
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Iso-pencillixanthone A from a marine-derived fungus reverses multidrug resistance in cervical cancer cells through down-regulating P-gp and re-activating apoptosis†
Li
Chen
a,
Xinxin
Li
a,
Miaomiao
Cheng
a,
Siyuan
Wang
a,
Qiuhong
Zheng
*b and
Qinying
Liu
*b
aInstitute of Biomedical and Pharmaceutical Technology, Fuzhou University, Fuzhou 350002, P. R. China
bFujian Provincial Key Laboratory of Tumor Biotherapy, Fujian Cancer Hospital, Fujian Medical University Cancer Hospital, Fuzhou 350014, P. R. China. E-mail: zqh2858@foxmail.com; liuqy@fjmu.edu.cn; Tel: +86-591-8366-0063
First published on 10th December 2018
Abstract
The occurrence of multidrug resistance (MDR) is highly associated with the overexpression of ATP-binding cassette (ABC) transporters, among which, P-glycoprotein (P-gp) plays one of the most important roles. Iso-pencillixanthone A (iso-PXA) is a compound isolated from the marine-derived fungus Penicillium oxalicum. No studies on the anti-tumor effect of this compound have been reported, except for a few focusing on its bactericidal properties. In this study, we found iso-PXA could stimulate P-gp ATPase activity and attenuate P-gp expression to increase the intracellular drug concentration in the cervical vincristine (VCR)-resistant cell line HeLa/VCR. Then, it increased ROS generation, depolarized MMP, promoted the release of cytochrome c from mitochondria, and further activated caspase-9, caspase-3 and PARP to induce cell apoptosis effectively through the intrinsic pathway. Caspase-8 medicated cleavage of Bid into the truncated form tBid partially initiated the mitochondrial apoptotic events. The elevation of the Bax/Bcl-2 ratio, the accumulation of FBW7 and the degradation of Mcl-1 accelerated the iso-PXA induced apoptotic process. The HeLa/VCR cell xenograft model again confirmed that iso-PXA had much better efficacy than vincristine in vivo. Taken together, these findings demonstrated that iso-PXA elicited remarkable anti-tumor and anti-MDR activity through inhibiting P-gp expression and function and re-activating the intrinsic apoptosis pathway in vitro and in vivo, suggesting it as a potential chemotherapeutic lead compound in the treatment of cervical MDR cancers.
1 Introduction
Cancer multidrug resistance (MDR) is a major reason for the failure of chemotherapy in the clinic. The induction of MDR in cancer cells results in lower levels of intracellular drug accumulation and lowered cytotoxicity towards the cancer cells. MDR can be correlated with a number of mechanisms, such as the overexpression of membrane transporter proteins, activation of DNA repair mechanisms, alteration of drug targets, and the disruption of cellular death signaling pathways.1,2 The overexpression of ATP-binding cassette (ABC) transporter proteins, such as P-glycoprotein (P-gp/MDR1/ABCB1), multidrug resistance protein 1 (MRP1/ABCC1), and breast cancer resistance proteins (BCRP/ABCG2), play a prominent role in MDR.3–5 In particular, P-gp, which could medicate drug extrusion, is one of the most important resistance factors. In addition, evasion of apoptosis, which is another crucial factor of chemo-resistance, has been proven to be related to these overexpressed proteins. Therefore, downregulation of these proteins' expression or activity would be powerful strategies for overcoming MDR.6,7The search for novel inhibitors of P-gp from marine natural products commenced some decades ago. Marine natural products have become one of the most interesting targets for drug development since they represent an attractive and diverse source of bioactive chemicals.8,9 Recently, several marine natural products or derivatives have been approved for clinical use, including sipholenol A, cytarabine, ISA/ISB, parguerenes, ET-743, and halaven,10–12 which brings hope for the discovery of a new generation of P-gp inhibitors.
Iso-pencillixanthone A (iso-PXA) is an efficient bioactive secondary metabolite isolated from marine fungi Penicillium oxalicum (P. oxalicum), but only the bactericidal property has been reported until now. In the present study, we investigated the anti-tumor and anti-MDR effects of iso-PXA on the vincristine (VCR)-resistant cell line HeLa/VCR in vitro and in vivo.
2 Experimental
2.1 Fungal materials
The fungus P. oxalicum was purified from marine sediments collected from the southeast coastal region of China. It was identified through ITS analysis by Sunbiotech Co., Ltd. The voucher specimen was preserved in the China Center for Type Culture Collection (No. M2013714). The producing strain was prepared on Martin medium and stored at 4 °C.2.2 Fermentation and extraction
The fungus was cultured under static conditions. Briefly, the liquid medium included NaCl (1.5%), maltose (2%), glucose (1%), mannitol (2%), monosodium glutamate (1%), MgSO4·7H2O (0.03%), KH2PO4 (0.05%) and yeast extract (0.3%). After culturing at 28 °C for 30 days (60 L), the broth was filtered to separate the supernatant from the mycelia. The mycelia were extracted three times with acetone solution (80%), which was concentrated to give an aqueous solution. The aqueous solution was extracted three times with EtOAc and concentrated to give the extract (36.5 g).2.3 Purification
The mycelia extract was purified on a Si gel column and divided into five fractions (from A to E). Fraction C (12.7 g) was further separated on a Sephadex column (LH-20, CH2Cl2/MeOH = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) to give four subfractions (C1 to C4). Subfraction C3 (4.9 g) was finally purified by HPLC (55% CH3CN and 0.1% TFA), yielding iso-PXA (510 mg).
:2) to give four subfractions (C1 to C4). Subfraction C3 (4.9 g) was finally purified by HPLC (55% CH3CN and 0.1% TFA), yielding iso-PXA (510 mg).
iso-PXA: yellow powder (CHCl3); [α]20D +105.6° (c 1, pyridine); [α]20D +3.1° (c 1, Me2CO); 1H and 13C NMR, see Table S1;† HRESIMS (m/z: 661.1550 [M + Na]+, calcd for C32H30NaO14, 661.1533). The detailed chart exhibiting the progress of extraction and purification for iso-PXA was shown as Fig. S8.†
2.4 Cytotoxicity assay
Cell viability was assessed by the WST-1 cell proliferation assay kit (Roche, Indianapolis, IN, USA), as previously described.13 Detailed method was recorded in ESI.†2.5 Real-time cell proliferation assay analysis
Cell proliferation was determined using the DP version of the xCELLigence real-time cell analyzer (RTCA) (ACEA, San Diego, CA, USA) as previously described.14 Detailed method was recorded in ESI.†2.6 Cell cycle analysis
The cell cycle assays were performed by flow cytometry (Beckman Coulter FC500, Brea, CA, USA) as we previously described.13 Detailed method was recorded in ESI.†2.7 AO/EB staining assay
Cells (1 × 105 cells per well) were pre-incubated with iso-PXA or VCR for 48 h and then stained with a 1:1 mixture of AO (100 μg mL−1) and EB (100 μg mL−1) for 1 min in the dark. The cell morphology was observed under a fluorescence microscope (Ti-E, Nikon, Tokyo, Japan).
2.8 Annexin V-FITC/PI double staining
Cells (1 × 105 cells per well) were pre-incubated with iso-PXA or VCR for 48 h. Apoptosis was detected by an Annexin V-FITC/PI apoptosis detection kit (BD Biosciences, San Jose, CA), according to the manufacturer's instructions. Briefly, after the desired treatments, cells were collected and washed twice with binding buffer and then re-suspended at a concentration of 1 × 106 cells per mL in binding buffer. 500 μL of the cell suspension was mixed with 5 μL of Annexin V-FITC and 5 μL of PI. After being incubated at room temperature for 15 min, apoptotic cells were determined by flow cytometry (Beckman Coulter FC500, Brea, CA, USA).2.9 Reactive oxygen species (ROS) production
A Reactive Oxygen Species Assay Kit (Beijing Ding Guo Chang Sheng Biotechnology Co., Ltd., Beijing, China) was used according to the manufacturer's instructions. After treatment, the cells were collected and incubated with DCFH-DA at 37 °C for 30 min in the dark. The cells were then visualized with a fluorescence microscope (Ti-E, Nikon, Tokyo, Japan).2.10 Mitochondrial transmembrane potential (ΔΨm) measurement
A Mitochondrial Membrane Potential Assay Kit (Beijing Ding Guo Chang Sheng Biotechnology Co., Ltd., Beijing, China) was used according to the manufacturer's instructions. After treatment, the cells were incubated with JC-1 at 37 °C for 30 min in the dark. The green fluorescence intensity was then measured by flow cytometry (Beckman Coulter FC500, Brea, CA, USA).2.11 Western blotting analysis
Western blotting was performed as we previously described.14 Detailed method was recorded in ESI.†2.12 RT-qPCR
Total RNA was isolated from cells with TRIzol reagent (Life Technologies, Carlsbad, CA, USA). The mRNA was synthesized to cDNA with the Goscript™ Reverse TranscriPTAse System (Promega, Wisconsin, USA). Quantitative PCR results were detected using a Light Cycler 480 II (Roche, Switzerland) with the miScript SYBR Green PCR Kit (QIAGEN, Hilden, Germany). The oligonucleotide primer pairs used in the present study are listed in Table S5.†2.13 Rh123 accumulation analysis
Cells (1 × 105 cells per well) were pretreated with iso-PXA and VCR for 6 h and then incubated with 10 μM of Rh123 at room temperature for 120 min in the dark. After washing off unbound dye with ice-cold PBS, the cells were resuspended and kept in the dark until detection by flow cytometry (Beckman Coulter FC500, Brea, CA, USA).2.14 ABCB1-mediated acetoxymethyl ester of calcein (calcein AM) efflux assay
MDCK/MDR1 cells (1 × 106 cells per well) were pretreated with iso-PXA and cyclosporin A for 20 min and then incubated with calcein AM in 5% CO2 at 37 °C for 30 min in the dark. After washing off unbound dye with ice-cold wash solution and uptake buffer, the final washing buffer was left in each well, and the fluorescence values were read by an EnVision plate reader (Perkin Elmer Mediums, Shanghai, China).2.15 P-gp ATPase activity assay
The P-gp ATPase activity was assayed using a Pgp-Glo™ assay systems kit (Promega, Madison, WI, USA), according to the manufacturer's instructions. iso-PXA (0–10 μM) was incubated with recombinant human P-gp membranes (25 μg) in the presence of 5 mM MgATP at 37 °C for 40 min, using Na3VO4 as an inhibitor and VRP (0.5 mM) as a positive control. The luminescence of the samples was detected by a spectraMax M5 (Molecular Devices, Sunnyvale, CA, USA).2.16 Tumor xenograft model
To investigate the effects of iso-PXA on tumor growth in vivo, we established a xenograft tumor model.15,16 Briefly, 6 week-old female BALB/c nude mice (Center of Wu Experimental Animal, Fuzhou, China) were implanted with 1.2 × 107 resistant HeLa/VCR cells s.c. into the left or right flank. The length and width of the tumors were measured, and the tumor volume (mm3) was calculated by the formula (L × W2)/2. Drug treatment was initiated when the tumor size reached approximately 100 mm3. The nude mice with xenografts were randomly divided into five groups (n = 5 for each group). Dry iso-PXA was suspended in 1% carboxy methyl cellulose-Na (CMC-Na, purchased from Sinoharm Chemical Reagent Co., Ltd., China) for immediate use. The mice were treated with vehicle (1% CMC-Na), iso-PXA (2.5, 5, 10 mpk d−1, i. g.) or VCR (2.5 mpk d−1, i. g.) every other day. The weight of the mice and the tumor volumes were recorded with an electronic scale and a slide caliper, respectively, every other day until the mice were sacrificed on day 15, at which point the massive tumors were removed and pictures were taken. At the same time, tumors and normal tissues, such as the heart, liver, spleen, kidney and lung, were obtained by dissecting. The tumors and normal tissues were paraffin embedded and stained with hematoxylin and eosin (H&E). The tumor inhibition rate (%) = (mean tumor weight of control group − mean tumor weight of treated group)/mean tumor weight of control group × 100%. All animals used in the investigation were handled in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996) and approved by the Animal Care and Use Committee of Institute of Biomedical and Pharmaceutical Technology, Fuzhou University.2.17 Statistical analysis
All results are expressed as the mean ± SD of triplicate samples. A probability (P) value < 0.05 was considered to be statistically significant, and P < 0.01 was highly statistically significant. All statistical analyses were conducted using SPSS 19 software.3 Results
3.1 Purification and structural elucidation
The mycelia extract of P. oxalicum was consecutively separated on silica gel, Sephadex LH-20 and preparative HPLC columns to obtain the bioactive compound, which was purified as a yellow powder and analyzed to have a molecular formula of C32H30O14 by positive HRESIMS (m/z: 661.1550 [M + Na]+, calcd for C32H30O14, 661.1533) (Fig. S1†). The 1H and 13C NMR data (Fig. S2–S4 and Table S1†) revealed thirty-two carbon signals, including four methyls, two methylenes, eight methines and eighteen quaternary carbons. These signals appeared almost in pairs, which indicated that the compound may be an asymmetrical dimer. After careful analysis and comparison with the literature, it was found that these 1H and 13C NMR data were very similar with those of PXA.17 Further structural elucidation by analysis of COSY, HMBC and NOE correlations in the NMR data suggested that both compounds had the same planar structure and relative configuration (Fig. S5–S7†). However, the opposite specific rotation illustrated that they were enantiomers. This compound was finally identified as iso-PXA and is illustrated in Fig. 1. | ||
| Fig. 1 Chemical structure and COSY, HMBC and NOE correlations for iso-PXA. | ||
3.2 iso-PXA displays potent cytotoxic activity in various human cancer cell lines, including MDR cancer cell lines
iso-PXA was shown to have spectrum cytotoxic activity against various human cancer cell lines by WST-1 assay tests. The average IC50 values of iso-PXA to 21 cancer cell lines were 1.300 μM, while there was no severe toxicity to the normal tissue cell lines HFF-1 (IC50 = 26.73 μM) and LO2 (IC50 = 8.238 μM), indicating that iso-PXA has strong anticancer effects but is safe to for use (Fig. 2a and Table S2†). More interestingly, iso-PXA seemed to exhibit a stronger potency to induce cell death in drug resistant cancer cell lines (HeLa/VCR, HeLa/DDP, K562/A02, A549/DDP and MCF-7/ADR cells) compared to classical chemotherapeutic drugs, such as vincristine (VCR), adriamycin (ADR) and cisplatin (DDP) (Table S3†), although these drugs showed excellent inhibitory activity in relatively non-resistant cells. Moreover, both HeLa/VCR and HeLa/DDP were resistant to several types of chemotherapeutic drugs, such as VCR, DDP, doxorubicin (DOX) and paclitaxel (PTX), suggesting that they were MDR cancer cell lines. But interestingly, iso-PXA had an essentially equivalent potency toward HeLa/VCR cells with the resistance index (RI) of only 0.84 (Fig. 2d and S9), while the RI of iso-PXA was 2.29 in HeLa/DDP cells, indicating that HeLa/VCR is more sensitive to iso-PXA than HeLa/DDP. As a result, we chose HeLa/VCR for our further study. | ||
| Fig. 2 Anti-MDR activity of iso-PXA in vitro. (a) Cytotoxicity of iso-PXA on 21 types of human cancer cell lines and 2 types of human normal tissue cell lines was determined by WST-1 assay. Data are the means ± SD of at least three independent experiments. (b) Cytotoxicity of iso-PXA on HeLa/VCR and HeLa cells was determined by WST-1 assay. Data are the means ± SD of at least three independent experiments. (c) Cytotoxicity of iso-PXA on HeLa/VCR and HeLa cells was determined by RTCA software. (d) Average resistance index of iso-PXA and references on the basis of the cytotoxic activity of agents against HeLa/VCR and HeLa cells, and HeLa/DDP and HeLa cells. | ||
We further evaluated the effect of iso-PXA on the proliferation of HeLa/VCR and the parental HeLa cells. As shown in Fig. 2b and c, the cells were treated with gradient concentrations of iso-PXA, and the proliferation was then tested by WST-1 assay or recorded for a continuous 120 h by the xCELLigence RTCA system. Both results showed that iso-PXA had anti-proliferative potential in a dose- and time-dependent manner in HeLa/VCR and HeLa cells. Taken together, our data suggest that iso-PXA may have great anti-tumor and anti-MDR activity in vitro.
3.3 iso-PXA induces G2/M phase arrest in HeLa/VCR cells
We further analyzed the cell cycle distribution of HeLa/VCR and HeLa cells after treatment with iso-PXA or VCR by flow cytometry. As shown in Fig. 3a and b, after treatment with 1 μM VCR, the G2/M percentage only increased from 7.7% to 15.0% in HeLa/VCR cells, while it increased dramatically from 17.1% to 78.8% in HeLa cells, confirming that HeLa/VCR is resistant to VCR. When treated with 0.25, 0.5 or 1 μM of iso-PXA, the G2/M percentage increased, respectively, from 7.7% to 24.6%, 33.0% and 44.0% in HeLa/VCR cells, and increased from 17.1% to 33.2%, 41.1% and 47.2% in HeLa cells, showing no significant difference in the cell cycle distribution between HeLa/VCR and HeLa cells. However, it seems that iso-PXA can partially induce G2/M arrest, even though the effect is not as strong as VCR.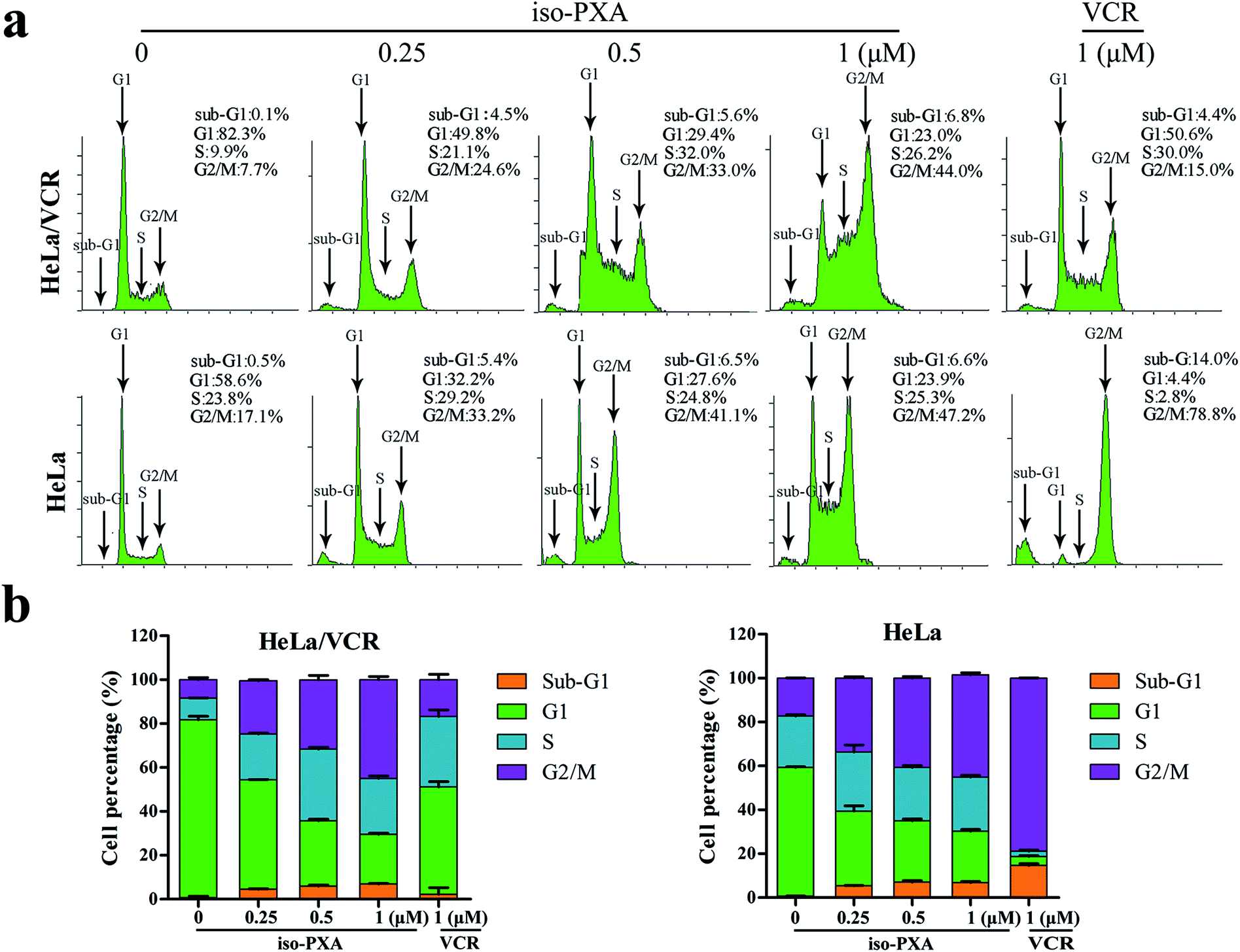 | ||
| Fig. 3 Cell cycle analysis by flow cytometry. (a) The dose effect of iso-PXA on the cell cycle distribution in HeLa/VCR and HeLa cells. HeLa/VCR and HeLa cells were treated with iso-PXA (0, 0.25, 0.5 and 1 μM) for 24 h and treated with VCR (1 μM) as a positive control. (b) The statistical data of the cell cycle distribution are shown. | ||
3.4 iso-PXA induces apoptosis in HeLa/VCR cells
Since the study of MDR cell lines and their corresponding sensitive strains showed that the MDR cells have resistance to apoptosis in animal experiment,18 we wanted to know whether iso-PXA had the ability to induce apoptosis in HeLa/VCR cells. First, we focused on observations of the morphological alterations induced by iso-PXA. As shown in Fig. 4a and b, when treated with 0.25, 0.5 and 1 μM of iso-PXA or 1 μM of VCR, the apoptosis rates were, respectively, 34.0%, 85.6%, 94.7%, and 33.5% in HeLa/VCR cells, and 25.0%, 90.0%, 95.1%, and 97.0% in HeLa cells, indicating that HeLa/VCR cells are sensitive to iso-PXA but not to VCR.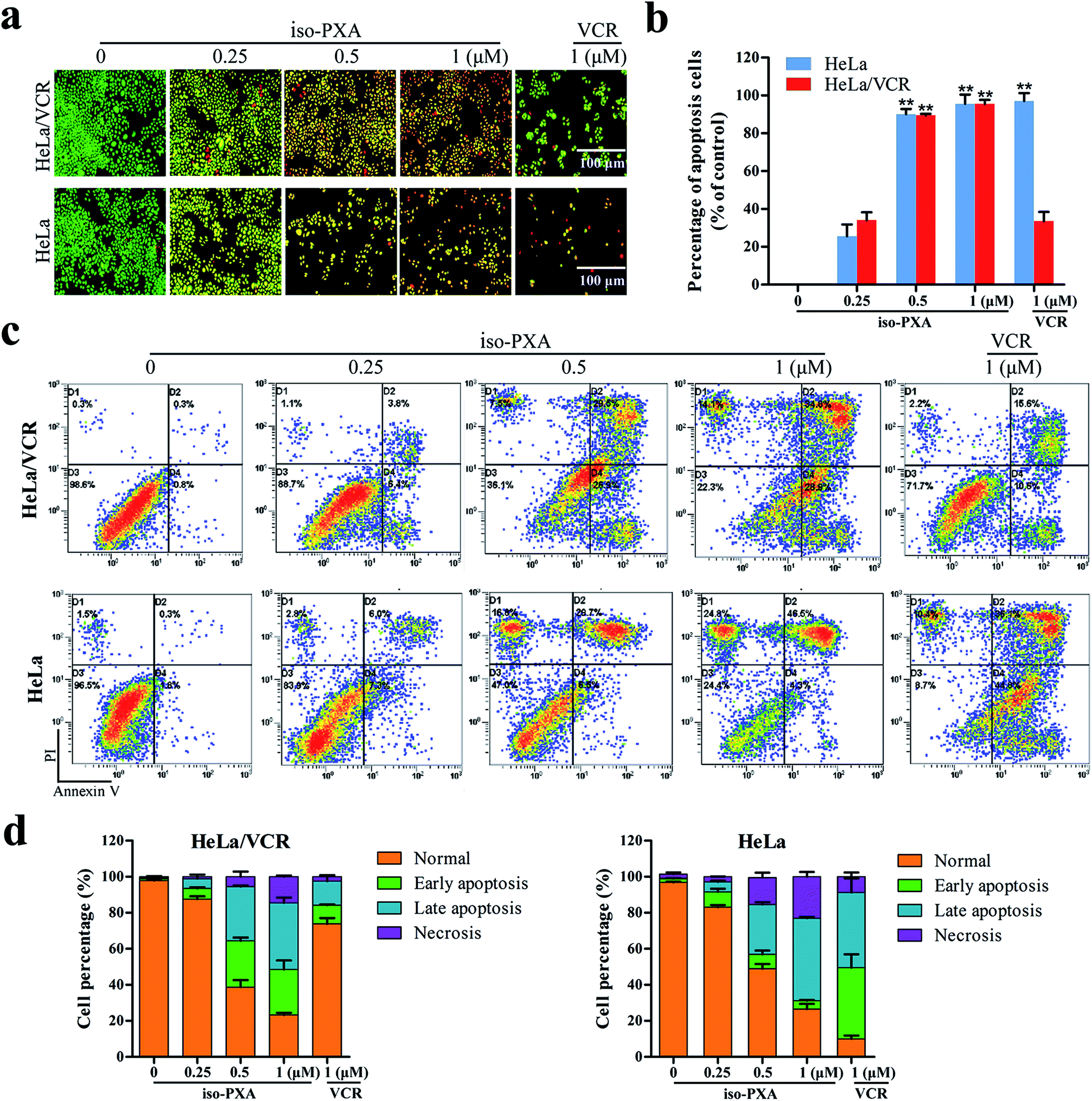 | ||
| Fig. 4 iso-PXA induces apoptosis in HeLa/VCR and HeLa cells. (a) AO/EB staining of HeLa/VCR and HeLa cells treated with iso-PXA (0, 0.25, 0.5 and 1 μM) and VCR (1 μM) for 48 h. (b) Densitometry of cell counts. (c) Annexin V/PI test of HeLa/VCR and HeLa cells treated with iso-PXA (0, 0.25, 0.5 and 1 μM) and VCR (1 μM) for 48 h by flow cytometry. (d) Densitometry of cell counts. Columns represent the means ± SD values of the apoptosis rate obtained from three individual experiments. *P < 0.05, **P < 0.01 HeLa/VCR vs. HeLa control. | ||
Second, iso-PXA-induced apoptosis was detected using an Annexin V-FITC/PI apoptosis detection kit. As shown in Fig. 4c, both HeLa/VCR and HeLa cells showed a high level of apoptotic-like death in a dose-dependent manner. After treatment with 1 μM VCR, the apoptosis rate merely raised from 1.1% to 26.2% in HeLa/VCR cells, while the apoptosis rate increased from 3.5% to 80.9% in HeLa cells, confirming that HeLa/VCR was resistant to VCR. When treated with 0.25, 0.5 and 1 μM of iso-PXA, the apoptosis rates raised, respectively, from 1.1% to 10.2%, 56.5%, and 63.7% in HeLa/VCR cells, and raised from 3.5% to 13.3%, 35.2%, and 50.8% in HeLa cells, showing the similar apoptosis potency of iso-PXA to HeLa/VCR and HeLa cells (Fig. 4d).
3.5 Intracellular ROS and depolarization MMP initiate iso-PXA-induced apoptosis
The generation of ROS exhibits a close correlation with tumor cell death.19 To identify the role of intracellular ROS in iso-PXA-induced apoptosis, we measured ROS levels by DCF probes in response to iso-PXA treatment (Fig. 5a and b). After treatment with 0.25, 0.5 and 1 μM of iso-PXA, the levels of ROS were improved, respectively, by 6.77-, 10.32-, and 15.47-fold in HeLa/VCR cells, and 5.82-, 7.02-, and 11.88-fold in HeLa cells, while the level of ROS was only improved by 4.70-fold in HeLa/VCR cells, but by 21.86-fold in HeLa cells after 1 μM VCR treatment. | ||
| Fig. 5 iso-PXA induced ROS production and mitochondrial dysfunction. (a) ROS production was detected by fluorescence microscopy after HeLa/VCR and HeLa cells were treated with iso-PXA (0, 0.25, 0.5 and 1 μM) and VCR (1 μM) for 24 h. (b) The statistical fluorescence data of the ROS production. (c) iso-PXA induced loss of mitochondrial membrane potential in HeLa/VCR and HeLa cells. Cells were treated with gradient concentrations of iso-PXA for 24 h, and the mitochondrial membrane potential was analyzed by flow cytometry after cells were stained with JC-1. (d) The statistical fluorescence data of the mitochondrial dysfunction. Columns represent the means ± SD values obtained from three individual experiments. *P < 0.05, **P < 0.01 HeLa/VCR vs. HeLa control. | ||
It is known that accumulation of intracellular ROS typically depolarizes the MMP.20 Thus, we further detected the MMP of HeLa/VCR cells after treatment with iso-PXA. The intracellular green fluorescence intensity was improved by 14.60-, 15.06-, and 19.17-fold in HeLa/VCR cells and by 5.87-, 8.86-, and 13.34-fold in HeLa cells after treatment with 0.25, 0.5 and 1 μM, respectively, of iso-PXA, while the mean fluorescence intensity was improved only by 1.99-fold in HeLa/VCR cells but by 14.57-fold in HeLa cells after 1 μM VCR treatment (Fig. 5c and d). This confirms that iso-PXA depolarized the MMP in a dose-dependent manner. These data indicate that the generation of ROS and the MMP depolarization initiate the apoptosis that is induced by iso-PXA in HeLa/VCR cells.
3.6 iso-PXA activates caspase and regulates the ratio of Bax/Bcl-2
The Bcl-2 family and caspase cascade are the main forms of mitochondria-mediated apoptosis.21 Caspases-8 and -9 are early stage initiators of cell apoptosis, while caspase-3 is a late-stage executor.22 Moreover, caspase-3 could cleave PARP and arrest its DNA damage repair activity. To clarify the underlying mechanisms of this process, we detected the expression of these proteins by western blot and RT-qPCR. As shown in Fig. 6a–d, iso-PXA upregulated the expression of cleaved caspase-9 and downregulated the expression of pro-PARP, pro-caspase-3, pro-caspase-8, and pro-caspase-9 in a dose-dependent manner in both HeLa/VCR and HeLa cells. Additionally, cyclin B1 proteins increased slightly with the increased concentration of iso-PXA. The expression of mRNA levels related to apoptosis was consistent with the protein levels (Fig. 6b and d) except for a slightly opposite trend in caspase-3. The release of mitochondrial inter-membrane proteins into the cytosol plays a crucial role in the activation of downstream caspases. Caspase-8 mediated cleavage of Bid into a pro-apoptotic activity, triggering mitochondrial apoptotic events. In addition, to further confirm how treatment with iso-PXA induces caspase-9 dependent apoptosis. Thus, we detected the activation of Bid and release of cytochrome c. As shown in Fig. 6e and f, the activity of Bid and the release of cytochrome c was significantly enhanced after the treatment of iso-PXA. Our data demonstrate that iso-PXA might induce apoptosis of HeLa/VCR cells through both a caspase-9-dependent signaling cascade. | ||
| Fig. 6 Expression of apoptosis-related proteins and genes in HeLa/VCR and HeLa cells treated with iso-PXA. (a) and (c) After cells were treated with different concentrations of iso-PXA for 24 h, the expression of apoptosis-related proteins was determined by western blot, with β-actin as a loading control. Band intensities of western blots were quantified. (b) and (d) The mRNA level of cells treated with 0, 0.25, 0.5 and 1 μM iso-PXA for 24 h by RT-qPCR. Quantitative gene expressions were normalized to GAPDH. (e) and (f) After cells were treated with different concentrations of iso-PXA for 24 h, the expression of Bid and cytochrome c was determined by western blot, with β-actin as a loading control. Band intensities of western blots were quantified. (g) and (i) Western blot analysis of the expression of Bax and Bcl-2 proteins in HeLa/VCR and HeLa cells treated with iso-PXA (0, 0.25, 0.5 and 1 μM) for 24 h. Band intensities of western blots were quantified. (h) and (j) The levels of apoptosis-related genes in HeLa/VCR and HeLa cells treated for 24 h were analyzed by RT-qPCR. GAPDH was used as a loading control. Bars represent the mean ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001 vs. the control group. | ||
We also detected expression of Bcl-2 and Bax, involved in the molecular switch-mediating mitochondrial apoptosis pathway. As shown in Fig. 6g–j, the protein expression of Bcl-2 was downregulated and Bax was upregulated after treatment with iso-PXA for 24 h, but the mRNA expression of Bcl-2 and Bax was opposite because iso-PXA induced apoptosis by regulating the apoptosis-related gene translation, rather than the gene transcription. Taken together, we considered that iso-PXA can induce mitochondria-related apoptosis in HeLa/VCR cells.
3.7 iso-PXA inhibits tumor growth in vivo
Based on the above results, we confirmed that iso-PXA had anti-tumor and anti-MDR effects in vitro. To further evaluate the efficacy of iso-PXA in vivo, we established HeLa/VCR xenografts in a nude mice model. As shown in Table S4,† the tumor inhibitory rate reached 54.7%, 77.8% and 89.1% after treatment with iso-PXA in gradient concentrations (2.5, 5, and 10 mpk, respectively), while the rate was only 37.9% in the VCR (2.5 mpk) treatment group, suggesting that iso-PXA significantly inhibited the MDR tumor growth in a dose- and time-dependent manner (Fig. 7c and e). In addition, the weight of the nude mice was almost unchanged after treatment with iso-PXA or VCR (Fig. 7d). H&E staining also showed that normal tissues such as the heart, liver, spleen, lung and kidney had no significant difference between the experimental and control groups (Fig. 7f), demonstrating that iso-PXA is safe to use at low concentrations.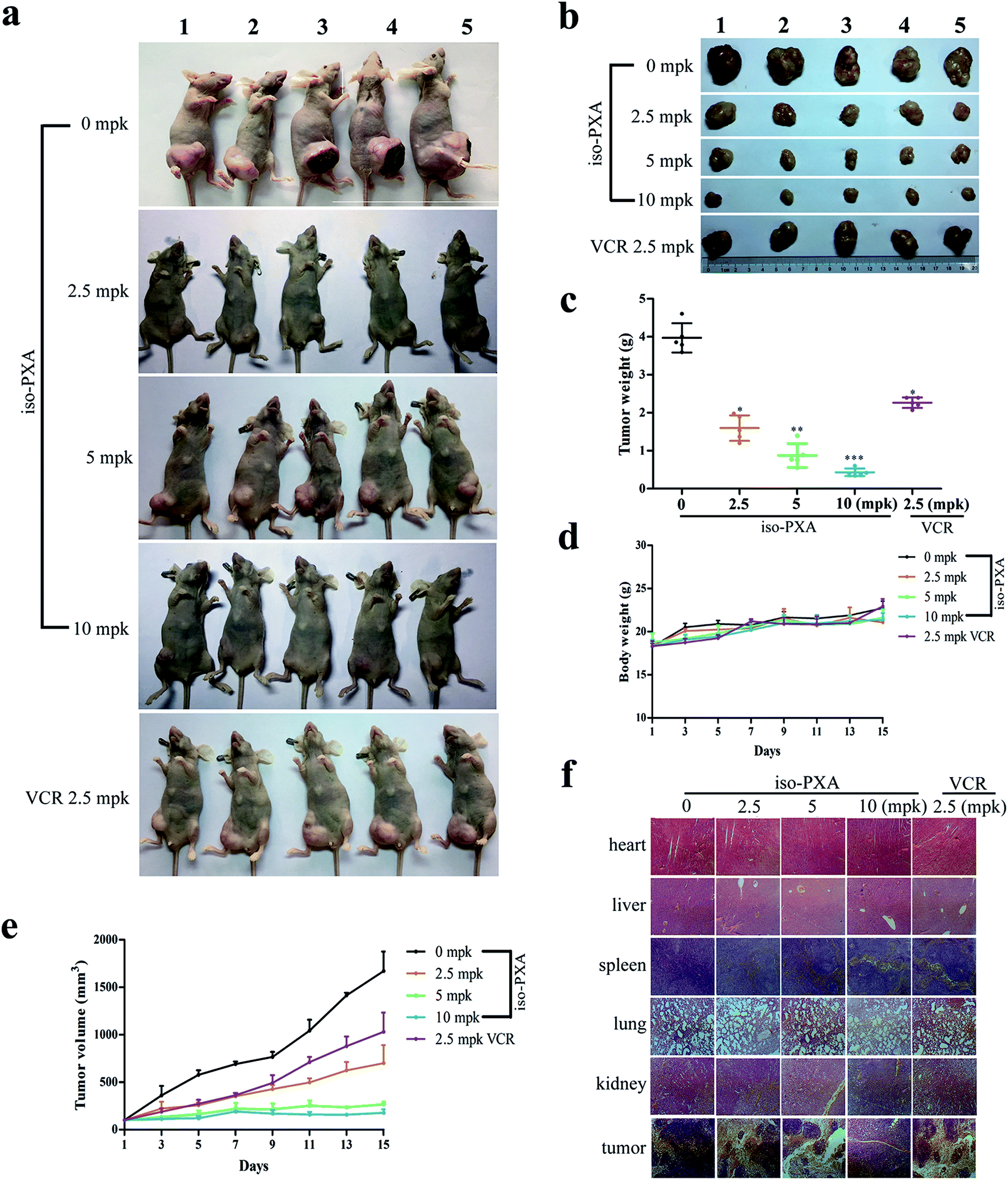 | ||
| Fig. 7 iso-PXA inhibits cancer growth in vivo. (a) and (b) Photographs of nude mice before and after the tumors were removed. (c) Tumor weight of all BALB/c nude mice treated with different concentrations of iso-PXA and VCR (as a positive control). (d) and (e) The tumor volume and mouse weight of each group were measured every two days. (f) H&E staining of tumors and normal tissues of BALB/c nude mice. | ||
3.8 iso-PXA is an inhibitor for efflux transporters
Many compounds are the substrates or inhibitors of P-gp and can be readily pumped out of cells by P-gp, leading to drug resistance.23,24 Western blot analysis showed that P-gp levels were high in HeLa/VCR cells, but it was not detectable in the parental HeLa cells (Fig. 8a). To determine whether iso-PXA was a substrate of P-gp-mediated efflux, we examined the P-gp ATPase activity stimulated by iso-PXA. As shown in Fig. 8b and c, the untreated vehicle control exhibited a small change in luminescence, reflecting the weak basal ATPase activity of P-gp. iso-PXA induced an obvious increase of P-gp ATPase activity when compared with the positive control verapamil (results were normalized relative to ATP consumption in the presence of Na3VO4, an inhibitor of P-gp ATPase activity), indicating that iso-PXA is a substrate of P-gp.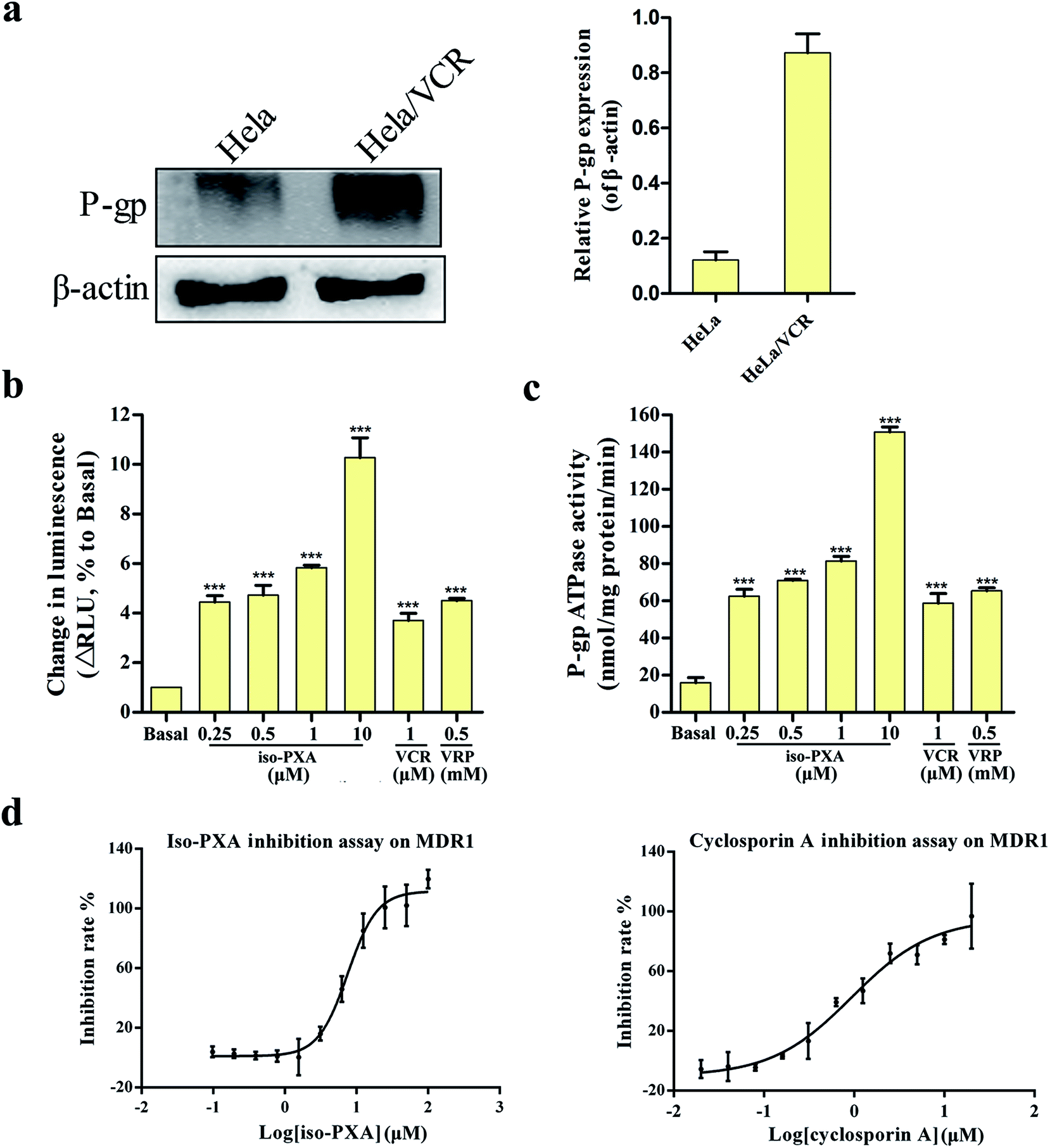 | ||
| Fig. 8 iso-PXA is an inhibitor for efflux transporters. (a) P-gp levels of HeLa/VCR and HeLa cells. (b) iso-PXA shows P-gp substrate characteristics similar to the positive control verapamil. (c) iso-PXA stimulates the ATPase activity of P-gp. The ATPase activity of P-gp was determined as described in the experimental procedures section. (d) Compound inhibition assay on MDR1. Bars represent the mean ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001 vs. the control group. | ||
We further determined whether iso-PXA was an inhibitor of P-gp by using the calcein AM (acetoxymethyl ester of calcein) assay. The ABCB1-mediated calcein AM efflux assay is typically used to detect drug interactions with multidrug resistance proteins in intact cells, since calcein AM is an excellent substrate of multidrug resistance transporter 1 (MDR1), P-gp, and multidrug resistance-associated protein (MRP1). The data showed that iso-PXA displayed a dose-dependent inhibition of ABCB1-mediated calcein AM efflux, similar to the positive control cyclosporine A, which indicated that iso-PXA was a P-gp inhibitor. However, the IC50 of iso-PXA (8.129 μM) was higher than cyclosporine A (1.044 μM) (Fig. 8d), suggesting that the inhibitory activity of iso-PXA might be weaker than the positive control.
3.9 iso-PXA acts as a P-gp modulator in HeLa/VCR cells via decreasing P-gp expression at the protein and mRNA levels
To further investigate whether the observed anti-MDR effect of iso-PXA was related to the P-gp transport function, the intracellular accumulation of Rh123, a classic fluorescent P-gp substrate, was examined by FACS after iso-PXA treatment in HeLa/VCR and HeLa cells. As shown in Fig. 9a and b, the intracellular Rh123 levels after iso-PXA treatment were significantly higher than after positive control VCR treatment in HeLa/VCR cells, while neither iso-PXA nor VCR treatment affected the intracellular accumulation of Rh123 in HeLa cells, which indicated that iso-PXA could block the efflux function of P-gp in HeLa/VCR cells. To elucidate the involved molecular mechanism, western blot analysis was used to determine how iso-PXA affected P-gp expression. The results showed that the expression of P-gp was significantly decreased by iso-PXA in a dose-dependent manner in HeLa/VCR cells and tumor tissues (Fig. 9c–f). Similarly, the corresponding mRNA levels were also reduced in a dose-dependent manner after iso-PXA treatment (Fig. 9g and h).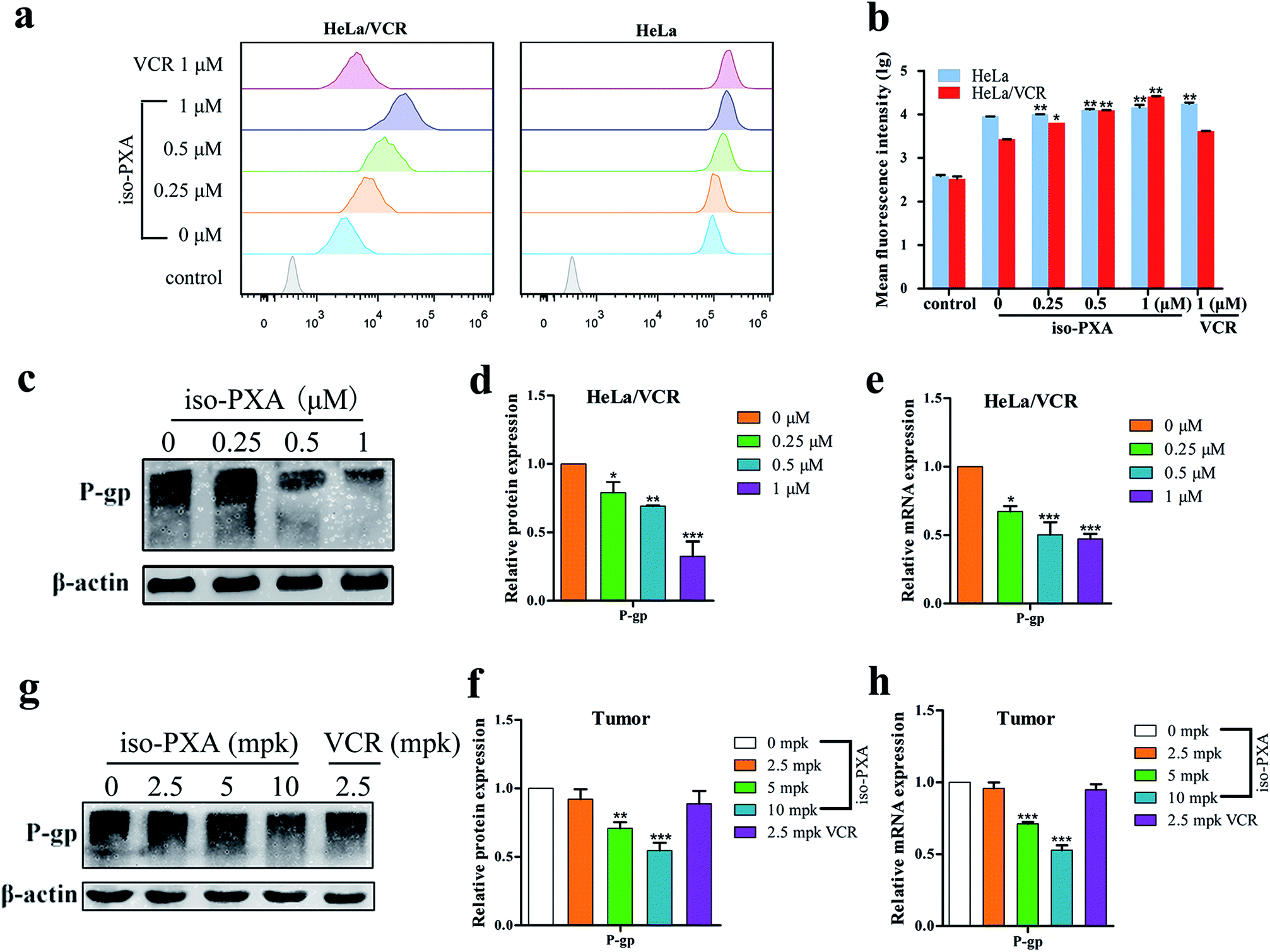 | ||
| Fig. 9 iso-PXA reverses MDR by downregulating P-gp expression and inhibiting drug efflux function. (a) and (b) Drug efflux pump function was detected by flow cytometry after cells were stained with Rh123, and statistical fluorescence intensity was analyzed. (c) and (d) After cells were treated with different concentrations of iso-PXA for 24 h, the expression of P-gp was determined by western blot and analyzed quantitatively, with β-actin as a loading control. (e) The level of the P-gp gene was analyzed by RT-qPCR. GAPDH was used as a loading control. (g) and (f) The expression of P-gp in tumor tissues was determined by western blot and analyzed quantitatively. (h) The P-gp gene level in tumor tissues by RT-qPCR. Bars represent the mean ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001 vs. the control group. | ||
3.10 iso-PXA increased FBW7 expression and decreased Mcl-1 expression in HeLa/VCR cells
In addition to P-gp, MAPK, PTEN, EGFR, β-catenin, survivin, JNK and FBW7 are also reported to be related to drug resistance. We screened these genes by RT-qPCR assay to find the probable mechanism involved in iso-PXA-mediated MDR reversion. We found that there was a significant alternation of the expression level of FBW7 after iso-PXA treatment (Fig. 10a). Previous studies have demonstrated that depletion of FBW7 leads to tyrosine kinase inhibitor (TKI) resistance through stabilization of the anti-apoptotic protein Mcl-1.25 Wertz26 also found that the degradation of Mcl-1 was blocked in patient-derived tumor cells that lacked FBW7 or had loss-of-function mutations in FBW7. By detecting the expression levels of FBW7 and Mcl-1 in HeLa/VCR cells, we found that the mRNA level of FBW7 was significantly upregulated, and Mcl-1 was notably downregulated after iso-PXA treatment compared to VCR treatment (Fig. 10b). The corresponding mRNA levels were also identical in vivo (Fig. 10c). We further detected the protein level of FBW7 and Mcl-1 in HeLa/VCR and HeLa cells by western blot before and after the treatment of iso-PXA and VCR. Similarly, the data showed that the treatment of iso-PXA increased the expression of FBW7 and decreased the expression of Mcl-1 both in HeLa/VCR and HeLa cells, however, the treatment of VCR only changed the level of FBW7 and Mcl-1 in HeLa cells, but not in HeLa/VCR cells (Fig. 10d). Thus, we speculate that iso-PXA may induce anti-MDR activity by targeting FBW7 as a strategy to overcome resistance. | ||
| Fig. 10 Expression of MDR-related genes in vitro and in vivo treated with iso-PXA. (a) The mRNA level of MDR-related genes in HeLa/VCR and HeLa cells treated for 24 h were analyzed by RT-qPCR. GAPDH was used as a loading control. (b) The mRNA level of FBW7 and its substrate Mcl-1 in HeLa/VCR and HeLa cells before and after the treatment of iso-PXA or VCR. (c) The mRNA level of FBW7 and Mcl-1 genes from tumor tissues were analyzed by RT-qPCR. (d) and (e) The protein level of FBW7 and Mcl-1 in HeLa/VCR and HeLa cells before and after the treatment of iso-PXA or VCR was determined by western blot and analyzed quantitatively. Cofilin was used as a loading control. Bars represent the mean ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001 vs. the control group. | ||
4 Discussion
Apoptosis has been a prevalent model to develop anti-cancer drugs, but apoptosis evasion usually results in tumor cells that are resistant to chemotherapy.27 Induction of apoptosis could an effective MDR reversal strategy in cancer therapy.28 For instance, isomahanine is able to induce apoptosis in multidrug-resistant human oral squamous cell carcinoma cells.29 Mitochondria are important to cell bioenergetics and play a central role in cellular apoptosis. Apoptosis mediated by mitochondria can trigger mitochondrial outer membrane permeabilization (MOMP), which has been proposed as a “point of no return” in the mitochondrial apoptotic pathway.30 The accumulation of ROS and the depolarization of the MMP may cause mitochondrial swelling and increase the permeability of the mitochondrial membrane, which induces intrinsic apoptosis. As expected, our data showed that iso-PXA led to increased ROS generation and loss of ΔΨm in both HeLa/VCR and HeLa cells (Fig. 5). Moreover, MOMP can be regulated by the Bcl-2 family, which consists of pro-apoptotic proteins (including Bax, Bak, Bad, Bid, and Bik) and anti-apoptotic proteins (including Bcl-2, Bcl-XL, and Mcl-1). Members of the Bcl-2 family can form homodimers or heterodimers, thereby functioning as agonists or antagonists of each other.31 Importantly, changes in the level of Bcl-2 and Bax are often related to chemotherapy resistance in many cancers. Here, iso-PXA significantly decreased the expression of Bcl-2 and increased the expression of Bax in HeLa/VCR cells, affirming that iso-PXA can inhibit apoptosis evasion and increase drug sensitivity in HeLa/VCR cells. In addition, the pro- and anti-apoptotic members of the Bcl-2 family also lead to the activation of caspases.32 Previous studies have shown that drug-induced cytotoxicity is related to the caspase family. In typical cell models, caspases are classified into initiators (for example caspase-8, -9) and effectors (caspase-3, -6, -7). Caspase-8 is a community of endogenous and exogenous pathways and is the best substitute for caspase-3.33 Drug-induced tumor cell apoptosis and drug resistance of tumor cells are both related to caspase-8. When caspase-8 is activated, apoptosis can be induced by two apoptotic signaling pathways. One pathway is that caspase-8 activates caspase-3 directly and induces apoptosis through an exogenous pathway. The other is that caspase-8 activates the endogenous pathway to induce apoptosis by activating Bid and mitochondrial connections. Caspase-8 activates caspase-9 by cleaving the BH3 domain of the apoptosis-induced protein BID.34 Activated caspase-9 further activates caspase-3, resulting in apoptosis. The activation of caspase-8 can also initiate the intracellular pathway.35 Caspase-9 is a participant in mitochondrial-dependent apoptosis. Active caspase-9 works as an initiating caspase by cleaving, and thus activating downstream executioner caspases, initiating apoptosis.36 Once activated, caspase-9 goes on to cleave caspase-3, -6, and -7, initiating the caspase cascade as they cleave several other cellular targets. PARP is the most important substrate of caspase-3, which can recruit the chromatin remodeler CHD2 to DNA damage and trigger the process of DNA damage repair.37 In this study, we detected the effect of iso-PXA on apoptosis pathways. We found that apoptosis of HeLa/VCR and HeLa cells induced by iso-PXA was dependent on the activation of the caspase-8-, caspase-9- and caspase-3-mediated apoptosis pathway (Fig. 6). This suggests that iso-PXA-induced apoptosis may be mainly due to the activation of caspase-9 through caspase-8 involved in the intrinsic pathway, rather than caspase-8 directly activating the caspase-3-mediated extrinsic pathway. Taken together, iso-PXA may reverse MDR of cancer cells by re-stimulating the apoptosis process.Many tumor cells have shown drug resistance due to the high expression of P-gp.38 As shown in Fig. 7, the protein expression level of P-gp in HeLa/VCR cells is 6.5-fold higher than in HeLa cells. In the present study, our results showed that HeLa/VCR cells were more resistant to VCR than were HeLa cells (Fig. 2 and Table S3†). However, there was no significant difference in the sensitivity of two cell lines to iso-PXA (Fig. 2b). P-gp plays a vital role in VCR resistance in HeLa/VCR cells by stimulating VCR efflux and delaying cell apoptosis. A previous study has reported that IXN can inhibit the efflux function of ABCB1 via increasing the intracellular accumulation of DOX and Rh123, both of which are ABCB1 substrates, to reverse MDR. The P-gp-mediated ATPase activity assay and ABCB1-mediated calcein AM assay results showed that iso-PXA increased both the ATPase activity and the inhibitory effect of MDR1 in a dose-dependent manner (Fig. 8), confirming that iso-PXA is a P-gp substrate or inhibitor. It has been widely reported that many agents play an important role in reversing MDR by inhibiting the function and expression of P-gp.39 As shown in Fig. 9a, iso-PXA significantly increases the intracellular accumulation of Rh123, which confirms that iso-PXA inhibits the efflux function of P-gp. A previous study also showed that dasatinib reversed MDR of breast cancer MCF-7 cells to ADR by downregulating P-gp expression.40 As expected, we revealed that iso-PXA significantly decreased the protein and mRNA expression levels of P-gp in vitro and in vivo (Fig. 9c–h). Thus, we presume that iso-PXA can inhibit P-gp function by decreasing P-gp expression and offer a competitive inhibition with P-gp substrates by reducing toxicity, enhancing efficacy of action and preventing the occurrence of P-gp-mediated cancer resistance.
Although P-gp-mediated multidrug resistance is the most common resistance mechanism, previous studies have also demonstrated that abnormal signal transduction pathways can lead to multidrug resistance of cells, such as the FBW7, ERK/MAPK, PTEN/PI3K/AKT, EGFR/HER2, Wnt/β-catenin and JNK/SAPK signaling pathways. The RT-qPCR results showed that iso-PXA sensitivity was not attributed to the basal activity of MAPK, PTEN, EGFR, β-catenin, survivin and JNK/SAPK genes, but the FBW7 gene was expressed at higher levels in HeLa/VCR cells when treated with iso-PXA (Fig. 10a). FBW7, also known as FBXW7 or CDC4, is a tumor suppressor that is frequently mutated in human cancers41 Intriguingly, Yokobori and colleagues42 observed that patients with low FBW7 expression levels presented with more progressive cancer and a significantly shorter survival. Ye M., et al.25 revealed an unexpected resistance mechanism, in which loss of FBW7 E3 ligase resulted in the accumulation of anti-apoptotic protein MCL-1, thereby enabling cancer cells to escape apoptosis following EGFR and ALK signaling inhibition. Tong J., et al.43 also suggested that FBW7 mutations mediate intrinsic and acquired resistance of colorectal cancers (CRCs) to targeted agents by blocking Mcl-1 degradation. The results showed that the mRNA level of FBW7 was significantly upregulated, and that of Mcl-1 was clearly downregulated after the treatment of iso-PXA both in HeLa/VCR cells and HeLa cells (Fig. 10b and c). However, VCR treatment only affected FBW7/Mcl-1 in HeLa cells, but not in HeLa/VCR cells (Fig. 10b and c). The protein level test by western blot was similar (Fig. 10d). Our results implied that iso-PXA may promote Mcl-1 degradation by targeting FBW7 as a strategy to overcome resistance. But it is still unclear whether P-gp and FBW7/Mcl-1 has any connection. The functional role of iso-PXA in regulating FBW7/Mcl-1 in detail still needs further study.
5 Conclusions
In conclusion, iso-PXA could overcome MDR in HeLa/VCR cells by decreasing P-gp expression, inhibiting P-gp function, and re-activating apoptosis through the intrinsic pathway under the regulation of Bax/Bcl-2 and FBW7/Mcl-1 (Fig. 11). Thus, iso-PXA may be a promising lead compound for patients bearing drug-resistant cancers. | ||
| Fig. 11 Schematic representation of iso-PXA induced MDR reversion and apoptosis activation. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Key Scientific Project of Ministry of Education in Fujian Province (JZ160419), the Fujian Provincial Science and Technology Innovation Joint Project (2017Y9075), the Natural Science Foundation of Fujian Province (2017J01855, 2017J01179 and 2016J01733), the Leading Project Foundation of Fujian Province (2018Y0015), and the “Young Top Creative Talents” of the Second Batch Special Support “Double Hundred Plan” of Fujian Province.Notes and references
- V. S. Jones, R. Y. Huang, L. P. Chen, Z. S. Chen, L. Fu and R. P. Huang, Biochim. Biophys. Acta, 2016, 1865, 255–265 CAS.
- C. Holohan, S. Van Schaeybroeck, D. B. Longley and P. G. Johnston, Nat. Rev. Cancer, 2013, 13, 714–726 CrossRef CAS PubMed.
- Q. Wu, Z. Yang, Y. Nie, Y. Shi and D. Fan, Cancer Lett., 2014, 347, 159–166 CrossRef CAS PubMed.
- M. S. Jin, M. L. Oldham, Q. Zhang and J. Chen, Nature, 2012, 490, 566–569 CrossRef CAS PubMed.
- J. I. Fletcher, M. Haber, M. J. Henderson and M. D. Norris, Nat. Rev. Cancer, 2010, 10, 147–156 CrossRef CAS PubMed.
- X. Wang, E. Wu, J. Wu, T. L. Wang, H. P. Hsieh and X. Liu, PLoS One, 2013, 8, e65686 CrossRef CAS PubMed.
- M. Cives, S. Ciavarella, F. M. Rizzo, M. De Matteo, F. Dammacco and F. Silvestris, Cell. Signalling, 2013, 25, 1108–1117 CrossRef CAS PubMed.
- G. Zhang, J. Li, T. Zhu, Q. Gu and D. Li, Curr. Opin. Biotechnol., 2016, 42, 13–23 CrossRef CAS PubMed.
- Y. Zhang, Y. K. Zhang, Y. J. Wang, S. G. Vispute, S. Jain, Y. Chen, J. Li, D. T. Youssef, K. A. El Sayed and Z. S. Chen, Mar. Drugs, 2015, 13, 2267–2286 CrossRef CAS PubMed.
- L. Cherigo, D. Lopez and S. Martinez-Luis, Mar. Drugs, 2015, 13, 2010–2029 CrossRef CAS PubMed.
- D. Lopez and S. Martinez-Luis, Mar. Drugs, 2014, 12, 525–546 CrossRef PubMed.
- J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. Munro and M. R. Prinsep, Nat. Prod. Rep., 2014, 31, 160–258 RSC.
- L. Chen, M. W. Gong, Z. F. Peng, T. Zhou, M. G. Ying, Q. H. Zheng, Q. Y. Liu and Q. Q. Zhang, Mar. Drugs, 2014, 12, 1939–1958 CrossRef PubMed.
- L. Chen, Y.-Y. Zhao, R.-F. Lan, L. Du, B.-S. Wang, T. Zhou, Y.-P. Li, Q.-Q. Zhang, M.-G. Ying, Q.-H. Zheng and Q.-Y. Liu, Tetrahedron, 2017, 73, 5900–5911 CrossRef CAS.
- H. Dong, X. Yang, J. Xie, L. Xiang, Y. Li, M. Ou, T. Chi, Z. Liu, S. Yu, Y. Gao, J. Chen, J. Shao and L. Jia, Biochem. Pharmacol., 2015, 93, 151–162 CrossRef CAS PubMed.
- J. Wang, Z. Jiang, L. Xiang, Y. Li, M. Ou, X. Yang, J. Shao, Y. Lu, L. Lin, J. Chen, Y. Dai and L. Jia, Sci. Rep., 2014, 4, 5006 CrossRef PubMed.
- T. Jiang, L. Tian, A. H. Guo, H. Z. Fu, Y. H. Pei and W. H. Lin, Yaoxue Xuebao, 2002, 37, 271–274 CAS.
- G. Xu, M. Cirilli, Y. Huang, R. L. Rich, D. G. Myszka and H. Wu, Nature, 2001, 410, 494–497 CrossRef CAS PubMed.
- M. Qiu, L. Chen, G. Tan, L. Ke, S. Zhang, H. Chen and J. Liu, Sci. Rep., 2015, 5, 15104 CrossRef CAS PubMed.
- M. S. Villaverde, A. M. Targovnik, M. V. Miranda, L. M. Finocchiaro and G. C. Glikin, Cytokine, 2016, 84, 47–55 CrossRef CAS PubMed.
- W. Liang, A. Cai, G. Chen, H. Xi, X. Wu, J. Cui, K. Zhang, X. Zhao, J. Yu, B. Wei and L. Chen, Sci. Rep., 2016, 6, 38267 CrossRef CAS PubMed.
- M. Olsson and B. Zhivotovsky, Cell Death Differ., 2011, 18, 1441–1449 CrossRef CAS PubMed.
- M. Liu, W. Zhang, W. Zhang, X. Zhou, M. Li and J. Miao, Toxins, 2017, 9, 208 CrossRef PubMed.
- Z. Bai, M. Gao, H. Zhang, Q. Guan, J. Xu, Y. Li, H. Qi, Z. Li, D. Zuo, W. Zhang and Y. Wu, Cancer Lett., 2017, 402, 81–92 CrossRef CAS PubMed.
- M. Ye, Y. Zhang, X. Zhang, J. Zhang, P. Jing, L. Cao, N. Li, X. Li, L. Yao, J. Zhang and J. Zhang, Cancer Res., 2017, 77, 3527–3539 CrossRef CAS PubMed.
- I. E. Wertz, S. Kusam, C. Lam, T. Okamoto, W. Sandoval, D. J. Anderson, E. Helgason, J. A. Ernst, M. Eby, J. Liu, L. D. Belmont, J. S. Kaminker, K. M. O'Rourke, K. Pujara, P. B. Kohli, A. R. Johnson, M. L. Chiu, J. R. Lill, P. K. Jackson, W. J. Fairbrother, S. Seshagiri, M. J. Ludlam, K. G. Leong, E. C. Dueber, H. Maecker, D. C. Huang and V. M. Dixit, Nature, 2011, 471, 110–114 CrossRef CAS PubMed.
- C. Thoma, Nat. Rev. Urol., 2016, 13, 631 CrossRef CAS PubMed.
- S. Basu, A. Ganguly, P. Chakraborty, R. Sen, K. Banerjee, M. Chatterjee, T. Efferth and S. K. Choudhuri, Biochimie, 2012, 94, 166–183 CrossRef CAS PubMed.
- T. Utaipan, A. Athipornchai, A. Suksamrarn, S. Chunsrivirot and W. Chunglok, Oncol. Rep., 2017, 37, 1243–1252 CrossRef CAS PubMed.
- S. W. Fesik and Y. Shi, Science, 2001, 294, 1477–1478 CrossRef CAS PubMed.
- L. M. Dejean, S. Y. Ryu, S. Martinez-Caballero, O. Teijido, P. M. Peixoto and K. W. Kinnally, Biochim. Biophys. Acta, 2010, 1797, 1231–1238 CrossRef CAS PubMed.
- A. Zhai, X. Zhu, X. Wang, R. Chen and H. Wang, Eur. J. Pharmacol., 2013, 713, 58–67 CrossRef CAS PubMed.
- S. Sharifi, J. Barar, M. S. Hejazi and N. Samadi, Asian Pac. J. Cancer Prev., 2014, 15, 8617–8622 CrossRef PubMed.
- C. Kantari and H. Walczak, Biochim. Biophys. Acta, 2011, 1813, 558–563 CrossRef CAS PubMed.
- K. Kim, M. J. Fisher, S. Q. Xu and W. S. el-Deiry, Clin. Cancer Res., 2000, 6, 335–346 CAS.
- D. R. McIlwain, T. Berger and T. W. Mak, Cold Spring Harbor Perspect. Biol., 2013, 5, a008656 Search PubMed.
- M. S. Luijsterburg, I. de Krijger, W. W. Wiegant, R. G. Shah, G. Smeenk, A. J. L. de Groot, A. Pines, A. C. O. Vertegaal, J. J. L. Jacobs, G. M. Shah and H. van Attikum, Mol. Cell, 2016, 61, 547–562 CrossRef CAS PubMed.
- R. Januchowski, K. Wojtowicz, P. Sujka-Kordowska, M. Andrzejewska and M. Zabel, BioMed Res. Int., 2013, 2013, 241763 Search PubMed.
- S. Ye, J. Zhang, J. Shen, Y. Gao, Y. Li, E. Choy, G. Cote, D. Harmon, H. Mankin, N. S. Gray, F. J. Hornicek and Z. Duan, Br. J. Pharmacol., 2016, 173, 613–626 CrossRef CAS PubMed.
- T. Chen, C. Wang, Q. Liu, Q. Meng, H. Sun, X. Huo, P. Sun, J. Peng, Z. Liu, X. Yang and K. Liu, Cancer Biol. Ther., 2015, 16, 106–114 CrossRef CAS PubMed.
- R. J. Davis, M. Welcker and B. E. Clurman, Cancer Cell, 2014, 26, 455–464 CrossRef CAS PubMed.
- T. Yokobori, Y. Yokoyama, A. Mogi, H. Endoh, B. Altan, T. Kosaka, E. Yamaki, T. Yajima, K. Tomizawa, Y. Azuma, R. Onozato, T. Miyazaki, S. Tanaka and H. Kuwano, Mol. Cancer Res., 2014, 12, 32–37 CrossRef CAS PubMed.
- J. Tong, S. Tan, F. Zou, J. Yu and L. Zhang, Oncogene, 2017, 36, 787–796 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09506j |
| This journal is © The Royal Society of Chemistry 2018 |