
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAromatic fluorocopolymers based on α-(difluoromethyl)styrene and styrene: synthesis, characterization, and thermal and surface properties†
Joanna Wolska a,
Justyna Walkowiak-Kulikowska*a,
Anna Szwajcaa,
Henryk Koroniaka and
Bruno Améduri*b
a,
Justyna Walkowiak-Kulikowska*a,
Anna Szwajcaa,
Henryk Koroniaka and
Bruno Améduri*b
aAdam Mickiewicz University, Faculty of Chemistry, Umultowska 89b, 61-614 Poznań, Poland. E-mail: Justyna.A.Walkowiak@amu.edu.pl
bInstitut Charles Gerhardt, Ingénierie et Architectures Macromoléculaires, UMR CNRS 5253, ENSCM, University of Montpellier, Palace Eugene Bataillon, 34095 Montpellier, France. E-mail: bruno.ameduri@enscm.fr
First published on 17th December 2018
Abstract
A study on the α-(difluoromethyl)styrene (DFMST) reactivity under conventional radical copolymerization conditions is presented. Although the homopolymerization of DFMST failed, its radical bulk copolymerization with styrene (ST) led to the synthesis of fluorinated aromatic polymers (FAPs). The resulting novel poly(DFMST-co-ST) copolymers were characterized by 1H, 19F and 13C NMR spectroscopies that evidenced the successful incorporation of DFMST units into copolymers and enabled the assessment of their respective molar percentages (10.4–48.2 mol%). The molar masses were in the range of 1900–17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 g mol−1. The bulkier CF2H group in the α-position induced the lower reactivity of the DFMST comonomer. ST and DFMST monomer reactivity ratios (rDFMST = 0.0 and rST = 0.70 ± 0.05 at 70 °C) were determined based on linear least-square methods. These values indicate that DFMST monomer is less reactive than ST, retards the polymerization rate, and thus reduces the molar masses. Moreover, the thermal properties (Tg, Td) of the resulting copolymers indicate that the presence of DFMST units incorporated into poly(ST) structure promotes an increase of the Tg values up to 109 °C and a slightly better thermal stability than that of poly(ST). Additionally, the thermal decomposition of poly(DFMST-co-ST) copolymer (10.4/89.6) was assessed by simultaneous thermal analysis coupled with Fourier-transform infrared spectroscopy and thermogravimetric analysis coupled with mass spectrometry showing that H2O, CO2, CO and styrene were released. The surface analysis was focused on the effects of the –CF2H group at the α-position of styrene comonomers on surface free energy of the copolymer films. Water and diiodomethane contact angle (CA) measurements confirmed that these copolymers (Mn = 2300–17200 g mol−1) are not exactly the same as polystyrenes (Mn = 2100–21600 g mol−1) in the solid state. The CA hysteresis for poly(ST) (6–8°) and poly(DFMST-co-ST) copolymers (3–5°) reflected these differences even more accurately.
200 g mol−1. The bulkier CF2H group in the α-position induced the lower reactivity of the DFMST comonomer. ST and DFMST monomer reactivity ratios (rDFMST = 0.0 and rST = 0.70 ± 0.05 at 70 °C) were determined based on linear least-square methods. These values indicate that DFMST monomer is less reactive than ST, retards the polymerization rate, and thus reduces the molar masses. Moreover, the thermal properties (Tg, Td) of the resulting copolymers indicate that the presence of DFMST units incorporated into poly(ST) structure promotes an increase of the Tg values up to 109 °C and a slightly better thermal stability than that of poly(ST). Additionally, the thermal decomposition of poly(DFMST-co-ST) copolymer (10.4/89.6) was assessed by simultaneous thermal analysis coupled with Fourier-transform infrared spectroscopy and thermogravimetric analysis coupled with mass spectrometry showing that H2O, CO2, CO and styrene were released. The surface analysis was focused on the effects of the –CF2H group at the α-position of styrene comonomers on surface free energy of the copolymer films. Water and diiodomethane contact angle (CA) measurements confirmed that these copolymers (Mn = 2300–17200 g mol−1) are not exactly the same as polystyrenes (Mn = 2100–21600 g mol−1) in the solid state. The CA hysteresis for poly(ST) (6–8°) and poly(DFMST-co-ST) copolymers (3–5°) reflected these differences even more accurately.
1. Introduction
Fluoropolymers are attractive niche materials that offer combination of outstanding properties. Due to the high value of the dissociation energy of carbon–fluorine bond (485 kJ mol−1), fluoropolymers exhibit high thermal stability and chemical inertness to acids, alkalis, various types of solvents and oils. Moreover, low polarizability of the C–F bond translates the hydrophobic character (low moisture uptake) of the polymers. In addition, the low surface energy of fluorine helps in oil-repellency resulting in increased resistance to wear and abrasion.1–4 For these reasons, the fluorine-containing polymers have found many applications in various fields including buildings, automotive, aircraft, aerospace industries, aeronautics, energy, chemistry, textile, microelectronics, optics and biomedical applications.5–9 However, some perfluorinated materials suffer from poor solubility in common organic solvents, while others cannot be melted or display very high melting points which hinder their processing. To overcome these issues, such materials can be obtained by copolymerization of commercially available halogenated or hydrocarbon monomers.10 Such processes could lead to the formation of fluoropolymers possessing peculiar properties which appreciably enhance their processability, especially solubility, heat and thermal resistance, hydrophobicity as well as adhesion and surface properties. The fluorinated aromatic polymers (FAPs)11 attempt to face up to current requirements for improvements and thereby seem to be an interesting generation of fluoropolymers. The incorporation of aromatic moieties into polymeric chain is a promising concept for processible products with abrasion resistance and significantly enhanced mechanical strength.12 On the other hand, the incorporation of fluorinated units into aromatic polymer chain may enhance some other physico-chemical properties.13In the past decades, much attention has been focused on the synthesis and (co)polymerization of styrenic monomers either bearing fluorine atoms/fluorinated substituents on the aromatic ring14–16 or at its external double bond.17 In 1949, Cohen et al.18 developed a first efficient synthesis of α,β,β-trifluoro-styrene (TFS), whereas Prober,19 Narita et al.20,21 and Steck and Stone22 studied its reactivity in copolymerizations with various olefins. Furthermore, Stone et al.23 described the preparation of materials based on such a monomer bearing phosphonic acid. On the other hand, Smith and Babb24 pioneered the synthetic route toward fluorinated aromatic monomer, 4-[(α,β,β-trifluorovinyl)oxy]bromobenzene (TFVOBB) while Souzy et al.25 presented its copolymerization reactions with various fluoroolefins: vinylidene fluoride (VDF), hexafluoropropene (HFP), perfluoromethyl vinyl ether (PMVE), and chlorotrifluoroethylene (CTFE). Furthermore, these latter authors proved that copolymerizations of TFVOBB with CTFE or VDF led to the formation of poly(TFVOBB-co-CTFE) and poly(TFVOBB-co-VDF) copolymers in low yields. Moreover, its copolymerizations either with HFP or PMVE were completely unsuccessful. Hence, to enhance the reactivity of VDF in a VDF/TFVOBB copolymerization, a fluorinated termonomer was introduced, such as HFP, PMVE, or CTFE which allowed the production of original fluorinated terpolymers bearing bromoaromatic side-groups. Additionally, modification of TFVOBB with sulfonic acid group led to attractive fluoropolymers for fuel cell membranes.26 More recently, Walkowiak-Kulikowska et al.27 reported the radical copolymerization of α-(trifluoromethyl)styrene (TFMST) with various fluoroolefins (VDF; CTFE; 3,3,3-trifluoropropene, TFP; 1H,1H,2H,2H-perfluoro-1-decyl vinyl ether) and proved that reaction was inhibited and did not lead even to any oligomers. In this case, only the synergic effect resulting from the reactivity of CTFE in combination with VDF enabled to a successful incorporation of TFMST aromatic units leading to poly(VDF-ter-CTFE-ter-TFMST) terpolymers.
Only a few examples of radical copolymerization of fluoroolefins with fluorinated α-methylstyrene monomers have been reported, so far. Among them, the copolymer based on α-(fluoromethyl)styrene (FMST) and CTFE, as the first aromatic copolymer with an allyl fluoride functionality, was achieved by Kostov et al.28 Fluorinated α-methylstyrenes (F-STs) are known to be difficult to polymerize under radical conditions although the hydrocarbon analogue such as α-methylstyrene is easily polymerizable under cationic polymerization conditions.21 Furthermore, Kyulavska et al.29 reported unexpected alternating radical copolymerization of CTFE with 3-isopropenyl-α,α′-dimethylbenzyl isocyanate. This indicates that the fluorinated alkyl group linked to a vinyl function significantly influences the reactivity of the monomers.21,30 The radical copolymerizations of FMST with methyl methacrylate (MMA) or styrene (ST), reported by Baldwin and Reed,31 proved that the fluorinated aromatic monomer retarded the rate of polymerization of both hydrocarbons. On the other hand, the reports on the reactivity of TFMST have mainly been focused on the reason why the fluoromonomer is unable to form homopolymer under radical initiation. Ito et al.32 investigated the initiation reaction of TFMST while Narita21,33 discussed the propagation step, which gave evidence on the failure to yield the homopolymer. In spite of its high e and Q values (0.90 and 0.43, respectively33), TFMST is reluctant to undergo radical and anionic homopolymerization. Ueda and Ito34 and then Walkowiak-Kulikowska et al.,35 who reported the kinetics of radical copolymerization of TFMST with styrene, determined the monomer reactivity ratios: rTFMST = 0.00, rST = 0.60 at 60 °C and rTFMST = 0.00, rST = 0.64 at 70 °C, respectively, which confirmed that TFMST did not self-propagate. The latter authors also presented successful iodine transfer copolymerization of both fluorinated monomers (FMST and TFMST) that led to the synthesis of well-defined FAPs,36 although TFMST retards the polymerization rates and significantly increases the reaction time.
To the best of our knowledge, studies on the radical copolymerization of α-(difluoromethyl)styrene has never been reported, so far. Similarly to monofluoro- and trifluoromethyl groups, the difluoromethyl function (–CF2H) is another fluorinated alkyl moiety of great interest since it plays critical roles as a lipophilic isostere of hydroxyl group as well as a hydrogen bond donor.37–39 However, synthesis of such molecules is not straightforward as in case of mono- and trifluorinated analogues.40 Therefore, studies on their properties and possible applications seem to be still an unexplored field, worth to investigate.40–42 Previously, we presented the cost-effective and grams scale synthesis of DFMST.43 Since our objective is to examine the influence of the fluorine content of styrenic monomers on both reactivity and copolymer composition, it was of interest to thoroughly study the behaviour of DFMST in radical copolymerization. Furthermore, the resulting novel FAPs possessing difluoromethyl styrenic units were evaluated in terms of its thermal and surface properties.
2. Experimental procedures
2.1. Materials
Styrene (ST, 99.9% containing 4-tert-butylcatechol as inhibitor from Sigma Aldrich) was washed with sodium hydroxide solution, rinsed deionized water, dried, and distilled under reduced pressure before use. The radical initiator, α,α′-azobis(isobutyronitrile) (AIBN, ≥98% from Sigma Aldrich), was purified by recrystallization from methanol before use. The common organic solvents (anhydrous tetrahydrofuran, THF; diethyl ether, Et2O, anhydrous dichloromethane, DCM, anhydrous toluene from Sigma-Aldrich), and following reagents: octadecyltrichlorosilane (OTS, ≥96% from Sigma-Aldrich), sulphuric acid (H2SO4, 96% POCH), and hydrogen peroxide (H2O2, 30% POCH) were used as received. Deuterated chloroform (CDCl3) for NMR characterizations were purchased from Apollo Scientific (purity > 99.8%).2.2. Measurements
Nuclear Magnetic Resonance spectra (1H, 13C and 19F NMR) were recorded on a Bruker Ultrashield 400 instrument. The purity of α-(difluoromethyl)styrene (DFMST) and the composition of copolymers (i.e. the molar contents of comonomers) were determined by 1H and 19F NMR spectroscopy. Deuterated chloroform (CDCl3) was used as solvent. The NMR spectra were calibrated using an internal reference: TMS (1H), CDCl3 (13C) or CFCl3 (19F). The experimental conditions for recording 1H and 19F NMR spectra were as follows: flip angle 90° (or 30°), acquisition time 4.1 s (or 0.7 s), pulse delay 2 s (or 5 s), number of scans 16 (or 256), and a pulse width of 5 μs for 19F NMR. Chemical shifts (δ) are quoted in parts per million (ppm) and coupling constants (J) are measured in hertz (Hz). In the figures and discussion below, the letters s, d, t, q and m stand for singlet, doublet, triplet, quintet and multiplet, respectively.Gel Permeation Chromatography (GPC) measurements were conducted using an Agilent 1260 Infinity equipped with RI detector and its corresponding software (Agilent Software GPC/SEC-1260 GPC set). The system uses Phenogel (10 μm linear (2) 300 × 7.8 mm) column (100 < Mw < 10000000 g mol−1) with THF as the eluent with a flow rate of 1.0 mL min−1 at room temperature. Tetrahydrofuran was used as solvent and monodispersed poly(styrene) standards (1000 < Mw < 3500000) were used for conventional calibration.
Thermogravimetric analysis (TGA) experiments of copolymers obtained in high conversion polymerizations were performed with a TGA 4000 apparatus from Perkin Elmer, under a nitrogen atmosphere, at the heating rate of 10 °C min−1 from room temperature up to a maximum of 900 °C. The sample size varied between 5 and 10 mg.
Differential scanning calorimetry (DSC) analyses of copolymers obtained in high conversion experiments were carried out with a DSC 8500 apparatus from Perkin Elmer, under a nitrogen atmosphere, at a heating rate of 10 °C min−1. The temperature range was from 60 to 140 °C. The sample size varied between 5 and 10 mg. The glass transition temperatures (Tg) were reported at the inflection point of the heat capacity drop during the second heating run.
Simultaneous Thermal Analysis-Fourier Transform-Infrared Spectroscopy (STA/FTIR) of materials obtained by high conversion experiments were carried out on STA 6000 apparatus from Perkin Elmer coupled with Frontier FTIR spectrometer from Perkin Elmer using a TL 8000 transfer line held at 300 °C with a nitrogen flow of 80 mL min−1 at atmospheric pressure. The sample size was ca. 10 mg. The analysis temperature range (30–800 °C) was scanned at rate of 10 °C min−1. Gas phase FTIR spectra were recorded with wave number ranging between 500–4000 cm−1 at a resolution of 4 cm−1. The data processing was performed in Spectrum, Timebase and Pyris softwares. Three-dimensional absorbance spectrum correlated to the time (min) of process and the wave number (cm−1) was obtained. The 3D-spectrum was taken from tested sample and results have been discussed based on eight 2D-spectra selected at the characteristic temperatures (e.g. Td5, Td10, Td30, Td50, Td90 and in the end of degradation process).
Thermogravimetric Analysis coupled with Mass Spectrometry (TGA/MS) of selected polymer obtained by high conversion experiment was carried out on TGA1/MS Clarus 680 SQ8 apparatus from Perkin Elmer in helium at flow rate of 40.0 mL min−1. The samples (10 mg) were heated in a platinum crucible from 300 °C up to 700 °C at a heating rate of 10 °C min−1. The thermal decomposition products were analyzed in the range of 10–250 m/z. The data processing was performed in Turbomass v. 6.1.0 software.
Advancing and Receding Contact Angle (ARCA) measurements. The surface dynamic wetting angles i.e. advancing and receding drop contact angles (°, ±1°) of the OTS functionalized glass surfaces were measured using the OCA 15+ contact angle measurement system using (2 μL drop) Ellipse Fitting function. The OTS functionalized glass surfaces were prepared as follows: the 2 cm × 2 cm samples of the glass (RVFM) wafers were cleaned by treatment with piranha solution (H2SO4:H2O2 1:1, 15 min at 85 °C) and immersed in 1 mM solution of OTS in anhydrous toluene at room temperature for 2 h. After this time the samples were rinsed with toluene and dichloromethane, dried with nitrogen, annealed at 30 °C in a nitrogen atmosphere for 1 h, and sonicated in dichloromethane solution. The presented series of poly(ST) and poly(DFMST-co-ST) films were obtained by dipping freshly prepared OTS/glass samples in a 3 wt% tetrahydrofuran solution of fluorinated copolymers, acquired from high conversion polymerizations, in inert atmosphere using glovebox.35,36 The final values of CAs were averages of at least five measurements which were taken over different areas on the same sample.
2.3. Synthesis of α-(difluoromethyl)styrene (DFMST)
Non-commercial fluorinated monomer, α-(difluoromethyl)-styrene (DFMST), was synthesized according to literature procedure (ESI, Section 1†).432.4. Radical homopolymerization of α-(difluoromethyl)-styrene (DFMST)
The bulk radical homopolymerization of DFMST was carried out at 70 °C for 20 hours in sealed Schlenk tubes degassed by five freeze–thaw cycles until no bubbling occurred. α,α′-Azobis-(isobutyronitrile) (AIBN) was used as the radical initiator (1 mol% relative to all comonomers). Afterwise, the tube was cooled to room temperature, opened, and the content was analyzed. Part of the mixture was characterized by 1H and 19F NMR spectroscopies, while the remaining part of the mixture was then dissolved in THF (1 mL/1 g of monomer) and precipitated from cold pentane (50 mL/1 g of monomer), but whatever the conditions, all experiments failed and did not lead to any poly(DFMST).2.5. Radical copolymerization of α-(difluoromethyl)styrene (DFMST) with styrene (ST)
The bulk radical copolymerization of styrene (ST) with α-(difluoromethyl)styrene (DFMST) was carried out at 70 °C for 20 hours in sealed Schlenk tubes and degassed as above. AIBN was used as the initiator (1 mol% relative to all comonomers). Afterwise, the tube was then cooled to room temperature, opened, and the content was analyzed. Part of the mixture was characterized by 1H and 19F NMR spectroscopies, while the remaining part of the mixture was then dissolved in THF (1 mL/1 g of monomers) and precipitated from cold pentane (50 mL/1 g of monomers). After purification, the copolymers, obtained as a white powders, were dried until constant weight under vacuum at 80 °C to remove any traces of the solvent and then analyzed. Conversions were determined by 1H and 19F NMR spectroscopies combined with gel permeation chromatography (GPC).Average molar masses were ascertained by GPC. In order to assess the copolymer compositions, the following equations have been employed (eqn (1) and 2):
| mol%ST = [(IA,B,B′ − IA′)/3]/([(IA,B,B′ − IA′)/3] + IA′) | (1) |
| mol%DFMST = (IA′)/([(IA,B,B′ − IA′)/3] + IA′) | (2) |
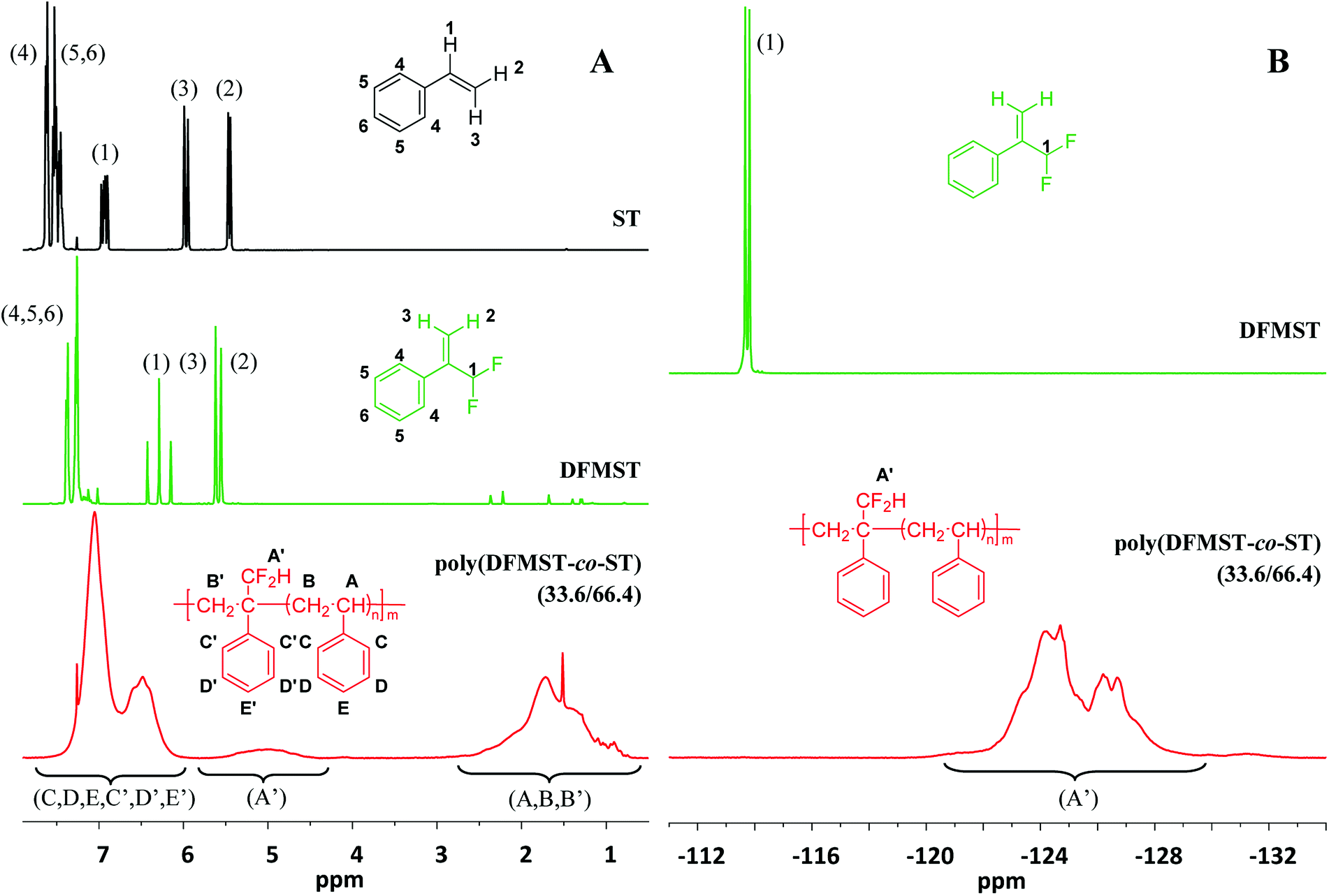 | ||
Fig. 1 Comparison of the 1H and 19F NMR spectra (recorded in CDCl3) of monomers (ST and DFMST) and the poly(DFMST-co-ST) copolymer. Left-hand cascade (A): 1H NMR spectra of styrene (ST,  ), α-(difluoromethyl)styrene (DFMST, ), α-(difluoromethyl)styrene (DFMST,  ), and poly(DFMST-co-ST) copolymer (33.6/66.4, Mn = 5400 g mol−1, entry 4, Table 1, ), and poly(DFMST-co-ST) copolymer (33.6/66.4, Mn = 5400 g mol−1, entry 4, Table 1,  ), respectively; right-hand cascade (B): 19F NMR spectra of α-(difluoromethyl)styrene (DFMST, ), respectively; right-hand cascade (B): 19F NMR spectra of α-(difluoromethyl)styrene (DFMST,  ) and poly(DFMST-co-ST) copolymer (33.6/66.4, Mn = 5400 g mol−1, entry 4, Table 1, ) and poly(DFMST-co-ST) copolymer (33.6/66.4, Mn = 5400 g mol−1, entry 4, Table 1,  ). ). | ||
The kinetics of radical copolymerizations that enabled to determine the reactivity ratios of comonomers are detailed in the ESI (Section 3†). For Mayo–Lewis (ML),44 Fineman–Ross (FR),45 Inverted Fineman–Ross (IFR),45 Yezrielev–Brokhina–Roskin (YBR)46 and Kelen–Tüdős (KT)47 methods, the copolymerizations were achieved at low monomer conversion (10–20%, detailed in ESI, Table S3†), whereas to determine reactivity ratios using Extended Kelen–Tüdős (EKT)48 method, the final (high) conversions were taken under consideration. Two separate sets of experiments were performed in order to define reactivity ratios of comonomers at low and high conversions, respectively. Materials obtained only in high conversion polymerizations were subjected for further thermal and surface properties analyses.
3. Results and discussion
3.1. Synthesis of α-(difluoromethyl)styrene (DFMST)
α-(Difluoromethyl)styrene (DFMST) was successful synthesized according to literature procedure.43 In the synthesis of targeted monomer the following three-step route (Scheme 1) was employed: (i) a base-induced difluoromethylation coupling with PhSO2CF2H as selective “CF2H−” equivalent allowed to introduce difluoromethyl function, (ii) the reductive desulfonylation and subsequent (iii) dehydration. The synthetic route was implemented for grams scale preparation of the DFMST in 56% overall yield.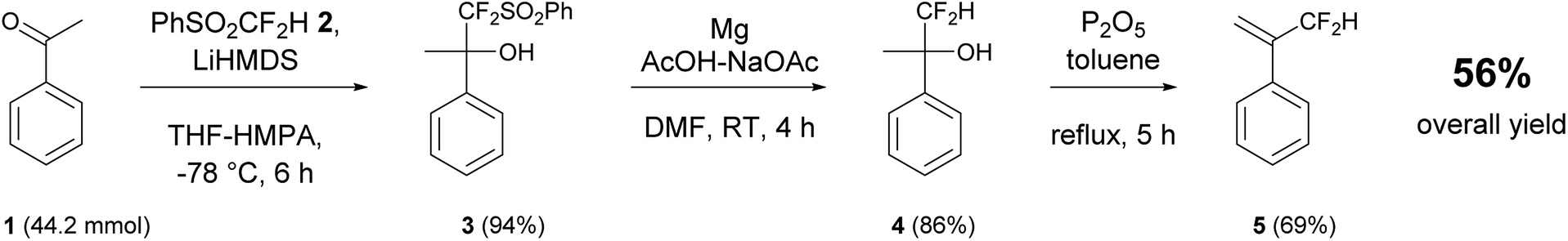 | ||
| Scheme 1 Preparation of α-(difluoromethyl)styrene (DFMST) by nucleophilic difluoromethylation of acetophenone 1 with difluoromethyl-phenyl sulfone 2. | ||
3.2. Radical homopolymerization of DFMST
All approaches to obtain poly(DFMST) homopolymers containing difluoromethyl functionalized aromatic units failed, and did not even lead to any oligomers, totally inhibiting reaction system (Table S1 in the ESI†). The F-STs often behave as captodative monomers since they bear both an electron-withdrawing group (EWG) (such as CF2H function in DFMST) and an electron-donating group (EDG) such as phenyl ring.27,49 The presence of such characteristic groups causes that the DFMST exhibits a peculiar reactivity.3.3. Radical copolymerization of DFMST with styrene
Several conventional radical bulk copolymerizations of ST with DFMST were initiated by α,α′-azobis(isobutyronitrile) (AIBN) at 70 °C for 20 hours as illustrated in Scheme 2. The results are listed in Table 1. The copolymerization reactions allowed us to obtain novel poly(DFMST-co-ST) copolymers of different molar masses (ranging from 1900 to 17200 g mol−1, with DFMST mol% varying between 10.4 and 48.2, Table 1), which were all thoroughly characterized by 1H, 19F and 13C NMR spectroscopies, as detailed in the ESI (Fig. S1–S3†). Fig. 1 illustrates the comparison of the 1H NMR spectra (cascade (A)) of ST and DFMST monomers with that of the poly(DFMST-co-ST) copolymer (33.6/66.4) and of the 19F NMR spectra (cascade (B)) of the DFMST monomer with the poly(DFMST-co-ST) copolymer (33.6/66.4). The 1H NMR spectrum of the poly(DFMST-co-ST) copolymer ( ) exhibits broad signals centred at 1.65, 5.05 and 6.81 ppm attributed to the secondary and tertiary aliphatic backbone protons of DFMST and ST units (HB′, HB and HA), side chain primary proton of the –CF2H group of DFMST (HA′), and aromatic protons of ST and DFMST units (HC, HD, HE, HC′, HD′ and HE′), respectively. Most probably, due to the absence of vinyl moieties, the signals of the aromatic protons in the copolymer underwent a high field shift to 6.81 ppm in comparison with the HAr multiplets centred at 7.48, 7.55, 7.64 and 7.40, 7.51 ppm observed in the NMR spectra of ST and DFMST monomers, respectively. The 19F NMR spectrum of DFMST monomer (
) exhibits broad signals centred at 1.65, 5.05 and 6.81 ppm attributed to the secondary and tertiary aliphatic backbone protons of DFMST and ST units (HB′, HB and HA), side chain primary proton of the –CF2H group of DFMST (HA′), and aromatic protons of ST and DFMST units (HC, HD, HE, HC′, HD′ and HE′), respectively. Most probably, due to the absence of vinyl moieties, the signals of the aromatic protons in the copolymer underwent a high field shift to 6.81 ppm in comparison with the HAr multiplets centred at 7.48, 7.55, 7.64 and 7.40, 7.51 ppm observed in the NMR spectra of ST and DFMST monomers, respectively. The 19F NMR spectrum of DFMST monomer ( ) exhibits a characteristic doublet at −113.6 ppm (2JF–H = 55.3 Hz) assigned to the –CF2H moiety of DFMST monomer, whereas that of the poly(DFMST-co-ST) copolymer (
) exhibits a characteristic doublet at −113.6 ppm (2JF–H = 55.3 Hz) assigned to the –CF2H moiety of DFMST monomer, whereas that of the poly(DFMST-co-ST) copolymer ( ) displays a broad high field shifted multiplet at −125.5 ppm attributed to the –CF2H moiety attached to the polymer chain.
) displays a broad high field shifted multiplet at −125.5 ppm attributed to the –CF2H moiety attached to the polymer chain.
 | ||
| Scheme 2 Bulk radical copolymerizations of ST with DFMST initiated by AIBN at 70 °C (τ1/2 = 5.1 h).50,51 | ||
| Entry | Molar ratio [mol%] | Conversionb [%] | Yield [wt%] | Mnc [g mol−1] | Mwc [g mol−1] | Đb | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| In feed | In copolymera | |||||||||
| DFMST | ST | DFMST | ST | DFMST | ST | |||||
| a Determined by 1H NMR spectroscopy.b Uncorrected and approximate values calculated based on relative molecular weights determined by GPC (THF, RI) with polystyrene standards.c Average molar masses (Mn, Mw) and dispersities (Đ) assessed from GPC (THF, RI) with poly(styrene) standards. | ||||||||||
| 1 | 10.0 | 90.0 | 10.4 | 89.6 | 84.0 | 81.0 | 88 | 17200 |
53000 |
3.0 |
| 2 | 20.0 | 80.0 | 20.3 | 79.7 | 54.7 | 53.7 | 55 | 12300 |
25500 |
2.0 |
| 3 | 30.0 | 70.0 | 29.2 | 70.8 | 44.6 | 46.4 | 50 | 7800 | 14500 |
1.9 |
| 4 | 40.0 | 60.0 | 33.6 | 66.4 | 30.6 | 40.3 | 42 | 5400 | 8600 | 1.6 |
| 5 | 50.0 | 50.0 | 39.9 | 60.1 | 24.8 | 38.3 | 31 | 2700 | 4000 | 1.5 |
| 6 | 60.0 | 40.0 | 43.9 | 56.1 | 8.4 | 16.0 | 11 | 2300 | 4600 | 2.0 |
| 7 | 80.0 | 20.0 | 48.2 | 51.8 | 0.9 | 4.0 | 1 | 1900 | 2400 | 1.2 |
The integrals of the characteristic proton signals (Fig. S4–S10 in the ESI†) assigned to the aliphatic backbone of ST and DFMST (signals centred at 1.59 ppm, marked as A, B and B′) and to the difluoromethyl moiety of DFMST (signal centred at 5.00 ppm, marked as A′) base units incorporated into the copolymers allowed us to determine the content of the ST and DFMST comonomers in the copolymers (Table S2 in the ESI†) by using eqn (1) and (2) (see Section 2.5).
Combinative NMR and GPC analysis enabled us to ascertain the conversion of comonomers involved in radical copolymerizations (Table 1). As expected, increasing DFMST content in the feed induced a significant decrease of the efficiency of the copolymerizations with styrene i.e. the monomer conversion, the yield, as well as the molar masses of fluorocopolymers. The copolymer composition curve for the copolymerization of DFMST with ST, presented in Fig. 2, demonstrates that (i) the DFMST in the copolymer is always lower than in the feed and (ii) a high DFMST feed concentration (80 mol%) resulted in the formation of a copolymer with an almost 1:1 alternation.
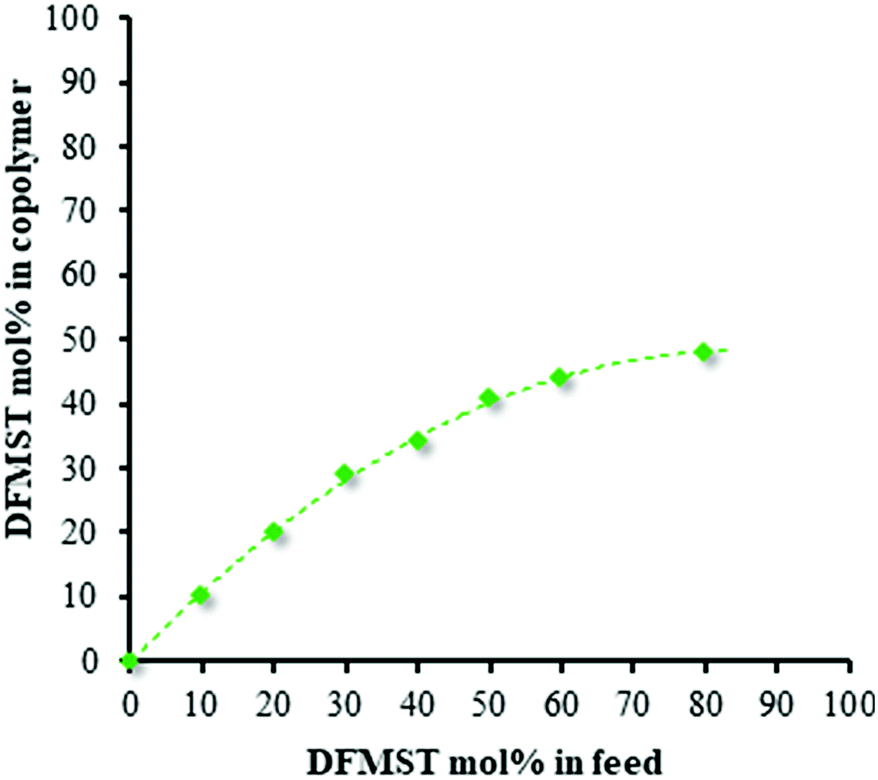 | ||
| Fig. 2 DFMST copolymer content vs. DFMST monomer in feed in the radical copolymerization of DFMST with styrene initiated by AIBN at 70 °C. | ||
However, copolymers containing DFMST–ST units greater than 49% cannot be obtained under these conditions, confirming the non-homopolymerizability of such a fluorinated monomer.
3.4. Determination of the monomer reactivity ratios of DFMST and ST
Monomer reactivity ratios are important quantitative values to predict the copolymer composition and to understand the kinetics and mechanistic aspects of copolymerization. Among well-known procedures useful to determine monomer reactivity ratio, the linear least-square (LLS) methods of Mayo–Lewis (ML),44 Fineman–Ross (FR),45 Inverted Fineman–Ross (IFR),45 Yezrielev–Brokhina–Roskin (YBR)46 and Kelen–Tüdős (KT)47 were chosen for the calculation of monomer reactivity ratios at low conversion. In addition, the Extended Kelen–Tüdős (EKT),48 involving more complex calculation, can be applied to higher conversion without significant systematic errors. The details of the calculation are given at the ESI† and the results are listed in Table 2.| LLS methods | rDFMST | rST | 1/rST |
|---|---|---|---|
| a Low conversions.b High conversions. | |||
| Mayo–Lewis | 0.00 | 0.72 ± 0.30 | 1.39 |
| Fineman–Ross | 0.00 | 0.71 ± 0.07 | 1.41 |
| Inverted Fineman–Ross | 0.00 | 0.74 ± 0.14 | 1.35 |
| Yezrielev–Brokhina–Roskin | 0.00 | 0.64 ± 0.30 | 1.56 |
| Kelen–Tüdős | 0.00 | 0.74 ± 0.05 | 1.35 |
| Extended Kelen–Tüdősa | 0.00 | 0.74 ± 0.02 | 1.35 |
| Extended Kelen–Tüdősb | 0.00 | 0.69 ± 0.04 | 1.45 |
In order to determine the monomer reactivity ratios in the kinetic stage, the monomer conversions in all copolymerizations were maintained below 20% (Table S3 in ESI†). Since the values of calculated reactivity ratios for DFMST were often negative by conventional methods, the rDFMST = 0.00 were presumed for further calculations. Moreover, two following features i.e. rDFMSTrST < 1 and rDFMST < rST indicate the statistic distribution of monomers in the resulting fluorocopolymers. The respective propagating chain terminated with DFMST prefers to add onto ST rather than another DFMST monomer involved in the reaction. Hence, the R–CH2(Ph)(CF2H)C˙ macroradical is reluctant to react with DFMST and the monomer is approximately 1.4 times more reactive than ST toward polystyrene R–CH2(Ph)CH˙ radical (1/rST ≈ 1.4). Additionally, R–CH2(Ph)(CF2H)C˙ radical adds onto ST monomer much faster than its corresponding polymer R–CH2(Ph)CH˙ radical, thereby preventing the formation of long PST blocks in the copolymer chain. The high reactivity of ST and low reactivity of DFMST towards the difluorinated R–CH2(Ph)(CF2H)C˙ macroradical also explains why the formation of copolymers with DFMST units greater than 50 mol% cannot be obtained under radical conditions for such a copolymerization system.
Hence, all the observations confirm that DFMST monomer is unreactive under radical polymerization conditions. Similar findings with regard to FMST and TFMST derivatives were previously reported.35 However, FMST seems to be slightly more reactive than DFMST and TFMST (rFMST = 0.08, rST = 0.72;35 rDFMST = 0.00, rST = 0.69; rTFMST = 0.00, rST = 0.64 all assessed at 70 °C,35 calculated using EKT method for high conversion). Moreover, similar behaviour of DFMST and TFMST in radical conditions allows to sort F-STs reactivities in the following decreasing order: FMST > DFMST ≈ TFMST. On the other hand, satisfactory amounts of DFMST units incorporated in the copolymers, indicate that such a monomer is as active comonomer rather than a radical scavenger or transfer agent, though retarding the rate of ST polymerization.31 The observed effect of retardation has already been thoroughly discussed in a previous report on FMST/ST and TFMST/ST copolymerizations.35 Similarly, in case of DFMST/ST system, both the electronic and steric factors that influence the stability or/and reactivity of tertiary R–CH2(Ph)(CF2H)C˙ radical formed in the process, should be considered. We also suspect that the presence of relatively labile proton in –CF2H group may result in chain transfer reactions to monomer or/and polymer.52,53 These side reactions may disturb the propagation process significantly effecting in formation of materials with low-molar masses and relatively high dispersities. However, the proton lability in the CF2H moiety may create an opportunity for post-modification allowing for new functional polymers synthesis.54 Additionally, a comparison of reactivity ratios for ST calculated from EKT method at high conversions (rDFMST = 0.00 and rST = 0.69 at 70 °C) vs. low conversions (rDFMST = 0.00 and rST = 0.74 at 70 °C) indicates that with an increase of the conversion the chain transfer reactions may occur more frequently.
Although the F-STs generally delay the polymerization rates and increase unacceptably the polymerization time, ST monomer seems slightly more reactive in copolymerization with DFMST than with TFMST. Nevertheless, this deficiency can be overcome by restraining the insertion of fluorinated monomer feed (up to 30 mol%). In this way, the resulting fluorinated copolymers that exhibit low molar masses could be obtained in satisfactory yields (Table 1).
3.5. Thermal properties of poly(DFMST-co-ST) copolymers by TGA and DSC studies
The thermal characteristics of the poly(DFMST-co-ST) copolymers were determined by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) to assess their temperature at 10% weight loss (Td10) and the glass transition temperature (Tg) of copolymers, respectively. The results are compared to those of polystyrene homopolymers and summarized in Fig. 3 and Table 3.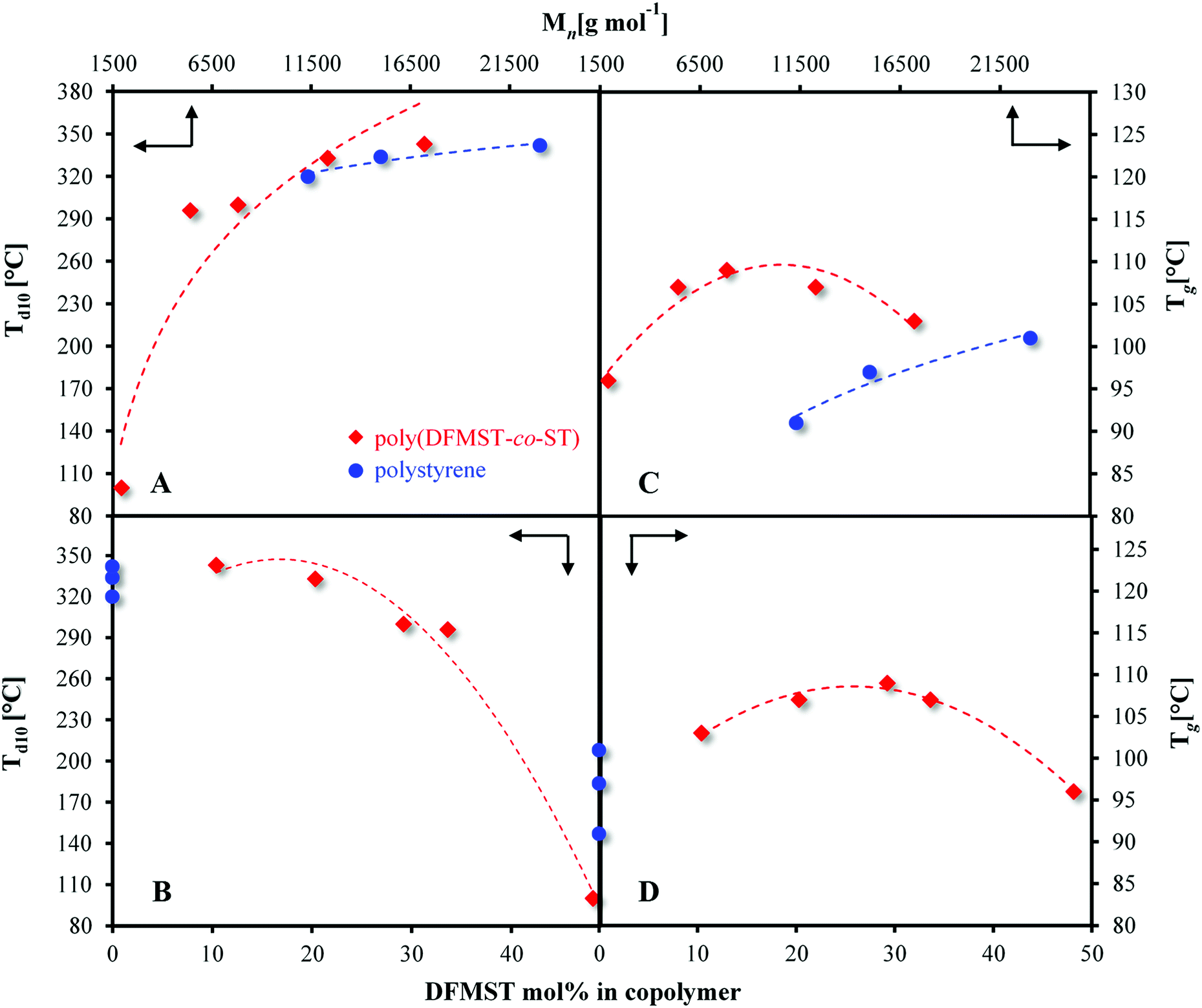 | ||
| Fig. 3 Thermal stabilities of the polymers [polystyrene, poly(DFMST-co-ST)]; (A) 10 wt% loss decomposition temperatures (Td10) versus number-average molar masses (Mns); (B) 10 wt% loss decomposition temperatures (Td10) versus the molar content of DFMST units incorporated into polymers; (C) glass transition temperatures (Tg) versus number-average molar masses (Mns); (D) glass transition temperatures (Tg) versus the molar content of DFMST units incorporated into polymers; in graphs (B) and (D) on the ordinates, for comparative purposes, Td10 and Tg temperatures of respective polystyrene homopolymers are included; relative and approximate average molar masses were determined by GPC (THF, RI) with polystyrene standards. | ||
| mol% in polymera | Reactivity ratios EKT method for high conv.b | Mnc [g mol−1] | Đc | Tgd [°C] | Td5d [°C] | Td10 [°C] | Td30d [°C] | Td50d [°C] | ||
|---|---|---|---|---|---|---|---|---|---|---|
| F-ST | ST | |||||||||
| a Determined by 1H and/or 19F NMR spectroscopies.b For DFMST–ST copolymerization system detailed in ESI and for FMST/TFMST–ST copolymerization system detailed in ref. 32 and its ESI.c Number-average molar masses (Mn) and dispersities (Đ) assessed from GPC (THF, RI) with poly(styrene) standards.d TGA and DSC analyses were performed under nitrogen atmosphere. | ||||||||||
| Poly(FMST-co-ST) | 10.6 | 89.4 | rFMST = 0.08 ± 0.02, rST = 0.72 ± 0.04 | 23700 |
3.66 | 102 | 315 | 337 | 365 | 381 |
| 20.2 | 79.8 | 18900 |
3.07 | 104 | 303 | 323 | 356 | 375 | ||
| 30.0 | 70.0 | 6600 | 1.88 | 111 | 249 | 302 | 351 | 371 | ||
| 31.0 | 69.0 | 4800 | 1.77 | 113 | 269 | 294 | 339 | 362 | ||
| 44.7 | 55.3 | 2900 | 1.93 | 106 | 231 | 266 | 349 | 372 | ||
| 49.7 | 50.3 | 1500 | 1.35 | N/A | 164 | 190 | 273 | 319 | ||
| Poly(DFMST-co-ST) | 10.4 | 89.6 | rDFMST = 0.00, rST = 0.69 ± 0.04 | 17200 |
3.0 | 103 | 320 | 343 | 372 | 386 |
| 20.3 | 79.7 | 12300 |
2.0 | 107 | 309 | 333 | 366 | 379 | ||
| 29.2 | 70.8 | 7800 | 1.9 | 109 | 279 | 300 | 348 | 362 | ||
| 33.6 | 66.4 | 5400 | 1.6 | 106 | 274 | 269 | 351 | 378 | ||
| 39.9 | 60.1 | 2700 | 1.5 | 105 | 263 | 279 | 325 | 371 | ||
| 43.9 | 56.1 | 2300 | 2.0 | 105 | 248 | 267 | 321 | 367 | ||
| 48.2 | 51.8 | 1900 | 1.2 | 96 | 90 | 100 | 251 | 346 | ||
| Poly(TFMST-co-ST) | 10.6 | 89.4 | rTFMST = 0.00, rST = 0.61 ± 0.01 | 14600 |
1.70 | 109 | 308 | 334 | 380 | 402 |
| 20.8 | 79.2 | 10800 |
1.51 | 110 | 302 | 321 | 358 | 378 | ||
| 28.7 | 71.3 | 8900 | 1.45 | 112 | 279 | 296 | 340 | 371 | ||
| 43.7 | 56.3 | 6500 | 1.36 | 114 | 269 | 279 | 318 | 356 | ||
| 46.9 | 53.1 | 2600 | 1.45 | N/A | N/A | N/A | N/A | N/A | ||
| 48.3 | 51.7 | 1500 | 1.58 | N/A | N/A | N/A | N/A | N/A | ||
| PST | — | 100 | — | 23000 |
1.71 | 101 | 318 | 342 | 373 | 384 |
| — | 100 | 15000 |
1.55 | 97 | 311 | 334 | 372 | 385 | ||
| — | 100 | 11300 |
1.38 | 96 | 304 | 320 | 349 | 363 | ||
Resulting aromatic fluorocopolymers containing up to 20 mol% of DFMST exhibited Td10 higher than 330 °C (Fig. 3A, B and Table 3). Moreover, the fluorinated copolymers with 10 and 20 mol% of DFMST units were more stable with a Td10 of 343 °C and 333 °C, respectively, versus 334 °C and 320 °C for the corresponding polystyrene homopolymers with comparable number-average molar masses (Table 3). The styrene monomers, such as DFMST, which possess substituents other than a labile proton at α-position to the aromatic ring allow to produce more stable materials.9,11 As expected, the higher the molar masses of polymers, the better its thermal stability. The trends were confirmed by both Td10s of poly(FMST-co-ST) and poly(TFMST-co-ST) copolymers35 and, as expected, of poly(DFMST-co-ST). Furthermore, with an increase of DFMST units incorporated in the copolymer, a substantial decrease in the molar masses was observed. Interestingly, poly(DFMST-co-ST) copolymers exhibited enhanced thermal properties in comparison to poly(FMST-co-ST) copolymers with comparable molar masses.35 Surprisingly, poly(DFMST-co-ST) copolymers displayed Td10s similar to respective poly(TFMST-co-ST) copolymers, thus indicating that the presence of such substituents (–CF2H, –CF3) at α-position of styrenic units significantly reduces the degradation process of the resulting fluorocopolymers.35,55
The Tgs of copolymers containing DFMST, evaluated by DSC (Table 3 and Fig. S19 in the ESI†), are plotted versus molar masses (Fig. 3C) and DFMST polymer contents (Fig. 3D). The Tgs increased up to 109 °C, with the decreasing molar masses of the examined polymers, up to 7800 g mol−1 only, as well as with the increasing DFMST content (up to 30 mol%). Whereas the DFMST mol% exceeded 30 mol% in copolymer, the Tgs values decreased, which was also due to the decreasing the molar masses of the polymers. The comparison of the Tgs values for the poly(FMST-co-ST) and poly(TFMST-co-ST) copolymers, previously described35 with Tgs for the obtained poly(DFMST-co-ST), showed that the latter one exhibited the lowest Tg values. However, in all cases and whatever the molar masses, the Tgs were slightly higher than those of the corresponding PST, which may indicate that the introduction of a reasonable amount of difluorinated monomer promotes an increase in the Tgs of the resulting copolymers by the presence of bulky –CF2H side groups.
3.6. Evolved gas analysis (EGA)
The degradation of copolymers may be elucidated by various approaches. One of the more accessible routes is a study of the generated volatile products by Evolved Gas Analysis (EGA). The EGA is a qualitative and quantitative analytical technique in which the volatile products released by a polymer during its decomposition processes are determined as a function of controlled temperature variation.56 The thermal decomposition of poly(DFMST-co-ST) copolymer (10.4/89.6) was investigated using simultaneous thermal analysis coupled with Fourier-transform infrared spectroscopy (STA/FTIR) and thermogravimetric analysis coupled with mass spectrometry (TGA/MS).![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O asymmetric stretching vibrations, indicating the presence of CO2 production, which is the main evolved gas during decomposition of poly(DFMST-co-ST) copolymer. The peak is very broad and exhibits two maxima. Interestingly, the carbon dioxide response was observed over extended timeframe. The CO2 started to release at 95–105 °C and the maximum intensity of its evolution was observed in the range of 380–420 °C. In addition, the evolution of water is evidenced by the peak between 3550 and 3800 cm−1 corresponding to O–H stretch. The release of H2O gas began at about 100 °C and reached a maximum peak at 450 °C. Two small absorption bands between 2100 and 2200 cm−1 are attributed to the presence of CO as further evolved gas. The C–H stretching vibration broad bands observed at 3010–3100 cm−1 are assigned to the presence of unsaturated hydrocarbon gases.61 Actually, the thermal degradation or depolymerization of polystyrene can occur by random scission process and may produce various aromatic substances, e.g. toluene, styrene, α-methylstyrene or styrene oligomers.61–63 Consequently, the frequency in FTIR spectra confirmed the production of such aromatic compounds. The thorough identification of the aromatic gases is presented in Section 3.6.2. Furthermore, the C–H stretching vibration band observed below 3000 cm−1 are assigned to the presence of saturated hydrocarbon gases, mainly CH4. The gas phase FTIR spectra also displayed absorbance band at 1100 cm−1 attributed to C–F stretch vibration indicating the presence of DFMST unit incorporated in polymer network.64 Additionally, the FTIR spectra of gases evolved from the thermal degradation of poly(DFMST-co-ST) copolymer (10.4/89.6), selected at the following temperatures: 320 °C (Td5), 343 °C (Td10), 372 °C (Td30), 386 °C (Td50), 421 °C (Td90), 450 °C and 480 °C, are presented in Fig. S21 in the ESI† scission reactions.56–59 In our case, the presence of the oxygen-information. The results clearly confirmed that CO2 was the main product that evolved during the entire thermal degradation pathway. Moreover, the water response was also observed over extended timeframe but in relatively smaller amount than that of carbon dioxide. The aromatic compounds were also found amongst the gaseous products (see Section 3.6.2) and thus TGA/MS was used to identify the unsaturated hydrocarbon gases which were observed in the aromatic region between 3010 and 3100 cm−1 in the FTIR spectra (Fig. 4).
O asymmetric stretching vibrations, indicating the presence of CO2 production, which is the main evolved gas during decomposition of poly(DFMST-co-ST) copolymer. The peak is very broad and exhibits two maxima. Interestingly, the carbon dioxide response was observed over extended timeframe. The CO2 started to release at 95–105 °C and the maximum intensity of its evolution was observed in the range of 380–420 °C. In addition, the evolution of water is evidenced by the peak between 3550 and 3800 cm−1 corresponding to O–H stretch. The release of H2O gas began at about 100 °C and reached a maximum peak at 450 °C. Two small absorption bands between 2100 and 2200 cm−1 are attributed to the presence of CO as further evolved gas. The C–H stretching vibration broad bands observed at 3010–3100 cm−1 are assigned to the presence of unsaturated hydrocarbon gases.61 Actually, the thermal degradation or depolymerization of polystyrene can occur by random scission process and may produce various aromatic substances, e.g. toluene, styrene, α-methylstyrene or styrene oligomers.61–63 Consequently, the frequency in FTIR spectra confirmed the production of such aromatic compounds. The thorough identification of the aromatic gases is presented in Section 3.6.2. Furthermore, the C–H stretching vibration band observed below 3000 cm−1 are assigned to the presence of saturated hydrocarbon gases, mainly CH4. The gas phase FTIR spectra also displayed absorbance band at 1100 cm−1 attributed to C–F stretch vibration indicating the presence of DFMST unit incorporated in polymer network.64 Additionally, the FTIR spectra of gases evolved from the thermal degradation of poly(DFMST-co-ST) copolymer (10.4/89.6), selected at the following temperatures: 320 °C (Td5), 343 °C (Td10), 372 °C (Td30), 386 °C (Td50), 421 °C (Td90), 450 °C and 480 °C, are presented in Fig. S21 in the ESI† scission reactions.56–59 In our case, the presence of the oxygen-information. The results clearly confirmed that CO2 was the main product that evolved during the entire thermal degradation pathway. Moreover, the water response was also observed over extended timeframe but in relatively smaller amount than that of carbon dioxide. The aromatic compounds were also found amongst the gaseous products (see Section 3.6.2) and thus TGA/MS was used to identify the unsaturated hydrocarbon gases which were observed in the aromatic region between 3010 and 3100 cm−1 in the FTIR spectra (Fig. 4).
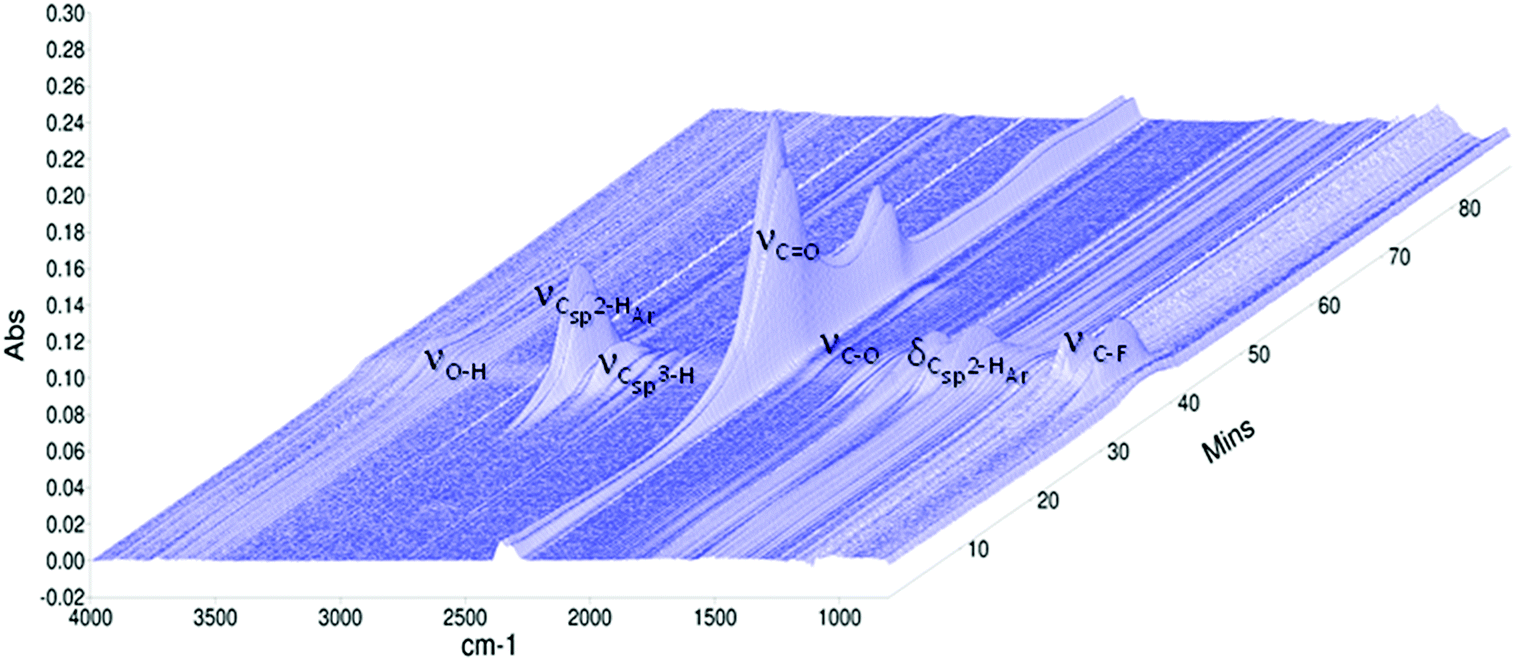 | ||
| Fig. 4 3D wavenumber-dependent FTIR absorbance of the poly(DFMST-co-ST) copolymer (10.4/89.6) as a function of time in the course of its thermal decomposition at 30–800 °C. | ||
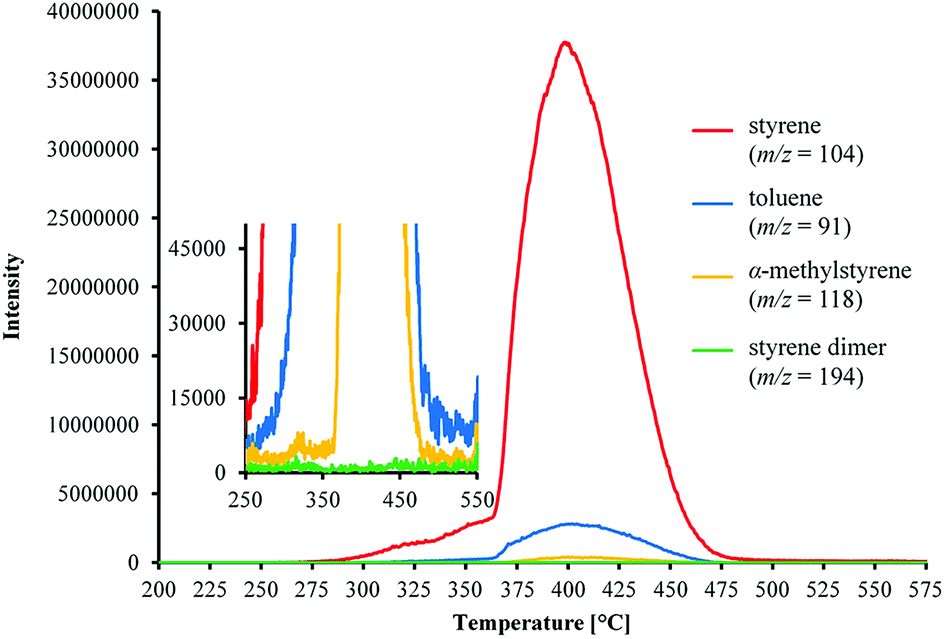 | ||
| Fig. 5 Evolution of m/z signals of volatile aromatic products from the thermal decomposition of poly(DFMST-co-ST) copolymer (10.4/89.6) from 200 to 575 °C. | ||
The results of TGA/MS analysis are displayed in Fig. 5 showing the evolution of generated aromatic compounds via the m/z mass spectra versus temperature. Moreover, the characteristic peaks of mass spectrum of evolved aromatic gases from the poly(DFMST-co-ST) copolymer (10.4/89.6) decomposition were observed (Fig. S22 in ESI†). The mass spectra in Fig. S22† of the evolved gases were captured at 370, 400 and 450 °C. The mass to charge ratios (m/z) detected as main products of poly(DFMST-co-ST) copolymer (10.4/89.6) depolymerization were assigned to styrene (m/z = 104), toluene (m/z = 91), α-methylstyrene (m/z = 118) and styrene dimer (m/z = 194). Moreover, the DFMST monomer was also observed among gaseous products of the fluorocopolymer thermal decomposition (m/z = 154).
3.7. Contact angle and surface energy of the fluorinated copolymers
The surface reorganization has been one of the key analyzed properties of the copolymers in recent years.66–70 Subsequently, to examine such a characteristic of copolymers, the effect of the comonomer structure on the surface energy is studied. To the best of our knowledge, there is a number of reports about the influence of wettability and oil repellency of fluorinated copolymers, depending on their structure.66,67,71 The advancing and receding contact angles of two liquids (polar and nonpolar) on the copolymer layers and the contact angle hysteresis (CAH) allowed us to get insight on the surface energy of these new fluorocopolymers. These layers were prepared via deposition of poly(DFMST-co-ST) copolymers on the glass surface beforehand modified by octadecyltrichlorosilane (OTS) according to a previous publication.36 Consequently, the copolymers were adsorbed from THF solution on a nonpolar substrate (glass/OTS). The comparison of advancing water and diiodomethane contact angles, AWCA and ADIMCA, obtained with H2O and CH2I2 is presented in Table 3.The first conclusion is that the diiodomethane contact angles are lower than the WCAs on a polar modified glass substrates. Fig. 6 displays the influence of the number of styrene moieties in polymers and copolymers on AWCA. The results of studies confirm our assumptions: the increasing number of styrene moieties in polymers and number of styrene moieties and DFMST molar ratio in copolymers (Fig. 6 and Table 3) give the changes in the contact angles. The similar dependence of CA on Mn of the copolymer was observed in a previous study,35 where we used the same method of application copolymer films on the modified glass surface.35,36
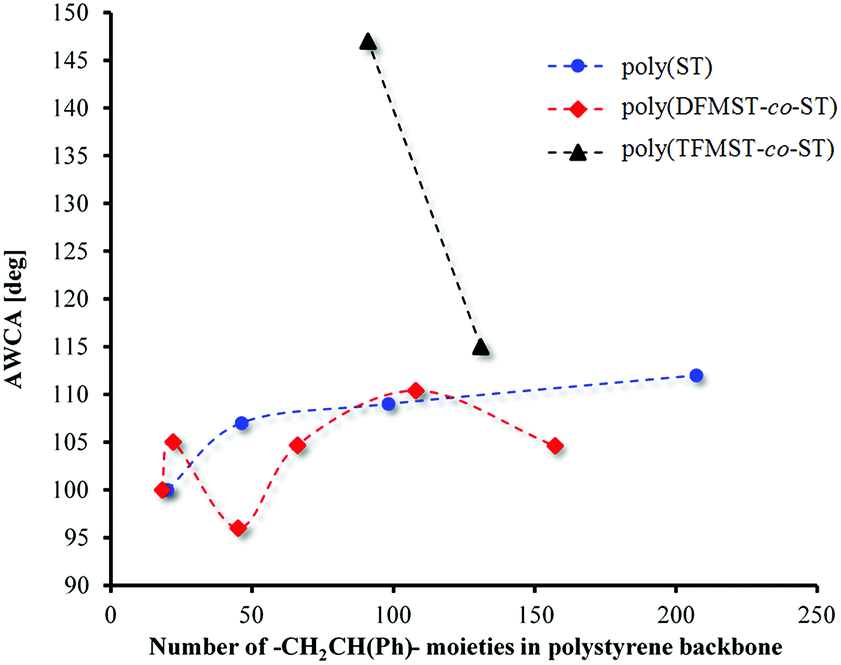 | ||
| Fig. 6 Advancing water contact angles as function of the number of styrene moieties in polymers and copolymers. | ||
As displayed in Fig. 6, the fluorine atoms in –CF2H group in these copolymers changed the hydrophobicity of the covered surface in an irregular way, while the H atom at the same position in polystyrenes did not supply a remarkable change in the approximately linear dependence. To overcome that issue, the following premise was considered: this irregularity is caused by separation of ST microblocks in fluorinated DFMST of copolymers. The fluorinated units can separate polystyrene microphases as in the PST-b-polybutadiene-b-PST block copolymer and causes a change in the wettability of the material by grafting hydrophilic units onto a hydrophobic backbone.72 To make our point stronger, the correlation of AWCA for polystyrenes and poly(TFMST-co-ST)copolymer was required (Fig. 6). As expected, compared with previous results,36 the CA values decrease significantly when CF3 is replaced by CF2H. The obtained CA results are in satisfactory agreement with those of previous studies on the perfluorohexylethyl(meth)acrylate/n-alkyl (meth)acrylate copolymers.66 Gu et al.66 found that the groups (H or CH3) at α-position of (meth)acrylate significantly influenced the contact angle, while the increase of side-chain lengths in the two types of copolymers did not.
To better describe the effect of the polymer surface properties, the CA hysteresis (CAH) was also determined. It is to be expected that the presented CAH of CA on styrene polymers and copolymers will be the effect of the structural differences among the samples.73,74 The obtained results show that CAH of highly polar water differs for polystyrene and fluorinated copolymers based on α-(difluoromethyl)styrene and styrene (Table 4).
| Molar ratios (mol%) in (co)polymers and polymers | Mn [g·mol−1] | No. –CH2CH(Ph)– in (co)polymers and polymersa | ACA (CAH) [deg] H2O/CH2I2 (°, ±1°) | |
|---|---|---|---|---|
| a The –CH2CH(Ph)– moieties are assumed as styrene backbone: in polystyrenes and also in fluorinated copolymers, calculated on the basis of styrene and α-(difluoro-methyl)styrene/α-(trifluoromethyl)styrene units. For poly(ST) homopolymers calculated from following formula; #ST = Mn/MST, where MST stands for the molar mass of ST (104.15 g mol−1). For poly(DFMST-co-ST) copolymers calculated from respective formula detailed in ESI, Table S2. | ||||
| Poly(TFMST-co-ST) copolymers35 | ||||
| ST | TFMST | |||
| 89.4 | 10.6 | 14600 |
131 | 147(2)/90(1) |
| 79.2 | 20.8 | 10800 |
91 | 115(3)/75(1) |
|
||||
| Poly(DFMST-co-ST) copolymers | ||||
| ST | DFMST | |||
| 89.6 | 10.4 | 17200 |
157 | 105(3)/70(1) |
| 79.7 | 20.3 | 12300 |
108 | 110(3)/82(1) |
| 70.8 | 29.2 | 7800 | 66 | 105(4)/79(1) |
| 66.4 | 33.6 | 5400 | 45 | 96(4)/72(1) |
| 60.1 | 39.9 | 2700 | 22 | 105(5)/80(1) |
| 56.1 | 43.9 | 2300 | 18 | 100(5)/73(1) |
|
||||
| Poly(ST) | ||||
| ST | DFMST | |||
| 100 | — | 21600 |
207 | 112(8)/62 |
| 100 | — | 10200 |
98 | 109(7)/64 |
| 100 | — | 4800 | 46 | 107(7)/67 |
| 100 | — | 2100 | 20 | 100(6)/72 |
The average value ranges from 6–8 degrees on polystyrene film while it is much fewer (3–5 degrees) on the fluorinated copolymers. In the literature, for the polystyrene surfaces, higher CAH values were obtained for example 11–12° (ref. 75) for oxygen plasma treated PS. The CAH increase points out that water droplet penetrates deeper into the polystyrene layers structure than into α-(difluoromethyl)styrene ones. Additionally, the CAH on the same copolymers films differs significantly for polar H2O and nonpolar CH2I2. In all cases, the diiodomethane CAHs are smaller than WCA one and below 1°. Considering these results, we expected that the water CAH values of homopolymers increase with increasing number of styrene moieties in polymer and the Mn, much more than hysteresis values of copolymers do. The differences in the CAH values reflect different strengths of interface interaction because of the nature of both these liquids.74 The surface organization is another feature to be considered for hysteresis. In the studied system, there might be a different orientation of phenyl rings of the polymer and copolymer surface in contact with the organic layer and in contact with air.
This change interferes with the surface energy of the copolymers. This information can be obtained from the calculated surface free energies (SFE = γtots) using the CAH approach (eqn (3)):74
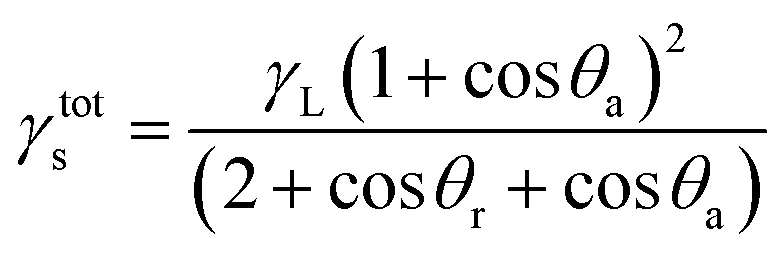 | (3) |
This is one of the methods of calculating the SFE value of polymeric materials.73,76 The values obtained on the basis of the formula depend on the type of liquids used. The calculated SFE values (liquid surface tension γL = 72.8 (water) or 50.8 (diiodomethane))77 from CAH of H2O and CH2I2 for six poly(DFMST-co-ST) copolymers and four poly(ST) layers deposited on organic film/glass surface are plotted in Fig. 7.
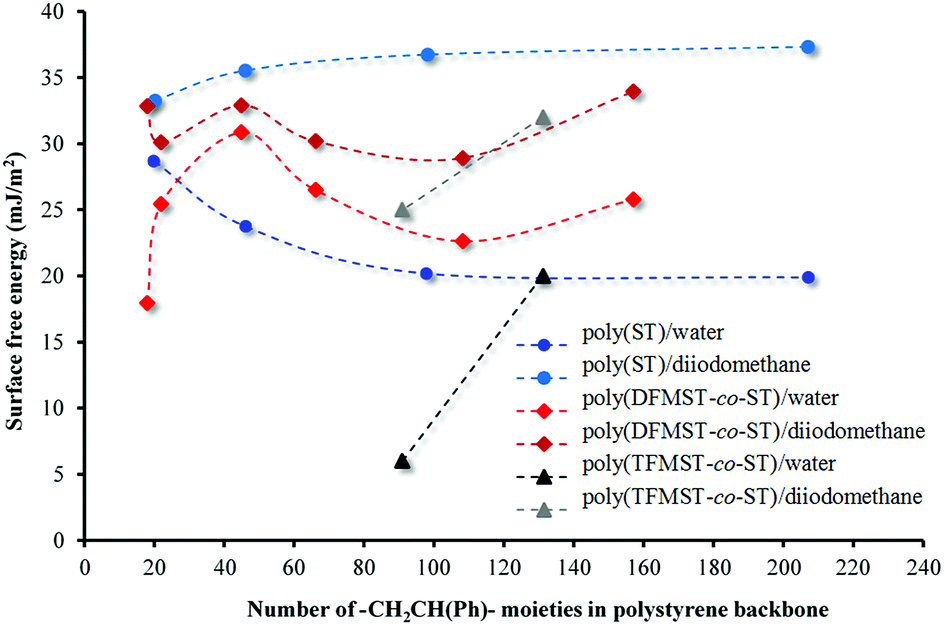 | ||
| Fig. 7 Surface free energy of poly(DFMST-co-ST), poly(TFMST-co-ST) and poly(ST) layers deposited on organic film/glass from contact angle hysteresis of water and diiodomethane. | ||
The result is contrary to expectations because the polymers containing even small fluorinated comonomers amounts still have lower surface energies compared to that of polystyrene.78 The results of our experiments show that the copolymers containing CF2H groups have the surface energy of about 25 mN m−1 (H2O) and 31 mN m−1 (CH2I2), while the polystyrene series have lower surface energy, i.e. about 20 mN m−1 and 35 mN m−1 (CH2I2).75 Only for copolymers containing 39.9 and 43.9 mol% of DFMST, the lowering of the surface energy is observed. This is in agreement with literature report78 and indicates adsorption of the DFMST segments at the surface.
The difluoromethyl group at the α-position of styrene comonomer results in the significant increase of SFE. These data provide the information about the SFE changes taking places with increasing the number of –CH2CH(Ph)– moieties. To the best of our knowledge, the literature on the calculations made with the same method based on CAH has not been found. In general, the SFE decreases in the following order, CH2 > CH3 > CF2 > CF2H > CF3.79 Our results confirm such a tendency. The SFE values determined for polystyrene films are practically constant on all the four series. This means that, in this case, the non-bonded interactions, i.e. London dispersion forces, are similar. Such a regular relationship, which is number of –CH2CH(Ph)– moieties dependent, is not observed for fluorinated copolymers. Controlling the surface energies affords not only verification over the surface of a material but also let us know about the orientation of the micro- and nanostructures in thin polymer films.80 Usually, the surface reorganization of the fluorinated chains is directly associated with the Tg or Tm of these copolymers.66 However, no clear relationship can be found in our case (see Table S4 in the ESI†) beyond the evident difference in Tg values of polystyrene and poly(DFMST-co-ST) copolymers.
4. Conclusion
Novel poly(DFMST-co-ST) copolymers were synthesized by conventional bulk radical copolymerizations of ST with DFMST. Detailed NMR spectroscopic characterizations with GPC analyses enabled us to determine the DFMST and ST monomer conversions, the composition, and the molar masses of the resulting copolymers, respectively. The DFMST incorporation into the copolymers ranged between 10.4 and 48.2 mol%. Moreover, a decrease in the efficiency of the copolymerization (with an increase of the DFMST feed), as well as the kinetic studies, confirmed that DFMST retards the polymerization rate of ST. The low DFMST reactivity and the retarding effect in radical polymerizations may result from the stability of the tertiary R–CH2C·(CF2H)(Ph) macroradical formed when a growing polymer attacks DFMST monomer. As DFMST can be regarded as a captodative species, the sterically hindered R–CH2(Ph)(CF2H)C˙ macroradical is stabilized by the phenyl ring and destabilized by the electron withdrawing –CF2H group. The combined steric and destabilizing effects reduced the ability of the macroradical to add onto another monomer unit and consequently decreased the propagation rate. As a result, the retardation of the polymerization rate, as well as an increase in the polymerization time, was observed. As expected, the assessment of the thermal properties pointed out that the incorporation of a small amount of difluorinated monomer units into the polystyrene increased both the Tgs significantly and the thermal stability slightly. Thermal degradation process of selected poly(DFMST-co-ST) copolymer, investigated by STA/FTIR and TG/MS analyses, identified water and carbon dioxide as the main volatile components, combined with unsaturated (mainly styrene monomer) and saturated hydrocarbon gases. On the basis of TG/MS technique, it was demonstrated that depolymerization reaction predominated the thermal degradation of poly(DFMST-co-ST) copolymers and styrene monomers were the dominant aromatic gases. Toluene, α-methylstyrene, dimer of styrene and α-(difluoromethyl)-styrene were also detected in evolved unsaturated hydrocarbons. The effect of α-difluoromethyl group in styrene comonomers caused poly(DFMST-co-ST) copolymer having a higher Tg value and a lower contact angle hysteresis than poly(ST). The surface free energy of fluorinated copolymers was influenced by the group –CF2H in α-position of comonomers due to its surface reorganization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Centre for Advanced Technologies, Adam Mickiewicz University, Poznań (CAT-AMU) for providing specialized analytical equipment and the National Science Centre Poland (HARMONIA 9, UMO-2017/26/M/ST5/00437) as well as French Fluorine Network (GIS) for supporting the research.References
- G. Hougham, P. E. Cassidy, K. Johns and J. Davidson, Fluoropolymers: Synthesis and Applications, Plenum Press, New York, 1999 Search PubMed.
- B. Ameduri and B. Boutevin, Well-Architectured Fluoropolymers: Synthesis, Properties and Applications, Elsevier Ltd., Amsterdam, 2004 Search PubMed.
- D. M. Smith, S. T. Iacono and S. S. Iyer, Handbook of Fluoropolymer Science and Technology, Wiley, Hoboken, New Jersey, 2014 Search PubMed.
- B. M. Ameduri, Chem.–Eur. J., 2018 DOI:10.1002/chem.201802708.
- W. Liu, Y. Koike and Y. Okamoto, Macromolecules, 2005, 38, 9466–9473 CrossRef CAS.
- J. G. Drobny, Technology of Fluoropolymers, CRC Press, Boca Raton, Florida, 2nd edn, 2008 Search PubMed.
- T. Soulestin, V. Ladmiral, F. D. Dos Santos and B. Améduri, Prog. Polym. Sci., 2017, 72, 16–60 CrossRef CAS.
- J. Walkowiak-Kulikowska, J. Wolska and H. Koroniak, Polymers Application in Proton Exchange Membranes for Fuel Cells (PEMFCs), in Polymer Engineering, ed. B. Tylkowski, K. Wieszczycka and R. Jastrząb, DeGruyter, Berlin, 2017, pp. 293–347 Search PubMed.
- V. F. Cardoso, D. M. Correia, C. Ribeiro, M. M. Fernandes and S. Lanceros-Méndez, Polymers, 2018, 10, 1–26 CrossRef.
- H. Dai, G.-Z. Yin, F.-J. Zhao, Z.-H. Bian, Y.-J. Xu, W.-B. Zhang, X.-R. Miao and H. Li, Polymer, 2017, 113, 46–52 CrossRef CAS.
- S. Banerjee, Handbook of Specialty Fluorinated Polymers, Elsevier Inc., Amsterdam, 2015 Search PubMed.
- S. M. Bhatt, US Pat., US 2005/0245693 A1, 2005.
- D. M. Lemal, J. Org. Chem., 2004, 69, 1–11 CrossRef CAS PubMed.
- M. Wiacek, S. Jurczyk, M. Kurcok, H. Janeczek and E. Schab-Balcerzak, Polym. Eng. Sci., 2014, 54, 1170–1181 CrossRef CAS.
- M. Wiacek, D. Wesolek, S. Rojewski, K. Bujnowicz and E. Schab-Balcerzak, Polym. Int., 2014, 63, 1982–1990 CrossRef CAS.
- M. Wiacek, D. Wesolek, S. Rojewski, K. Bujnowicz, S. Jurczyk, M. Kurcok and E. Schab-Balcerzak, J. Appl. Polym. Sci., 2015, 132, 42839 CrossRef.
- R. Souzy, B. Ameduri and B. Boutevin, Prog. Polym. Sci., 2004, 29, 75–106 CrossRef CAS.
- S. G. Cohen, H. T. Wolosinski and P. J. Scheuer, J. Am. Chem. Soc., 1949, 71, 3439–3440 CrossRef CAS.
- M. Prober, J. Am. Chem. Soc., 1953, 75, 968–973 CrossRef CAS.
- T. Narita, T. Hagiwara, H. Hamana, K. Shibasaki and I. Hiruta, J. Fluorine Chem., 1995, 71, 151–153 CrossRef CAS.
- T. Narita, Macromol. Rapid Commun., 2000, 21, 613–627 CrossRef CAS.
- A. E. Steck and C. Stone, US Pat., no. 5 834 523, 1998.
- C. Stone, T. S. Daynard, L.-Q. Hu, C. Mah and A. E. Steck, J. New Mater. Electrochem. Syst., 2000, 3, 43–50 CAS.
- D. W. Smith and D. A. Babb, Macromolecules, 1996, 29, 852–860 CrossRef CAS.
- R. Souzy, B. Ameduri and B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5077–5097 CrossRef CAS.
- R. Souzy, B. Ameduri, B. Boutevin, P. Capron, D. Marsacq and G. Gebel, Fuel Cells, 2005, 5, 383–397 CrossRef CAS.
- J. Walkowiak-Kulikowska, F. Boschet, G. Kostov, V. Gouverneur and B. Ameduri, Eur. Polym. J., 2016, 84, 612–621 CrossRef CAS.
- G. Kostov, M. Tredwell, V. Gouverneur and B. Ameduri, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3843–3850 CrossRef CAS.
- M. Kyulavska, G. Kostov, B. Ameduri and R. Mateva, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2681–2697 CrossRef CAS.
- T. Narita, J. Fluorine Chem., 2010, 131, 812–828 CrossRef CAS.
- M. G. Baldwin and S. F. Reed, J. Polym. Sci., Part A: Polym. Chem., 1968, 6, 2627–2635 CrossRef CAS.
- H. Ito, A. F. Renaldo and M. Ueda, Macromolecules, 1989, 22, 45–51 CrossRef CAS.
- T. Narita, Prog. Polym. Sci., 1999, 24, 1095–1148 CrossRef CAS.
- M. Ueda and H. Ito, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 89–98 CrossRef CAS.
- J. Walkowiak-Kulikowska, A. Szwajca, V. Gouverneur and B. Ameduri, Polym. Chem., 2017, 8, 6558–6569 RSC.
- J. Walkowiak-Kulikowska, A. Szwajca, F. Boschet, V. Gouverneur and B. Ameduri, Macromolecules, 2014, 47, 8634–8644 CrossRef CAS.
- B. Zheng, S. V. D'Andrea, L.-Q. Sun, A. X. Wang, T. Chen, P. Hrnciar, J. Friborg, P. Falk, D. Hernandez and F. Yu, ACS Med. Chem. Lett., 2018, 9, 143–148 CrossRef CAS PubMed.
- Y. Zafrani, D. Yeffet, G. Sod-Moriah, A. Berliner, D. Amir, D. Marciano, E. Gershonov and S. Saphier, J. Med. Chem., 2017, 60, 797–804 CrossRef CAS PubMed.
- G. K. S. Prakash, J. Hu, Y. Wang and G. A. Olah, Eur. J. Org. Chem., 2005, 11, 2218–2223 CrossRef.
- J. Walkowiak, T. Martinez Del Campo, B. Ameduri and V. Gouverneur, Synthesis, 2010, 11, 1883–1890 Search PubMed.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- D. J. Leng, C. M. Black and G. Pattison, Org. Biomol. Chem., 2016, 14, 1531–1535 RSC.
- J. Walkowiak-Kulikowska, J. Kanigowska and H. Koroniak, J. Fluorine Chem., 2015, 179, 175–178 CrossRef CAS.
- F. R. Mayo and F. M. Lewis, J. Am. Chem. Soc., 1944, 66, 1594–1601 CrossRef CAS.
- M. Fineman and S. D. Ross, J. Polym. Sci., 1949, 5, 259–262 CrossRef.
- A. I. Yezrielev, E. L. Brokhina and Y. S. Roskin, Polym. Sci. U.S.S.R., 1969, 11, 1894–1907 CrossRef.
- T. Kelen and F. Tüdős, J. Macromol. Sci., Part A: Pure Appl.Chem., 1975, A9, 1–27 CAS.
- F. Tüdős, T. Kelen, T. Foldes-Berezsnich and B. Turcsanyi, J. Macromol. Sci., Part A: Pure Appl.Chem., 1976, A10, 1513–1540 CrossRef.
- K. Miyashita, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1994, 27, 1093–1098 CrossRef CAS.
- J. P. van Hook and A. V. Tobolsky, J. Am. Chem. Soc., 1958, 80, 779–782 CrossRef CAS.
- C. G. Overberger, H. Biletch, A. B. Finestone, J. Lilker and J. Herbert, J. Am. Chem. Soc., 1952, 2661, 2078–2082 Search PubMed.
- C. D. Sessler, M. Rahm, S. Becker, J. M. Goldberg, F. Wang and S. J. Lippard, J. Am. Chem. Soc., 2017, 139, 9325–9332 CrossRef CAS PubMed.
- W. B. Nilsson and G. O. Pritchard, Int. J. Chem. Kinet., 1982, 14, 299–323 CrossRef CAS.
- S. Fustero, A. Simón-Fuentes, P. Barrio and G. Haufe, Chem. Rev., 2015, 115, 871–930 CrossRef CAS PubMed.
- J. Yu, B. Yi, D. Xing, F. Liu, Z. Shao, Y. Fu and H. Zhang, Phys. Chem. Chem. Phys., 2003, 5, 611–615 RSC.
- K. Candelier, J. Dibdiakova, G. Volle and P. Rousset, Thermochim. Acta, 2016, 644, 33–42 CrossRef CAS.
- N. H. Giron and M. C. Celina, Polym. Degrad. Stab., 2017, 145, 93–101 CrossRef CAS.
- E. Granada, P. Eguía, J. A. Vilan, J. A. Comesaña and R. Comesaña, Renewable Energy, 2012, 41, 416–421 CrossRef CAS.
- L. Odochian, C. Moldoveanu and D. Maftei, Thermochim. Acta, 2014, 598, 28–35 CrossRef CAS.
- P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Wiley, Weinheim, 2013 Search PubMed.
- M. Suzuki and C. A. Wilkie, Polym. Degrad. Stab., 1995, 47, 217–221 CrossRef CAS.
- C. A. Wilkie, Polym. Degrad. Stab., 1999, 66, 301–306 CrossRef CAS.
- Y. Yang, J. Yang, W.-M. Wu, J. Zhao, Y. Song, L. Gao, R. Yang and L. Jiang, Environ. Sci. Technol., 2015, 49, 12080–12086 CrossRef CAS PubMed.
- J. Coates, Interpretation of Infrared Spectra, A Practical Approach, in Encyclopedia of Analytical Chemistry, John Wiley & Sons, Hoboken, New Jersey, 2006, pp. 1–23 Search PubMed.
- L. Jiao and J. A. Sun, Procedia Eng., 2014, 71, 622–628 CrossRef CAS.
- Z. Gu, M. Zhang, J. He and P. Ni, Colloids Surf., A, 2016, 502, 159–167 CrossRef CAS.
- S. Saïdi, F. Guittard, C. Guimon and S. Géribaldi, Eur. Polym. J., 2006, 42, 702–710 CrossRef.
- K. Honda, M. Morita, O. Sakata, S. Sasaki and A. Takahara, Macromolecules, 2010, 43, 454–460 CrossRef CAS.
- Y. Oda, A. Horinouchi, D. Kawaguchi, H. Matsuno, S. Kanaoka, S. Aoshima and K. Tanaka, Langmuir, 2014, 30, 1215–1219 CrossRef CAS PubMed.
- S. Sugimoto, Y. Oda, T. Hirata, R. Matsuyama, H. Matsuno and K. Tanaka, Polym. Chem., 2017, 8, 505–510 RSC.
- I. Yamamoto, Fluoroalkyl Acrylate Polymers and Their Applications, in Fluorinated Polymers: Volume 2: Application, ed. B. Ameduri and H. Sawada, Royal Society of Chemistry, 2017, pp. 32–53 Search PubMed.
- C. L. Higginbotham and J. E. Kennedy, Biomed. Eng., Trends Mater. Sci., 2011, 465–488 Search PubMed.
- E. Chibowski, Adv. Colloid Interface Sci., 2007, 133, 51–59 CrossRef CAS PubMed.
- E. Chibowski and M. Jurak, Colloid Polym. Sci., 2013, 291, 391–399 CrossRef CAS PubMed.
- E. Occhiello, M. Morra, F. Garbassi, D. Johnson and P. Humphrey, Appl. Surf. Sci., 1991, 47, 235–242 CrossRef CAS.
- E. Chibowski, Adv. Colloid Interface Sci., 2003, 103, 149–172 CrossRef CAS PubMed.
- C. J. Oss, W. Wu, A. Docoslis and R. F. Giese, Colloids Surf., B, 2001, 20, 87–91 CrossRef PubMed.
- J. Hopken and M. Moller, Macromolecules, 1992, 25, 1461–1467 CrossRef CAS.
- W. A. Zisman, Relation of the Equilibrium Contact Angle to Liquid and Solid Constitution, in Contact Angle, Wettability, and Adhesion, ed. M. F. Fowkes, American Chemical Society, Washington, 1964, pp. 1–51 Search PubMed.
- D. Y. Ryu, K. Shin, E. Drockenmuller, C. J. Hawker and T. P. Russell, Science, 2005, 308, 236–239 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09340g |
| This journal is © The Royal Society of Chemistry 2018 |