
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly conjugated architectures and labile reaction intermediates from coupling between 10π electron-deficient heteroaromatics and sym-trihydroxy- or triamino-benzene derivatives†
Gabriele Micheletti *a,
Carla Bogaa,
Silvia Cinoa,
Silvia Bordonia and
Elena Chugunovabc
*a,
Carla Bogaa,
Silvia Cinoa,
Silvia Bordonia and
Elena Chugunovabc
aDepartment of Industrial Chemistry ‘Toso Montanari’, Alma Mater Studiorum Università di Bologna, Viale Del Risorgimento, 4, 40136 Bologna, Italy. E-mail: gabriele.micheletti3@unibo.it
bArbuzov Institute of Organic and Physical Chemistry, FRC Kazan Scientific Center of RAS, Akad. Arbuzovst. 8, Kazan, Tatarstan 420088, Russia
cKazan Federal University, 18 Kremlyovskaya St., Kazan, Tatarstan 420008, Russia
First published on 13th December 2018
Abstract
The C–C coupling reaction between neutral aromatic carbon nucleophiles such as 1,3,5-triamino- and 1,3,5-trihydroxy-benzene derivatives and 7-chloro-4,6-dinitrobenzofuroxan (Cl-DNBF) or 7-chloro-4,6-dinitrobenzofurazan (Cl-DNBZ) gave in high yield the corresponding conjugated structures, which are of potential interest in the materials field. When chlorine is absent on the electrophile, as in 4,6-dinitrobenzofurazan (DNBZ), the reaction with 1,3,5-triaminobenzene derivatives produces Wheland–Meisenheimer (WM) zwitterionic intermediates, intercepted and fully characterized via NMR at low temperature, whereas stable Meisenheimer complexes (M) were isolated in the reaction of phloroglucinol with 1,3,5-trimethoxybenzene. The latter also gave exclusively M complexes when reacted with 4,6-dinitrobenzofuroxan (DNBF), or 4,6-dinitrotetrazolopyridine (DNTP). The detection of WM or M intermediates can be rationalized by invoking the substituent stabilization ability of the positive charge in WM intermediates, on going from neutral carbon nucleophiles 1,3,5-triaminobenzenes to 1,3,5-trihydroxybenzene derivatives. Furthermore, variable-temperature NMR experiments on the M intermediates gave insights into the rotational barrier about the newly formed C–C bond.
Introduction
Benzofurazan and benzofuroxan derivatives are heterocyclic compounds, which possess interesting properties useful for different applications in many applied fields. For example, they exhibit a broad spectrum of biological activities including antibacterial, antifungal, antileukemic, and acaricide activities. Benzofuroxans are also of great interest as NO donors1–4 and have found applications as dyes, fluorescent biosensors and high energy materials.5,6Due to the 10π-electron ring heteroaromatic structure,7 they behave also as electrophiles and they have been classified as superelectrophilic heteroaromatic compounds8,9 when one or more nitro groups are bound to the carbocyclic ring. For this peculiarity, they are of great interest from the mechanistic point of view. For instance, the 4,6-dinitrobenzofuroxan (DNBF) ring is able to stabilize the negative charge in the Meisenheimer complexes.7,10–12 Furthermore, when DNBF was combined with neutral aromatic nucleophilic species, such as triaminobenzene derivatives 1–3, the detection and characterization of WM intermediates (Scheme 1), simultaneously Wheland type on the nucleophilic moiety and Meisenheimer on the electrophilic one, was achieved for the first time.13
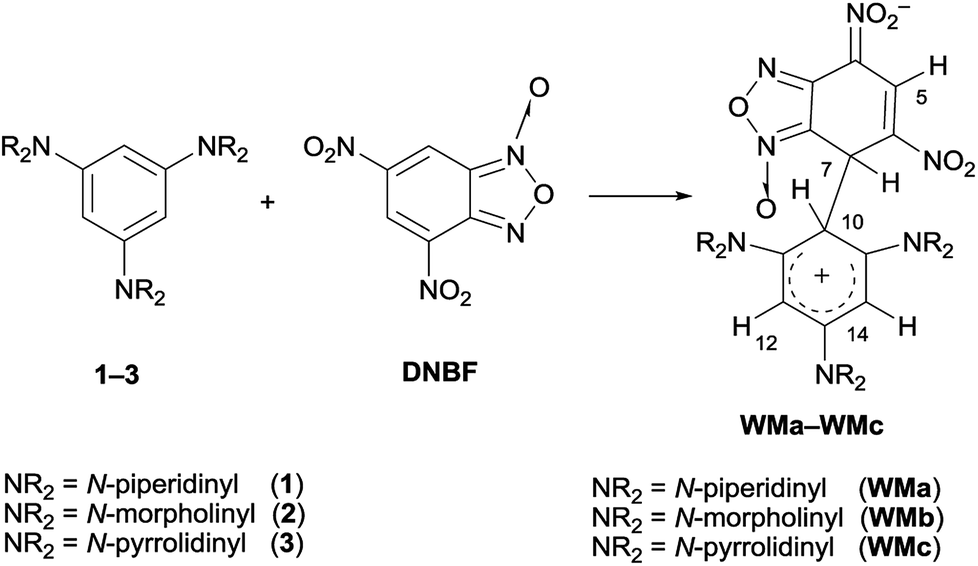 | ||
| Scheme 1 Formation of Wheland–Meisenheimer complexes (WMa–WMc) from the reaction between 1,3,5-tris(N,N-dialkylamino)benzenes and DNBF. | ||
Analogous behaviour was further observed with 4,6-dinitrotetrazolopyridine (DNTP),14 which acts as an electrophile stronger than DNBF (EMayr DNBF = −5.06 (ref. 8) EMayr DNTP = −4.67 (ref. 16)), according to the Mayr's electrophilicity scale.15 The species DNFB and DNTP gave rise to WM complexes, not only with triaminobenzene derivatives 1–3 (ref. 13 and 14) but also with 2-aminothiazole derivatives.17,18 Thus, the zwitterionic intermediates are stable enough to be characterized by NMR spectroscopy, owing to the lack of good leaving groups on the electrophilic moiety, that makes difficult the evolution to the substitution products by hydride departure. In the case of the coupling between 2,4-dipyrrolidinylthiazole and DNBF or DNTP, the stability of the WM intermediates was so evident to permit the first structural determination of WM complexes through X-ray diffraction.18 The access to structures with a scaffold, bearing both electron-donor and electron-acceptor moiety, is of great interest in the applied fields, such as solar energy conversion19 or optoelectronic devices.20 An efficient procedure to obtain such conjugated structures can be SEAr/SNAr reactions between aromatic electrophiles as 7-chloro-4,6-dinitrobenzofuroxan (Cl-DNBF) or 7-chloro-4,6-dinitrobenzofurazan (Cl-DNBZ) and aromatic nucleophiles. The compounds Cl-DNBF and Cl-DNBZ have been reported to react with 2,4,6-trinitroaniline21,22 and 1,3-diaminobenzenes,23 but never with sym-triaminobenzenes 1–3. As described by the pioneering works of Effenberger, they have been coupled with a variety of electrophiles as proton,24–32 halogens,33,34 acyl-,35–37 alkyl-,38 and aryl-halides39 giving Wheland complexes, whose high stability has been ascribed to the great ability of the three amino substituents to stabilize the positive charge on the carbocyclic ring. Recently, the coupling of triaminobenzenes with DNBF,13 DNTP,14 proton,40 aryldiazonium salts,41–45 trinitrothiophene46 and chloro-nitrobenzofurazanes47 produced novel intermediates of the aromatic substitution reaction, together, sometimes, with unexpected species.
Based on the above findings and by considering the importance to find novel conjugated structures, we planned to investigate the reactivity of 7-chloro-4,6-dinitrobenzofurazan (Cl-DNBZ) and 7-chloro-4,6-dinitrobenzofuroxan (Cl-DNBF) with 1,3,5-triaminobenzene derivatives and 1,3,5-trihydroxybenzene derivatives (phloroglucinol and 1,3,5-trimethoxybenzene). Thus, we extended our previous studies on SNAr/SEAr reaction intermediates to the reactions of 1,3,5-triaminobenzenes 1–3 with DNBZ and to the less nucleophilic phloroglucinol or 1,3,5-trimethoxybenzene with DNBZ, DNBF or DNTP.
Results and discussion
For the sake of simplicity, this part has been divided in sub-headings. The first part focused on the construction of novel aromatic architectures by coupling Cl-DNBF and Cl-DNBZ with 1,3,5-triaminobenzenes, 1,3,5-trihydroxybenzene, and 1,3,5-trimethoxybenzene. The second section is dealing with the 1,3,5-triaminobenzenes 1–3 coupled with DNBZ and 1,3,5-trihydroxybenzene (12) and 1,3,5-trimethoxybenzene (13) coupled with DNBZ, DNBF, and DNTP.Reactions of 7-chloro-4,6-dinitrobenzofuroxan (4) and 7-chloro-4,6-dinitrobenzofurazan (5) with neutral carbon aromatic nucleophiles
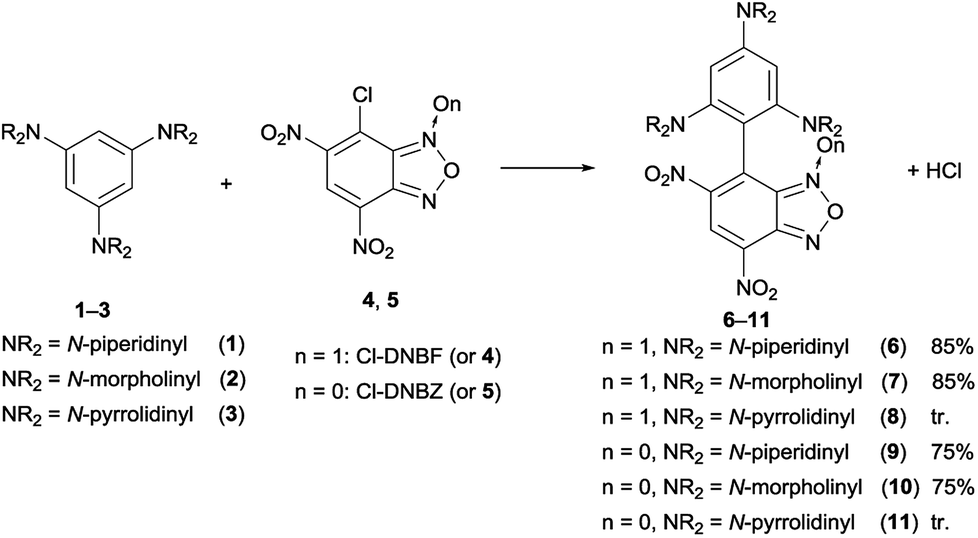 | ||
| Scheme 2 Reactions between 1,3,5-triaminobenzene derivatives 1–3 and 7-chloro-4,6-dinitrobenzofuroxan (4) (in CHCl3) or 7-chloro-4,6-dinitrobenzofurazan (5) (in CH3CN). | ||
When NaHCO3 was used as a base, the solution promptly turns to deep green and the crude was subjected to work-up after about 1 h; the new substitution products 6 and 7 obtained in 85% yield after purification by chromatography on column chromatography on silica gel were fully characterized. They resulted to be stable as solids, whereas decompose after 24 h in CDCl3 or CD3CN solution, as indicated by the presence of 1H NMR unidentified signals. On the contrary, their salts were stable for several days, as confirmed by adding an equimolar amount of picric acid to a deuterochloroform solutions of 6 and monitoring the 1H NMR signals of picrate (6H+picrate) for seven days.
In the 1H NMR spectrum of the crude reaction mixture between 1,3,5-tris(N-pyrrolidinyl)benzene (3) and 4, the presence of signals belonging to unidentified species, likely due to the high reactivity of the reactants, was detected, and only traces of 8 were obtained after chromatographic purification.
The reactions of 7-chloro-4,6-dinitrobenzofurazan (5, Cl-DNBZ) with 1–3 (Scheme 2) were run in acetonitrile by using two-fold molar excess of nucleophile. After 1 h the solvent was removed and the new products 9 and 10 (derived from the coupling of 5 with 1 and 2, respectively) were isolated in high yield. In the case of the reaction between 3 and 5, the coupling gave species 11 which was observed exclusively if the reaction was run in the NMR spectroscopy tube.
Therefore, the reaction courses were monitored in the absence of a base: the 1H NMR spectrum of mixtures of equimolar amount of Cl-DNBF and 1 (or 2), in CDCl3 at 25 °C, evidenced signals consistent only with those of the salt 6H+Cl– (or 7H+Cl−) (Fig. 1). The reaction between 4-chloro-7-nitrobenzofurazan (ClNBZ) and 1 or 2 has previously been47 reported affording the related substitution product in 60% or 18%, respectively, in the case of 1 or 2 after 96 h. The increased electrophilicity of Cl-DNBF, due to the presence of the additional EWG nitro group with respect to ClNBZ, is likely responsible of the higher conversion and shorter reaction time.
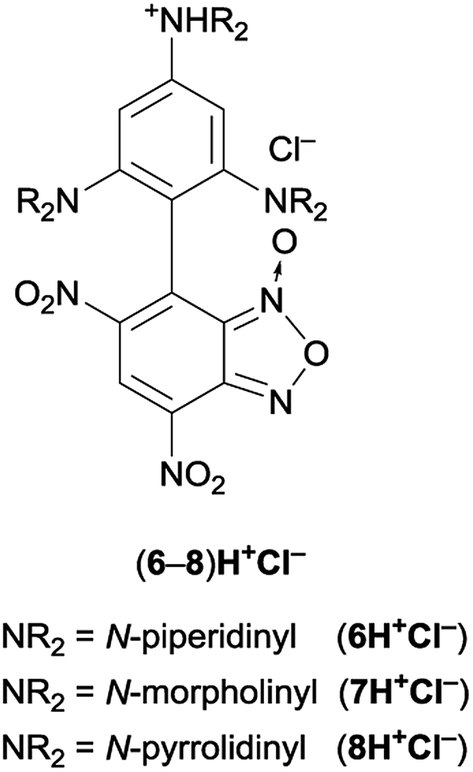 | ||
| Fig. 1 Salts derived from the reaction between equimolar amount of 1–3 and 4. | ||
The comparison of the results obtained by 7-chloro-4,6-dinitrobenzofuroxan with those obtained with 4-chloro-7-nitrobenzofurazan instead of 4-chloro-7-nitrobenzofuroxan is permitted taking into account that Cl-DNBF48 and Cl-DNBZ8 have the same Mayr's electrophilicity value (EMayr = −6.11).
Finally, we carried out the reaction between 1–3 and 4 by direct mixing of the reagents in the NMR spectroscopy tube at low temperature to eventually trap reaction intermediates. Previously, the coupling between 1–3 and 4,6-dinitrobenzofuroxan (DNBF) allowed the NMR detection at low temperature of the first Wheland–Meisenheimer complexes (WM1–3 in Scheme 1).13 Although we were aware that the occurrence of a chlorine atom in the 7-chloro-4,6-dinitrobenzofuroxan, causing good leaving group in SNAr, makes hard the challenge to intercept the zwitterionic WM species (Scheme 3), the ability of amino group in stabilizing the positive charge enables the intent to spectroscopically intercept the W intermediate (Scheme 3). It has to be noted that similar stable intermediates have been previously observed in the reaction between 3 and ClNBZ.47
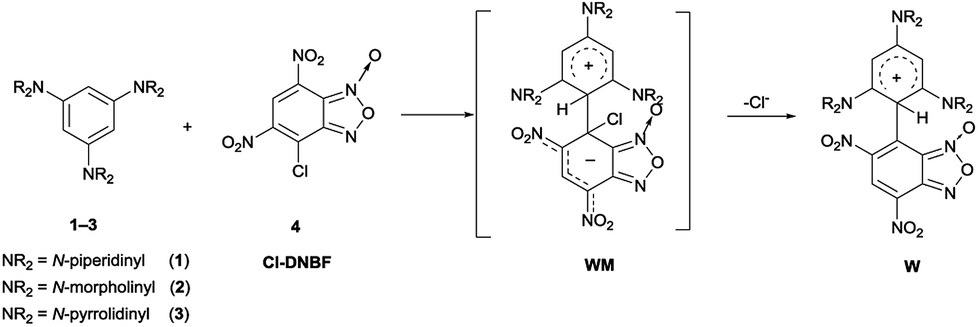 | ||
| Scheme 3 Possible intermediates (WM and W) from the reactions between 1–3 and 4. | ||
First, our strategy pursued to select 4 to be reacted with 3, since it is well known that the latter is able to stabilize the positive charge of Wheland intermediates. In this case, the 1H NMR spectrum, recorded in CD2Cl2 at −70 °C, as the reactants were mixed, showed a set of signals compatible with those of the salt 8H+Cl– (Fig. 1). In particular, a broad singlet at δ = 11.35 ppm was ascribed to the NH signal and the presence of a singlet at δ = 8.9 ppm and of two singlets at δ = 6.02 and 5.96 ppm (integrating 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) was attributed to the proton belonging to the benzofuroxanyl moiety and to the aromatic protons of the triaminobenzene moiety, respectively.
:1:1) was attributed to the proton belonging to the benzofuroxanyl moiety and to the aromatic protons of the triaminobenzene moiety, respectively.
The latter resulted not equivalent due to the restricted rotation around the new C–C bond formed, as also confirmed by the splitting of the signals of the methylene hydrogen atoms of the pyrrolidinyl moiety in the aliphatic region. On raising the temperature, the two singlets belonging to the hydrogen atoms on the aminobenzene moiety and those of the pyrrolidinyl groups, gradually broadened until coalescence at about −43 °C; above this value they gradually sharpened. The spectrum exhibited further signals increasing the temperature from +2 °C to +25 °C, indicating a possible evolution to unidentified compounds, explaining the failure of our previous attempts to isolate 8.
An analogous behavior was observed when the reaction between 2 and 4 was carried out at −88 °C in CD2Cl2: after reaction mixture the 1H NMR spectrum showed disappearance of the reactant signals and appearance of new signals at δ = 8.98, 7.52, and 6.39 ppm in the aromatic region integrating 1:1:1, respectively, attributable to the salt 7H+Cl−. The morpholinyl signals, at this temperature, were distinct due to the restricted rotation around the new formed C–C bond. On gradually raising the temperature, as observed in the previous case, the signals belonging to the aminobenzene moiety gradually sharpened, and the coalescence situation was reached around −37 °C. The phenomenon was reversible on gradually lowering the temperature from 25 °C until −88 °C, since the final spectrum was equal to that obtained after mixing the two reagents at the same temperature. When the sample has been extracted from the NMR probe, a solid was observed on the bottom of the NMR tube; after filtration, it was dissolved in CD3CN and analyzed through 1H, 13C NMR and DEPT experiments and its spectral data were consistent with those of structure 7H+ (Fig. 1). This solid was percolated through Al2O3 eluting with acetonitrile and the 1H NMR spectrum of the residue was in agreement with that of the neutral form 7.
:70 relative % molar ratio of residue reactants (4 or 12) and the coupling product 14. The hydrogen relative signal integration between compounds 4 and 14, became approximately 1:4, after 5 h and the conversion appeared almost complete after 24 h. Analogous behavior was observed for the reaction between 5 and 12, yielding 16.
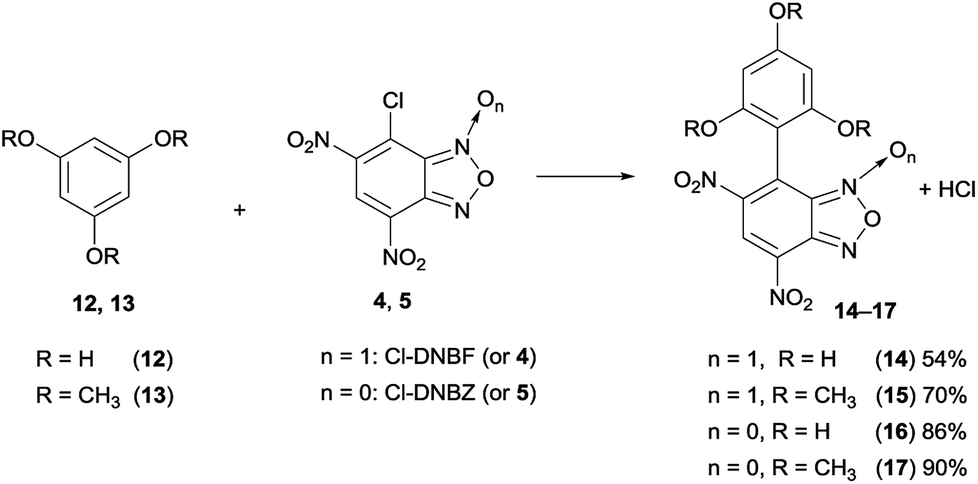 | ||
| Scheme 4 Reactions between 12 and 13 and 7-chloro-4,6-dinitrobenzofuroxan (4) or 7-chloro-4,6-dinitrobenzofurazan (5), yields are after purification by FC (more details in Experimental section). | ||
The reaction between 1,3,5-trimethoxybenzene (13) and 4 (Scheme 4) was carried out under the analogous conditions used for phloroglucinol: after 15 min, the spectrum showed a 86:14 relative percent molar ratio between the starting materials and the coupling product 15; then the ratio increased, until the conversion reached 84% after 24 h. The spectral data of compound 15 (17), obtained in 70% (90%) yield after purification by silica gel flash chromatography is in agreement with those reported in the literature.49
The data obtained by monitoring the reactions by NMR clearly reveal that the reaction of 4 with phloroglucinol is more facile than that with 13. This observation can be due to higher electron donor effect of hydroxyl, with respect the methoxy group (σOH+ = −0.92, σOMe+ = −0.78)50 and to the occurrence of tautomeric equilibria involving phloroglucinol, as recently investigated.51 Further, the reaction carried out between 12 and 4-chloro-7-nitrobenzofurazan (ClNBZ) did not occur, even if in refluxing acetonitrile, in contrast with the already reported results between ClNBZ and 1–3; these findings indicate that the lower nucleophilicity of trihydroxybenzene derivatives with respect to triaminobenzenes requires the presence of a further nitro group to permit the aromatic substitution reactions.
Reactions between neutral carbon aromatic nucleophiles and 10π electron-deficient heteroaromatics without good leaving groups
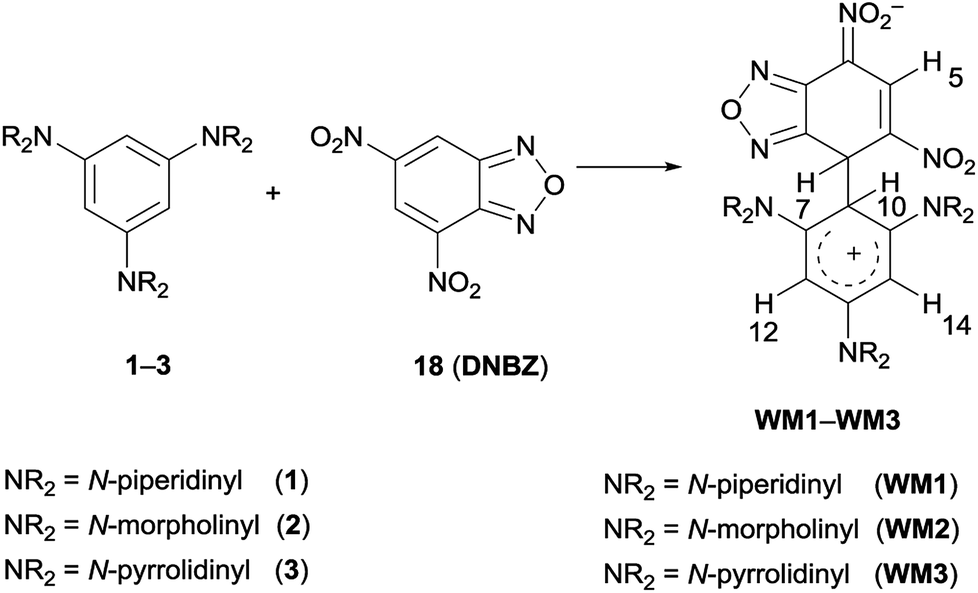 | ||
| Scheme 5 WM intermediates from the C–C coupling between 1–3 and DNBZ. | ||
Particularly diagnostic was the presence of a single signal owing to hydrogen atom of the electrophilic moiety (H-5 in Scheme 5, e.g. at 9.09 ppm for WM1) together with four signals in the δ 5.8–4.0 interval.
Two of them were ascribed to H-7 and H-10 (two doublets at 5.00 and 4.82 ppm, J = 2.6 Hz, for WM1, respectively), which were shielded with respect to the corresponding starting signals due to the sp2 to sp3 hybridisation change of the relative carbon atom. Analogously, the anisochronous signals of H-12 and H-14 (i.e. 5.54 and 4.93 ppm for 1H NMR of WM1) in both the 1H and 13C NMR spectra are attributable to the restricted rotation about C-2-C-10 bond.52,53 The same effect was observed for the atoms belonging to the amino rings. The signals, related to H-7, H-10, H-12, H-14 and amino moiety, gradually broadened on increasing the temperature, as a result of an exchange process. The signals coalesce at −5 °C for WM1 and at −37 °C for WM2, then gradually sharpened. In case of WM1 in CD2Cl2 or WM3 in CDCl3, the coalescence occurred at +3 °C/+6 °C respectively, and involved H-10, H-12, and H-14 signals, whereas the sharp H-7 signal appeared independent from the temperature, recalling the observed phenomenon, which involved coupling between 1–3 and DNBF13 or DNTP14 Warming the solution from −54 °C to room temperature and cooling again, a spectrum identical to the starting one was obtained indicating the occurrence of a reversible dynamic process.
Furthermore, the coupling between DNBZ and 4-(3,5-di(piperidin-1-yl)phenyl)morpholine (19) (Scheme 6) was run in the same conditions. The 1H NMR spectrum of the reaction mixture, recorded at −54 °C, showed a set of signals compatible with those of the two distinct WM4 and WM5 zwitterionic intermediates, as shown in Scheme 6, reflecting the same behavior observed for WM1–WM3.13,14
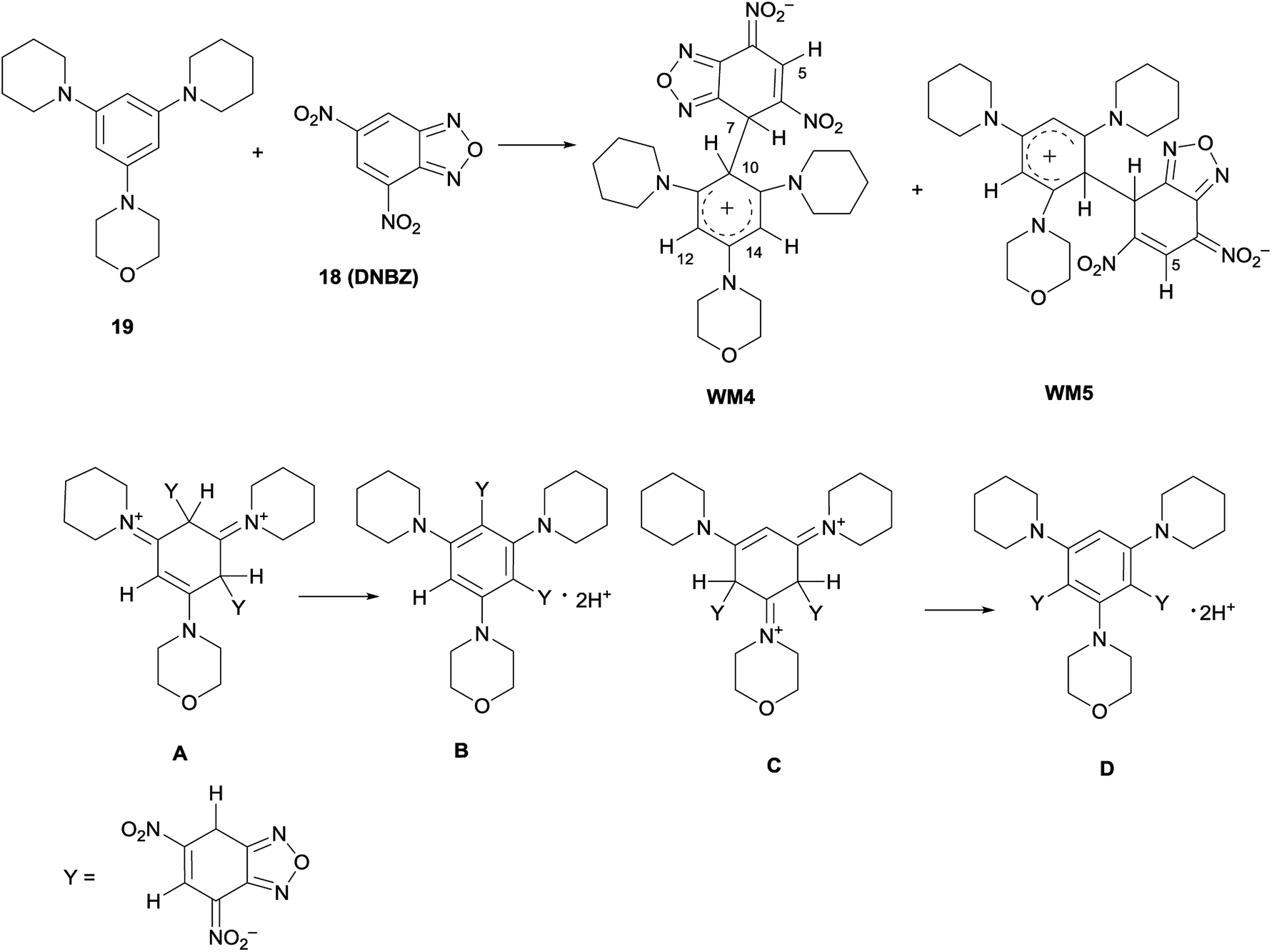 | ||
| Scheme 6 Zwitterionic reaction intermediates WM4 and WM5 formed from 18 and 19 and structure of other possible intermediates. | ||
In principle, also a double attack might occur to give structures A- or C-like, that might evolve to di-Meisenheimer species B- or D-like. Significant examples of similar anionic diadducts have been actually reported by coupling between ambient nucleophiles with DNBF54a or trinitrobenzene.54b,c In current case, the signals and their relative integration in the 1H NMR spectrum, together with the absence of those belonging to unreacted 19, permit to exclude the presence of the above structures A–D.
The structure of the intermediates WM1–WM5 was confirmed also by 13C NMR spectroscopy and/or direct proton to carbon (g-HSQC sequence) correlation experiments (Fig. S26–S35†). In the case of the morpholinyl derivative WM2, the 1H NMR signals belonging to H-7, H-10, H-12, H-14, and to the morpholinyl moiety, appeared broad in the spectrum recorded in CDCl3 at −54 °C. Thus, the reaction was carried out in CD2Cl2 at −80 °C to achieve a better resolution; in this case the coalescence was reached at −33 °C. By comparison, the reaction between 1 and 18 was also carried out in CD2Cl2 at −80 °C. As previously reported for WMa-c,13,14 the signal belonging to H-7 atom remained sharp on varying the temperature. Furthermore, WM1–WM5 resulted to be unstable when kept in CDCl3 at 25 °C overnight, since the solution presents several 1H NMR signals, belonging to unidentified compounds.
Reactions of 4,6-dinitrobenzofuroxan (DNBF), 4,6-dinitrotetrazolopyridine (DNTP), and 4,6-dinitrobenzofurazan (DNBZ) with 1,3,5-trihydroxybenzene (12) and 1,3,5-trimethoxybenzene (13)
Based on previous and current findings, which have shown the formation of WM intermediates at low temperature between 1–3 and 4,6-dinitrobenzofuroxan (DNBF),13 4,6-dinitrotetrazolopyridine (DNTP),43 or 4,6-dinitrobenzofurazan (DNBZ, see above section), we planned to investigate the behavior of the above electrophiles with 12 and 13. Each nucleophile/electrophile combination was realized by adding the two reagents directly in the NMR spectroscopy tube at low temperature (−35 °C in CD3CN, or −80 °C in acetone-d6 for the DNBZ/12 couple) and by 1H NMR monitoring. In all cases, the Meisenheimer M1–M6 complexes (Scheme 7) were observed and characterized by 1H NMR, 13C NMR, DEPT and/or g-HSQC experiments. In most cases, due to the poor stability in solution at room temperature of these intermediates, the 13C NMR spectrum was recorded at low temperature.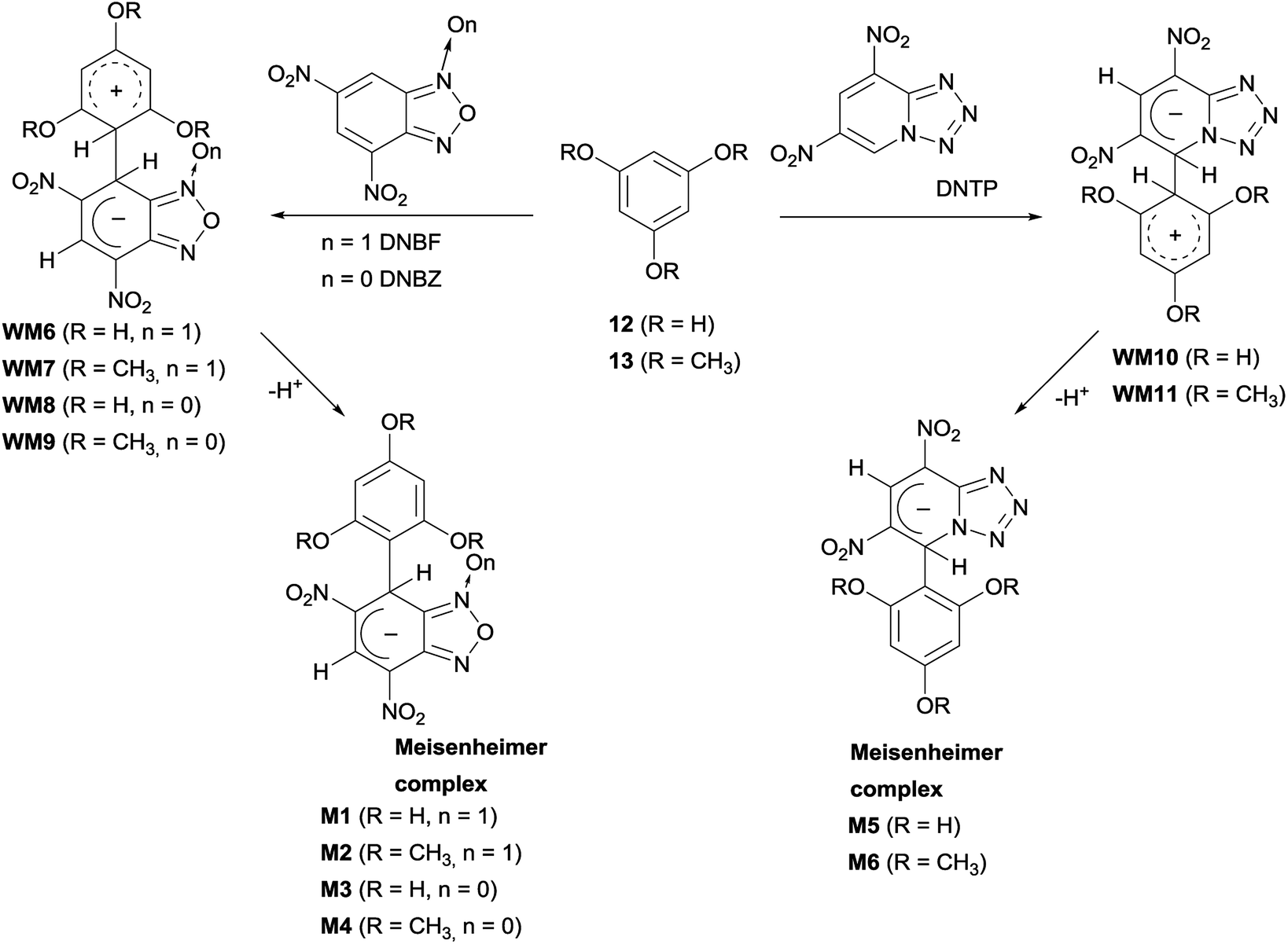 | ||
| Scheme 7 Reaction intermediates from the reactions of 12 and 13 with DNBF, DNBZ, and DNTP. | ||
Although the first step of the SNAr/SEAr reaction consists in the formation of the WM intermediate, the detection of only the M1–M6 intermediates by NMR measurements at low temperature indicates that WM6–WM11 species, derived from the coupling with 12 or 13 (Scheme 7), are less stable than WM1–WM5 (Schemes 5 and 6). The presence of hydroxy- or methoxy- substituents on the carbocyclic ring of 12 and 13 causes very elusive species on the NMR time-scale detection due to the minor stabilization of the Wheland moiety positive charge by hydroxy (or methoxy) substituents with respect to the amino substituents. On the other hand, the M1–M6 anionic intermediates resulted stable enough, due to the ability to stabilize the negative charge by the moieties derived from DNBF, DNBZ, and DNTP.
Moreover, the 1H NMR spectra recorded at low temperature showed non-equivalence of the hydrogen atoms belonging to the nucleophilic moiety of M1–M6 and, in case of M2, M4, and M6 to methyl groups also. This may be ascribed to rotational constraints at low temperature, about the C–C bond, formed between the nucleophilic and the electrophilic moiety. Coalescence phenomenon was observed on raising temperature, then the signals gradually sharpened. From the proton NMR spectra of M1–M6, recorded under dynamic conditions, the free activation energy (ΔG#) was derived by means of the Eyring equation.56 The Fig. 2 shows the line shape simulation for M2 and the ΔG# values obtained are reported in Table 1.
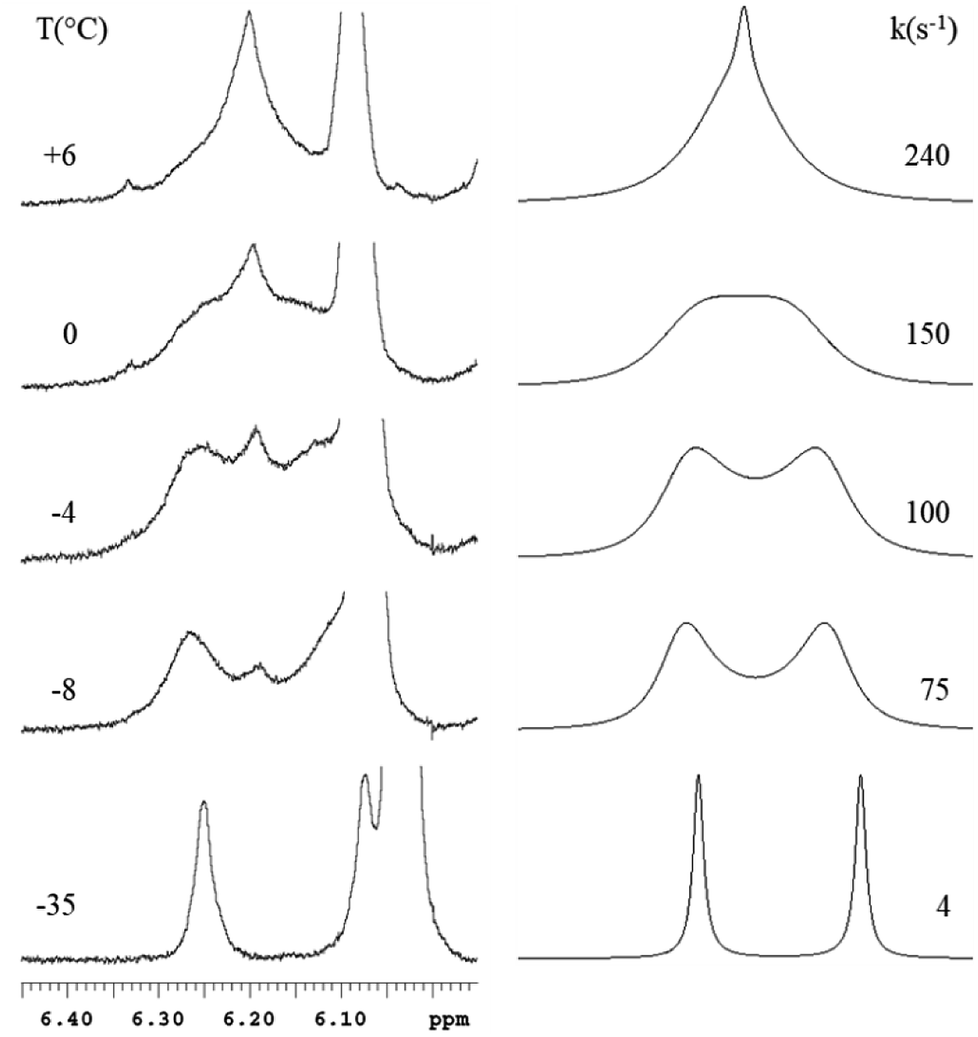 | ||
| Fig. 2 Left: experimental variable temperature 1H NMR spectra in CDCl3 of M2. Right: line-shape simulation obtained with the rate constant indicated. The reported real spectra results partially hidden by the signal of residual starting material. | ||
| ΔG# (kcal mol−1) | Solvent | |
|---|---|---|
| M1 | 13.2 ± 0.2 | CD3CN |
| M2 | 13.2 ± 0.2 | CD3CN |
| M3 | 13.9 ± 0.2 | CD3COCD3 |
| M4 | 14.1 ± 0.2 | CD3CN |
| M5 | 12.4 ± 0.2 | CD3CN |
| M6 | 12.5 ± 0.2 | CD3CN |
The ΔG# values obtained are in line with those commonly reported for rotational processes.57 In particular, slightly different values have been found on changing the electrophile, but appear almost the same by altering the nucleophilic moiety; thus indicating that the rotational constraints are independent from the hydroxy- or methoxy- substituents on the nucleophilic moiety.
Concerning the formation of the σ-adducts, the M2 and M4 have previously been obtained by Terrier and coll. in DMSO.55 In the case of M2, the authors observed the spontaneous oxidation to 15 in 50% yield in the same solvent. In the current case, adducts M2 and M4 have instead been prepared in CD3CN solution, and M2 undergo departure of the hydride ion to be completed in about three days. Evolution to 15 was confirmed by spectral data comparison of authentic 15 obtained as shown in Scheme 4. Analogous behavior has been observed for the reaction of 13 with DNBZ. These findings agree with those previously reported55 about the facile oxidation of some DNBF and DNBZ σ-adducts and provide further examples of formal hydride displacement from an aromatic ring.
Conclusions
New compounds, interesting for several potential applications, have been obtained in good yield by aromatic substitution reaction of triamino- and trihydroxybenzene derivatives with 7-chloro-4,6-dinitrobenzofuroxan (Cl-DNBF) and 7-chloro-4,6-dinitrobenzofurazan (Cl-DNBZ).Novel WM intermediates from the reaction between 1,3,5-triaminobenzenes and 4,6-dinitrobenzofurazan (DNBZ) have been detected and characterized by NMR.
Contrarily to what observed by reacting 1,3,5-triaminobenzene derivatives and 4,6-dinitrobenzofuroxan, 4,6-dinitrobenzofurazan, or 4,6-dinitrotetrazolopyridine, the reactions between the latter electrophiles and 1,3,5-trihydroxybenzene (or 1,3,5-trimethoxybenzene), carried out directly at low temperature in the NMR tube, did not evidence Wheland–Meisenheimer intermediates, but solely Meisenheimer complexes M1–M6. The different behaviour with respect to that of triaminobenzene derivatives may be devoted to the lower ability to stabilize positive charge on the nucleophilic moiety of WM intermediates in the hydroxy- or methoxy-substituted benzene species. Further, the low temperature NMR dynamic behaviour of M1–M6 species showed non-equivalent signals, which, increasing the temperature, gradually broadened to a coalescence situation, until they sharpen again. The observed spectra allowed determine the free activation energy of the restricted rotational process at low temperature.
Experimental section
The 1H and 13C NMR spectra were recorded with a Mercury 400 and Inova 600 (Varian, Palo Alto USA) spectrometers operating at 400, or 600 MHz (for 1H NMR) and 100.56, or 150.80 MHz (for 13C NMR), respectively. J values are given in hertz (Hz). Signal multiplicities were established by DEPT experiments. Chemical shifts were referenced to the solvent [(δ = 5.31 and 53.8 ppm for CD2Cl2), (δ = 1.96 and 118.2 ppm for CD3CN), (δ = 2.50 and 39.5 ppm for CD3SOCD3), (δ = 2.10 and 30.2 ppm for CD3COCD3), and (δ = 7.26 and 77.0 ppm for CDCl3) for 1H and 13C NMR, respectively].The variable-temperature NMR spectra and 2D low temperature spectra were recorded on Mercury 400 or Inova 600 instruments with a direct PFG Probe. The temperatures were calibrated by substituting the sample with a precision Cu/Ni thermocouple before the measurements. Complete fitting of dynamic NMR line shapes was carried out using a PC version of the DNMR-6 program.58
ESI-MS spectra were recorded with a WATERS 2Q 4000 instrument. Chromatographic purifications were carried out on silica gel (0.037–0.063 mm, Merck) columns at medium pressure. Thin layer chromatography (TLC) was performed on silica gel 60 F254 coated aluminum foils (Fluka). Melting points were measured on a Stuart SMP3 apparatus and are uncorrected. Solvents were commercial materials (Aldrich or VWR International) if not specified. 1,3,5-Tris(dialkylamino)benzenes 1–3 were prepared as described previously.13 7-Chloro-4,6-dinitrobenzofuroxan (Cl-DNBF, 4),3b DNBF,12 DNBZ,12 and DNTP14 were synthesized and purified as described in the literature.
Synthesis of 7-chloro-4,6-dinitrobenzofurazan (5)
In a three-necked round bottom flask, equipped with thermometer, dropping funnel, and kept in an ice bath, 0.5 g of 4-chloro-7-nitrobenzofurazan (ClNBZ) were introduced and dissolved in conc. H2SO4 96% (7 mL). The solution was magnetically stirred then a solution of conc. HNO3 65% (0.75 mL) conc. H2SO4 96% (2 mL) was slowly drop-wise added. The reaction temperature was maintained at 0 °C during the addition of the sulfonitric mixture. Then the mixture was increased until 40 °C and kept at this temperature for 12 h. The reaction mixture was put into a becker with ice and, once the ice was fused, the solid obtained was isolated by filtration on Buchner funnel, washed with water until the pH of filtrate was neutral, and dried in a desiccator. Chemico-physical data of the solid agreed with literature data.59General procedure for the synthesis of compounds 6–8 and related salts
To a solution of 1,3,5-triaminobenzene derivative (6 × 10−5 mol) in CDCl3 (10 mL), 1.3 eq. of sodium bicarbonate and an equimolar amount of 7-chloro-4,6-dinitrobenzofuroxan (4) were added at room temperature. The mixture was magnetically stirred and the reaction progress was monitored through TLC and 1H NMR analysis. After solvent removal, the reaction products were purified by column chromatography on silica gel (FC). Compound 6 was obtained using as eluent first a 6/4 diethyl ether/light petroleum and then diethyl ether; in the case of compound 7, the eluent was diethyl ether/ethyl acetate (1/1). In the case of the reaction between 3 and 4, due to the formation of many compounds, only traces of a compound which 1H NMR spectrum, reported below, is compatible with that of 8, were obtained.The reaction between equimolar amount of 1 and 4 was carried out also in CDCl3 directly in the NMR spectroscopy tube without base and the corresponding salt 6H+Cl– was obtained. Picrate salt of 6 (6H+picrate) was obtained by adding to 6 equimolar amount of picric acid. The reaction between equimolar amount (7.7 × 10−5 mol) of 1 and 4 dissolved in CH2Cl2 (6 mL) was carried out also at room temperature: immediately the colour of the solution became dark green and after one night a green solid precipitated and was isolated by filtration. Its chemico-physical data agreed with those of compound 6H+Cl–. A solution of this solid was percolated through Al2O3 (eluent: CH3CN) and the 1H NMR spectrum of the residue was equal to that of the neutral form 6. Analogous behavior was observed for the reaction between 2 and 4. Chemico-physical data of the obtained compounds are reported below.
Variable temperature NMR experiments for the reaction between 1–3 and 5. Typical procedure
Cl-DNBF (4, 0.038 mmol) was dissolved in CD2Cl2 (1 mL) and introduced in a NMR spectroscopy tube, that was inserted in the NMR probe. When the probe temperature reached −88 °C, an equimolar amount of 1,3,5-tris(morpholinyl)benzene (2) was added to the solution, that became blue then dark green, and the 1H NMR spectrum of the resulting solution was quickly recorded. The system was monitored with times and the spectra were recorded at different temperatures until 25 °C was reached. The spectra showed the presence of signals ascribable to the salt 7H+Cl−. At low temperature the signals in the spectrum showed non-equivalence of the hydrogen atoms belonging to the carbocyclic ring; on raising the temperature the signals broadened until to reach a coalescence situation. After, the peaks became sharp and a re-cooling produced again their broadening until to their splitting as observed after the reagents mixing at low temperature. The systems were monitored until no further change could be detected in the recorded spectra.General procedure for the synthesis of compounds 9–11
To a solution of 1,3,5-triaminobenzene derivative (0.14 mmol) in CD3CN (10 mL), 7-chloro-4,6-dinitrobenzofurazan (5, 0.07 mmol) was added at room temperature. The mixture was magnetically stirred and the reaction progress was monitored through TLC and 1H NMR analysis. After solvent removal, the reaction products were purified by column chromatography on silica gel (FC). Compound 9 was obtained using dichloromethane as eluent; in the case of compound 10, the eluent was dichloromethane/ethyl acetate (from 10/1 until 8/2). In the case of the reaction between 3 and 5, due to its easy decomposition to many unidentifiable compounds, compound 11 was only characterized by 1H NMR of the crude reaction mixture after solvent removal.![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2), 26.2 (NCH2H2), 24.9 (NCH2CH2H2), 24.5 (NCH2CH2H2). MS (ES+) m/z: 536 [M + H]+,558 [M + Na]+; HRMS calc. for C27H34N7O5+: 536.2616; found: 536.2618.
H2), 26.2 (NCH2H2), 24.9 (NCH2CH2H2), 24.5 (NCH2CH2H2). MS (ES+) m/z: 536 [M + H]+,558 [M + Na]+; HRMS calc. for C27H34N7O5+: 536.2616; found: 536.2618.General procedure for the synthesis of compounds 14–17
To a magnetically stirred solution of 12 or 13 (2 × 10−4 mol) dissolved in CD3CN (5 mL and 10 mL, respectively), an equimolar amount of 7-chloro-4,6-dinitrobenzofuroxan (4) or 7-chloro-4,6-dinitrobenzofuroxan (5), was added at room temperature. 1H-NMR analysis and TLC were used to monitor the reaction progress. Compound 14 was purified by precipitation from diethyl ether and light petroleum. Compound 15 was purified by column chromatography on silica gel (FC), using as eluent diethyl ether/light petroleum (7:3). Compound 16 was isolated from the concentrated crude after treatment with a little amount of CH2Cl2 and removal of the unreacted phloroglucinol by filtration. Compound 17 was purified by column chromatography on silica gel (FC), using as eluent diethyl ether/light petroleum (2/8) then CH2Cl2/ethyl acetate 9/1. Although the reactions carried out in NMR spectroscopy tube showed conversion almost quantitative, the yields in isolated product were lower likely due to the purification methods, but we did not focus our efforts on yields optimization.
All the products were characterized by usual spectroscopic methods. Chemico-physical data are reported below.
Formation and detection of Wheland–Meisenheimer intermediates WM1–WM5
A solution of 1,3,5-triaminobenzene derivative (0.025 mmol) was dissolved in CDCl3 or CD2Cl2 (1 mL) and introduced in a NMR spectroscopy tube that was inserted in the NMR probe. When the probe temperature reached −54 °C (in CDCl3) or −80 °C (CD2Cl2), an equimolar amount of DNBZ (5.17 mg, 0.025 mmol) was added to the solution, that became dark red-coloured, and the 1H NMR spectrum was quickly recorded. The system was monitored at various times and at different temperatures until 25 °C. Immediately after the mixing, the spectrum showed the appearance of new signals, ascribed to zwitterionic intermediates WM, as confirmed by 13C NMR, DEPT or g-HSQC experiments. On raising the temperature, signals belonging to WM gradually broadened and the return to the previous temperature lead to the re-appearance of sharp signals. Complexes WM1–WM5 resulted not stable for long time in solution, as noted when they were kept at room temperature for a night to record the 13C NMR spectra: the day after, the spectrum was complicated by the presence of new signals belonging to unidentified compounds. In the case of formation of WM4 and WM5 the integration of the signals revealed that the two species were formed in 42:58 percent relative molar ratio but we were not able to assign the signals of one or the other of these structures, thus, below, we describe the signals as belonging to the major (Maj) or the minor species (Min). The names of complexes WM1–WM5 have been created from ChemDraw Professional 17.1 version by drawing the canonical resonance form bearing the negative charge of the Meisenheimer moiety situated on the nitro group in para position with respect to the new bond formed and the positive charge of the Wheland moiety situated on the nitrogen atom of the amino substituent in para position to the new bond formed.
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2), 2.05–1.87 (m, 8H, NCH2C2). 13C NMR (CDCl3, 100.6 MHz, −54 °C), δ (ppm): 158.2, 155.4, 155.2, 150.7, 134.1 (CH), 118.7, 113.8, 109.3, 104.8, 88.8 (CH), 87.6 (CH), 49.4 (CH2), 49.2 (CH2), 48.87 (CH2), 48.86 (CH2), 48.76 (CH2), 48.4 (CH2), 45.1 (CH, C-10), 41.0, (CH, C-7), 26.0 (CH2), 25.7 (CH2), 24.83 (CH2), 24.79 (CH2), 24.7 (CH2), 24.6 (CH2).
2), 2.05–1.87 (m, 8H, NCH2C2). 13C NMR (CDCl3, 100.6 MHz, −54 °C), δ (ppm): 158.2, 155.4, 155.2, 150.7, 134.1 (CH), 118.7, 113.8, 109.3, 104.8, 88.8 (CH), 87.6 (CH), 49.4 (CH2), 49.2 (CH2), 48.87 (CH2), 48.86 (CH2), 48.76 (CH2), 48.4 (CH2), 45.1 (CH, C-10), 41.0, (CH, C-7), 26.0 (CH2), 25.7 (CH2), 24.83 (CH2), 24.79 (CH2), 24.7 (CH2), 24.6 (CH2).1H NMR (400 MHz, CDCl3, −54 °C), δ (ppm): 9.12 (s, 1H Min, H-5), 9.08 (s, 1H Maj, H-5), 5.61 (br.s., 1H Maj, H-12 or H-14), 5.56 (br.s., 1H Min, H-12 or H-14), 5.04 (s., 2H Maj + Min, H-7), 4.98 (br.s., 2H Maj + Min, H-12 or H-14), 4.84 (s, 1H Maj, H-10), 4.76 (s, 1H Min, H-10), 4.23–2.81 (m.s, 16H Maj + Min, OCH2 and NCH2), 2.00–1.31 (m.s, 12H Maj + Min, NCH2C2, NCH2CH2C2). g-HSQC (CDCl3, −54 °C): 1H–13C correlations: 136.5–9.12, 136.3–9.08, 93.2–5.61, 91.3–5.56, 44.1–5.04, 91.9–4.98, 89.9–4.98, 40.5–4.84, 39.9–4.76, and 66.5–3.65, 66.4–3.62, 66.2–3.83, 65.3–3.62, 50.1–3.24, 50.0–3.09, 49.97–3.54, 49.87–3.85, 48.3–2.91, 26.8–1.77, 26.6–1.63, 24.3–1.60, 24.2–1.84, 26.5–1.45.
Study on the formation of σ-complexes M1–M6 by NMR spectroscopy
4,6-Dinitrobenzofuroxan (DNBF), 4,6-dinitrobenzofurazan (DNBZ), or 4,6-dinitrotetrazolopyridine (DNTP) (∼4.4 × 10−5 mol) was dissolved in CD3CN (0.3 mL) and the solution was cooled at −35 °C. This solution was added to an equimolar amount of compound 12 (or 13) dissolved in CD3CN (0.7 mL) and directly prepared in the NMR spectroscopy tube at −35 °C. The 1H NMR spectra were recorded immediately after the mixing of the reagents and successively at intervals of 5–10 °C, from −35 °C to room temperature. At low temperature the spectrum showed non-equivalence of the hydrogen atoms belonging to the carbocyclic ring; on raising the temperature the signals broadened until to reach a coalescence situation. After, the peaks became sharp and a re-cooling produced again their broadening until to their splitting, as observed after the reagents mixing at low temperature. Complexes M1 and M3 were obtained in about 90% yield (calculated from 1H NMR spectrum) while M2 and M4 are formed in CD3CN in about 50% yield and, if kept in solution for three days or one night, respectively, evolved to the relevant substitution product 15 or 17, as ascertained by comparison with authentic samples. 13C NMR spectrum of M4 has not been reported because, recorded in acetonitrile during one night, it showed presence of signals belonging to the starting nucleophile and the substitution product 17. The combination DNBZ/12 was carried out in CD3COCD3 from −80 °C to +25 °C and also in this case the 1H NMR spectrum recorded after having collected 13C NMR spectrum overnight showed presence of new signals belonging to unidentified compounds; the 13C NMR signals reported below for M3 have been selected and tentatively ascribed. Reactions of 12 and 13 with DNBF were also carried out at 25 °C in DMSO-d6. Herein are reported NMR data obtained for complexes M1–M6; see ESI† section for further details.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful, for the financial support, to Alma Mater Studiorum – Università di Bologna (RFO funds). Many thanks to Prof. Daniele Casarini for line shape simulation, Dr Leonardo Iannuzzo, Dr Francesco Giarratano and Mr Luca Zuppiroli for mass spectra.Notes and references
- G. N. Nikonov and S. Bobrov, 1,2,5-Oxadiazoles, in Comprehensive Heterocyclic Chemistry III, ed. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven and R. J. K.Taylor, Elsevier, 2008, Vol. 5, p. 315 Search PubMed.
- (a) A. Gasco and A. J. Boulton, Adv. Heterocycl. Chem., 1981, 29, 251 CrossRef CAS; (b) P. B. Ghosh, B. Terna and M. W. Whitehouse, Med. Res. Rev., 1981, 2, 158 Search PubMed.
- (a) H. Cerecetto and W. Porcal, Mini-Rev. Med. Chem., 2005, 5, 57 CrossRef CAS; (b) E. Chugunova, C. Boga, I. Sazykin, S. Cino, G. Micheletti, A. Mazzanti, M. Sazykina, A. Burilov, L. Khmelevtsova and N. Kostina, Eur. J. Med. Chem., 2015, 93, 349 CrossRef CAS.
- C. Medana, A. Di Stilo, S. Visentin, R. Fruttero, A. Gasco, D. Ghigo and A. Bosia, Pharm. Res., 1999, 16, 956 CrossRef CAS.
- (a) J. F. Callan, A. P. de Silva, J. Ferguson, A. J. M. Huxley and A. M. O'Brien, Tetrahedron, 2004, 60, 11125 CrossRef CAS; (b) F. Davì and D. Rinaldi, J. Elastoplast., 2015, 120, 197 CrossRef.
- C. K. Lowe-Ma, R. A. Nissan and W. S. Wilson, J. Org. Chem., 1990, 55, 3760 CrossRef.
- (a) F. Terrier, Nucleophilic Aromatic Displacement, ed. H. Feuer, VCH, New York, 1991 Search PubMed; (b) F. Terrier, Modern Nucleophilic Aromatic Substitution, John Wiley & Sons, New York, 2013 CrossRef.
- F. Terrier, S. Lakhdar, T. Boubaker and R. Goumont, J. Org. Chem., 2005, 70, 6242 CrossRef CAS.
- S. Lakhdar, R. Goumont, F. Terrier, T. Boubaker, J. M. Dust and E. Buncel, Org. Biomol. Chem., 2007, 5, 1744 RSC.
- F. Terrier, Chem. Rev., 1982, 82, 77 CrossRef CAS.
- E. Buncel, J. M. Dust and F. Terrier, Chem. Rev., 1995, 95, 2261 CrossRef CAS.
- C. Boga and L. Forlani, J. Chem. Soc., Perkin Trans. 2, 2001, 1408 RSC.
- C. Boga, E. Del Vecchio, L. Forlani, A. Mazzanti and P. E. Todesco, Angew. Chem., Int. Ed., 2005, 44, 3285 CrossRef CAS.
- C. Boga, E. Del Vecchio, L. Forlani, A. Mazzanti, C. Menchen Lario, P. E. Todesco and S. Tozzi, J. Org. Chem., 2009, 74, 5568 CrossRef CAS.
- (a) H. Mayr and M. Patz, Angew. Chem., Int. Ed. Engl., 1994, 33, 938 CrossRef; (b) H. Mayr, B. Kempf and A. R. Ofial, Acc. Chem. Res., 2003, 36, 66 CrossRef CAS; (c) H. Mayr, M. Patz, M. F. Gotta and A. R. Ofial, Pure Appl. Chem., 1998, 70, 1993 CAS; (d) H. Mayr, T. Bug, M. F. Gotta, N. Hering, B. Irrgang, B. Janker, B. Kempf, R. Loos, A. R. Ofial, G. Remmenikov and N. Schimmel, J. Am. Chem. Soc., 2001, 123, 9500 CrossRef CAS; (e) H. Mayr and A. R. Ofial, Acc. Chem. Res., 2016, 49, 952 CrossRef CAS and ref. therein; (f) H. Mayr, Tetrahedron, 2015, 71, 5095 CrossRef CAS.
- (a) F. Terrier, S. Lakhdar, R. Goumont, T. Boubaker and E. Buncel, Chem. Commun., 2004, 2586 RSC; (b) S. Lakhdar, R. Goumont, F. Terrier, T. Boubaker, J. M. Dust and E. Buncel, Org. Biomol. Chem., 2007, 5, 1744 RSC.
- C. Boga, E. Del Vecchio, L. Forlani, R. Goumont, F. Terrier and S. Tozzi, Chem.–Eur. J., 2007, 13, 9600 CrossRef CAS.
- L. Forlani, C. Boga, A. Mazzanti and N. Zanna, Eur. J. Org. Chem., 2012, 6, 1123 CrossRef.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245 CrossRef CAS.
- A. K. S. Mehilal, R. B. Salunk and N. Sikder, New J. Chem., 2001, 25, 1549 RSC.
- A. K. S. Mehilal, R. B. Salunke and N. Sikder, J. Energ. Mater., 2002, 20, 39 CrossRef CAS.
- G. Micheletti, S. Bordoni, E. Chugunova and C. Boga, Molecules, 2017, 22, 684 CrossRef.
- F. Effenberger and R. Niess, Angew. Chem., Int. Ed. Engl., 1967, 6, 1067 CrossRef CAS.
- F. Effenberger, Acc. Chem. Res., 1989, 22, 27 CrossRef CAS.
- F. Effenberger and R. Niess, Chem. Ber., 1968, 101, 3787 CrossRef CAS.
- T. Yamaoka, H. Hosoya and S. Nagakura, Tetrahedron, 1968, 24, 6203 CrossRef.
- T. Yamaoka, H. Hosoya and S. Nagakura, Tetrahedron, 1970, 26, 4125 CrossRef CAS.
- W. Knoche, W. Sachs and S. Vogel, Bull. Soc. Chim. Fr., 1988, 377 CAS.
- W. Knoche, W. W. Schoeller, R. Schomaecker and S. Vogel, J. Am. Chem. Soc., 1988, 110, 7484 CrossRef CAS.
- W. Sachs, W. Knoche and S. Herrmann, J. Chem. Soc., Perkin Trans. 2, 1991, 2, 701 RSC.
- D. T. Glatzhofer, D. Allen and R. W. Taylor, J. Org. Chem., 1990, 55, 6229 CrossRef CAS.
- P. Menzel and F. Effenberger, Angew. Chem., Int. Ed. Engl., 1972, 11, 922 CrossRef CAS.
- F. Effenberger, P. Menzel and W. Seufert, Chem. Ber., 1979, 112, 1660 CrossRef CAS.
- R. Niess, K. Nagel and F. Effenberger, Tetrahedron Lett., 1968, 4265 CrossRef CAS.
- F. Effenberger, K. E. Mack, K. Nagel and R. Niess, Chem. Ber., 1977, 110, 165 CrossRef CAS.
- P. Fkher, K. E. Mack, E. Mhsner and F. Effenberger, Chem. Ber., 1977, 110, 181 CrossRef.
- P. Menzel and F. Effenberger, Angew. Chem., Int. Ed. Engl., 1975, 14, 62 CrossRef.
- F. Effenberger, W. Agster, P. Fischer, K. H. Jogun, J. J. Stezowski, E. Daltrozzo and G. Kollmannsberger-von Nell, J. Org. Chem., 1983, 48, 4649 CrossRef CAS.
- C. Boga, L. Forlani, S. Tozzi, E. Del Vecchio, A. Mazzanti, M. Monari and N. Zanna, Curr. Org. Chem., 2014, 18, 512 CrossRef CAS.
- C. Boga, E. Del Vecchio and L. Forlani, Eur. J. Org. Chem., 2004, 1567 CrossRef CAS.
- C. Boga, E. Del Vecchio, L. Forlani and S. Tozzi, J. Org. Chem., 2007, 72, 8741 CrossRef CAS.
- L. Forlani, C. Boga, E. Del Vecchio, A. L. T. D. Ngobo and S. Tozzi, J. Phys. Org. Chem., 2007, 20, 201 CrossRef CAS.
- C. Boga, E. Del Vecchio, S. Tozzi, L. Forlani, M. Monari, G. Micheletti and N. Zanna, ARKIVOC, 2014,(iv), 51 Search PubMed.
- E. Del Vecchio, C. Boga, L. Forlani, S. Tozzi, G. Micheletti and S. Cino, J. Org. Chem., 2015, 80, 2216 CrossRef CAS PubMed.
- C. Boga, G. Micheletti, S. Cino, S. Fazzini, L. Forlani, N. Zanna and D. Spinelli, Org. Biomol. Chem., 2016, 14, 4267 RSC.
- G. Micheletti, C. Boga, M. Pafundi, S. Pollicino and N. Zanna, Org. Biomol. Chem., 2016, 14, 768 RSC.
- S. Lakhdar, M. Westermaier, F. Terrier, R. Goumont, T. Boubaker, A. R. Ofial and H. Mayr, J. Org. Chem., 2006, 71, 9088 CrossRef CAS.
- Y. P. Semenyuk, P. G. Morozov, O. N. Burov, M. E. Kletskii, A. V. Lisovin, S. V. Kurbatov and F. Terrier, Tetrahedron, 2016, 72, 2254 CrossRef CAS.
- O. Exner, Advances in Linear Free Energy Relationship, ed. N. B. Chapman and J. Shorter, Plenum Press, London, 1972 Search PubMed.
- G. Micheletti, C. Boga, L. Forlani, E. Del Vecchio, N. Zanna, A. Mazzanti and M. Monari, Eur. J. Org. Chem., 2017, 964 CrossRef CAS.
- W. B. Jennings, Chem. Rev., 1975, 75, 307 CrossRef CAS.
- K. Mislow and M. Raban, Top. Stereochem., 1967, 1, 1 CAS.
- (a) J.-C. Hallé, M.-P. Simonnin, M.-J. Pouet and F. Terrier, Tetrahedron Lett., 1983, 24, 2255 CrossRef; (b) J.-C. Hallé, M.-J. Pouet, M.-P. Simonnin, F. Debleds and F. Terrier, Can. J. Chem., 1982, 60, 1988 CrossRef; (c) M.-P. Simonnin, J.-C. Hallé, F. Terrier and M.-J. Pouet, Can. J. Chem., 1985, 63, 866 CrossRef CAS.
- (a) F. Terrier, M.-J. Pouet, J.-C. Halle, E. Kizilian and E. Buncel, J. Phys. Org. Chem., 1998, 11, 707 CrossRef CAS; (b) J. C. Halle, M. J. Pouet, M. P. Simonnin and F. Terrier, Tetrahedron Lett., 1985, 26, 1307 CrossRef CAS.
- J. Sandstrom, Dynamic NMR Spectroscopy, Academic Press, London, 1982, p. 99 Search PubMed.
- Dynamic Nuclear Magnetic Resonance Spectroscopy, ed. L. M. Jackman and F. A. Cotton, Academic Press, New York, 1975 Search PubMed.
- QCPE program no. 633, Indiana University, Bloomington, IN, USA Search PubMed.
- A. Tatarov, S. Kurbatov, G. Borodkin, R. Goumont and F. Terrier, Tetrahedron, 2010, 66, 995 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09460h |
| This journal is © The Royal Society of Chemistry 2018 |