
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLncRNA CASC2 inhibits autophagy and promotes apoptosis in non-small cell lung cancer cells via regulating the miR-214/TRIM16 axis
Qian Lia,
Kai Chena,
Rong Donga and
Hengxiao Lu*b
aDepartment of Respiratory, People's Hospital of Rizhao, 276800, China
bDepartment of Thoracic Surgery, Weifang People's Hospital, No.151, Guangwen Street, Kuiwen District, Weifang, 261041, China. E-mail: xheng45698@sina.com; Tel: +86-0536-8192133
First published on 5th December 2018
Abstract
Background: Dysregulated long noncoding RNAs (lncRNAs) have been frequently observed in various cancers including non-small cell lung cancer (NSCLC) and are closely associated with cancer progression. Previous studies also found that low expression of lncRNA cancer susceptibility candidate 2 (CASC2) functioned as a tumor suppressor in NSCLC. Our study aimed to explore the detailed molecular mechanism of CASC2 involved in NSCLC progression. Methods: The expressions of CASC2, tripartite motif-containing protein 16 (TRIM16) and miR-214 in NSCLC tissues and cells were detected by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) or western blot. Flow cytometry analysis was performed to evaluate apoptosis. Autophagy was assessed using green fluorescent protein microtubule-associated protein 1 light chain 3α (GFP-LC3) puncta analysis, acridine orange (AO) staining and western blot. Luciferase reporter assay, RNA immunoprecipitation (RIP), RNA pull-down and immunofluorescence staining were employed to explore the association between CASC2, TRIM16 and miR-214. Results: CASC2 and TRIM16 expressions were significantly downregulated and miR-214 expression was dramatically upregulated in NSCLC tissues and cells. Overexpression of CASC2 induced apoptosis and inhibited autophagy in NSCLC cells. miR-214 was bound to CASC2 and its knockdown reversed the regulatory effect of CASC2 inhibition on apoptosis and autophagy in NSCLC cells. Moreover, TRIM16 was validated as a target of miR-214 and its interference attenuated miR-214 knockdown-mediated promotion of apoptosis and inhibition of autophagy. Besides, CASC2 enhanced TRIM16 expression through functioning as a competing endogenous RNA (ceRNA) for miR-214 in NSCLC cells. Conclusion: lncRNA CASC2 inhibited autophagy and promoted apoptosis in NSCLC cells via regulating the miR-214/TRIM16 axis, shedding light on the mechanism underlying NSCLC carcinogenesis.
1. Introduction
Lung cancer, one of the most aggressive malignancies, is the leading cause of cancer-associated deaths all over the world, accounting for almost 610![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 deaths of the 2814200 total cancer deaths in 2015.1 As the most common type of lung cancer, non-small cell lung cancer (NSCLC) accounts for nearly 85% of all lung cancer cases.2 Despite the tremendous improvements in early diagnosis and clinical treatment of NSCLC over the past decades, the prognosis of NSCLC patients remains unsatisfactory, with a lower than 15% 5 year overall survival rate of NSCLC patients.3 Thus, it is imperatively needed to better understand the mechanisms involved in the development and progression of NSCLC and to develop novel therapeutic approaches for the treatment of NSCLC patients.
200 deaths of the 2814200 total cancer deaths in 2015.1 As the most common type of lung cancer, non-small cell lung cancer (NSCLC) accounts for nearly 85% of all lung cancer cases.2 Despite the tremendous improvements in early diagnosis and clinical treatment of NSCLC over the past decades, the prognosis of NSCLC patients remains unsatisfactory, with a lower than 15% 5 year overall survival rate of NSCLC patients.3 Thus, it is imperatively needed to better understand the mechanisms involved in the development and progression of NSCLC and to develop novel therapeutic approaches for the treatment of NSCLC patients.
Emerging evidence has documented the roles of noncoding RNAs (ncRNAs), including long ncRNAs (lncRNAs) and microRNAs (miRNAs), in malignant transformation as well as cancer development.4 LncRNAs are a class of recently recognized ncRNA transcripts longer than 200 nucleotides, which play functional roles in regulating and controlling various cellular processes, including cell proliferation, differentiation, apoptosis, autophagy and migration.5 It has been shown that dysregulated lncRNAs are frequently observed in various cancers including NSCLC and are closely associated with cancer progression, acting as oncogenic or tumor-suppressive genes.6 LncRNA cancer susceptibility candidate 2 (CASC2), located at chromosome 10q26, was firstly identified as a downregulated gene in human endometrial cancer.7 Moreover, CASC2 has been reported to play a tumor-suppressive role in regulating cell proliferation and metastasis in various cancers, such as hepatocellular carcinoma, breast cancer and bladder cancer.8–10 Notably, previous studies also found that low expression of CASC2 functioned as a tumor suppressor in NSCLC.11 However, the detailed molecular mechanism of CASC2 involved in NSCLC progression remains to be further explored.
miRNAs are endogenous, highly conserved ncRNAs with 18–25 nucleotides in length that post-transcriptionally regulate gene expression by binding to the 3′ untranslated regions (UTRs) of their targets.12 miRNAs have been reported to play crucial regulatory roles in various physiological and pathological processes including cell differentiation, proliferation, apoptosis, and metastasis.13 Moreover, convincing evidence has demonstrated that aberrantly expressed miRNAs contribute to the initiation and development of human cancers.14 miR-214, a vertebrate-specific family of miRNA precursor, has been demonstrated to be deregulated in malignancies and shows contrasting roles as oncogenic or tumor suppressor.15 It has been previously demonstrated that miR-214 expression was elevated in lung cancer and promoted the progression of lung cancer by acting as an oncogene.16,17 Recently, competing endogenous RNA (ceRNA) hypothesis suggests that lncRNAs could function as miRNA sponges to prevent miRNAs from binding to their regulatory targets, thus participating in the pathogenesis and development of cancers.18 Nevertheless, whether CASC2 could interact with miR-214 to regulate the progression of NSCLC remains largely unknown.
In the present study, we investigated the effects of CASC2 on apoptosis and autophagy of NSCLC cells and explored its underlying molecular mechanism.
2. Materials and methods
2.1 Patients and tissue samples
A total of 21 pairs of NSCLC tissues and adjacent normal tissues were collected from patients who had undergone surgery between 2016 and 2017 in respiratory medical of Rizhao People's Hospital. No patient had received radiotherapy, chemotherapy or other systemic treatments before surgery resection. NSCLC patients were diagnosed by two experienced pathologists according to the World Health Organization criteria.19 All tissue samples were snap-frozen immediately into liquid nitrogen following collection and stored at −80 °C for use. Our study was approved by the Ethic Committee of People's Hospital of Rizhao in accordance with the Declaration of Helsinki Principles. Informed consents were obtained from all participants.2.2 Cell culture and transfection
Human NSCLC cell lines (A549 and H1299) and human bronchial epithelial cell line (16HBE) were obtained from the Institute of Biochemistry and Cell Biology of the Chinese Academy of Sciences (Shanghai, China). All cells were propagated in Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Inc.), 100 U ml−1 penicillin G/streptomycin (Sigma, St. Louis, MO, USA) at 37 °C in a humidified atmosphere containing 5% CO2.pcDNA-CASC2 (CASC2), pcDNA-tripartite motif-containing protein (TRIM) 16 (TRIM16), pcDNA empty vector (pcDNA), siRNA specifically targeting CASC2 (si-CASC2), siRNA specifically targeting TRIM16 (si-TRIM16), siRNA negative control (si-NC), miR-214 mimic (miR-214), mimic negative control (miR-NC), miR-214 inhibitor (anti-miR-214), inhibitor negative control (anti-miR-NC) were synthesized by GenePharma Co. Ltd., (Shanghai, China). A549 and H1299 cells were seeded into 6-well plates at a density of 2 × 106 cells per well and transfected with these above plasmids or nucleotides at 70–80% confluence using lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol.
2.3 Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated from tissue samples or treated cells with TRIzol® reagent (Invitrogen) and RNA concentration was determined using a Nanodrop® ND-1000 spectrophotometer (Thermo Fisher Scientific, Inc.). For the detection of miR-214 expression, total RNA was reversely transcribed into complementary DNA (cDNA) using a TaqMan® MicroRNA Reverse Transcription kit (Applied Biosystems, Foster City, CA, USA) and qPCR was performed using the TaqMan MicroRNA Assay kit (Applied Biosystems), with U6 small nuclear RNA (snRNA) as an internal control. For the measurement of CASC2 and TRIM16 mRNA expressions, cDNA was synthesized from total RNA using PrimeScript™ RT Reagent Kit (Promega, Madison, WI, USA) (Takara, Dalian, China) and a SYBR Green qPCR Master Mix (Takara) was used to the mRNA level, with GAPDH as an endogenous control. All PCR reaction was conducted on an Applied Biosystems 7500 thermocycler (Thermo Fisher Scientific, Inc.). The relative expression levels were calculated using the 2−ΔΔCt method.2.4 Green fluorescent protein microtubule-associated protein 1 light chain 3α (GFP-LC3) analysis
The transfected A549 and H1299 cells were seeded into 6-well plates and co-transfected with GFP-LC3-expressing plasmids (Hanbio Co., LTD, China), and CASC2, TRIM16, pcDNA, anti-miR-214, anti-miR-NC, si-NC + anti-miR-214, si-CASC2 + anti-miR-214, miR-NC + TRIM16, or miR-214 + TRIM16 using Lipofectamine (Invitrogen). Then GFP-LC3-positive punctate cells were visualized by confocal microscope (magnification, ×100; Olympus, Tokyo, Japan) and the percentage of GFP-LC3-positive cells were calculated. Cells with five or more intense GFP-LC3 puncta were considered as autophagic.2.5 Acridine orange (AO) staining
Autophagy is a lysosomal degradation pathway for cytoplasmic material. The formation of autophagic vesicles was measured by AO staining. The transfected A549 and H1299 cells were washed with PBS and then stained with 1 μg ml−1 AO (Sigma) for 20 min at 37 °C in the dark. Subsequently, cells were observed under a confocal microscope. The autophagic lysosomes displayed as orange/red fluorescent cytoplasmic vesicles, but the cytoplasm and nucleolus were green because of the difference of acidity.2.6 Western blot analysis
Total protein was extracted from treated cells with RIPA buffer (Wanlei Biotechnology, Shanghai, China) containing a protease inhibitor cocktail (Roche, Mannheim, Germany). The protein samples (30 μg per lane) were loaded onto 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels and electro-transferred onto polyvinylidene difluoride membranes (PVDF) (Millipore, Billerica, MA, USA). Then, the membranes were saturated with 5% non-fat milk in Tris-buffered saline with Tween (TBST) for 2 h at room temperature and separately incubated at 4 °C overnight with primary antibodies against p62 (1:1000 dilution; Cell Signaling Technology, Danvers, MA, USA), autophagy related protein-5 (Atg-5) (1:1000 dilution; Cell Signaling Technology), LC3 (1:1000 dilution; Abcam, Cambridge, MA, USA), TRIM16 (1:500 dilution; Abcam) and GAPDH (1:2000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), followed by horseradish peroxidase (HRP)-conjugated secondary antibody IgG (1:2000 dilution; Cell Signaling Technology). The protein signals were visualized using an enhanced chemiluminescence detection system (Thermo Fisher Scientific, Inc.), with GAPDH as a loading control.
2.7 Apoptosis analysis by flow cytometry
Cell apoptosis was assessed using Annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection kit (BD Biosciences, Franklin Lakes, NJ, USA). A549 and H1299 cells were seeded into 6-well plates overnight and transfected with CASC2, TRIM16, pcDNA, anti-miR-214, anti-miR-NC, si-NC + anti-miR-214, si-CASC2 + anti-miR-214, miR-NC + TRIM16, or miR-214 + TRIM16. Following 48 h of transfection, A549 and H1299 cells were collected and digested with trypsin, washed with ice-cold PBS and resuspended in 200 μl binding buffer. Then cells were labeled with 10 μl annexin V-FITC and 5 μl propidium iodide (PI) for in the dark 15 min at room temperature. The cell apoptotic rates were analyzed by flow cytometry using a flow cytometer (FACScan; BD Biosciences).2.8 Luciferase reporter assay
A wild-type (WT) fragment of CASC2 harboring the miR-214 binding site and its mutated (MUT) seed sequence were purchased from Guangzhou RiboBio Co., Ltd. (Guangzhou, China) and cloned into the pMirGLO reporter vector (Promega, Madison, WI, USA), namely CASC2-WT and CASC2-MUT. Similarly, the luciferase reporter plasmids containing the WT or MUT 3′UTR of TRIM16 were also constructed and named as TRIM16-WT and TRIM16-MUT. For the luciferase reporter assay, 293T cells were seeded into 24-well plates and co-transfected with 100 ng WT or MUT luciferase reporter, together with 20 ng Renilla luciferase vector (Promega) and 100 nM miR-214 or miR-NC using lipofectamine 2000 (Invitrogen). At 48 h post-transfection, the luciferase activity was measured using the dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions.2.9 RNA immunoprecipitation (RIP)
The EZ-Magna RIP Kit (Millipore) was applied to conduct the RIP assay. A549 cells at 80% confluence were collected and lysed in complete RIP lysis buffer. Then, the supernatant of RIP lysate was incubated with RIP buffer containing magnetic beads conjugated with human anti-Ago2 or anti-Ago1 antibodies (Cell Signaling Technology) or negative control IgG (Cell Signaling Technology). After incubated at 4 °C overnight, samples were incubated with proteinase K with shaking to digest proteins and the coprecipitated RNA was isolated and subjected to RT-qPCR analysis.2.10 RNA pull-down assay with biotinylated CASC2
The biotinylated DNA probe complementary to CASC2 was amplified by PCR using T7-containing primer (Roche, Basel, Switzerland) and then inserted into GV394 (Genechem, Shanghai, China). The resultant plasmids DNAs were linearized using restriction enzyme XhoI. Biotin-labeled RNAs were transcribed with Biotin RNA Labeling Mix (Roche) and T7 RNA polymerase (Takara). The products were treated with RNase-free DNase I (Roche) and purified with the RNeasy Mini Kit (Qiagen, Valencia, CA, USA). RNA were extracted for RT-qPCR analysis.2.11 RNA pull-down assay with biotinylated miR-214
A549 cells were transfected with biotinylated miR-214 (bio-miR-214-WT or bio-miR-214-MUT), collected 48 h after transfection and lysed in 1 ml of lysis buffer (Promega). The cell lysates were incubated with M-280 streptavidin magnetic beads (Invitrogen) and bound RNAs were purified using TRIzol reagent (Invitrogen) for further RT-qPCR analysis.2.12 Immunofluorescence staining
A549 cells were seeded into 24-well plates on slide overnight and transfected with miR-214, anti-miR-214, or matched controls. After 48 h of transfection, the cells were fixed with 4% paraformaldehyde for 10 min at room temperature, followed by permeabilization with 0.1% Triton X-100 for 10 min. After being blocked with 5% bovine serum albumin for 30 min at 37 °C, cells were incubated overnight at 4 °C with primary antibodies against TRIM16 (1:500 dilution; Abcam) and then incubated with CY3-conjugated secondary antibodies IgG (1:1000 dilution; Boster, Wuhan, China) for 1 h at 37 °C. The nucleus was stained with 4′,6-diamidino-2-phenylindole (DAPI; Sigma) for 15 min. The fluorescence images were obtained using a LSM 5 Pa Laser Scanning Microscope (Zeiss Germany, Oberkochen, Germany) and merged by Image-Pro Plus (Media Cybernetics, Bethesda, MD, USA).
2.13 Statistical analysis
All data were showed as mean ± standard deviation (SD). All statistical analyses were performed using SPSS 17.0 software (SPSS, Chicago, IL, USA). Statistical differences were analyzed by two-tailed Student's t-test or one-way analysis of variance (ANOVA). A value of P less than 0.05 was considered to be statistically significant.3. Results
3.1 CASC2 overexpression induced apoptosis and impeded autophagy in NSCLC cells
The expression profile of CASC2 in 21 paired NSCLC tissues and adjacent normal tissues were initially examined by RT-qPCR. The result disclosed that CASC2 expression was aberrantly downregulated in NSCLC tissues in comparison with normal tissues (Fig. 1A). Moreover, the expression of CASC2 was determined in NSCLC cell lines and human bronchial epithelial cell line (16HBE). As compared with 16HBE cells, A549 and H1299 cells exhibited evidently reduction of CASC2 expression (Fig. 1B). To characterize the effects of CASC2 on apoptosis and autophagy in NSCLC, A549 and H1299 cells were transfected with CASC2 or pcDNA. As displayed in Fig. 1C, CASC2 expression was distinctly enhanced by CASC2 transfection in A549 and H1299 cells with respect to pcDNA group. Flow cytometry analysis presented that the apoptotic rate of A549 and H1299 cells transfected with CASC2 was greatly enhanced versus that in pcDNA-introduced cells (Fig. 1D). During the process of autophagy, LC3 is recruited to the autophagosomes and thus fluorescent GFP-LC3 spots can be used to monitor the autophagic flux.20 As shown in Fig. 1E, the percentage of GFP-LC3 positive cells were substantially reduced following CASC2 overexpression in A549 and H1299 cells. Moreover, western blot analysis revealed that p62 level was notably increased but Atg-5 expression and the ratio of LC3-II/LC3-I were markedly diminished in the CASC2-overexpressing group in A549 and H1299 cells with respect to pcDNA group (Fig. 1F). Besides, the formation of autophagic vesicles was measured in A549 and H1299 cells by AO staining. Results showed that addition of CASC2 decreased the bright orange/red vacuoles in A549 and H1299 cells (Fig. 1G). These results demonstrated that CASC2 functioned as a tumor suppressor in NSCLC cells by promoting apoptosis and blocking autophagy.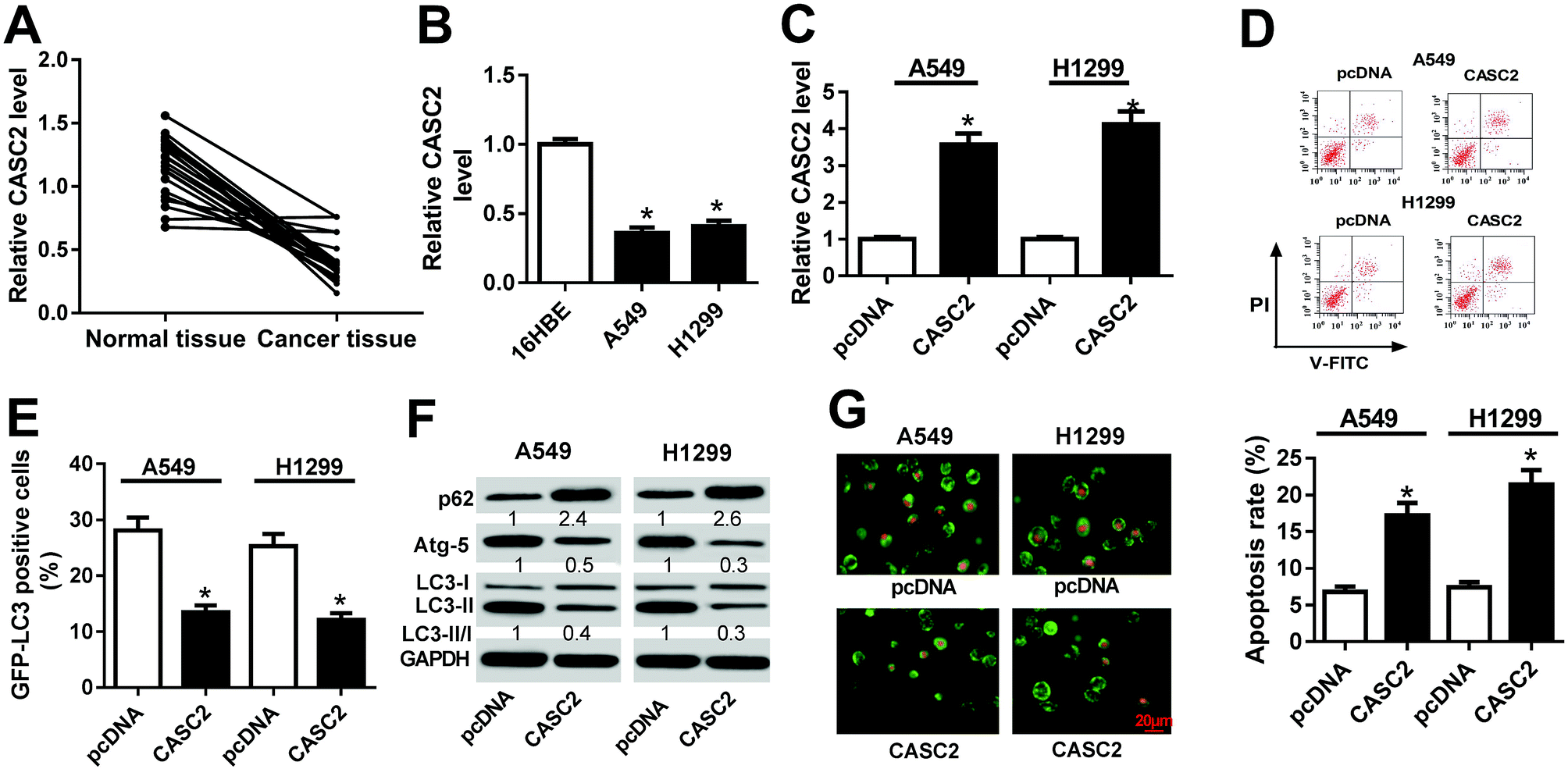 | ||
| Fig. 1 CASC2 overexpression induced apoptosis and impeded autophagy in NSCLC cells. (A) The expression of CASC2 in 21 NSCLC tissues and adjacent normal tissues was detected by RT-qPCR. (B) The expression of CASC2 in human bronchial epithelial cell line (16HBE) and NSCLC cell lines (A549 and H1299) was detected by RT-qPCR. (C) The expression of CASC2 in A549 and H1299 transfected with CASC2 or pcDNA was measured by RT-qPCR. (D) Flow cytometry analysis was conducted to evaluate the apoptosis of A549 and H1299 cells after transfection with CASC2 or pcDNA. (E) GFP-LC3 analysis was performed to monitor the autophagic flux in CASC2 or pcDNA-introduced A549 and H1299 cells. (F) Western blot was performed to examine the protein levels of p62, Atg-5, LC3-I and LC3-II in A549 and H1299 cells with CASC2 or pcDNA transfection. (G) AO staining was conducted to analyze the formation of autophagic vacuoles in A549 and H1299 cells with CASC2 or pcDNA transfection. *P < 0.05. | ||
3.2 CASC2 directly interacted with miR-214 in NSCLC cells
To explore the underlying mechanism of CASC2 functioning in NSCLC cells, bioinformatics analyses were performed to predict the potential targets of CASC2. The prediction analysis showed that CASC2 contained the potential binding sites of miR-214 (Fig. 2A). To confirm the prediction, we cloned the WT or MUT fragment of CASC2 containing the predicted binding sites into the luciferase reporter vectors and transfected into 293T cells. The luciferase reporter assay demonstrated that ectopic expression of miR-214 led to a significant decrease of luciferase activity in 293T cells transfected with CASC2-WT, but not in those transfected with CASC2-MUT (Fig. 2B). Moreover, RIP assay revealed that CASC2 and miR-214 were remarkably enriched in the Ago2 pellet in contrast to IgG pellet (Fig. 2C). To further confirm the direct interaction between CASC2 and miR-214, RNA pull-down assay with biotin-labeled miR-214 was performed to capture CASC2 using M-280 streptavidin magnetic beads from 549 cells transfected with bio-miR-214-WT or bio-miR-214-MUT. As shown in Fig. 2D, CASC2 was pulled down in the cell lysates of 549 cells transfected with bio-miR-214-WT compared to those transfected with biotin-labeled scrambled controls (bio-NC), whereas miR-214-MUT with mutant binding sites of CASC2 resulted in the inability of miR-214 to pull down CASC2. In addition, the inverse pull-down system using a biotin-labeled specific CASC2 probe was performed to confirm whether CASC2 could pull down miR-214 and the results demonstrated that miR-214 was specifically enriched in the CASC2 pulled down pellet compared with biotin-labeled scrambled controls (Fig. 2D). Therefore, these results demonstrated that CASC2 could direct bind to miR-214 in NSCLC cells.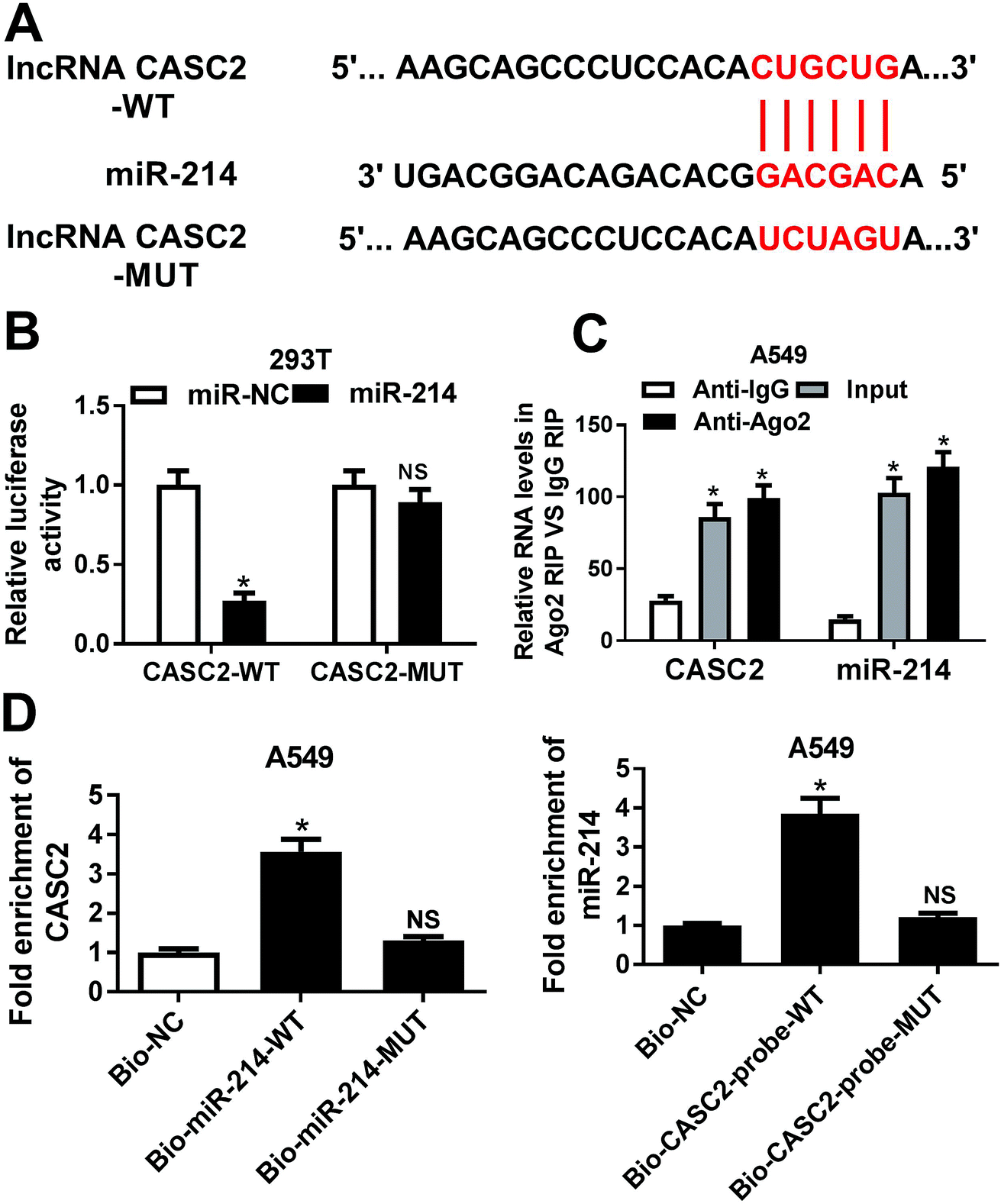 | ||
| Fig. 2 The interaction between CASC2 and miR-214 in NSCLC cells. (A) Sequence alignment showing the binding sites between CASC2 and miR-214. (B) Luciferase activity was measured by luciferase reporter assay in 293T cells co-transfected with CASC2-WT or CASC2-MUT and miR-214 or miR-NC. (C) Amount of CASC2 and miR-214 bound to Ago2 or IgG detected by RT-qPCR after RIP. (D) A549 cells were transfected with bio-miR-214-WT, bio-miR-214-MUT, bio-NC. At 48 h post-transfection, cells were collected and the level of CASC2 in the precipitated samples was detected by RT-qPCR. miR-214 expression in the cell lysate of A549 cells pulled down by biotinylated CASC2 probe was detected by RT-qPCR. *P < 0.05. | ||
3.3 CASC2 knockdown partially reversed miR-214 inhibition-mediated promotion of apoptosis and suppression of autophagy in NSCLC cells
RT-qPCR results hinted that miR-214 expression was significantly increased in 21 NSCLC tissues and NSCLC cells (A549 and H1299 cells) compared with normal tissues and 16HBE cells, respectively (Fig. 3A and B). To explore the regulatory effects of CASC2 on the functional roles of miR-214 in NSCLC progression, rescue experiments were performed in A549 and H1299 cells by transfecting with anti-miR-214, anti-miR-NC, or combined with si-CASC2 or si-NC. Flow cytometry analysis proved that A549 and H1299 cells transfected with anti-miR-214 showed an evident increase of the apoptotic rates compared with anti-miR-NC-transfected cells, which was effectively abolished following CASC2 knockdown (Fig. 3C). GFP-LC3 analysis revealed that suppression of miR-214 successfully reduced the percentage of GFP-LC3 positive cells in A549 and H1299 cells, while CASC2 depletion remarkably restored the reduction of the percentage of GFP-LC3 positive cells induced by anti-miR-214 (Fig. 3D). Consistently, western blot analysis manifested that miR-214 inhibitor apparently augmented the protein level of p62, an index of autophagic degradation, but declined the expression of Atg-5 and ratio of LC3-II/LC3-I, useful markers of autophagy, in A549 and H1299 cells relative to anti-miR-NC group, which was partially overturned by CASC2 silencing (Fig. 3E). Therefore, these data suggested that CASC2 knockdown partially reversed miR-214 inhibition-mediated promotion of apoptosis and suppression of autophagy in NSCLC cells.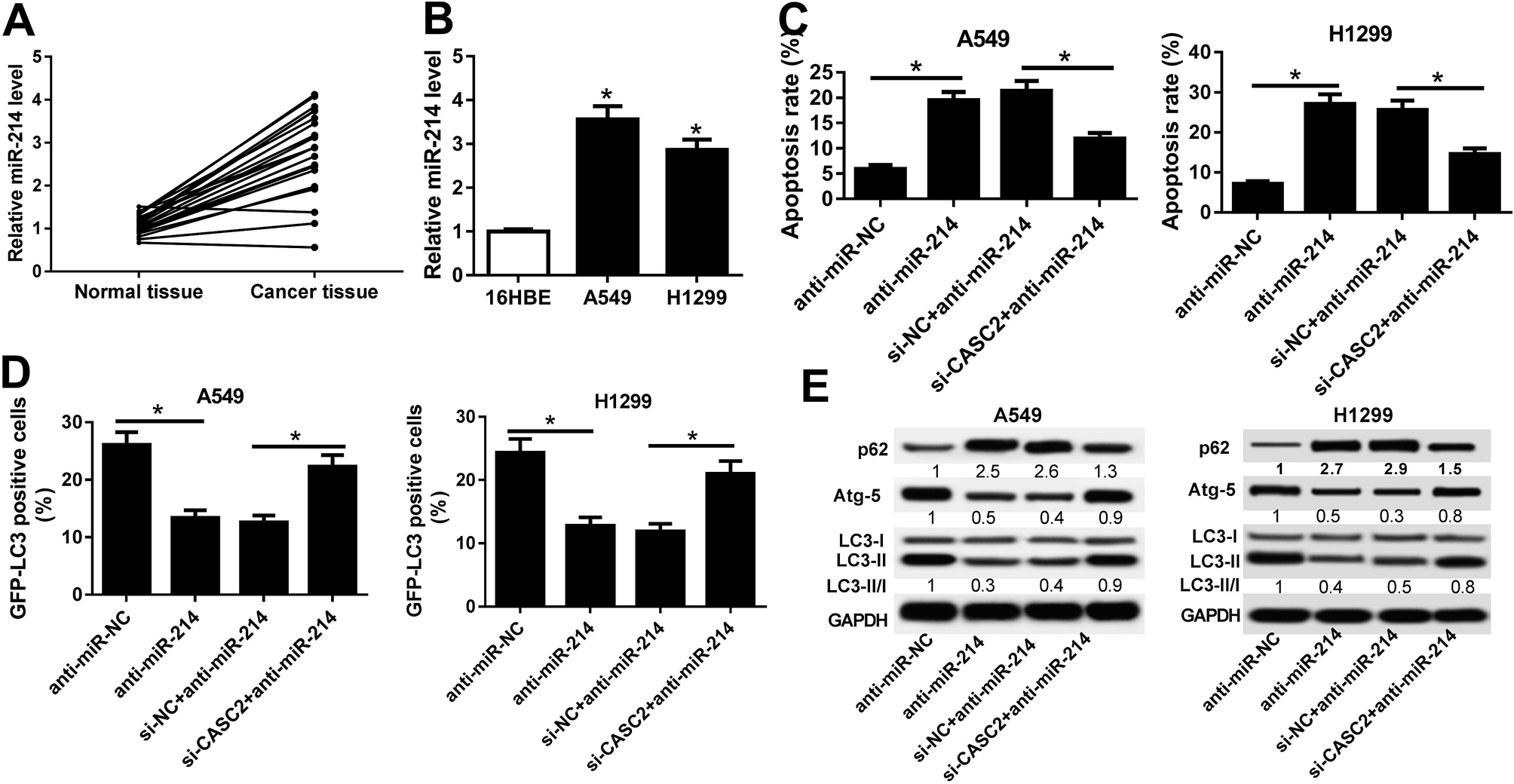 | ||
| Fig. 3 CASC2 knockdown partially reversed miR-214 inhibition-mediated promotion of apoptosis and suppression of autophagy in NSCLC cells. (A) RT-qPCR analysis of the expressions of miR-214 in 21 NSCLC tissues and adjacent normal tissues. (B) RT-qPCR analysis of the expressions of miR-214 in NSCLC cell lines (A549 and H1299) and 16HBE cells. (C) Flow cytometry analysis was employed to assess apoptosis of A549 and H1299 cells transfected with anti-miR-214, anti-miR-NC, or combined with si-CASC2 or si-NC. (D) GFP-LC3 analysis was performed to detect the percentage of GFP-LC3 positive cells in A549 and H1299 cells transfected with anti-miR-214, anti-miR-NC, or combined with si-CASC2 or si-NC. (E) Western blot was performed to determine the protein levels of p62, Atg-5, LC3-I and LC3-II in A549 and H1299 cells transfected with anti-miR-214, anti-miR-NC, or combined with si-CASC2 or si-NC. *P < 0.05. | ||
3.4 TRIM16 was a direct target of miR-214 in NSCLC cells
Bioinformatics analysis showed that TRIM16 was a potential target of miR-214, as shown in Fig. 4A. TRIM6 was found to play an important role in different types of cancers, including NSCLC.21 To confirm the direct binding between miR-214 and TRIM16, luciferase reporter plasmids containing the WT or MUT 3′UTR of TRIM16 were constructed. The subsequent luciferase reporter assay demonstrated that co-transfection of miR-214 and TRIM16-WT significantly reduced the luciferase activity in 293T cells, while the luciferase activity showed little change after co-transfection of miR-214 and TRIM16-MUT (Fig. 4B). Moreover, RIP assay showed that the mRNA of TRIP16 could be specifically recruited to the miRNP complexes isolated using anti-Ago1 antibody after miR-214 overexpression in A549 cells (Fig. 4C), confirming the authentic binding between miR-214 and TRIM16. Western blot further revealed that up-regulation of miR-214 caused a remarkable decrease of TRIM16 expression, while its knockdown led to an obvious increase of TRIM16 level in A549 cells, suggesting that miR-214 negatively regulated TRIM16 expression. Similar results were obtained from immunofluorescence staining (Fig. 4E). Collectively, these data implicated that TRIM16 was a direct target of miR-214 in NSCLC cells.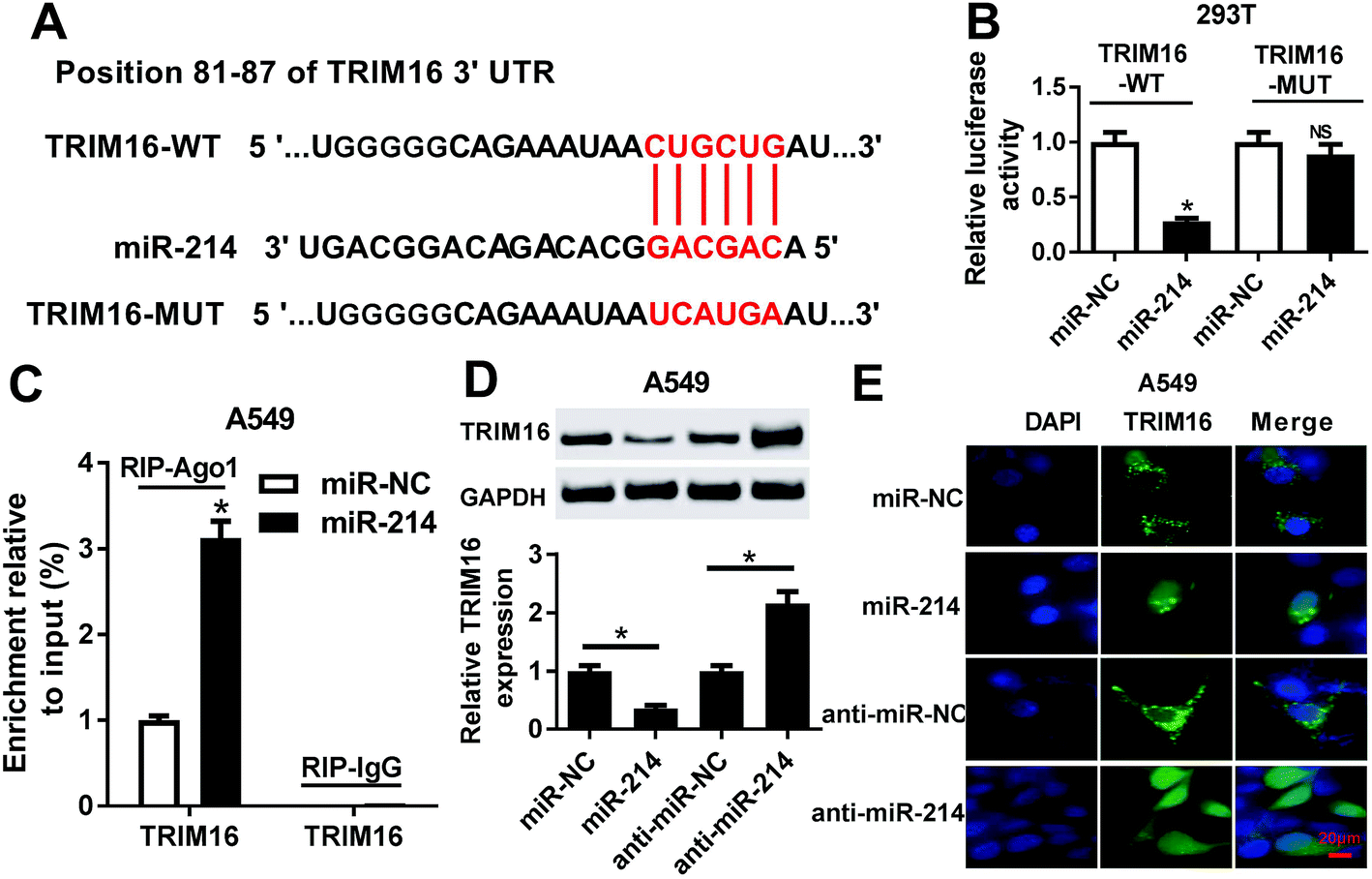 | ||
| Fig. 4 The interaction between miR-214 and TRIM16 in NSCLC cells. (A) The predicted binding sites of miR-214 in the 3′UTR of TRIM16. (B) Luciferase activity was detected by luciferase reporter assay in 293T cells after co-transfection of miR-214 or miR-NC and TRIM16-WT or TRIM16-MUT. (C) Anti-Ago1 RIP assay was performed to confirm the association between miR-214 and TRIM16. (D and E) Expression and localization of TRIM16 in A549 cells transfected with miR-214, anti-miR-214, or matched controls by western blot and immunofluorescence staining. *P < 0.05. | ||
3.5 TRIM16 knockdown significantly overturned anti-miR-214-mediated promotion of apoptosis and inhibition of autophagy in NSCLC cells
Next, we analyzed the expression of TRIM16 in NSCLC tissues and cells. We found that TRIM16 was remarkably downregulated at mRNA and protein levels in NSCLC tissues and cell lines (A549 and H1299) in comparison with respective controls (Fig. 5A and B). Then, rescue experiments were performed to explore the effects of anti-miR-214 or combined with TRIM16 silencing on apoptosis and autophagy in NSCLC cells. Flow cytometry analysis manifested that anti-miR-214 effectively induced apoptosis in A549 and H1299 cells, while reintroduction of si-TRIM16 partially receded the increase of apoptotic rates induced by anti-miR-214 (Fig. 5C). In addition, ectopic expression of anti-miR-214-mediated suppression of the number of GFP-LC3 positive punctate cells was notably abrogated following the transfection of si-TRIM16 (Fig. 5D). Also, the increased level of p62 and decreased expression of Atg-5 and ratio of LC3-II/LC3-I were observed in anti-miR-214-transfected A549 and H1299 cells, which were remarkably reversed by co-transfection of anti-miR-214 and si-TRIM16 (Fig. 5E). Collectively, these findings indicated that TRIM16 depletion significantly overturned anti-miR-214-mediated promotion of apoptosis and inhibition of autophagy in NSCLC cells.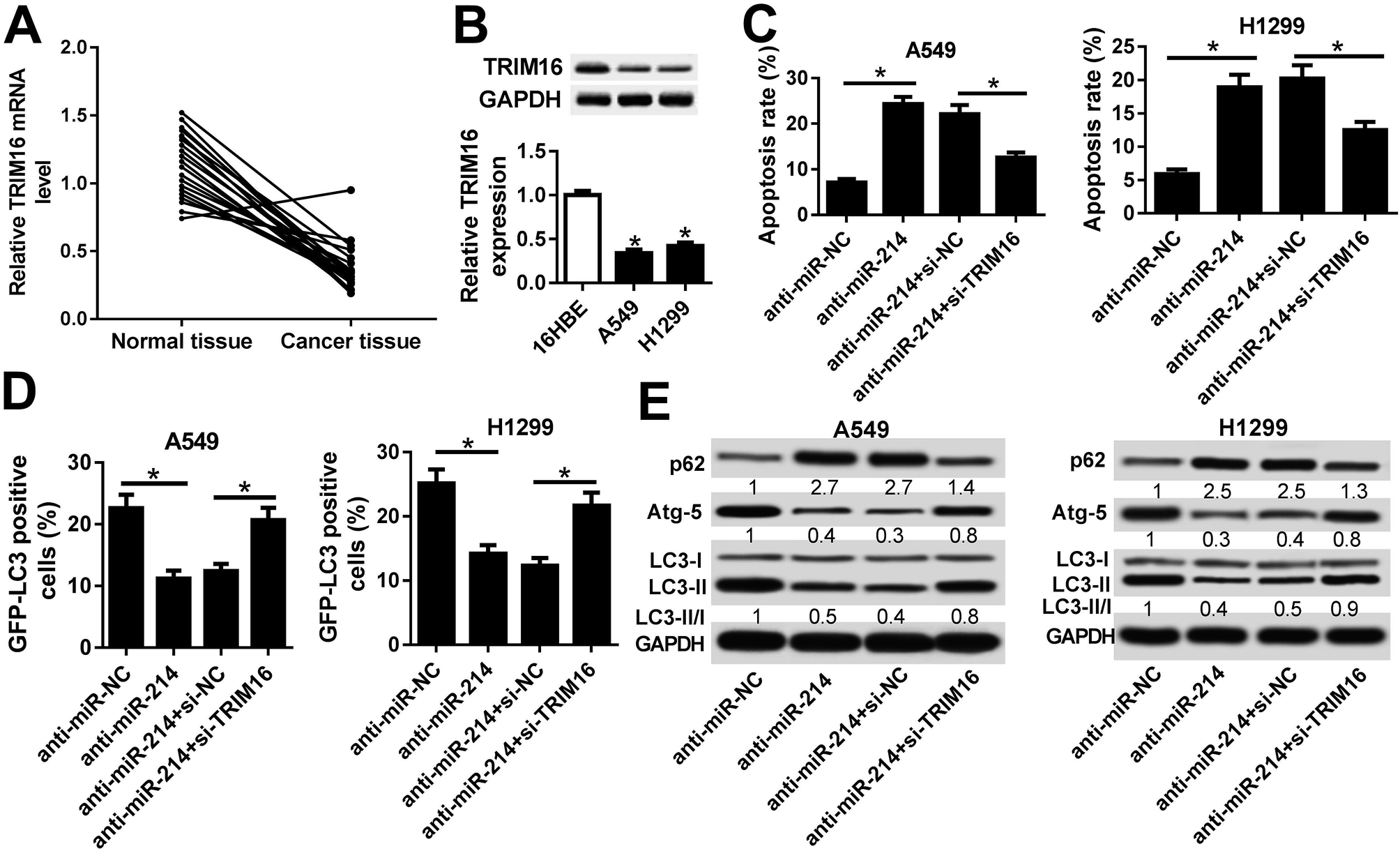 | ||
| Fig. 5 TRIM16 knockdown significantly overturned anti-miR-214-mediated promotion of apoptosis and inhibition of autophagy in NSCLC cells. (A) The expression of TRIM16 mRNA in 21 NSCLC tissues and adjacent normal tissues was examined by RT-qPCR. (B) The protein level of TRIM16 in 16HBE, A549, and H1299 cells was determined by western blot. (C) Apoptosis of A549 and H1299 cells transfected with anti-miR-214 or anti-miR-NC along with si-NC or si-TRIM16 was evaluated by flow cytometry analysis. (D) A549 and H1299 cells were transfected with anti-miR-214 or anti-miR-NC along with si-NC or si-TRIM16 and GFP-LC3-positive punctate cells were visualized by confocal microscope and the percentage of GFP-LC3-positive cells were calculated. (E) Western blot was applied to detect the protein levels of p62, Atg-5, LC3-I and LC3-II in A549 and H1299 cells transfected with anti-miR-214 or anti-miR-NC along with si-NC or si-TRIM16. *P < 0.05. | ||
3.6 CASC2 positively regulated TRIM16 expression by acting as a sponge of miR-214
Of note, spearman correlation analysis found that miR-214 expression was negatively correlated with CASC2 and TRIM16 expression in NSCLC tissues (Fig. 6A and B). Moreover, a positive correlation between CASC2 and miR-214 was observed in NSCLC tissues (Fig. 6C). Luciferase reporter assay demonstrated that miR-214-mediated suppression of the luciferase activity of reporter plasmids carrying the WT fragment of TRIM16 3′UTR was dramatically recuperated after overexpressing CASC2 in 293T cells (Fig. 6D). The regulatory effects of CASC2 or combined with miR-214 on the expression of TRIM16 in A549 and H1299 cells were further studied. As shown in Fig. 6E and F, ectopic expression of CASC2 notably enhanced TRIM16 level in A549 and H1299 cells, while this effect was greatly attenuated increased expression of miR-214. Therefore, these results demonstrated that CASC2 positively regulated TRIM16 expression by suppressing miR-214 expression in NSCLC cells.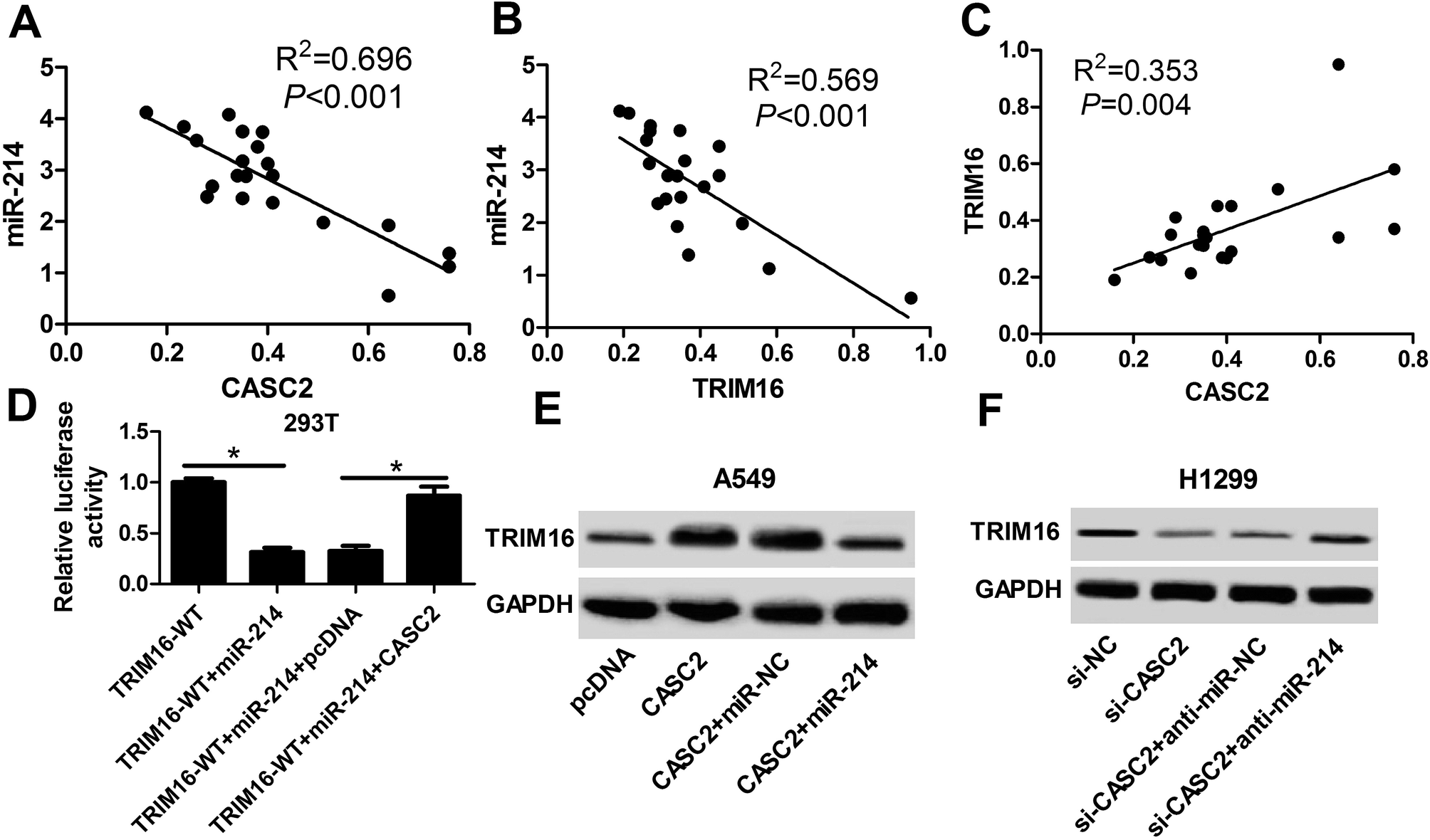 | ||
| Fig. 6 CASC2 positively regulated TRIM16 expression by suppressing miR-214 expression in NSCLC cells. (A) The negative correlation between miR-214 and CASC2 expressions in NSCLC tissues. (B) The negative correlation between miR-214 and TRIM16 expressions in NSCLC tissues. (C) The positive correlation between TRIM16 and miR-214 expressions in NSCLC tissues. (D) After 293T cells were co-transfected with miR-214, miR-214 + pcDNA, or miR-214 + CASC2 and TRIM16-WT or TRIM16-MUT, luciferase activity was measured by luciferase reporter assay. (E and F) Western blot was conducted to detect the protein level of TRIM16 in A549 and H1299 cells after transfection with CASC2, pcDNA, CASC2 + miR-214, CASC2 + miR-NC. *P < 0.05. | ||
4. Discussion
Previous evidence has suggested the critical functions of lncRNAs in regulating the tumorigenesis and development of malignancies including NSCLC.22 However, more researches are needed to better understand the underlying molecular mechanisms by which lncRNAs function in tumors. In our study, we showed that CASC2 and TRIM16 were downregulated but miR-214 was upregulated in NSCLC tissues and cells. CASC2 restoration induced apoptosis and inhibited autophagy in NSCLC cells. Notably, we found that CASC2 could positively regulate TRIM16 by sponging miR-214 in NSCLC cells. Moreover, rescue experiments demonstrated that CASC2 and TRIM16 knockdown significantly reversed anti-miR-214-mediated promotion of apoptosis and suppression of autophagy in NSCLC cells. These results suggested that CASC2 induced apoptosis and inhibited autophagy in regulating the miR-214/TRIM16 axis in NSCLC cells.CASC2 is a novel lncRNA transcript which is frequently downregulated and plays a tumor suppressor role in the progression of human malignancies.23 Pei et al. revealed that downregulation of CASC2 promoted cell proliferation as well as metastasis and promoted early apoptosis of bladder cancer cells by activation of Wnt/β-catenin signal pathway.10 Xiong et al. found that low expression of CASC2 indicated poor prognosis and its overexpression inhibited the proliferation and arrested cell cycle at Go/G1 stage in thyroid cancer cells.24 Gan et al. reported that CASC2 expression was downregulated in hepatocellular carcinoma (HCC) cases and cell lines and overexpression of CASC2 decreased HCC cell proliferation, migration and invasion as well as promoted apoptosis via inactivating the mitogen-activated protein kinase signaling pathway.25 More importantly, He et al. demonstrated that low expression of CASC2 served as an independent predictor for overall survival of NSCLC and overexpression of CASC2 significantly inhibited NSCLC cell proliferation.11 In accordance with the previous study, we demonstrated that CASC2 expression was downregulated in NSCLC tissues and cells, and its overexpression triggered apoptosis and impeded autophagy in NSCLC cells, suggesting the tumor suppressive role of CASC2 in NSCLC cells.
A growing body of evidence suggested that lncRNAs play as a “Sponge” of miRNA to segregate miRNAs away from their target mRNA. Previous studies have reported this novel regulatory mechanism of CASC2 in several tumors. For instance, Ba et al. revealed that ectopic expression of CASC2 suppressed the osteosarcoma cell growth and invasion through suppressing miR-181a expression and thereby enhancing the expression of RAS association domain family member 6 (RASSF6).26 Wang et al. reported that CASC2 positively modulated F-box and WD repeat domain containing 7 (FBXW7) expression by functioning as a ceRNA for miR-367 in HCC cells.27 In this study, luciferase reporter assay, RIP, and RNA pull down assay demonstrated that CASC2 directly interacted with miR-214 in NSCLC cells. There is striking evidence that miR-214 is often dysregulated in multiple cancers and its specific roles vary depending on the types of cancers. It has been shown that miR-214 was upregulated and contributed to tumor progression in melanoma,28 nasopharyngeal carcinoma29 and lung cancer.16,17 By contrast, miR-214 was reported to be downregulated and exerted a tumor suppressive role in other tumors such as cervical cancer,30 breast cancer31 and HCC.32 Our study demonstrated that miR-214 was aberrantly upregulated in NSCLC tissues and cells, consistently with the previous studies.16,17 Rescue experiments demonstrated that CASC2 knockdown partially reversed miR-214 inhibition-mediated promotion of apoptosis and suppression of autophagy in NSCLC cells, suggesting that CASC2 knockdown exerted its function in NSCLC cells by upregulating miR-214.
TRIM16 (also known as estrogen-responsive B-box protein, EBBP), a member of TRIM family, is identified as a positive transcriptional regulator of the retinoic acid receptor β2 in retinoid-treated cancer cells.33 TRIM16 has been shown to be implicated in the pathogenic mechanism of various tumors by acting as a tumor suppressor.34 Previous studies reported that TRIM16 expression acted as a tumor suppressor, affecting neuritic differentiation, cell migration, and replication through directly interacting with cytoplasmic vimentin and nuclear E2F1 in neuroblastoma cells.34 Additionally, TRIM16 expression was downregulated in distant metastatic prostate cancer tissues and overexpression of TRIM16 inhibited the migration, invasion and epithelial-to-mesenchymal transition (EMT) process of prostate cancer by inhibiting the Snail signaling pathway.35 Moreover, TRIM16 expression was found to be markedly decreased in NSCLC and significantly promoted EMT and metastasis in NSCLC both in vitro and in vivo by activating the sonic hedgehog pathway.36 Consistently, our study confirmed the downregulation of TRIM16 in NSCLC tissues and cells. Besides, TRIM16 was identified as a direct target of miR-214 in NSCLC cells by luciferase reporter assay and RIP. TRIM16 knockdown strikingly attenuated anti-miR-214-mediated promotion of apoptosis and inhibition of autophagy in NSCLC cells, suggesting that anti-miR-214 promoted apoptosis and inhibited autophagy in NSCLC cells by upregulating TRIM16. Furthermore, we found that CASC2 positively regulated TRIM16 expression by acting as a sponge of miR-214. Therefore, we concluded that CASC2 regulated apoptosis and autophagy in NSCLC cells by upregulating TRIM16 through acting as a sponge of miR-214.
In summary, our study indicated that CASC2 and TRIM16 were significantly downregulated and miR-214 was dramatically upregulated in NSCLC tissues and cells. Moreover, we provided the first evidence that CASC2 promoted apoptosis and impeded autophagy in NSCLC cells by regulating the miR-214/TRIM16 axis, suggesting that targeting CASC2/miR-214/TRIM16 regulatory network may be a novel therapeutic application in the treatment of NSCLC.
Author's contributions
This work was designed and conceived by Qian li and Kai Chen. The experiment procedures and data analysis were carried out by Rong Dong, Hengxiao Lu, Qian Li, and Kai Chen. The manuscript was prepared by Qian Li and Hengxiao Lu.Conflicts of interest
There is no conflict of interest regarding the publication of this paper.References
- W. Chen, R. Zheng, P. D. Baade, S. Zhang, H. Zeng, F. Bray, A. Jemal, X. Q. Yu and J. He, Ca-Cancer J. Clin., 2016, 66, 115–132 CrossRef PubMed.
- L. A. Torre, F. Bray, R. L. Siegel, J. Ferlay, J. Lortet-Tieulent and A. Jemal, Ca-Cancer J. Clin., 2015, 65, 87–108 CrossRef PubMed.
- A. Fassina, R. Cappellesso and M. Fassan, Chest, 2011, 140, 1305–1311 CrossRef CAS PubMed.
- M. C. Tsai, R. C. Spitale and H. Y. Chang, Cancer Res., 2011, 71, 3–7 CrossRef CAS PubMed.
- X. Shi, M. Sun, H. Liu, Y. Yao and Y. Song, Cancer Lett., 2013, 339, 159–166 CrossRef CAS PubMed.
- S. W. Cheetham, F. Gruhl, J. S. Mattick and M. E. Dinger, Br. J. Cancer, 2013, 108, 2419–2425 CrossRef CAS PubMed.
- P. Baldinu, A. Cossu, A. Manca, M. P. Satta, M. C. Sini, C. Rozzo, S. Dessole, P. Cherchi, F. Gianfrancesco, A. Pintus, A. Carboni, A. Deiana, F. Tanda and G. Palmieri, Hum. Mutat., 2004, 23, 318–326 CrossRef CAS PubMed.
- J. C. Fan, F. Zeng, Y. G. Le and L. Xin, J. Cell. Biochem., 2018, 119, 6391–6397 CrossRef CAS PubMed.
- Y. Zhang, M. Zhu, Y. Sun, W. Li, Y. Wang and W. Yu, Oncol. Res., 2018 DOI:10.3727/096504018X15199531937158.
- Z. Pei, X. Du, Y. Song, L. Fan, F. Li, Y. Gao, R. Wu, Y. Chen, W. Li, H. Zhou, Y. Yang and J. Zeng, Oncotarget, 2017, 8, 18145–18153 Search PubMed.
- X. He, Z. Liu, J. Su, J. Yang, D. Yin, L. Han, W. De and R. Guo, Tumour Biol., 2016, 37, 9503–9510 CrossRef CAS PubMed.
- D. P. Bartel, Cell, 2009, 136, 215–233 CrossRef CAS PubMed.
- C. M. Croce and G. A. Calin, Cell, 2005, 122, 6–7 CrossRef CAS PubMed.
- L. Zhang, J. Huang, N. Yang, J. Greshock, M. S. Megraw, A. Giannakakis, S. Liang, T. L. Naylor, A. Barchetti, M. R. Ward, G. Yao, A. Medina, A. O'Brien-Jenkins, D. Katsaros, A. Hatzigeorgiou, P. A. Gimotty, B. L. Weber and G. Coukos, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9136–9141 CrossRef CAS PubMed.
- E. Penna, F. Orso and D. Taverna, J. Invest. Dermatol., 2015, 135, 960–969 CrossRef CAS PubMed.
- J. Liao, J. Lin, D. Lin, C. Zou, J. Kurata, R. Lin, Z. He and Y. Su, Sci. Rep., 2017, 7, 781 CrossRef PubMed.
- H. Long, Z. Wang, J. Chen, T. Xiang, Q. Li, X. Diao and B. Zhu, Oncotarget, 2015, 6, 38705–38718 Search PubMed.
- L. Bossi and N. Figueroa-Bossi, Nat. Rev. Microbiol., 2016, 14, 775–784 CrossRef CAS PubMed.
- S. Lantuejoul, I. Rouquette, E. Brambilla and W. D. Travis, Ann. Pathol., 2016, 36, 5–14 CrossRef PubMed.
- Y. Kabeya, N. Mizushima, T. Ueno, A. Yamamoto, T. Kirisako, T. Noda, E. Kominami, Y. Ohsumi and T. Yoshimori, EMBO J., 2000, 19, 5720–5728 CrossRef CAS PubMed.
- P. Y. Kim, A. S. Rahmanto, O. Tan, M. D. Norris, M. Haber, G. M. Marshall and B. B. Cheung, Apoptosis, 2013, 18, 639–651 CrossRef CAS PubMed.
- A. Roth and S. Diederichs, Curr. Top. Microbiol. Immunol., 2016, 394, 57–110 Search PubMed.
- G. Palmieri, P. Paliogiannis, M. C. Sini, A. Manca, G. Palomba, V. Doneddu, F. Tanda, M. R. Pascale and A. Cossu, Crit. Rev. Oncol. Hematol., 2017, 111, 31–38 CrossRef PubMed.
- X. Xiong, H. Zhu and X. Chen, Biomed. Pharmacother., 2017, 93, 391–397 CrossRef CAS PubMed.
- Y. Gan, N. Han, X. He, J. Yu, M. Zhang, Y. Zhou, H. Liang, J. Deng, Y. Zheng, W. Ge, Z. Long and X. Xu, Tumour Biol., 2017, 39, 101042831770622 CrossRef PubMed.
- Z. Ba, L. Gu, S. Hao, X. Wang, Z. Cheng and G. Nie, Cell Proliferation, 2018, 51 DOI:10.1111/cpr.12409.
- Y. Wang, Z. Liu, B. Yao, Q. Li, L. Wang, C. Wang, C. Dou, M. Xu, Q. Liu and K. Tu, Mol. Cancer, 2017, 16, 123 CrossRef PubMed.
- E. Penna, F. Orso, D. Cimino, E. Tenaglia, A. Lembo, E. Quaglino, L. Poliseno, A. Haimovic, S. Osella-Abate, C. De Pitta, E. Pinatel, M. B. Stadler, P. Provero, M. G. Bernengo, I. Osman and D. Taverna, EMBO J., 2011, 30, 1990–2007 CrossRef CAS PubMed.
- Z. C. Zhang, Y. Y. Li, H. Y. Wang, S. Fu, X. P. Wang, M. S. Zeng, Y. X. Zeng and J. Y. Shao, PLoS One, 2014, 9, e86149 CrossRef PubMed.
- R. Q. Peng, H. Y. Wan, H. F. Li, M. Liu, X. Li and H. Tang, J. Biol. Chem., 2012, 287, 14301–14309 CrossRef CAS PubMed.
- A. Derfoul, A. H. Juan, M. J. Difilippantonio, N. Palanisamy, T. Ried and V. Sartorelli, Carcinogenesis, 2011, 32, 1607–1614 CrossRef CAS PubMed.
- T. C. Shih, Y. J. Tien, C. J. Wen, T. S. Yeh, M. C. Yu, C. H. Huang, Y. S. Lee, T. C. Yen and S. Y. Hsieh, J. Hepatol., 2012, 57, 584–591 CrossRef CAS PubMed.
- B. B. Cheung, J. Koach, O. Tan, P. Kim, J. L. Bell, C. D'Andreti, S. Sutton, A. Malyukova, E. Sekyere, M. Norris, M. Haber, M. Kavallaris, A. M. Cunningham, C. Proby, I. Leigh, J. S. Wilmott, C. L. Cooper, G. M. Halliday, R. A. Scolyer and G. M. Marshall, J. Pathol., 2012, 226, 451–462 CrossRef CAS PubMed.
- G. M. Marshall, J. L. Bell, J. Koach, O. Tan, P. Kim, A. Malyukova, W. Thomas, E. O. Sekyere, T. Liu, A. M. Cunningham, V. Tobias, M. D. Norris, M. Haber, M. Kavallaris and B. B. Cheung, Oncogene, 2010, 29, 6172–6183 CrossRef CAS PubMed.
- L. Qi, Z. Lu, Y. H. Sun, H. T. Song and W. K. Xu, Int. J. Mol. Med., 2016, 38, 1734–1742 CrossRef CAS PubMed.
- X. Huo, S. Li, T. Shi, A. Suo, Z. Ruan and Y. Yao, Biochem. Biophys. Res. Commun., 2015, 460, 1021–1028 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |