = −1.24, DNA Kb = 3 × 105 M−1) that shows in vitro anti-cancer activity31 and toxicity has been established in murine models.32 Here, we present a detailed characterisation of the cellular response to DNA damage generated by Ru1 in p53-deficient oesophageal cancer cells and explore the complex in combination with IR.
P = −1.24, DNA Kb = 3 × 105 M−1) that shows in vitro anti-cancer activity31 and toxicity has been established in murine models.32 Here, we present a detailed characterisation of the cellular response to DNA damage generated by Ru1 in p53-deficient oesophageal cancer cells and explore the complex in combination with IR.
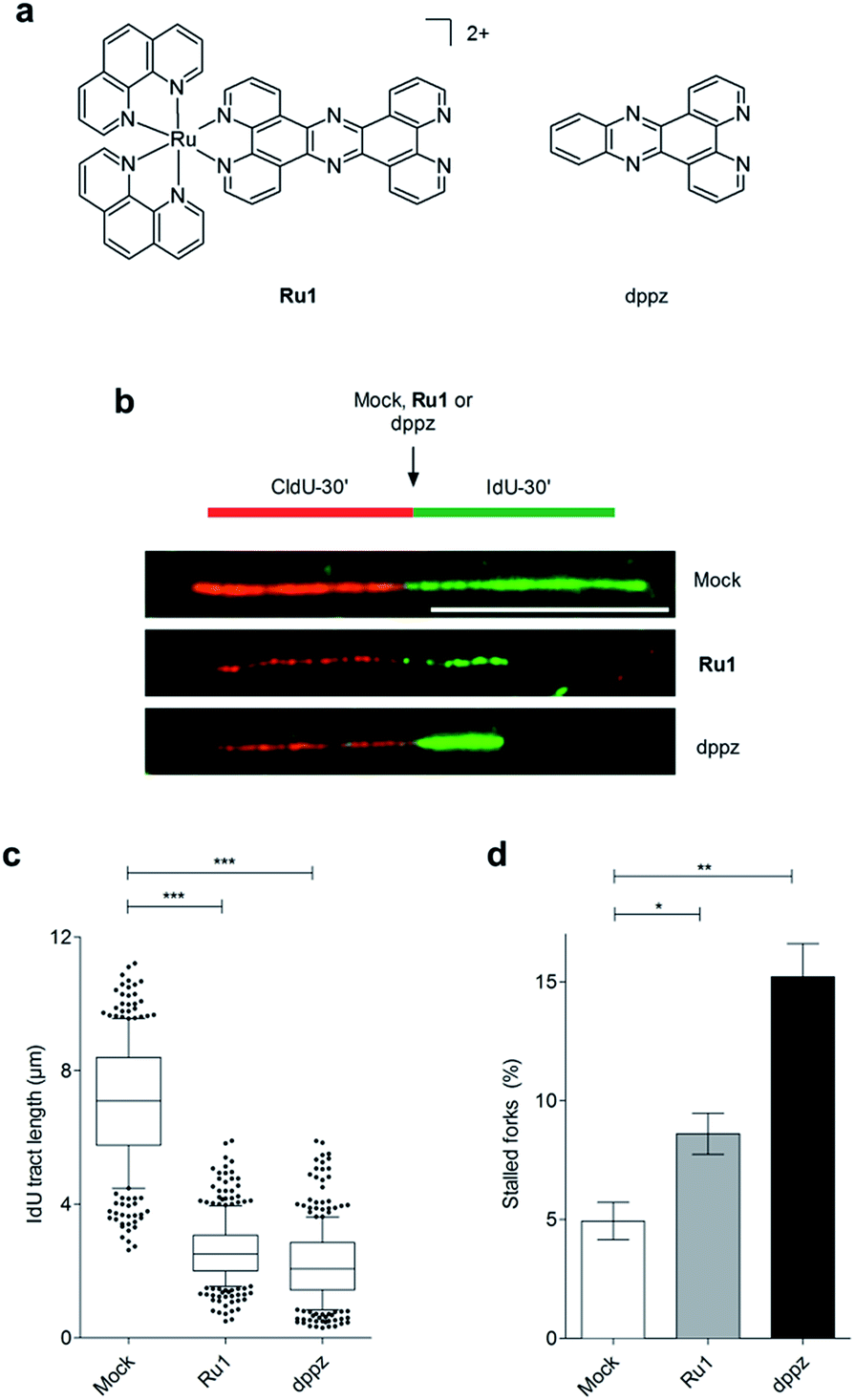 | ||
| Fig. 1 (a) Structures of Ru1 and dppz. Ru1 was used as the chloride salt and as a mixture of Λ and Δ enantiomers. (b) Representative images of DNA replication fibres in OE21 oesophageal cancer cells. CldU (first pulse, 30 min) incorporation is shown in red, incorporated IdU (second pulse, 30 min) in green. The second nucleotide (IdU) was either mock-treated or incubated in the presence of Ru1 or dppz to determine impact on DNA replication fork progression. Scale bar = 10 μm. (c) Quantification of IdU-labelled tract length in the presence of Ru1 (21 μM) or dppz (7 μM). Tract length was determined for >100 forks per condition. Whisker box plots show mean values and data within the 10–90 percentile. (d) Quantification of completely stalled forks (i.e. CldU tract only) for Ru1 or dppz treatment as in (b). Fork stalling was quantified for >300 forks per condition and experiment. The experiment was repeated three times. Data were analysed using unpaired two-tailed t test (*P < 0.1, **P < 0.01, ***P < 0.005). | ||
Results and discussion
Nuclear localisation of Ru1
Previous confocal laser scanning microscopy (CLSM) and transmission electron microscopy (TEM) studies indicated that Ru1 targets nuclear DNA.31 ICP-MS (inductively coupled plasma mass spectrometry) analysis confirmed high (>70%) cellular ruthenium content in isolated nuclear fractions of Ru1-treated oesophageal cancer cells (Fig. S1 in the ESI†) while visualisation of intracellular MLCT (metal to ligand charge-transfer) emission of Ru1 in OE21 oesophageal squamous cell carcinoma (ESCC) cells showed strong overlap with the DNA dye DAPI (Fig. S2†). In comparison, OE21 cells treated with [Ru(phen)2(dppz)]2+ (logP = −1.48 (ref. 18)) possessed a 13-fold lower average cellular Ru content and a 28-fold reduction in nuclear Ru content than cells treated with Ru1 (Fig. S1a†). In addition to this, minimal nuclear MLCT emission in [Ru(phen)2(dppz)]2+-treated cells was evident by CLSM (Fig. S2†). These results indicate substantially greater cellular uptake and enhanced nuclear targeting are demonstrated by Ru1 over [Ru(phen)2(dppz)]2+.
Ru1 stalls replication fork progression
On the basis of our recent discovery that the multi-intercalator [Ru(dppz)2(PIP)]2+ (PIP = 2-(phenyl)imidazo[4,5-f][1,10]phenanthroline) is able to stall replication forks,23 the ability of Ru1 to similarly affect DNA replication was examined by DNA fibre assay. By sequential incorporation of halogenated nucleotides and immunofluorescence staining, this technique allows visualisation of the progression of individual replicating DNA strands.33,34 Accordingly, OE21 cells were pulse-labelled with the thymidine analogue CIdU for 30 min before treatment with Ru1 and concomitant labelling with a second thymidine analogue (IdU) for an additional 30 min, thereby allowing examination of the direct impact of Ru1 upon replication fork progression (Fig. 1b). Strikingly, the addition of 21 μM Ru1 (the 24 h IC50 concentration, Table 1) in this manner resulted in a large decrease in median IdU tract length in DNA fibres, indicating extensive replication fork stalling generated due to the inclusion of the complex (Fig. 1c). This was accompanied by a 1.7-fold increase in completely stalled/terminated replication forks (CldU tract only – Fig. 1d). This marked impact on replication fork progression indicates pronounced and rapid DNA replication inhibition is generated directly by the addition of Ru1; an unprecedented result for a substitutionally inert metal complex that binds DNA solely by mono-intercalation.| OE21 | OE33 | FLO1 | hSAEC1-KT | |||||
|---|---|---|---|---|---|---|---|---|
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |
| a Determined by MTT assay (24 or 72 h constant exposure). Data mean of two independent experiments ± S.D. N.D. = not done. | ||||||||
| Ru1 | 21 ± 4.2 | 2.9 ± 1.5 | 44.5 ± 3.5 | 27 ± 2.8 | 42 ± 8 | 11.5 ± 2.1 | 78.5 ± 1.3 | 22 ± 2 |
| dppz | 6.5 ± 2.1 | 3.3 ± 0.4 | 6.6 ± 2 | 5.7 ± 0.9 | 7.5 ± 0.8 | 1.9 ± 1.1 | >10 | >10 |
| Cisplatin | 22.6 ± 3.6 | 6.3 ± 0.6 | 11 ± 1.4 | 3.25 ± 1 | 26.5 ± 0.7 | 4.7 ± 0.7 | 21.5 ± 5 | 15.8 ± 2 |
| [Ru(phen)2(dppz)]2+ | >100 | >100 | >100 | >100 | >100 | 34.0 ± 5.7 | N.D. | N.D. |
DNA damage response (DDR) activation by Ru1
To examine DNA damage response (DDR) cell signalling activation in response to lesions generated by Ru1, we carried out western blot analyses of protein extracts of OE21 cells using antibodies against the phosphorylated (activated) forms of several DDR signalling proteins. In Ru1-treated cells, an increased level of Chk1 phosphorylation at Ser345 and phospho-ATR (at Ser428) (ATR = ataxia telangiectasia and Rad3-related protein) at 3 h onwards indicated activation of ATR/Chk1 signalling (Fig. 2a), in agreement with generation of replication stress as a result of stalled replication forks.8 An increased level of phospho-Chk2 (Thr68) and γH2AX (Histone H2AX phosphorylated at Ser139), both of which are generated following DNA double-strand damage,8 was also apparent, the level of each increased with Ru1 exposure time (Fig. 2a). Comparable temporal DDR activation induced by Ru1 was observed in wild type p53-containing MCF7 breast cancer cells, showing that DDR activation occurs independently of p53 status (Fig. S3†). These results show Ru1 induces both replication stress and double-strand break (DSB) DDR pathway activation; a hallmark of replication fork collapse.35 | ||
| Fig. 2 (a) DNA damage response (DDR) activation by Ru1. Whole-cell extracts of OE21 cells treated with Ru1 (20 μM) for 1, 3, 8 or 24 h were immunoblotted for activated (phosphorylated, p) p-Chk1 (Ser345), p-Chk2 (Thr68), p-ATR (Ser428) or γH2AX (pH2AX at Ser139), as indicated. Total Chk1 and Chk2 protein levels independent of phosphorylation status are shown. α-Tubulin or β-actin were used as loading controls. (b) Relative expression of pChk1 and pChk2 in OE21 cells treated with equipotent (IC50) concentrations of cisplatin (23 μM), dppz (7 μM) or Ru1 (21 μM) for 3 h, as determined by immunoblotting and quantified by densitometry (bottom panels). Phosphorylated protein levels relative to total protein and normalised to control are presented. γH2AX/β-actin ratio also provided. (c) Impact of Ru1, dppz, [Ru(phen)2(dppz)]2+ or cisplatin on viability of OE21 cells, as determined by MTT assay (24 h incubation time). Mean of quadruplicates ± S.D. and representative of two independent experiments. (d) Immunoblotting of γH2AX levels in OE21 cells after 24 h treatment with cisplatin (Cis), Ru1 or dppz (IC50 and IC70 concentrations). [Ru(phen)2(dppz)]2+ treatment also included. β-Actin was used as a loading control. * 0.25% DMSO. | ||
Impact of dppz on replication fork progression
Although X-ray crystal structures have confirmed that DNA intercalation of RPCs is achieved primarily through the insertion of extended coordinated polypyridyl ligand(s) between base pairs,11–16 it is unknown whether this effect drives bioactivity. As poor solubility prevented the use of free tpphz, the cellular response to free (non-coordinated) dppz (Fig. 1a) – a close structural analogue of tpphz and the prototypical polypyridyl intercalating ligand – was also examined. As seen in Fig. 1b–d, replication fork progression in OE21 cells was impaired after the addition of 7 μM dppz, as indicated by a substantial decrease in IdU tract length and large (three-fold) increase in levels of stalled forks compared to mock-treated. The similarity of the impact on replication fork progression for dppz and Ru1 is consistent with the notion that polypyridyl ligand intercalation is responsible for replication inhibition. However, approximately two-fold greater levels of stalled forks are generated by dppz than Ru1, indicating a more potent replication block generated by the organic ligand (Fig. 1d). Examining DDR activation due to dppz treatment, high pChk1 levels and decreased levels of the DSB damage marker γH2AX and pChk2 activation in comparison to Ru1 treatment were apparent, indicating reduced DSB damage pathway activation by dppz (Fig. 2b and d).In comparison to Ru1 and dppz, [Ru(phen)2(dppz)]2+ demonstrated substantially reduced impact on cell proliferation along with no evidence of DDR activation (Fig. 2c and d). This may be explained by the reduced cellular uptake of [Ru(phen)2(dppz)]2+ compared to Ru1 (vide supra). The low bioactivity and poor nuclear targeting of [Ru(phen)2(dppz)]2+ are in agreement with other studies.18,32
Global response to Ru1-induced DNA damage
Depending upon the precise lesion generated, the cellular response to unrepaired DNA damage is typically either cell-cycle arrest or cell death.36 High levels of γH2AX foci are retained for a substantial time after Ru1 treatment of OE21 cells (Fig. 3a and b), indicating prolonged DDR activation generated by the complex and the persistence of unrepaired DNA. Examining impact on cell-cycle progression, treatment of cycling OE21 cells with Ru1 induced a two-fold increase in G2/M phase cells compared to control (Fig. 3c). A distinct cell-cycle response was induced in response to dppz where instead cells accumulated in G1 or early S phase (Fig. 3c). In contrast to cisplatin treatment, limited evidence of annexin-V-positive cells, no observable karyorrhexis and reduced cleaved caspase 3 expression was observed in OE21 cells treated with Ru1 (Fig. 3d, S4 and S5†), indicating low levels of apoptosis. Despite this, a loss of proliferative capacity as a result of Ru1 treatment was shown by clonogenic survival assay (Fig. 3e). Furthermore, numerous growth-arrested Ru1-treated OE21 cells appeared “flattened” with enlarged nuclei (Fig. S5†), a characteristic phenotype of senescence.37 These results are therefore consistent with permanent growth arrest being the primary response to Ru1-generated DNA damage in OE21 cells; although Trypan blue staining indicates ∼20% necrotic cell death to accompany this outcome (Fig. S5a†). | ||
| Fig. 3 (a) Representative CLSM images of OE21 cells treated with Ru1 (20 μM, 24 h) visualised at 0, 1, 6 and 24 h after complex removal. Immunofluorescence staining with anti-γH2AX antibody (white) provides visualisation of DSB damage. DNA (DAPI) staining included for reference. Scale bars = 20 μm. (b) Quantification of γH2AX foci/nucleus for OE21 cells treated as in (a). Data mean ± S.D. of four or five micrographs where a minimum of 20 nuclei were counted per image. (c) Impact of Ru1 (20 μM, 24 h) or dppz (7 μM, 24 h) on cell-cycle distribution of OE21 cells, as determined by flow cytometric analysis of DNA content. DNA content in Ru1-treated cells was quantified using the MLCT emission of Ru1 (see Experimental section). Data are mean of three independent experiments ± S.D. (d) AnnexinV staining of OE21 cells treated with cisplatin (23 μM), Ru1 (21 μM) or dppz (7 μM) for 24 h, as determined by flow cytometry. Average of two independent experiments ± S.D. (e) Clonogenic survival of OE21 cells treated with Ru1, cisplatin or dppz (24 h incubation time). Average of triplicates ± S.D. | ||
The targeting of genomic DNA without generating high levels of apoptosis by Ru1 is a distinct cellular response compared to cisplatin. While at first this appears surprising, Ru1 and cisplatin possess different DNA binding modalities (cisplatin forms covalent Pt–DNA adducts while Ru1 binds non-covalently via intercalation) and growth arrest in response to DNA-damage is common, particularly in cells that lack functional p53 and thus possess a reduced capacity to activate apoptosis.36 It is also evident that Ru1 demonstrates a different mechanism of action compared to structurally-related hydrophobic mitochondrial-targeting RPCs, which induce reactive oxygen species (ROS)-mediated apoptosis.38–40 As the hydrophilic Ru1 instead targets nuclear DNA, this illustrates the role of organelle targeting in determining RPC bioactivity.
The differential DDR activation and cell-cycle deregulation exhibited by Ru1 and dppz are also noteworthy. This may be rationalised by the greater levels of stalled forks generated by dppz resulting in a potent G1-S block. For Ru1, fork collapse instead results in slowed progression through S-phase and the accumulation of DSB damage, culminating in G2/M checkpoint activation. This indicates the RuII metal centre influences bioactivity and is not solely a “carrier” for the hydrophobic polypyridyl intercalating ligand. More detailed structural binding studies of Ru1 and dppz with DNA could be highly illuminating and provide a molecular basis for these observations.
Ru1 generates metaphase chromosome non-attachment
Close inspection revealed Ru1 induced chromosome misalignment in metaphase OE21 cells (Fig. 4a), possibly as a consequence of non-attachment of sister chromatids to spindle microtubules. To test this, z-stack confocal analysis and 3D reconstruction confirmed dispersion of condensed chromosomes in Ru1-treated populations with evidence of complete failure of attachment to the mitotic spindle (Fig. 4b and S6, ESI Videos 1 and 2†). Misalignment was a consequence of failure of any kinetochore attachment as no monotelic, syntelic, or merotelic figures were observed. These mitotic aberrations accounted for ∼8% of the total cell population at the IC50 concentration of Ru1 compared with 1.2% for control cells (Fig. 4c). Examining the impact of Ru1 on m-phase progression, an increase in prophase, prometaphase and metaphase cells was observed with an accompanying decrease in anaphase, telophase and cytokinesis populations (Fig. 4d and e). Increased levels of chromatin-associated activated (phosphorylated) p44/42 MAPK (mitogen-activated protein kinases) of aberrant metaphases indicated activation of the spindle assembly checkpoint (SAC) by Ru1 (Fig. 4f and S7†) and confirming the absence of merotelic attachment as the latter do not activate the SAC.41–43 In addition, treatment with Ru1 resulted in a large increase in cells containing multiple micronuclei (Fig. 4g and S8†), indicating that despite activation of the SAC, a significant proportion of these cells fail to maintain SAC-induced mitotic arrest and progress through mitosis; such observations are a documented consequence of fragmented or detached chromosomes generated during mitosis.44 These results are therefore consistent with DNA damage accumulated during mitosis causing errors in chromosome segregation and spindle attachment failure, thereby resulting in SAC activation and delayed metaphase-to-anaphase transition.45 This phenotype has been observed with other DNA-damaging agents, but these studies have always required genetic knockdown or co-treatment with a Chk1 inhibitor to abrogate the G2 checkpoint to achieve this outcome.46–48 It is therefore significant that Ru1 generates this effect applied as a single-agent.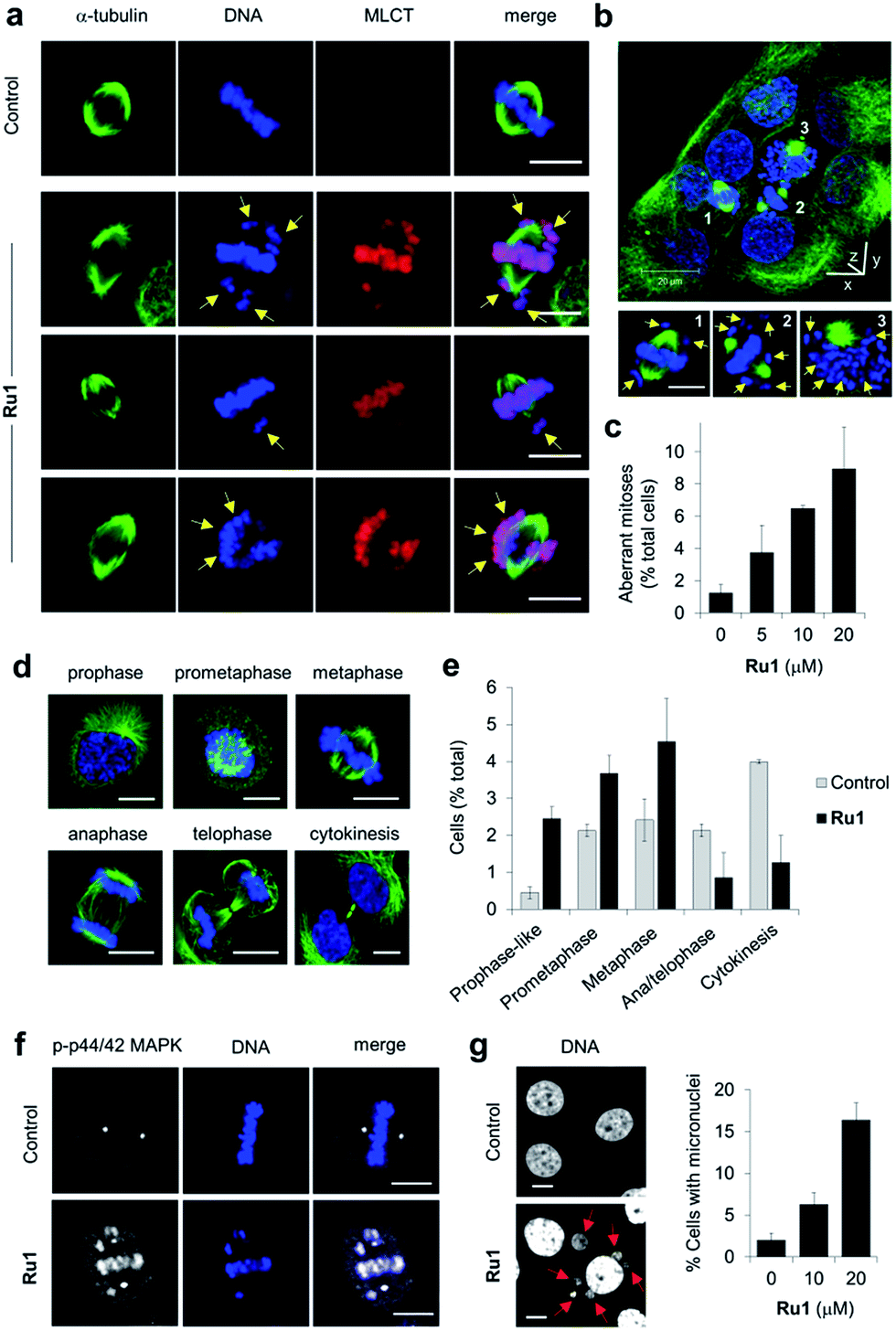 | ||
| Fig. 4 (a) Misaligned metaphase chromosomes (yellow arrows) in OE21 cells treated with Ru1 (20 μM, 4 h). After fixation, cells were stained for α-tubulin (green) and DNA (DAPI, blue). MLCT (metal to ligand charge-transfer) emission of Ru1 included. (b) 3D representation of Ru1-treated OE21 cells (20 μM, 24 h) prepared from z-stack images. Non-aligned chromosomes in labelled cells shown by yellow arrows. (c) Quantification of aberrant mitoses in Ru1-treated OE21 cells (24 h). Data average of two independent experiments ± S.D., >200 cells were counted for each condition. (d) CLSM images of mitotic stages of OE21 cells. (e) Quantification of mitotic sub-populations in Ru1-treated OE21 cells (20 μM, 24 h). Data are expressed as a percentage of total cell population. Data average of two independent experiments ± S.D., >200 cells counted per experiment. (f) Localisation of phospho(p)-p44/p42 MAP kinase in metaphase OE21 cells treated with Ru1 (20 μM, 24 h), as determined by immunofluorescence (AlexaFluor594, white). DNA staining (DAPI, blue) included for reference. (g) Formation of micronuclei (red arrows) in OE21 cells treated with Ru1 (20 μM, 24 h). Data average of two independent experiments ± S.D. where >100 cells were counted for each condition. Scale bars = 10 μm. | ||
Ru1 demonstrates distinct cancer cell-selectivity
The dual-mode mechanism of action of Ru1 implies that rapidly-proliferating cells with an elevated mitotic index and/or mutations of mitotic checkpoint genes (characteristics of many oesophageal cancers49,50) would be more susceptible to treatment. Accordingly, a panel of p53-deficient oesophageal cancer cell lines defective in Aurora kinase function49,50 were treated with Ru1 and relative potency assessed by MTT assay, which measures both cytotoxic and growth inhibitory (cytostatic) effects.51 Immortalised human small airway epithelial hSAEC1-KT cells, which are p53-functional, have a slow growth rate and low mitotic index,52 were used as a control. Comparable cellular and nuclear uptake of Ru1 in the three cancer cell lines was observed (Fig. S1†), however, derived half-inhibitory (IC50) concentrations indicated that the relative potency of Ru1 was greatest towards OE21 cells (Table 1 and Fig. S9†), which possess the highest mitotic index and growth rate (Table S1†). Notably, Ru1 demonstrates a two-fold greater potency than cisplatin in OE21 cells after 72 h incubation (Table 1). Encouragingly, reduced activity of Ru1 towards normal hSAEC1-KT cells was observed. In contrast, cisplatin showed comparable activity towards the cancer cell lines and consistent apoptosis induction across all cell lines tested, including hSAEC1-KT cells (Table 1 and Fig. S4b†). Dppz showed consistent activity towards the three cancer cell lines (Table 1) while the topoisomerase inhibitor doxorubicin demonstrated a three-fold reduction in potency towards OE21 cells compared to OE33 or FLO-1 cells after 72 h exposure (Table S2†). These results indicate the cell-selectivity profile demonstrated by Ru1 is not a general outcome for DNA-damaging agents. The observation that dppz demonstrated comparable or greater potency than Ru1 and substantially greater effects than [Ru(phen)2(dppz)]2+ is particularly striking as several RPC PDT candidates containing dppz-derived ligands exert bioactivity by light-activated ligand dissociation and it is often assumed their activity is due to the metal centre coordinating to DNA.26,27,53,54 These results are therefore consistent with the hypothesis that “uncaged” ligands play a significant role in these cases.54Mutagenicity of Ru1
DNA cross-linking agents such as cisplatin are often mutagenic.55 Although increased micronuclei formation indicates Ru1 induces genotoxic stress, this effect was most apparent in OE21 cells (Table S3†), a cell line highly susceptible to genotoxic insult due to chromosomal instability and Aurora kinase B dysfunction.49 Employing the hypoxanthine phosphorybosyl transferase (HPRT) forward mutation assay in V79 Chinese hamster cells we show that although Ru1 does increase mutation frequency above control, the rate is approximately two-fold lower than for cisplatin (Table 2).Ru1 radiosensitizes cancer cells via DNA damage enhancement
Many DNA-targeting drugs are employed as clinical radiosensitizers, as they induce specific DNA lesions and/or target cell-cycle progression to achieve synergy with IR targeted to cancer cells.1,2 To determine whether Ru1 functions as a radiosensitizer, three oesophageal cancer cell lines were pre-treated with sub-IC50 concentrations of Ru1 (2 μM) for 24 h before irradiation (0–8 Gy IR; 137Cs-γ-rays; dose rate = 0.81 Gy min−1) and relative cytotoxicity assessed by clonogenic survival. Fig. 5a shows that Ru1 decreases the surviving fraction (S.F.) of all cell lines in combined treatment compared to radiation alone. Single-agent Ru1 at this low concentration had a negligible impact on clonogenic survival (S.F. values > 0.87, Table S4†), thereby demonstrating the synergistic combination of Ru1 with IR. Resultant dose modification factors (DMF) at surviving fractions of 0.1 were 1.19–1.31 for Ru1: a comparable level of radiosensitization to treatment of the same cell lines with sub-cytotoxic doses of cisplatin (DMFs at 0.1 = 1.05–1.44). The concentration required for radiosensitization by Ru1 offers a significant improvement over previous work using the multi-intercalator [Ru(dppz)2(PIP)2+]23 and is similar to more cytotoxic Ru(arene)-halide monocationic complexes, which – like cisplatin – rely upon metal-centred reactivity and the formation of coordination bonds with DNA.56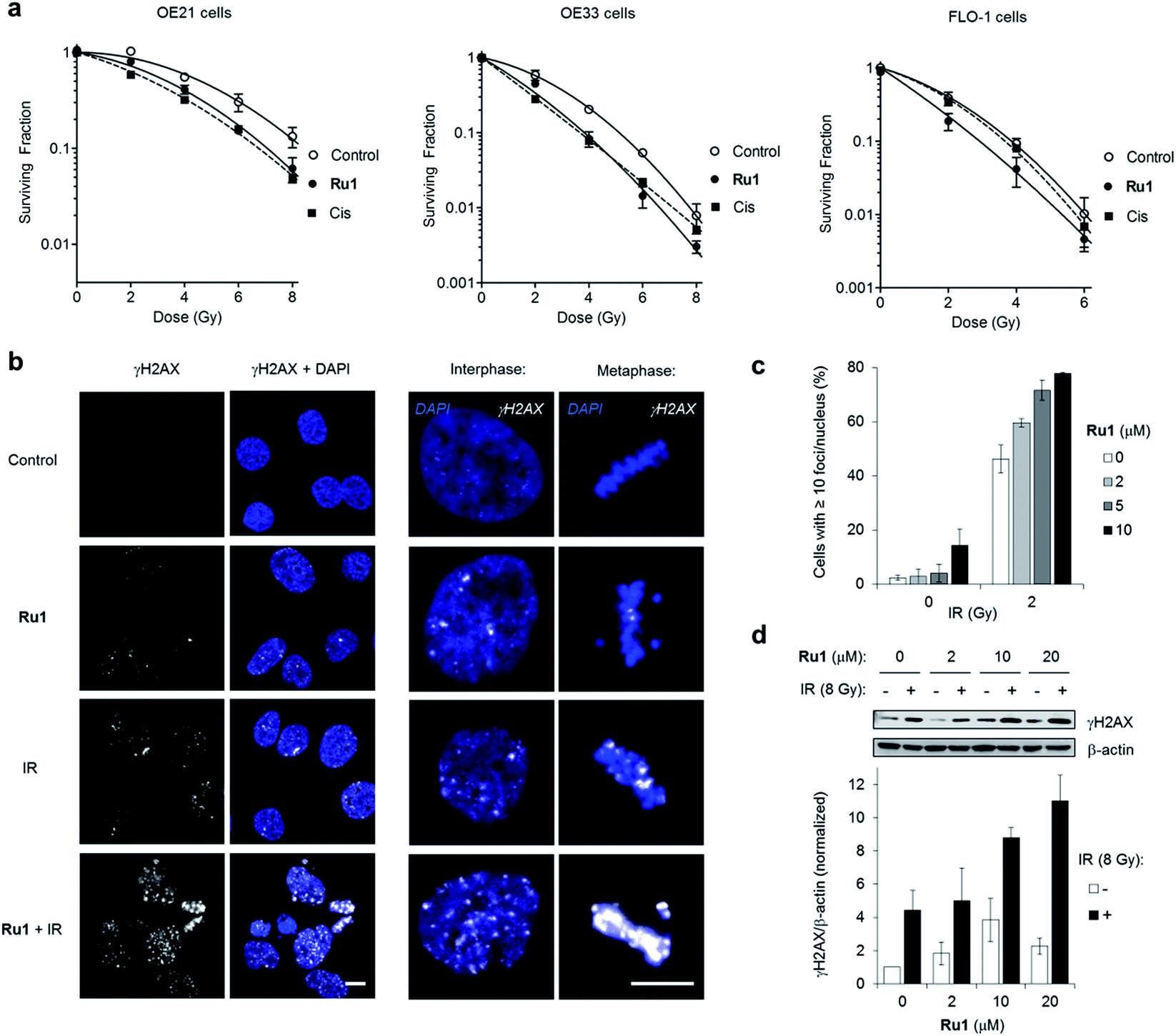 | ||
| Fig. 5 (a) Clonogenic survival of OE21, OE33 or FLO-1 cells pre-treated with Ru1 (2 μM, 24 h) before irradiation with 0–8Gy 137Cs-γ-rays. Mean ± SEM of two or three independent experiments. Data were fit to a second order polynomial function (R2 values > 0.99). Data for sub-cytotoxic doses of cisplatin (Cis) (500 nM OE21 and OE33, 300 nM FLO-1 cells) included (dashed lines). (b) CLSM images of OE21 cells treated with Ru1 (10 μM, 24 h), IR (2 Gy), or both, where IR was applied at the end of Ru1 treatment. Samples were fixed 1 h after irradiation. Immunofluorescence staining with anti-γH2AX antibody (white) provides visualisation of DSB damage. DNA (DAPI) staining included for reference. Scale bars = 10 μm. (c) Quantification of γH2AX foci/nucleus for OE21 cells treated as in (b). Data mean of two independent experiments ± S.D. (d) Immunoblotting (top) and corresponding densitometry (bottom) of γH2AX levels in OE21 cells treated with Ru1 (24 h) with or without 8 Gy IR after treatment. Whole-cell extracts were prepared 1 h after irradiation and γH2AX levels were measured relative to β-actin loading controls by densitometry. Data normalised to untreated cells and are the mean ± S.D. of two independent experiments. | ||
DNA replication inhibitors often act as radiosensitizers by enhancing IR-induced cytotoxic DSB damage.57,58 Accordingly, levels of the DSB marker γH2AX59 in cell cultures pre-treated with Ru1 before exposure to IR were examined. As shown in Fig. 5b and c, OE21 cells treated with Ru1 before IR (2 Gy) demonstrated a marked increase in γH2AX foci compared to single-agent treatment groups. The large increase in γH2AX foci in concomitant Ru1 and IR treatment indicated a substantial increase in IR-induced DSB formation compared to either treatment in isolation. A large increase in γH2AX levels is sustained even at higher doses of IR (8 Gy) and Ru1 (10 or 20 μM) (Fig. 5d), implying a large therapeutic window exists to combine the effects of Ru1 and IR.
Compared to work employing RPCs as photosensitizers, where in situ singlet oxygen or cytotoxic species formation is required for phototoxicity,25 radiosensitization often requires a complimentary cellular mechanism of action to enhance the DNA-damaging effects of IR.57,58 Considering many small molecules that inhibit DNA replication such as gemcitabine and 5FU (fluorouracil) are potent radiosensitizers,58 it seems likely that the DNA-targeting properties of Ru1 and subsequent impact upon replication fork progression play a significant role in its radiosensitizing effects. Furthermore, G2/M phase cells are documented to be the most sensitive to IR-induced DNA damage,1,2 and so the G2 arrest and metaphase block generated in response to Ru1 – induced DNA damage would also be predicted to increase radiosensitivity; a concept supported by the high levels of γH2AX visible in Ru1-treated metaphase cells exposed to IR (Fig. 5b). In addition to these effects, work employing cisplatin has also indicated DNA repair inhibition contributes to radiosenstization,4 and it will be interesting to measure DNA repair kinetics in Ru1-radiosensitized cells. Finally, although this work has established DNA as the primary target of Ru1, the peripheral coordination site on the tpphz ligand may additionally chelate metal ions.60,61 In agreement with this principle, the addition of ten-fold excess Zn2+ and Fe2+ ions to free or DNA-bound Ru1 resulted in a clear decrease in MLCT emission of the complex (Fig. S10†). Moreover, this effect was reversed by the addition of the metal chelator EDTA (ethylenediaminetetraacetic acid) for Zn2+ binding but, interestingly, not Fe2+, thereby demonstrating a high affinity of Ru1 for ferrous iron. These observations raise the possibility that Ru1 functions as a metal ion chelator in addition to binding DNA. While the possibility of intracellular chelation impacting bioactivity cannot be discounted, this effect would not be predicted to interfere with intercalative DNA binding60,61 and the rapid block of replication fork progression by Ru1 and DDR activation timeframe are consistent with results obtained using non-chelating DNA-binding compounds such as [Ru(dppz)2(PIP)2+].23,62 However, this may be of relevance to other effects caused by Ru1 as iron chelators have been indicated to demonstrate radiosensitizing properties.63,64 Future work will explore these concepts.
Conclusions
In summary, we present a detailed characterisation of the cellular response to [Ru(phen)2(tpphz)]2+ (Ru1) in p53-deficient oesophageal cancer cells, finding this ruthenium(II) metallo-intercalator induces a potent replication block accompanied by replication stress and DSB damage repair pathway activation, without triggering apoptosis. To our knowledge, this is the first example of a substitutionally inert ruthenium(II) mono-intercalator demonstrated to function as a replication inhibitor. In parallel to this, metaphase chromosome attachment is impaired by Ru1. This multi-mode mechanism of action results in growth inhibition of highly proliferative oesophageal cancer cells with elevated mitotic indices. Finally, efficient radiosensitization through synergistic DNA damage enhancement illustrates the efficacy of Ru1 in combination with IR, where the lower mutagenicity and reduced cytotoxicity of Ru1 compared to cisplatin would be predicted to be advantageous in its use alongside radiotherapy.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
M. R. G. and K. A. V. received financial support from Cancer Research UK (C5255/A15935) and a Medical Research Council Confidence in Concept award. K. R. acknowledges the Medical Research Council programme grant (MC_PC_12001/1) and a University of Oxford Goodger Scholarship to S. H. P. J. is the recipient of an EPSRC Prize Fellowship and C. S. acknowledges the EPSRC for an industrial research secondment award. We also acknowledge the Leverhulme Trust (M. G. W. and J. A. T.). We thank P. Holdship and F. Larner for ICP-MS analysis, G. Brown and E. Abbott for assistance and the reviewers for their useful suggestions.Notes and references
- T. Y. Seiwert, J. K. Salama and E. E. Vokes, Nat. Clin. Pract. Oncol., 2007, 4, 86 CrossRef CAS PubMed.
- M. R. Gill, N. Falzone, Y. Du and K. A. Vallis, Lancet Oncol., 2017, 18, e414 CrossRef CAS PubMed.
- Z. H. Siddik, Oncogene, 2003, 22, 7265 CrossRef CAS PubMed.
- C. R. Sears, S. A. Cooney, H. Chin-Sinex, M. S. Mendonca and J. J. Turchi, DNA Repair, 2016, 40, 35 CrossRef CAS PubMed.
- E. C. Smyth, J. Lagergren, R. C. Fitzgerald, F. Lordick, M. A. Shah, P. Lagergren and D. Cunningham, Nat. Rev. Dis. Primers., 2017, 3, 17048 CrossRef PubMed.
- L. Kleinberg and A. A. Forastiere, J. Clin. Oncol., 2007, 25, 4110 CrossRef CAS PubMed.
- J. M. Findlay, F. Castro-Giner, S. Makino, E. Rayner, C. Kartsonaki, W. Cross, M. Kovac, D. Ulahannan, C. Palles, R. S. Gillies, T. P. MacGregor, D. Church, N. D. Maynard, F. Buffa, J.-B. Cazier, T. A. Graham, L.-M. Wang, R. A. Sharma, M. Middleton and I. Tomlinson, Nat. Commun., 2016, 7, 11111 CrossRef CAS PubMed.
- M. J. O'Connor, Mol. Cell, 2015, 60, 547 CrossRef PubMed.
- A. E. Friedman, J. C. Chambron, J. P. Sauvage, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1990, 112, 4960 CrossRef CAS.
- B. M. Zeglis, V. C. Pierre and J. K. Barton, Chem. Commun., 2007, 4565 RSC.
- J. P. Hall, K. O'Sullivan, A. Naseer, J. A. Smith, J. M. Kelly and C. J. Cardin, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17610 CrossRef CAS PubMed.
- H. Niyazi, J. P. Hall, K. O'Sullivan, G. Winter, T. Sorenson, J. M. Kelly and C. J. Cardin, Nat. Chem., 2012, 4, 621 CrossRef CAS PubMed.
- H. Song, J. T. Kaiser and J. K. Barton, Nat. Chem., 2012, 4, 615 CrossRef CAS PubMed.
- J. P. Hall, F. E. Poynton, P. M. Keane, S. P. Gurung, J. A. Brazier, D. J. Cardin, G. Winter, T. Gunnlaugsson, I. V. Sazanovich, M. Towrie, C. J. Cardin, J. M. Kelly and S. J. Quinn, Nat. Chem., 2015, 7, 961 CrossRef CAS PubMed.
- J. P. Hall, P. M. Keane, H. Beer, K. Buchner, G. Winter, T. L. Sorensen, D. J. Cardin, J. A. Brazier and C. J. Cardin, Nucleic Acids Res., 2016, 44, 9472 CrossRef CAS PubMed.
- J. P. Hall, S. P. Gurung, J. Henle, P. Poidl, J. Andersson, P. Lincoln, G. Winter, T. Sorensen, D. J. Cardin, J. A. Brazier and C. J. Cardin, Chem.–Eur. J., 2017, 23, 4981 CrossRef CAS PubMed.
- P. M. Takahara, A. C. Rosenzweig, C. A. Frederick and S. J. Lippard, Nature, 1995, 377, 649 CrossRef CAS PubMed.
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2007, 129, 46 CrossRef CAS PubMed.
- M. R. Gill, J. Garcia-Lara, S. J. Foster, C. Smythe, G. Battaglia and J. A. Thomas, Nat. Chem., 2009, 1, 662 CrossRef CAS PubMed.
- M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179 RSC.
- (a) L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z.-S. Chen, Chem. Soc. Rev., 2017, 46, 5771 RSC; (b) A. Notaro and G. Gasser, Chem. Soc. Rev., 2017 10.1039/C7CS00356K.
- H. Huang, P. Zhang, B. Yu, Y. Chen, J. Wang, L. Ji and H. Chao, J. Med. Chem., 2014, 57, 8971 CrossRef CAS PubMed.
- M. R. Gill, S. N. Harun, S. Halder, R. A. Boghozian, K. Ramadan, H. Ahmad and K. A. Vallis, Sci. Rep., 2016, 6, 31973 CrossRef CAS PubMed.
- C. A. Griffith, A. Dayoub, T. Janaratne, N. Alatrash, K. Abayan, A. Mohamedi, Z. Breitbach, D. W. Armstrong and F. M. MacDonnell, Chem. Sci., 2017, 8, 3726 RSC.
- C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660 RSC.
- C. Mari, V. Pierroz, R. Rubbiani, M. Patra, J. Hess, B. Spingler, L. Oehninger, J. Schur, I. Ott, L. Salassa, S. Ferrari and G. Gasser, Chem.–Eur. J., 2014, 20, 14421 CrossRef CAS PubMed.
- V. Pierroz, R. Rubbiani, C. Gentili, M. Patra, C. Mari, G. Gasser and S. Ferrari, Chem. Sci., 2016, 7, 6115 RSC.
- S.-H. Liu, J.-H. Zhao, K.-K. Deng, Y. Wu, J.-W. Zhu, Q.-H. Liu, H.-H. Xu, H.-F. Wu, X.-Y. Li, J.-W. Wang and Q.-F. Guo, Spectrochim. Acta, Part A, 2015, 140, 202 CrossRef CAS PubMed.
- Z. Deng, L. Yu, W. Cao, W. Zheng and T. Chen, Chem. Commun., 2015, 51, 2637 RSC.
- M. Baumann, M. Krause, J. Overgaard, J. Debus, S. M. Bentzen, J. Daartz, C. Richter, D. Zips and T. Bortfeld, Nat. Rev. Cancer, 2016, 16, 234 CrossRef CAS PubMed.
- M. R. Gill, H. Derrat, C. Smythe, G. Battaglia and J. A. Thomas, ChemBioChem, 2011, 12, 877 CrossRef CAS PubMed.
- A. Yadav, T. Janaratne, A. Krishnan, S. S. Singhal, S. Yadav, A. S. Dayoub, D. L. Hawkins, S. Awasthi and F. M. MacDonnell, Mol. Cancer Ther., 2013, 12, 643 CrossRef CAS PubMed.
- C. J. Merrick, D. Jackson and J. F. X. Diffley, J. Biol. Chem., 2004, 279, 20067 CrossRef CAS PubMed.
- B. Vaz, M. Popovic, J. A. Newman, J. Fielden, H. Aitkenhead, S. Halder, A. N. Singh, I. Vendrell, R. Fischer, I. Torrecilla, N. Drobnitzky, R. Freire, D. J. Amor, P. J. Lockhart, B. M. Kessler, G. W. McKenna, O. Gileadi and K. Ramadan, Mol. Cell, 2016, 64, 704 CrossRef CAS PubMed.
- B. Müller, J. Blackburn, C. Feijoo, X. Zhao and C. Smythe, J. Cell Biol., 2007, 179, 1385 CrossRef.
- W. P. Roos, A. D. Thomas and B. Kaina, Nat. Rev. Cancer, 2016, 16, 20 CrossRef CAS PubMed.
- J. Campisi, Curr. Opin. Genet. Dev., 2011, 21, 107 CrossRef CAS PubMed.
- T. Chen, Y. Liu, W.-J. Zheng, J. Liu and Y.-S. Wong, Inorg. Chem., 2010, 49, 6366 CrossRef CAS PubMed.
- V. Pierroz, T. Joshi, A. Leonidova, C. Mari, J. Schur, I. Ott, L. Spiccia, S. Ferrari and G. Gasser, J. Am. Chem. Soc., 2012, 134, 20376 CrossRef CAS PubMed.
- J.-Q. Wang, P.-Y. Zhang, C. Qian, X.-J. Hou, L.-N. Ji and H. Chao, J. Biol. Inorg Chem., 2014, 19, 335 CrossRef CAS PubMed.
- J. Minshull, H. Sun, N. K. Tonks and A. W. Murray, Cell, 1994, 79, 475 CrossRef CAS PubMed.
- P. S. Shapiro, E. Vaisberg, A. J. Hunt, N. S. Tolwinski, A. M. Whalen, J. R. McIntosh and N. G. Ahn, J. Cell Biol., 1998, 142, 1533 CrossRef CAS PubMed.
- M. Zecevic, A. D. Catling, S. T. Eblen, L. Renzi, J. C. Hittle, T. J. Yen, G. J. Gorbsky and M. J. Weber, J. Cell Biol., 1998, 142, 1547 CrossRef CAS PubMed.
- A. J. Holland and D. W. Cleveland, Nat. Med., 2012, 18, 1630 CrossRef CAS PubMed.
- A. Mikhailov, R. W. Cole and C. L. Rieder, Curr. Biol., 2002, 12, 1797 CrossRef CAS PubMed.
- C. Vogel, A. Kienitz, R. Müller and H. Bastians, J. Biol. Chem., 2005, 280, 4025 CrossRef CAS PubMed.
- D. Maskey, S. Yousefi, I. Schmid, I. Zlobec, A. Perren, R. Friis and H.-U. Simon, Nat. Commun., 2013, 4, 2130 Search PubMed.
- S.-B. Koh, A. Courtin, R. J. Boyce, R. G. Boyle, F. M. Richards and D. I. Jodrell, Cancer Res., 2015, 75, 3583 CrossRef CAS PubMed.
- C. D. Fichter, C. Herz, C. Münch, O. G. Opitz, M. Werner and S. Lassmann, BMC Cell Biol., 2011, 12, 13 CrossRef CAS PubMed.
- V. Sehdev, D. Peng, M. Soutto, M. K. Washington, F. Revetta, J. Ecsedy, A. Zaika, T. T. Rau, R. Schneider-Stock, A. Belkhiri and W. El-Rifai, Mol. Cancer Ther., 2012, 11, 763 CrossRef CAS PubMed.
- V. Ramu, M. R. Gill, P. J. Jarman, D. Turton, J. A. Thomas, A. Das and C. Smythe, Chem.–Eur. J., 2015, 21, 9185 CrossRef CAS PubMed.
- R. D. Ramirez, S. Sheridan, L. Girard, M. Sato, Y. Kim, J. Pollack, M. Peyton, Y. Zou, J. M. Kurie, J. M. DiMaio, S. Milchgrub, A. L. Smith, R. F. Souza, L. Gilbey, X. Zhang, K. Gandia, M. B. Vaughan, W. E. Wright, A. F. Gazdar, J. W. Shay and J. D. Minna, Cancer Res., 2004, 64, 9027 CrossRef CAS PubMed.
- B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324 CrossRef CAS PubMed.
- J.-A. Cuello-Garibo, M. S. Meijer and S. Bonnet, Chem. Commun., 2017, 53, 6768 RSC.
- B. Szikriszt, Á. Póti, O. Pipek, M. Krzystanek, N. Kanu, J. Molnár, D. Ribli, Z. Szeltner, G. E. Tusnády, I. Csabai, Z. Szallasi, C. Swanton and D. Szüts, Genome Biol., 2016, 17, 99 CrossRef PubMed.
- R. Carter, A. Westhorpe, M. J. Romero, A. Habtemariam, C. R. Gallevo, Y. Bark, N. Menezes, P. J. Sadler and R. A. Sharma, Sci. Rep., 2016, 6, 20596 CrossRef CAS PubMed.
- P. Wardman, Clin. Oncol., 2007, 19, 397 CrossRef CAS PubMed.
- T. S. Lawrence, A. W. Blackstock and C. McGinn, Sem. Radiat. Oncol., 2003, 13, 13 CrossRef PubMed.
- E. P. Rogakou, D. R. Pilch, A. H. Orr, V. S. Ivanova and W. M. Bonner, J. Biol. Chem., 1998, 273, 5858 CrossRef CAS PubMed.
- S. A. Tysoe, R. Kopelman and D. Schelzig, Inorg. Chem., 1999, 38, 5196 CrossRef CAS.
- Y. Liu, A. Chouai, N. N. Degtyareva, D. A. Lutterman, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2005, 127, 10796 CrossRef CAS PubMed.
- R. Zellweger, D. Dalcher, K. Mutreja, M. Berti, J. A. Schmid, R. Herrador, A. Vindigni and M. Lopes, J. Cell Biol., 2015, 208, 563 CrossRef CAS PubMed.
- J. Turner, C. Koumenis, T. E. Kute, R. P. Planalp, M. W. Brechbiel, D. Beardsley, B. Cody, K. D. Brown, F. M. Torti and S. V. Torti, Blood, 2005, 106, 3191 CrossRef CAS PubMed.
- J. Tian, D. M. Peehl, W. Zheng and S. J. Knox, Cancer Lett., 2010, 298, 231 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, supplementary figures and tables. See DOI: 10.1039/c7sc03712k |
| This journal is © The Royal Society of Chemistry 2018 |