
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design and construction of an open source-based photometer and its applications in flow chemistry†
Gabriel
Glotz
ab and
C. Oliver
Kappe
 *ab
*ab
aCenter for Continuous Flow Synthesis and Processing (CC FLOW), Research Center Pharmaceutical Engineering GmbH (RCPE), Inffeldgasse 13, Graz, Austria
bInstitute of Chemistry, NAWI Graz, University of Graz, Heinrichstrasse 28, 8010 Graz, Austria. E-mail: oliver.kappe@uni-graz.at
First published on 31st May 2018
Abstract
The design and construction of a low-cost and compact USB photometer based on open source hardware and software is described. The device, featuring a 1 ms sample acquisition time, utilizes a single interchangeable light emitting diode (LED) as a light source in combination with photodiode detectors and a Texas Instruments LaunchPad as the main control unit. An appropriately constructed flow cell can be connected to a flow reactor made out of transparent polymeric material such as perfluoroalkoxy (PFA) tubing. The flow cell can be moved to different positions of the tube in order to take measurements at different residence times without having to disassemble the photometer or the flow reactor system. The bespoke photometer was used to determine the residence time distribution (RTD) of various microreactors/micromixers, as well as for measuring the reaction rate constant for the base-induced hydrolysis of p-nitrophenol acetate in a biphasic segmented flow system. Owing to the 1 ms sample acquisition time of the novel device the lengths of individual segments can be measured in real time.
Introduction
Advances in synthetic organic chemistry combined with a multitude of sophisticated analytical techniques capable of providing rapid information on the structure and/or concentration of reaction intermediates and products have revolutionized modern chemical and pharmaceutical manufacturing. Traditionally, pharmaceutical manufacturing is conducted mostly using well-established batch processes accompanied by off-line process analysis and process control. Although very useful as multipurpose reactors, such batch processes often suffer from poor reproducibility, have safety issues associated with process control and may lead to inconsistent product quality. For this reason, pharmaceutical companies and manufacturers are currently considering to convert batch to continuous processes. This transition requires the development of novel process analytical technology (PAT) tools for rapid and accurate real-time process analysis in order to ensure optimum product quality.1The benefits of flow chemistry and continuous manufacturing, in general, can be divided into two categories. First of all, there is much better heat and mass transfer in microreactors as compared to batch reactors, allowing for much wider operating windows and at the same time better process control.2 Secondly, the ability to use and generate hazardous, highly toxic or unstable reagents and intermediates, ideally on-site and on-demand, thereby increasing safety and allowing so-called “forbidden chemistries” to be developed.3 To fully exploit these benefits there is a need to know the chemical composition of the reaction mixture at any given moment and at any position inside the flow reactor. Only then, this information can be used in an efficient manner to generate feedback loops that control reactor performance.4 Among current examples of in-line analysis, spectroscopic techniques without doubt, play a very significant role. The use of NIR,5 IR,6 Raman,7 and NMR8 spectroscopy for the in-line monitoring of intermediates, impurities, and products in continuous processes has been extensively studied.
Even though UV-vis spectroscopy provides less structural information about specific species in the reaction mixture, it is often the method of choice for concentration measurements owing to a lower level of interference from the matrix, i.e., solvents and non-absorbing components in a particular wavelength range.9 The majority of published examples using UV-vis spectroscopy in continuous systems are related to measurements in biochemistry10 and analytical chemistry applying chemical sensors.11 In terms of flow chemistry applications UV-vis spectroscopy has been applied for, e.g., monitoring photocatalytic reactions,12 esterifications,13 oxidations,14 nucleophilic aromatic substitutions15 and for the online analysis of nanoparticles generated under segmented flow conditions.16
In addition, UV-vis spectroscopy is often the method of choice for the characterization of microreactors and micromixers in terms of their efficiency. This can be accomplished by monitoring the change in concentration of inert tracer molecules in time, using simple photometry. From these measurements, information regarding mixing performance,17 axial dispersion,18 flow regimes19 and residence time distribution (RTD)20,26 can be derived, providing a relatively straightforward access to a variety of dimensionless numbers in fluid mechanics (e.g., Reynolds and Bodenstein numbers).21
A clear advantage of using visible light absorption lies in the ready availability of equipment and the moderate cost of the required optical parts, making the overall measurement system accessible to many scientists in the field. To the best of our knowledge, the only commercial UV-vis spectrophotometer specifically dedicated to flow chemistry applications was recently introduced by Uniqsis (Flow-UV™).22 The system uses optical fibers for light transmission from the source through a flow cell and on to the detector.
Herein we describe the development and use of a compact and inexpensive photometer system that is based on open source hardware and software. The system was designed utilizing the Texas Instruments MSP430 LaunchPad platform23 as the main unit in combination with self-designed extension boards. These extension boards feature the detector circuitry, amplifiers and filters, LED drivers and an accompanying power supply for the complete system (Fig. 1). A light emitting diode (LED) was selected as the light source owing to its narrow emission spectra, long-term stability, and ease of implementation. Additionally, replacement of the LED affords flexibility, providing access to more than one wavelength range to be used for measurements. The source code for communication with the LaunchPad was written in the Energīa24 open source software featuring an integrated development environment (IDE) based on processing.25 The photometer was evaluated for the measurement of dye concentrations over time (microreactor characterization) and for determining the concentration of p-nitrophenol product in the biphasic hydrolysis of p-nitrophenol acetate (reaction kinetics).
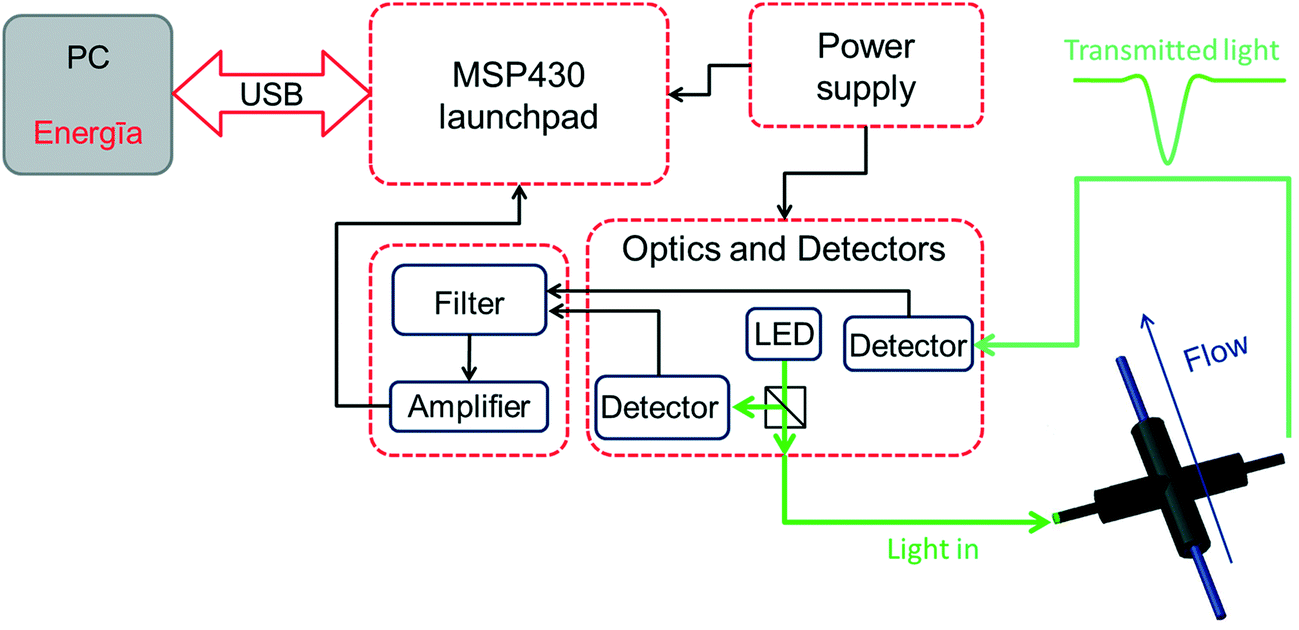 | ||
| Fig. 1 Diagram presenting the main components of the photometer system (additional details are presented in Fig. S1–S5†). | ||
Results and discussion
Photometer design
In order to meet the desired characteristics for an optical measurement system within a flow chemistry regime, certain boundary conditions had to be met. First of all, the system should utilize optical fibers for light transmission. Secondly, the light intensity should be high since the concentration of reagents in flow is usually in the range of 1 mol L−1. In addition, the system electronics should be able to handle the high light intensity, fast signal acquisition (in the order of ms) and be configurable according to a specific application. Apart from these boundary conditions, it appeared desirable to design a UV-vis flow measurement cell which is not fixed but could be moved along the flow path. In the particular case described herein, the flow cell was constructed from a cross mixer wrapped in aluminum foil and black tape to protect the cell from external light interfering with the experiments (Fig. 2a and b).26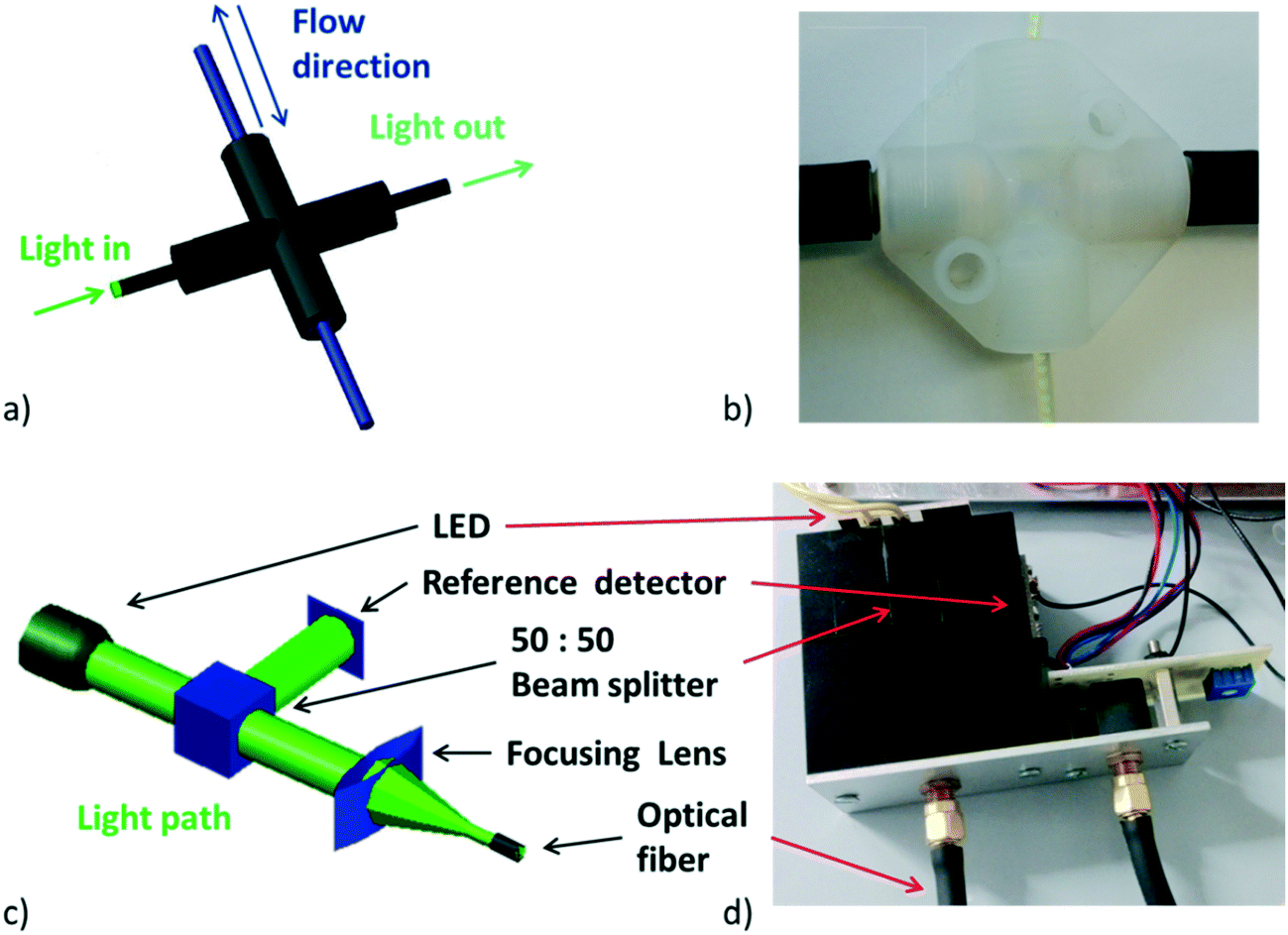 | ||
| Fig. 2 a) View of constructed flow cell, b) image of manufactured flow cell, c) 3D view of optical path, d) image of manufactured optical part. | ||
Having the boundary conditions mentioned above in mind, the light source was designed in a way that depending on the application and the absorption wavelength of the species of interest a single 1 or 3 W LED can easily be connected to a standard constant current LED driver module (Mean Well LDD-700L), with a feature that enables LED light intensity control by one digital signal. Light from the LED (see Fig. S6† for emission spectra of used LEDs) is split into two perpendicular light beams with the aid of a beam splitter cube (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 split ratio, Thorlabs), one for the reference detector and the second one for the application. The second light beam is focused on the optical fiber using a poly(methyl methacrylate) (PMMA) lens. Light is then focused on the perfluoroalkoxy (PFA) tube and the transmitted light ultimately passes through another optical fiber to the detector (Fig. 2c and d).
:50 split ratio, Thorlabs), one for the reference detector and the second one for the application. The second light beam is focused on the optical fiber using a poly(methyl methacrylate) (PMMA) lens. Light is then focused on the perfluoroalkoxy (PFA) tube and the transmitted light ultimately passes through another optical fiber to the detector (Fig. 2c and d).
Light intensity was measured using an OPT101 chip with integrated photodiode detector and transimpedance amplifier (Fig. 3a) configured for the gain of 0.1 with external resistors (0.1% tolerance, 25 ppm temperature coefficient). The second stage in the signal path is filtered through a low pass filter (4th order Sallen Key Butterworth) with a 1 kHz cutoff frequency (using TLV2141 IC-s as an active element). This is followed by summing of the reference and measured signal using a PGA205 zero-drift programmable gain instrumentation amplifier providing a low impedance output signal to the analog-to-digital converter (ADC), thus minimizing noise pickup in the signal path. The amplifier has the additional feature of adjusting the output offset voltage, which in turn provides the user with the ability to adjust the output of the system to zero for the blank measurements. In order to provide the system with some flexibility, digital gain control of the PGA205 amplifier is enabled, the gain is selected in the source code by a user and controlled by two digital signals directly from the MSP-EXP430G2 LaunchPad.
 | ||
| Fig. 3 a) OPT101 detector chip in 8DIL package, b) MSP-EXP430G2 LaunchPad. | ||
The MSP-EXP430G2 LaunchPad development kit (Fig. 3b), is an easy-to-use microcontroller development board for the low-power and low-cost MSP430G2x MCUs. It has onboard emulation for programming and debugging. Finally, the power supply for the system is designed to reduce the incoming 12 V/1 A power from the power adapter to the appropriate voltage for the other boards in the system and to drive the light source and fan for cooling of the LED. In Fig. 1 a schematic representation of the constructed photometer is given. The cost of the parts used for the construction of the photometer is ca. 360 USD (see Table S1† for the bill of materials).
Application 1: reactor characterization
The determination of residence time distribution (RTD) in a flow device is a valuable tool for the characterization of microreactors, micromixers, and flow reactors in general.21,26 We have therefore set out to evaluate the usefulness of our flow photometer system for establishing the RTD of several commercial microreactors/micromixers as well as of a standard 0.8 mm (i.d.) PFA tubular reactor. The experiments were performed by measuring the change of concentration of Rose Bengal dye (RB) over time at the exit of the microreactor or micromixer, providing a so-called C-curve.27A 1 W green LED was used as the light source having an emission maximum at 565 nm, close to the absorption maximum of RB dye at 559 nm (Fig. S7†). Two types of experiments were performed based on the internal volume of the microreactor or micromixer. For microreactors with an internal volume larger than 100 μL a pulse experiment was used, where a small volume of tracer dye (13 μL) is injected and the exiting concentration of the dye is measured. For micromixers with an internal volume smaller than 100 μL a step experiment was used, where the concentration of dye at the exit is measured after an instant change from the constant flow of water to the flow of dye. For both, step and pulse experiments, a 1 mmol L−1 solution of RB was employed using a six-way valve to manipulate solvent and dye flows (Fig. S8 and S9†).Initially, we have performed step experiments on two commercial glass micromixers: 1) a 26 μL Syrris Asia micromixer chip, and 2) a 4.64 μL Dolomite micromixer chip (see Table 1 for details). The experimental data (Fig. 4 and Fig. S15–S17†) clearly demonstrate the effectiveness of mixing as a function of flow rate through the micromixer. As expected,21,27 decreasing the flow rate from 1.00 mL min−1 to 0.25 mL min−1 causes a broader distribution of the tracer dye as a consequence of decreased mixing efficiency, along with an increased tailing of the dye as seen for the Dolomite micromixer chip using the step experiment (Fig. 4).
| Microreactor/micromixer | Internal volume (μL) | Chanel dimensions (μm) | Mixing geometry | Number of mixing stages | Material |
|---|---|---|---|---|---|
| a Depth × width of channels for both of two connected plates [ref. 30]. b Depth × width of channel. | |||||
| Dolomite micromixer chip | 4.64 | 125 × 350 and 50 × 125a | Split and recombine | 12 | Glass |
| Syrris Asia micromixer chip | 26 | 125 × 350 and 50 × 125a | Split and recombine | 12 | Glass |
| Syrris Asia microreactor | 250 | 250 × 400b | Double T contacting | 1 | Glass |
| Uniqsis microreactor | 1800 | 1700 × 1500 | Double chicane | Throughout channel | Glass |
| PFA tube | Variable | 800 i.d. (1600 o.d.) | N/A | N/A | PFA |
| 3D printed SS reactor | 1890 | 800 i.d. | Curvature based | N/A | 316 L stainless steel |
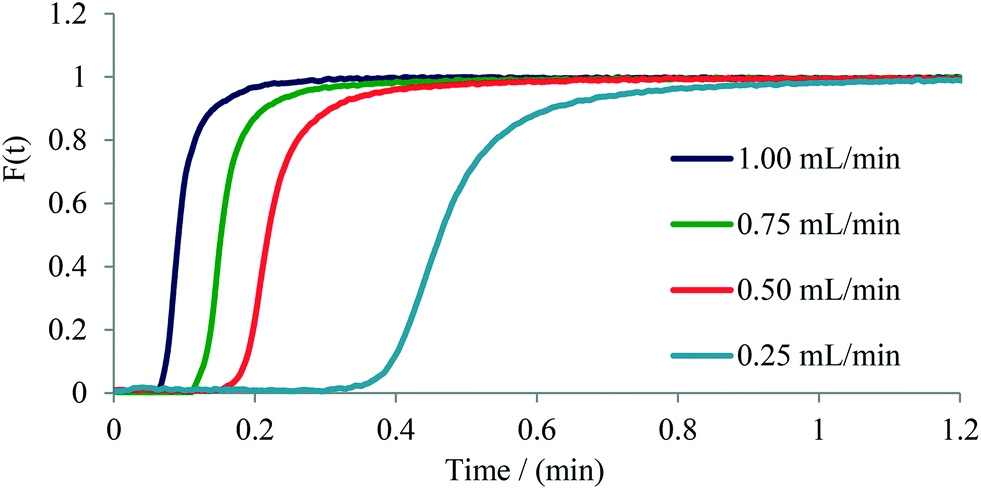 | ||
| Fig. 4 F-curve calculated for the Dolomite micromixer chip (4.64 μL) from a step experiment. | ||
Next, pulse experiments were performed on two commercially available glass microreactors and a tubular PFA reactor, namely on 1) a 250 μL Syrris microreactor, 2) a 1800 μL Uniqsis microreactor, and 3) a 0.8 mm i.d. PFA tube of variable length (Table 1). For these experiments a 13 μL dye pulse was injected into the reactor with the help of a six-way valve. The output was connected via a short (15 mm) PFA tube to the UV-vis flow cell (in the case of the tubular PFA reactor the UV-vis flow cell was 50 cm downstream from the six-way valve thus providing an internal volume of 250 μL). Experiments were conducted applying flow rates ranging from 0.25 mL min−1 to 1.00 mL min−1. In case of the 1.8 mL Uniqsis glass microreactor, the flow rate of 0.25 mL min−1 was not used since the dye was too diluted and thus the signal-to-noise ratio of the output signal was not high enough to be interpreted with confidence. The symmetrical shape of the curves indicates a good radial mixing performance in the microreactor, whereas an unsymmetrical shape indicates poor radial mixing and a parabolic shape of the flow profile within the reactor.26,27 Similar to the observations with micromixers (see above), an increase in tailing and width of the dye signal with decreasing flow rate was observed for the investigated microreactors. The only microreactor exhibiting a relatively symmetrical signal shape for all applied flow rates was the Uniqsis microreactor (Fig. 5), which can be attributed to the internal mixing structure within the reactor. The other microreactors exhibited classic parabolic flow profiles owing to the lack of mixing structures (i.e., for the PFA tube) or in cases where the mixing structure is located only at the mixing point of the input feeds and does not stretch through the channels of the microreactor (Syrris microreactor, see Fig. S10 to S14† for details). As expected, the tubular PFA reactor exhibited the highest axial diffusion and broadest distribution of the tracer dye. The completely unsymmetrical shape of the curves additionally indicates poor radial mixing, high axial diffusion and a significant tailing (Fig. S18–S22†).
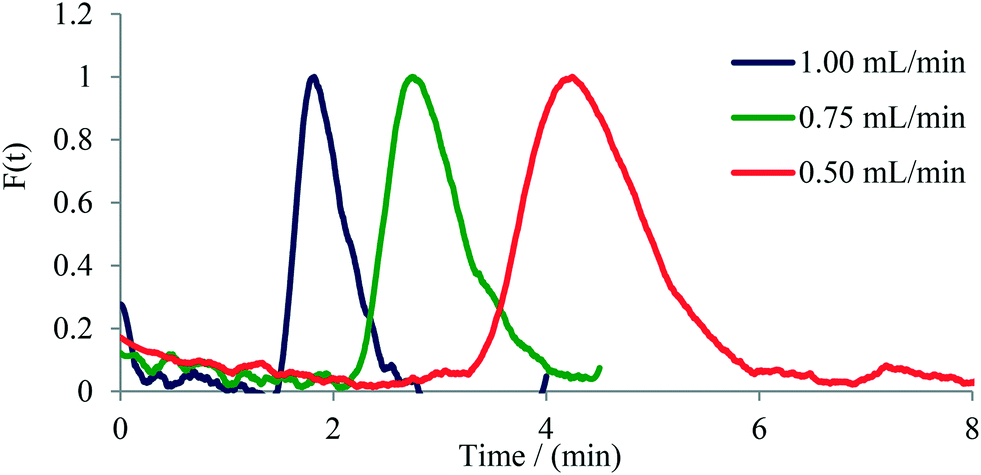 | ||
| Fig. 5 F-curve calculated for the Uniqsis (ca. 1.6 mL glass microreactor). | ||
We have recently published the design and fabrication of a 3D printed stainless steel flow reactor specifically made for a low-temperature two-step continuous deprotonation/difluoromethylation process.28 The reactor design and mixing geometry was optimized for this process with channels being slightly oval in the cross section, affording better surface quality after the 3D printing process. The 4 m in total of reaction channels (0.8 mm i.d., ca. 1.9 mL internal volume) are serpentined around a cooling element in zig-zag form providing advanced advective mixing by stretching and folding of the flow stream (Fig. S14†). Computational fluid dynamics was used to predict the mixing performace of the reactor.28 Measuring the RTD with a step experiment (RB dye) at 1 mL min−1 suggests moderate axial diffusion through the reactor (Fig. S23†).28
The disadvantage of the general method used herein for measuring RTD is the assumption that the input of the tracer dye is following an ideal Dirac function. When this is not the case the same flow cell should be positioned at the input as well as output of the micromixer/microreactor in order to correct for the input.29
Application 2: reaction kinetics
We next evaluated the performance of the flow photometer in monitoring the concentration of p-nitrophenol anion (p-NF) in the hydrolysis of p-nitrophenol acetate (p-NFA) with sodium hydroxide. When executed under biphasic conditions this transformation is often used as a model reaction in order to study e.g. the mixing performance/mass transfer of liquid–liquid biphasic reaction systems.31 In a tubular reactor one can typically observe a segmented flow pattern of the organic phase containing the p-NFA in CH2Cl2 and the aqueous sodium hydroxide phase.32 As the reaction progresses the concentration of the strongly yellow colored (400 nm absorption max) p-nitrophenol anion (p-NF) in the aqueous phase serves as a measure for conversion. Monitoring the concentration of p-NF at different positions along the PFA tubular reactor therefore enables determination of reaction kinetics. For this experiment, a 1 W LED with a maximum of emission at 400 nm was used as the light source with experiments performed at room temperature (25 °C). Prior to performing a hydrolysis experiment, a calibration diagram was obtained by measuring the absorption (as ADC value) of stock solutions of p-NF in 2 M sodium hydroxide solution (Fig. S25†) from which a response factor was calculated.For the hydrolysis reaction, two pumps were used to deliver the organic and aqueous solutions to a standard PEEK T mixer (17.5 μL, 1.25 mm i.d.) connected to a straight PFA tube as a reactor (0.8 mm i.d., 1.6 mm o.d., 120 cm total length) producing a biphasic reaction mixture (Fig. 6). One feed was composed of 2.0 mol L−1 sodium hydroxide solution in water (100 μL min−1) and the second one of 1.0 mol L−1 of a CH2Cl2 solution of p-NFA (50 μL min−1) producing a 4:1 ratio of OH ions to p-NFA. Measuring the absorbance at defined locations from the mixing point allowed us to measure the concentration of p-NF at different residence times (Fig. 6). The hydrolysis reaction was monitored at 14 different points, initially 4 cm downstream from the mixing point, then 10 cm from mixing and subsequently every 5 cm until no further increase in the concentration of p-NF was observed (70 cm as the last measurement point).
 | ||
| Fig. 6 Experimental setup for the hydrolysis of p-NFA with NaOH. The insert shows the formed biphasic reaction mixture. | ||
Since the reaction is biphasic, segmented flow is produced immediately after mixing which potentially can interfere with absorption measurements at slow sampling times because of light refraction at the layer boundary. Gratifyingly, this was not the case when using a 1 ms sample acquisition time at the used flow rates. At the desired measurement location, at least 200 data points were recorded and the upper signal value was averaged providing the concentration value for p-NF (Fig. 7a and b).
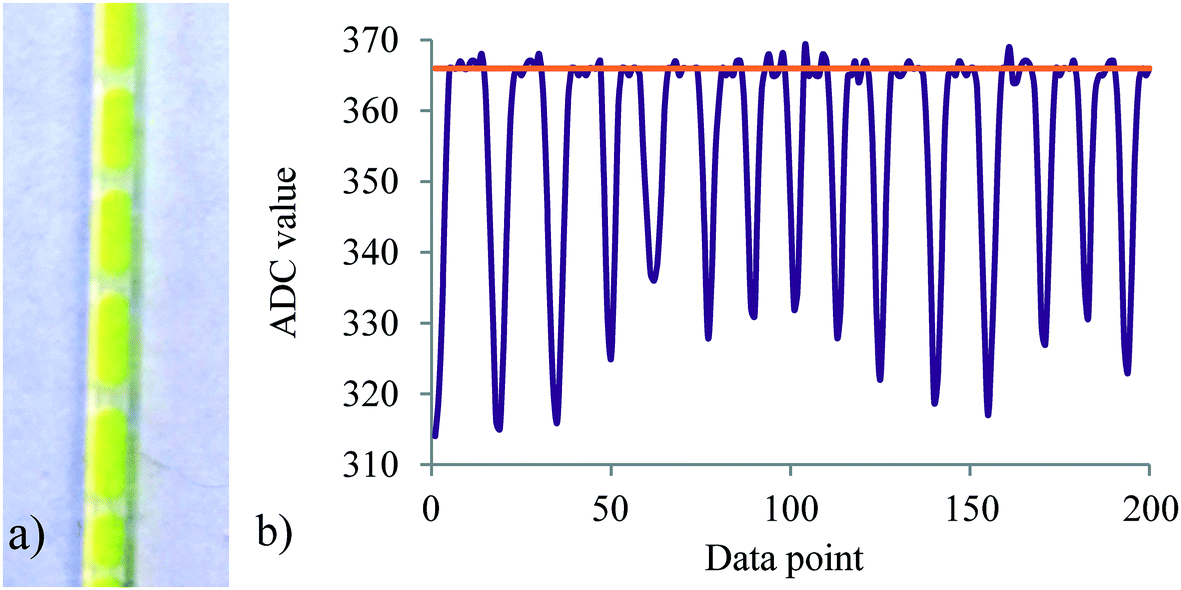 | ||
| Fig. 7 a) Image of the segmented flow pattern at the 30 cm point, b) obtained data at the 30 cm point with a data acquisition speed of 1 ms. The orange line presents the ADC value which is converted to p-NF concentration. | ||
This was followed by connecting concentrations at specific locations with residence times at the corresponding positions of the tubular reactor. The ADC values of absorbance were thus converted to p-NF concentrations using the data obtained from the calibration graph (Fig. S25†) and plotted vs. residence time (Fig. S26†). These concentrations were used to calculate the corresponding concentrations of p-NFA during the reaction applying [p-NFA]t = [p-NFA]0 − [p-NF]t. To show that the reaction is first order in [p-NFA] the Ln of [p-NFA]t was calculated and plotted vs. residence time (Fig. S27†). The slope of the plotted data was used to calculate the reaction rate constant to be 2.26 min−1 at 25 °C, assuming that the reaction is first order in p-NFA. The reaction has been studied under biphasic conditions in microreactors31–33 and under homogenous batch conditions,34 indicating a significant influence of the reaction conditions on the reaction rate constant. It is not surprising that the conversion of p-NFA is time dependent which is in turn inversely proportional to the flow rate. In addition, the reaction is dependent on segment size, since the smaller the segments the larger the interfacial surface area, resulting in better mass transfer.
The measurement of mass transfer between phases is an important tool in the characterization of a microreaction system. Mass transfer coefficients can be determined by knowing the lengths of the individual segments in the flow system.35 Since channel shape and structure influences mass transfer between immiscible phases in microreactors,36 such measurements are of importance for the rational design of microreactors. The design of the photometer described herein allows measuring the length of the segments in a biphasic system, being liquid–liquid or gas–liquid systems. This is due to the rapid sampling time of 1 ms and the difference in the refraction indexes of the two phases.37 The first derivative of the signal was calculated and summed with the average of the signal in order to determine the boundary between segments (Fig. S28 and S29†). Using eqn (1) where h is the length of the segment, F is the flow rate in mL min−1, t is time between data points in s and r2 is the square of the PFA tube radii, the length of the aqueous segments was calculated to be 3.5 mm. The calculation here is based on the assumption that segments have the same velocity as the total flow rate, since the small capillary numbers involved (<0.001) at which the liquid film surrounding the segment/segment body (if present) is negligibly thin.38
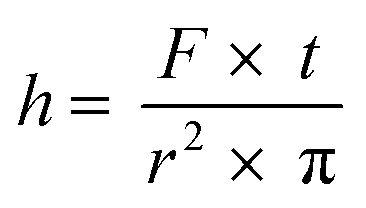 | (1) |
With the current photometer the flow cell can be readily moved along the PFA tube (see above) thus allowing the measurement of segment sizes at different residence times without disturbing the flow pattern during measurement. In principle, segment sizes and thus mass transfer coefficients can be derived for biphasic system where the sampling time is shorter than the time an individual segment spends in the optical path and the segment length is longer than the diameter of the optical fiber. Compared to the classical snapshot technique,39 this truly in-line measurement allows for real time monitoring of segment lengths and reactor performance with respect to mass transfer.
Conclusion
With an ever-increasing awareness of flow chemistry and process analytical technology tools, there is growing interest in the development of novel analytical tools to enable rapid and cost efficient in-line analysis of reactions under flow conditions. Herein we have presented the design of a flexible photometer system from readily available electronic components enabled by the MSP-EXP430G2 LaunchPad prototyping board and Energīa, an open source electronic platform accompanied by a large support community. This brings electronics and programming closer to the chemist and chemical engineer, allowing a faster development of novel process analytics, as seen from recently published studies.40 Our novel photometer system was used for concentration measurements at the outlet of glass micromixers and microreactors in order to perform residence time distribution measurements and for establishing the kinetics of chemical transformations in flow by performing concentration measurements at different residence times of the reactor. The latter approach is possible since in order to perform concentration measurements at different locations of the reactor the flow cell is simply moved up or down the PFA tubing. In summary, the novel system introduced herein appears to be an attractive tool for rapid and cost-efficient in-line concentration measurements, additionally enabling the determination of segment lengths in segmented flow regimes.Experimental
General information
Solvents and chemicals were obtained from standard commercial vendors and were used without any further purification unless otherwise noted. HPLC analysis (Shimadzu LC 20 AD) was carried out on a C18 reversed-phase analytical column (150 mm × 4.6 mm, particle size 5 μm) at 37 °C using mobile phases A (water/acetonitrile, 90:10 (v/v) + 0.1% TFA) and B (acetonitrile + 0.1% TFA) at a flow rate of 1.5 mL min−1. The following gradient was applied: linear increase from solution 30% B to 100% B in 5 min. GC-MS spectra were recorded using a Focus GC coupled with a DSQ II (EI, 70 eV). An HP5-MS column (30 m × 0.250 mm × 0.25 μm) was used with helium as carrier gas (1 mL min−1 constant flow). The injector temperature was set at 280 °C. After 1 min at 50 °C the temperature was increased in 25 °C min−1 steps up to 300 °C and kept at 300 °C for 4 min.
Photometer design
All the circuit board layouts were designed using the EAGLE 7.3.0 Light version, an open-access graphical software. CAD drawings of the Beam splitter cube and lens holder were done using the AutoCAD 2018 software. The Texas Instruments MSP430 LaunchPad platform and integrated circuits (IC) were purchased from Texas Instruments. LEDs, constant current LED driver modules, passive components and the required equipment for construction were purchased from a local electronics supplier. Data files for the printed circuit board assembly and CAD files for manufacturing the optics holder can be obtained on request from the authors.Step experiments
RTD experiments were performed using either step or pulse experiments. For the step experiments, a setup shown in Fig. S9† was used. The evaluated micromixers (Table 1) were connected via a PFA (1.6 mm o.d., 0.8 mm i.d.) tube (5 cm length) to the six-way valve. One feed was a 1 mM solution of RB dye in water, the second feed was pure water. Both feeds were pumped with the same flow rate (0.25, 0.50, 0.75 or 1.00 mL min−1) using continuous syringe pumps (Syrris Asia). Initially, the micromixer was purged with pure water and a baseline was recorded with the photometer. The measurement was started immediately upon changing from the water flow to the dye flow accomplished by switching the six-way-valve. The PFA tube was connected to the output of the micromixer and the flow cell was positioned as close as possible to the output. The timing of the measurements was set to every 1 ms for all experiments. After the values from one flow rate were recorded, flow rate values were changed on both pumps and the procedure was repeated.Pulse experiment
Pulse experiments were performed using the setup shown in Fig. S8.† The 13 μL loop was loaded with 1 mM solution of RB dye using the syringe. The microreactor was connected to the six-way valve using a 5 cm long PFA (1.6 mm o.d, 0.8 mm i.d.) tube. The flow rate on the syringe pump (Syrris Asia) was set (0.25, 0.50, 0.75 or 1.00 mL min−1), and the reactor was purged with pure water. The recording was started immediately upon switching the six-way-valve whereupon a 13 μL pulse of the dye was injected into the microreactor. The PFA tube was connected to the output of the microreactor, and the flow cell was positioned as close as possible to the output. After the values for one flow rate were recorded the flow rate values were changed on both pumps and the procedure was repeated. The timing of the measurements was depending on the evaluated microreactor and the used flow rate. For flow rates of 0.25 and 0.50 mL min−1 the recording time was set to every 10 ms and for flow rates of 0.75 and 1.00 mL min−1 timing was set to 1 ms. The exception was the Uniqsis microreactor (internal volume of 1.8 mL) where timing was set to 50 ms for the 0.50 and 0.75 mL min−1 runs and to 25 ms for the 1.00 mL min−1 experiment.Hydrolysis of p-nitrophenyl acetate
The flow setup consisted of two syringe pumps (Syrris Asia) to introduce a 2 M solution of sodium hydroxide and a 1 M solution of p-NFA in CH2Cl2 with flow rates of 100 μL min−1 and 50 μL min−1, respectively. Solutions were mixed in a standard T mixer (IDEX, 17.5 μL, 1.25 mm i.d.) to which a PFA tube (1.6 mm o.d, 0.8 mm i.d., 120 cm total length) was connected. The flow cell was positioned as close to the mixing point as possible (4 cm), see Fig. S24.† Initially, pure water and CH2Cl2 were mixed and a baseline recorded. Subsequently, the two reaction solutions were pumped and the recording was initiated. For each spatial point, 200 data points were recorded at 1 ms time intervals between data points. After recording at the length of 4 cm from the mixing point, the flow cell was moved along the PFA tube in increments of 5 cm and 200 data points where measured at each location. At the length of 70 cm from the mixing point, the measurements were finished. The output feed was analyzed to determine if any residual p-NFA was present. HPLC and GC-MS analysis confirmed complete conversion to p-nitrophenol and absence of any p-NFA in the CH2Cl2 layer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The CC FLOW project (Austrian Research Promotion Agency FFG No. 862766) is funded through the Austrian COMET Program by the Austrian Federal Ministry of Transport, Innovation, and Technology (BMVIT), the Austrian Federal Ministry of Science, Research and Economy (BMWFW) and by the State of Styria (Styrian Funding Agency SFG). The authors thank Dr. Heidrun Gruber-Wölfler and Manuel Maier (Institute for Process and Particle Engineering, Graz University of Technology) for their helpful advice.References
- K. A. Bakeev, Process Analytical Technology – Spectroscopic Tools and Implementation Strategies, John Wiley & Sons, 2nd edn, 2010 Search PubMed.
- (a) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef PubMed; (b) D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed; (c) R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2–25 CrossRef; (d) M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2015, 11, 1194–1219 CrossRef PubMed.
- (a) B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef PubMed; (b) M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC; (c) F. Fanelli, G. Parisi, L. Degennaro and R. Luisi, Beilstein J. Org. Chem., 2017, 13, 520–542 CrossRef PubMed.
- (a) J. Yue, J. C. Schouten and T. A. Nijhuis, Ind. Eng. Chem. Res., 2012, 51, 14583–14609 CrossRef; (b) S. V. Ley, D. E. Fitzpatrick, R. J. Ingham and R. M. Myers, Angew. Chemie., Int. Ed., 2015, 54, 3449–3464 CrossRef PubMed; (c) B. J. Reizman and K. F. Jensen, Acc. Chem. Res., 2016, 49, 1786–1796 CrossRef PubMed; (d) D. C. Fabry, E. Sugiono and M. Rueping, React. Chem. Eng., 2016, 1, 129–133 RSC; (e) D. E. Fitzpatrick and S. V. Ley, React. Chem. Eng., 2016, 1, 629–635 RSC; (f) V. Sans and L. Cronin, Chem. Soc. Rev., 2016, 45, 2032–2043 RSC; (g) C. A. Shukla and A. A. Kulkarni, Beilstein J. Org. Chem., 2017, 13, 960–987 CrossRef PubMed.
- (a) A. E. Cervera-Padrell, J. P. Nielsen, M. Jønch Pedersen, K. Müller Christensen, A. R. Mortensen, T. Skovby, K. Dam-Johansen, S. Kiil and K. V. Gernaey, Org. Process Res. Dev., 2012, 16, 901–914 CrossRef; (b) J. Wiss, M. Länzlinger and M. Wermuth, Org. Process Res. Dev., 2005, 9, 365–371 CrossRef; (c) J. Wiss and G. Ermini, Org. Process Res. Dev., 2006, 10, 1282–1286 CrossRef.
- (a) K. L. A. Chan, S. Gulati, J. B. Edel, A. J. de Mello and S. G. Kazarian, Lab Chip, 2009, 9, 2909 RSC; (b) A. Perro, G. Lebourdon, S. Henry, S. Lecomte, L. Servant and S. Marre, React. Chem. Eng., 2016, 1, 577–594 RSC; (c) J. S. Moore and K. F. Jensen, Angew. Chem., Int. Ed., 2014, 53, 470–473 CrossRef PubMed.
- (a) T. A. Hamlin and N. E. Leadbeater, Beilstein J. Org. Chem., 2013, 9, 1843–1852 CrossRef PubMed; (b) E. Cao, M. Sankar, S. Firth, K. F. Lam, D. Bethell, D. K. Knight, G. J. Hutchings, P. F. McMillan and A. Gavriilidis, Chem. Eng. J., 2011, 167, 734–743 CrossRef; (c) S. Mozharov, A. Nordon, D. Littlejohn, C. Wiles, P. Watts, P. Dallin and J. M. Girkin, J. Am. Chem. Soc., 2011, 133, 3601–3608 CrossRef PubMed.
- (a) J. Bart, A. J. Kolkman, A. J. O. De Vries, K. Koch, P. J. Nieuwland, H. J. W. G. Janssen, J. P. J. M. Van Bentum, K. A. M. Ampt, F. P. J. T. Rutjes, S. S. Wijmenga, H. J. G. E. Gardeniers and A. P. M. Kentgens, J. Am. Chem. Soc., 2009, 131, 5014–5015 CrossRef PubMed; (b) B. Ahmed-Omer, E. Sliwinski, J. P. Cerroti and S. V. Ley, Org. Process Res. Dev., 2016, 20, 1603–1614 CrossRef; (c) A. J. Oosthoek-de Vries, J. Bart, R. M. Tiggelaar, J. W. G. Janssen, P. J. M. van Bentum, H. J. G. E. Gardeniers and A. P. M. Kentgens, Anal. Chem., 2017, 89, 2296–2303 CrossRef PubMed; (d) M. V. Gomez and A. De La Hoz, Beilstein J. Org. Chem., 2017, 13, 285–300 CrossRef PubMed.
- (a) J. M. Hollas, Modern Spectroscopy, Wiley-VCH Verlag GmbH & Co. KGaA, 4th edn., 2003 Search PubMed; (b) B. Wardle, Principles and Applications of Photochemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2009 Search PubMed.
- (a) H. Bi, C. M. Duarte, M. Brito, V. Vilas-Boas, S. Cardoso and P. Freitas, Biosens. Bioelectron., 2016, 85, 568–572 CrossRef PubMed; (b) M. Bai, H. Huang, J. Hao, J. Zhang, H. Wu and B. Qu, Sci. Rep., 2015, 5, 10476 CrossRef PubMed.
- K. M. G. de Lima, Microchem. J., 2012, 103, 62–67 CrossRef; M. Sorouraddin, M. Saadati and H. Karimi, Sens. Actuators, B, 2013, 188, 73–77 CrossRef.
- (a) N. Wang, F. Tan, Y. Zhao, C. C. Tsoi, X. Fan, W. Yu and X. Zhang, Sci. Rep., 2016, 6, 28928 CrossRef PubMed; (b) N. Wang, X. Zhang, B. Chen, W. Song, N. Y. Chan and H. L. W. Chan, Lab Chip, 2012, 12, 3983–3990 RSC.
- (a) F. Pena-Pereira, I. Costas-Mora, V. Romero, I. Lavilla and C. Bendicho, Trends Anal. Chem., 2011, 30, 1637–1648 CrossRef; (b) G. J. Lichtenegger, V. Tursic, H. Kitzler, K. Obermaier, J. G. Khinast and H. Gruber-Wölfler, Chem. Ing. Tech., 2016, 88, 1518–1523 CrossRef.
- H. Bi, C. M. Duarte, M. Brito, V. Vilas-Boas, S. Cardoso and P. Freitas, Biosens. Bioelectron., 2016, 85, 568–572 CrossRef PubMed.
- F. Benito-Lopez, W. Verboom, M. Kakuta, J. G. E. Gardeniers, R. J. M. Egberink, E. R. Oosterbroek, A. van den Berg and D. N. Reinhoudt, Chem. Commun., 2005, 2857–2859 RSC.
- J. Yue, F. H. Falke, J. C. Schouten and T. A. Nijhuis, Lab Chip, 2013, 13, 4855–4863 RSC.
- J. Aubin, M. Ferrando and V. Jiricny, Chem. Eng. Sci., 2010, 65, 2065–2093 CrossRef.
- (a) M. Zheng and M. Mackley, Chem. Eng. Sci., 2008, 63, 1788–1799 CrossRef; (b) E. B. Nauman and A. Nigam, Ind. Eng. Chem. Res., 2005, 44, 5031–5035 CrossRef; (c) V. D. Dang, Ind. Eng. Chem. Fundam., 1984, 23, 326–331 CrossRef.
- N. Hawbaker, E. Wittgrove, B. Christensen, N. Sach and D. G. Blackmond, Org. Process Res. Dev., 2016, 20, 465–473 CrossRef.
- (a) V. Sans, N. Karbass, M. I. Burguete, E. García-Verdugo and S. V. Luis, RSC Adv., 2012, 2, 8721–8728 RSC; (b) P. Danckwerts, Chem. Eng. Sci., 1953, 2, 1–13 CrossRef; (c) S. R. L. Gobert, S. Kuhn, L. Braeken and L. C. J. Thomassen, Org. Process Res. Dev., 2017, 21, 531–542 CrossRef.
- O. Levenspiel, Chemical Reaction Engineering, John Wiley & Sons, 3rd edn, 1999 Search PubMed.
- http://www.uniqsis.com .
- http://www.ti.com/tool/MSP-EXP430G2 .
- http://energia.nu/ .
- https://processing.org/ .
- D. Bošković and S. Loebbecke, Chem. Eng. J., 2008, 135, 138–146 CrossRef.
- (a) P. V. Danckwerts, Chem. Eng. Sci., 1995, 50, 3857–3866 CrossRef; (b) S. Schwolow, J. Hollmann, B. Schenkel and T. Röder, Org. Process Res. Dev., 2012, 16, 1513–1522 CrossRef.
- B. Gutmann, M. Köckinger, G. Glotz, T. Ciaglia, E. Slama, M. Zadravec, S. Pfanner, M. C. Maier, H. Gruber-Wölfler and C. O. Kappe, React. Chem. Eng., 2017, 2, 919–927 Search PubMed.
- M. Günther, S. Schneider, J. Wagner, R. Gorges, T. Henkel, M. Kielpinski, J. Albert, R. Bierbaum and J. M. Köhler, Chem. Eng. J., 2004, 101, 373–378 CrossRef.
- https://marketing.blacktrace.com/acton/attachment/28584/f-02bc/1/-/-/-/-/3200401%20Micromixer%20Chip.pdf .
- (a) P. Plouffe, D. M. Roberge, J. Sieber, M. Bittel and A. Macchi, Chem. Eng. J., 2016, 285, 605–615 CrossRef; (b) P. Plouffe, D. M. Roberge and A. Macchi, Chem. Eng. J., 2016, 300, 9–19 CrossRef.
- B. Ahmed-Omer, D. Barrow and T. Wirth, Chem. Eng. J., 2008, 135, 280–283 CrossRef.
- B. Ahmed, D. Barrow and T. Wirth, Adv. Synth. Catal., 2006, 348, 1043–1048 CrossRef.
- (a) E. Sacher and K. J. Laidler, Can. J. Chem., 1964, 42, 2404–2409 CrossRef; (b) H. J. Goren, Eur. J. Biochem., 1974, 41, 263–272 CrossRef PubMed.
- (a) J. R. Burns and C. Ramshaw, Chem. Eng. Res. Des., 1999, 77, 206–211 CrossRef; (b) J. A. Wesselingh and A. M. Bollen, Chem. Eng. Res. Des., 1999, 77, 89–96 CrossRef.
- (a) P. Sobieszuk, P. Cygański and R. Pohorecki, Chem. Eng. Res. Des., 2010, 88, 263–269 CrossRef; (b) J. Tan, Y. C. Lu, J. H. Xu and G. S. Luo, Chem. Eng. J., 2012, 181–182, 229–235 CrossRef; (c) J. Jovanović, E. V. Rebrov, T. A. Nijhuis, M. T. Kreutzer, V. Hessel and J. C. Schouten, Ind. Eng. Chem. Res., 2012, 51, 1015–1026 CrossRef.
- (a) B. M. A. Wolffenbuttel, T. A. Nijhuis, A. Stankiewicz and J. A. Moulijn, Meas. Sci. Technol., 2002, 13, 1540–1544 CrossRef; (b) T. Kraus, A. Guenther, N. De Mas, M. A. Schmidt and K. F. Jensen, Exp. Fluids, 2004, 36, 819–832 CrossRef.
- P. Aussillous and D. Quere, Phys. Fluids, 2000, 12, 2367–2371 CrossRef.
- (a) B. Xu, W. Cai, X. Liu and X. Zhang, Chem. Eng. Res. Des., 2013, 91, 1203–1211 CrossRef; (b) J. Tan, Y. C. Lu, J. H. Xu and G. S. Luo, Chem. Eng. J., 2012, 181–182, 229–235 CrossRef; (c) J. H. Xu, J. Tan, S. W. Li and G. S. Luo, Chem. Eng. J., 2008, 141, 242–249 CrossRef.
- (a) B. Yang, M. C. Patsavas, R. H. Byrne and J. Ma, Mar. Chem., 2014, 160, 75–81 CrossRef; (b) J. Shen, D. Yang, Y. Liu, S. Qin, J. Zhang, J. Sun, C. Liu, C. Liu, X. Zhao, C. Chu and R. Liu, Org. Lett., 2014, 16, 350–353 CrossRef PubMed; (c) J. P. Grinias, J. T. Whit, E. D. Guetschow and R. T. Kennedy, J. Chem. Educ., 2016, 93, 1316–1319 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8re00070k |
| This journal is © The Royal Society of Chemistry 2018 |