
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence19F multiple-quantum coherence NMR spectroscopy for probing protein–ligand interactions†
Anna Zawadzka-Kazimierczuk *ab,
Mate Somlyaya,
Hanspeter Kaehligc,
George Iakobsond,
Petr Beierd and
Robert Konrat*a
*ab,
Mate Somlyaya,
Hanspeter Kaehligc,
George Iakobsond,
Petr Beierd and
Robert Konrat*a
aDepartment of Structural and Computational Biology, Max F. Perutz Laboratories, University of Vienna, Vienna Biocenter Campus 5, A-1030 Vienna, Austria
bBiological and Chemical Research Centre, Faculty of Chemistry, University of Warsaw, Żwirki i Wigury 101, 02-089 Warsaw, Poland
cInstitute of Organic Chemistry, University of Vienna, Währinger Strasse 38, A-1090 Vienna, Austria
dInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nam. 2, 160 00 Prague, Czech Republic. E-mail: anzaw@chem.uw.edu.pl; Robert.Konrat@univie.ac.at
First published on 5th December 2018
Abstract
A new 19F NMR method is presented which can be used to detect weak protein binding of small molecules with up to mM affinity. The method capitalizes on the synthetic availability of unique SF5 containing compounds and the generation of five-quantum coherences (5QC). Given the high sensitivity of 5QC relaxation to exchange events (i.e. reversible protein binding) fragments which bind to the target with weak affinity can be identified. The utility of the method in early stage drug discovery programs is demonstrated with applications to two model proteins, the neurotoxic NGAL and the prominent tumor target β-catenin.
Introduction
Fragment-based drug discovery (FBDD) has become a cornerstone of modern drug discovery programs. Several compounds have entered clinical trials1,2 and, as for now, two of them are already used for patient treatment.3,4 Particularly noteworthy is that FBDD strategies provide valid starting points for drug discovery even in cases where conventional high throughput screens (HTS) fail.5 During the course of a fragment-based drug discovery program weak binders in the 100–300 Da range are initially identified which are subsequently evolved in an iterative manner into larger compounds with higher binding affinities and better target selectivity. For the identification of initial hits several biophysical techniques exist, among them NMR spectroscopy is unique as it provides reliable quantitative binding information over a wide range of dissociation constants (KD). Subsequent fragment optimization either by fragment merging (linking different fragments) or fragment extension (introducing additional functional groups) exploits chemical information provided by structural studies using (mostly) X-ray crystallography and/or NMR spectroscopy.6 Additionally, lead compounds identified in early stages of the drug design process are sometimes used as reporter ligands for either screening additional chemical libraries or validate optimized follow-up hits.7Ligand-based NMR spectroscopy is particularly powerful in drug discovery to identify small molecule binders. First, contrary to methods based on observation of protein molecules, only very little protein material is required and even allows for the examination of ligand mixtures. Second, protein–ligand interaction is a dynamic process where the protein exchanges between the apo (ligand free) and the ligand-bound state. In case of weak interaction both states exist leading to averaging of NMR observables and weighted by their respective populations which in turn depend on the dissociation constant (KD) of the complex and the concentrations. In principle, monitoring and quantification of protein–ligand binding by NMR spectroscopy can be achieved by chemical shift measurements or nuclear Overhauser enhancement spectroscopy (NOESY). A large and diverse set of different NMR techniques exist which exploit these possibilities,8 among others saturation transfer difference (STD)9 or WaterLOGSY.10 Finally, in early stages of the drug design process ligand binding affinities are weak (μM KD's) leading to fast exchange between the free and bound state which can be efficiently probed by Carr–Purcell–Meiboom–Gill (CPMG) relaxation dispersion methodology.11 Furthermore, it has been shown that ligand-observed CPMG measurements can be effectively used not only for ligand screening but also to determine, for example, lifetimes of drug–target complexes.12
Among the various ligand-based methods, 19F NMR spectroscopy has gained significant attention due to various advantages: the NMR-active 19F isotope has high gyromagnetic ratio and 100% natural isotope abundance, assuring high sensitivity. Fluorine has a very large chemical shift dispersion that allows the investigation of large compound mixtures in a high throughput manner.7,13–17 NMR detection is straightforward as the spectra are devoid of background signals from solvents and proteins. Last and most importantly, the chemical shifts and the relaxation properties of the fluorine nuclei are extremely sensitive to small changes of the chemical environment (binding to target).18 Furthermore, fluorine incorporation into drugs is an established optimization strategy in medicinal chemistry.19,20 Insertion of a fluorine atom in a molecule can change the pKa, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, conformation and metabolism of a compound. Owing to these factors, 20% of all drugs contain at least one fluorine atom.
P, conformation and metabolism of a compound. Owing to these factors, 20% of all drugs contain at least one fluorine atom.
Particularly interesting in that respect is the pentafluorosulfanyl (SF5) group, which was introduced in small molecules first in 1960.21 It is a peculiar chemical group with octahedral geometry. The group consists of one axial and four equatorial fluorines, which give rise to a quintet and a doublet (J ≈ 150 Hz) in the 19F NMR spectra, respectively. The SF5 group is very often compared to the CF3 group: it is sterically demanding and highly electronegative (σp = +0.68 versus +0.54 for CF3). It has high dipole moment (μ = 3.4 D versus 2.6 for CF3) and high lipophilicity (logP = 2.55),22–25 which are usually opposing properties. The review of Welch and Savoie is an excellent summary of the various applications of SF5 compounds.26
In the present study we exploit this highly symmetric spin system for the generation of high order spin states (multiple quantum coherences) and explore the applicability to increase the sensitivity of 19F-based NMR screening methods. Specifically, we demonstrate that this novel 19F NMR methodology is able to detect weak affinity binders typically found in early stages of drug developmental programs. This could be applied in fragment screening where weak binders are used as spy ligands7 to probe additional chemical libraries and follow the chemical optimisation process.
Results and discussion
Our new method relies on the change of relaxation of the ligand upon binding to a protein. In case of a ligand reversibly binding to a protein target the observed relaxation (R′) is the average of relaxation of the ligand in a free form (RL) and bound form (RLP):| R′ = pLRL + pLPRLP | (1) |
However, there is another effect that contributes to the relaxation and stems from exchange contribution to the linewidth. In case of a fast two-site exchange process between free and bound state the exchange contribution is given by Rex = pLpLPΔω2/kex where kex is the exchange rate, Δω is resonance frequency difference between free and bound states. Depending on the chemical shift changes upon binding this can even become the dominating contribution to transverse 19F relaxation.27
Exchange processes, however, can only be detected by CPMG provided that populations and rates are within a narrow window for observation of relaxation dispersion experiments. Instead, measuring the decay of multiple quantum coherences has been proposed as an attractive alternative28 as these higher order coherences evolve as the sum of individual chemical shifts. For example, in case of symmetric spin systems the multiple-quantum coherence chemical shift difference, ΔωMQ depends on the coherence order n and the chemical shift difference observed for single quantum coherence, ΔωMQ = nΔω. In this case, the exchange contribution to the linewidth is given by Rex,MQ = pLpLPn2Δω2/kex. Thus, while the observed ligand relaxation rate in the presence of target can be small in case of single quantum coherences, the relaxation of multiple-quantum coherence could be sizeable. This effect has been already exploited in fluorine double-quantum relaxation measurements.29
Here we exploit the dependence of exchange contributions to the linewidth by n2Δω2 through the generation of five quantum coherences (5QC) in SF5-containing ligands and probe their binding to protein targets. The new experiment allows probing of protein ligand binding events involving smaller populations of the bound state than would be possible by existing single-quantum coherence techniques. The 19F 5Q pulse scheme for probing of 19F ligand binding to protein targets is illustrated in Fig. 1. In SF5 group axial and equatorial fluorines are magnetically non-equivalent and can thus be excited independently from each other using appropriate shaped pulses. Key to the pulse scheme is the efficient generation of five quantum coherence given by 16Fa+F1+F2+F3+F4+, where Fa indicates the axial 19F and 1, 2, 3 and 4 are labels for the equivalent equatorial fluorine spins. Here we adapted a strategy originally proposed by Kay and co-workers.28 Instead of directly generating 16FaxF1xF2xF3xF4x, which would be an unfavourable superposition of different 19F higher order terms we preserve most of the signal by selecting the following term 16FaxF1xF2yF3yF4y (as well as other symmetry-related linear combinations). In brief, this is achieved by generating a single anti-phase term 2FazF1x during the first Δ1 period (see Fig. 1), followed by conversion into 2FayF1x and evolution into 16FaxF1xF2zF3zF4z and after a π/2 pulse conversion into 16FaxF1xF2yF3yF4y (and corresponding terms for 2,3,4) which finally relaxes during the relaxation period Δ. After the relaxation period the signal is transferred back to the sensitive (magnetically equivalent) F4 group for detection. The efficiency of the magnetization transfer and generation of 5Q coherences was carefully checked using 2D NMR spectroscopy (see ESI†). Quality and efficacy of 5Q coherence generation was assessed based on peak position and lineshape of the cross peak and demonstrated that phase cycling for removal of undesirable coherence pathways was successful.
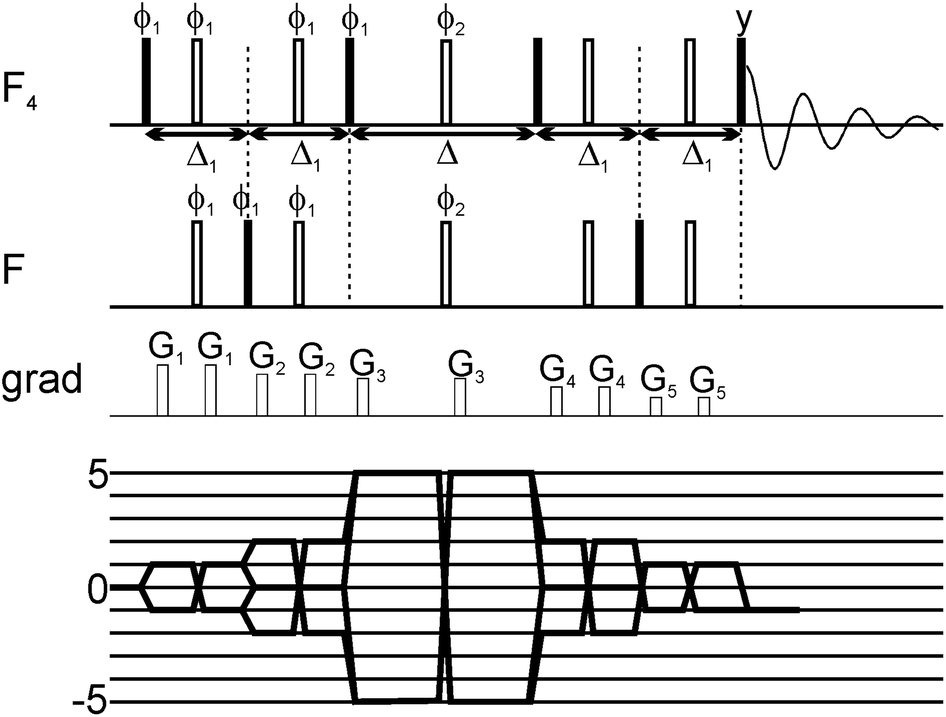 | ||
| Fig. 1 Scheme of the 5Q pulse sequence for T2 measurements of the SF5 system, together with coherence transfer pathway. All pulses were selective shaped pulses. The pulses acting on one type of fluorine nuclei (F or F4 group) were sinc-shaped; their length was set to 250 μs and offset was set on the frequency of the group. The pulses applied simultaneously on F and F4 groups were cosine-modulated sinc-shaped pulses; their length was 250 μs and offset was set in the middle between the two frequencies. the Δ1 delay was set to 6.28 ms and the relaxation delay Δ was incremented. The pulse phases were set to x, unless shown explicitly. On the phase ϕ1 a 10-step phase cycle was performed to select the coherence of ±5 order during the multiple-quantum period. Additionally, on the phase ϕ2 a 4-step phase cycle was performed. | ||
After establishing that 19F 5Q coherences can be efficiently created and as a first application of the new methodology we have studied the binding of small SF5-substituted small molecule compounds to proteins and investigated (quantitatively) relaxation changes of 1Q and 5Q coherences upon binding to protein targets. Two SF5-ligands were selected. 5-(pentafluoro-λ6-sulfanyl)-1,3-benzoxazole-2(3H)-thione is denoted below as ligand 1, and 2-bromo-4-(pentafluoro-λ6-sulfanyl)aniline is denoted as ligand 2 (see Fig. 2). Their binding to neutrophil gelatinase-associated lipocalin (NGAL) and β-catenin was studied by 19F NMR. In the 19F NMR spectrum of the SF5 group the four equatorial fluorines give rise to an intense doublet while the axial fluorine atom appears as a complex quintet. Due to sensitivity consideration we prefer to record the equatorial F4 doublet. Arguably it would be beneficial to observe the 19F signal under homonuclear decoupling conditions. In reality, however, homonuclear decoupling schemes can be challenging for the general user to set up and often do not provide significant sensitivity gains largely due to relaxation losses during acquisition and decoupler sideband interferences. Since we were aiming at a robust and easy to implement experimental set-up avoiding sophisticated parameter optimization we opted for a computational strategy to eliminate homonuclear scalar couplings. Specifically, we pursue a shifting-merging strategy. Details are explained in the ESI.†
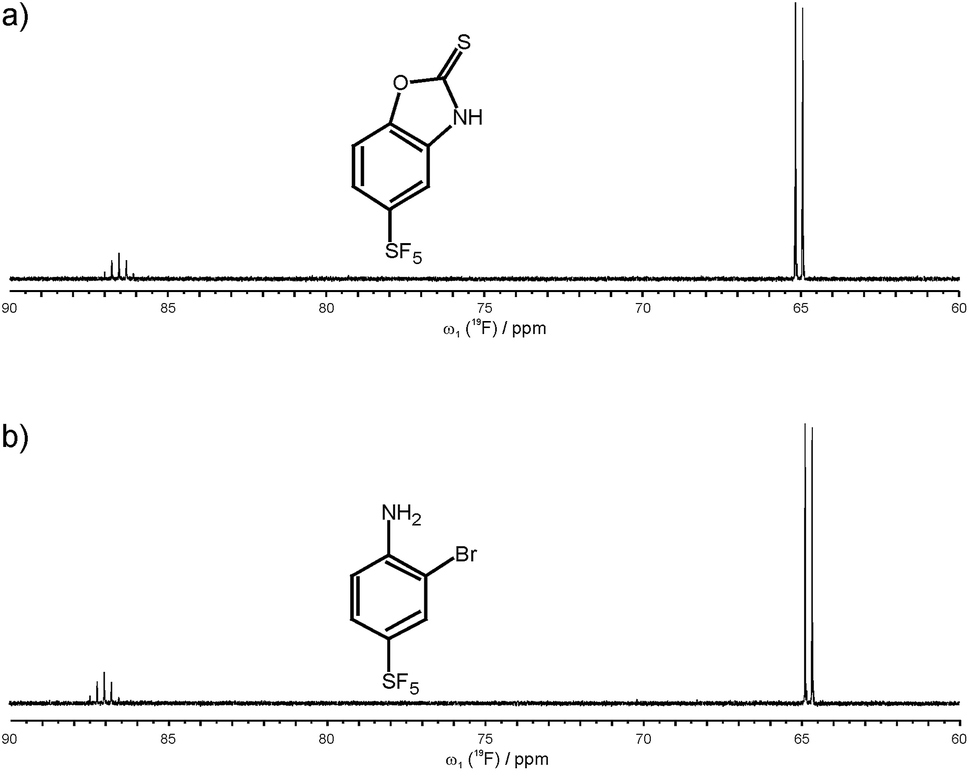 | ||
| Fig. 2 Chemical structure and 19F NMR spectrum of ligand 1, 5-(pentafluoro-λ6-sulfanyl)-1,3-benzoxazole-2(3H)-thione (a), and ligand 2, 2-bromo-4-(pentafluoro-λ6-sulfanyl)aniline (b). | ||
Decaying 1Q and 5Q spectra of the F4 group of ligand 1 at the absence and presence of protein are shown in Fig. 3. The dependence of peak intensity on relaxation delay, together with the exponentially decaying trends obtained for 1Q and 5Q relaxation under different conditions (protein-free vs. protein-bound) are shown in Fig. 4. The analysis of the curves reveals that the 5Q relaxation is more sensitive to protein binding than the relaxation of the 1Q term. As expected relaxation is more pronounced in the protein-bound state as a consequence of the larger correlation time of the bound state (protein complex) and due to exchange contributions to the 19F linewidth resulting from the reversible binding process. As we described above exchange contributions of multiple-quantum coherences scale with the square of the coherence order. In case of 5Q coherences sizeable differential contributions can thus be expected.
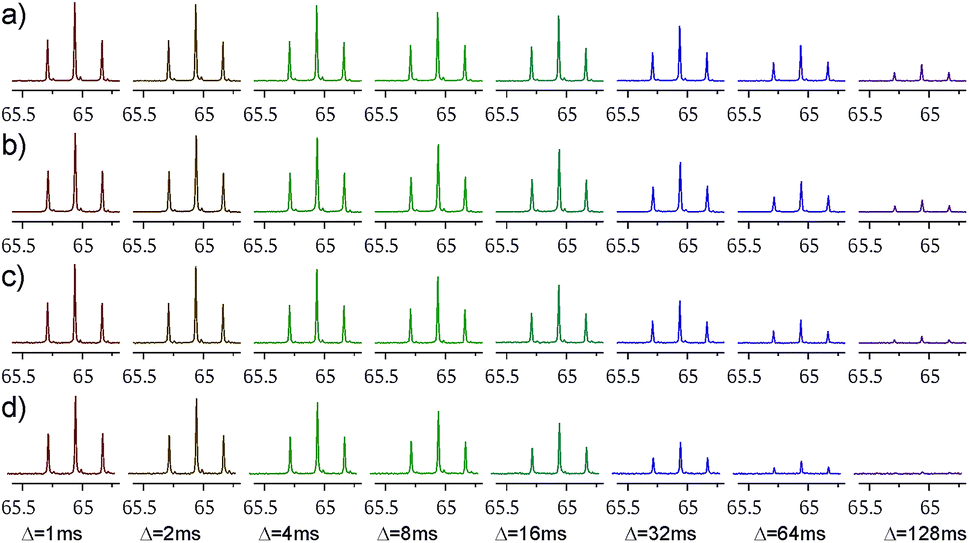 | ||
| Fig. 3 Decaying spectra of the F4 group signal (after the merging procedure), with increasing relaxation delay Δ. panels (a) and (c) show the ligand 1 alone (at 1 mM concentration), while panels (b) and (d) ligand 1 with 8 μM of NGAL protein. Panels (a) and (b) show decays of 1Q coherence, while panels (c) and (d) show decays of 5Q coherence. | ||
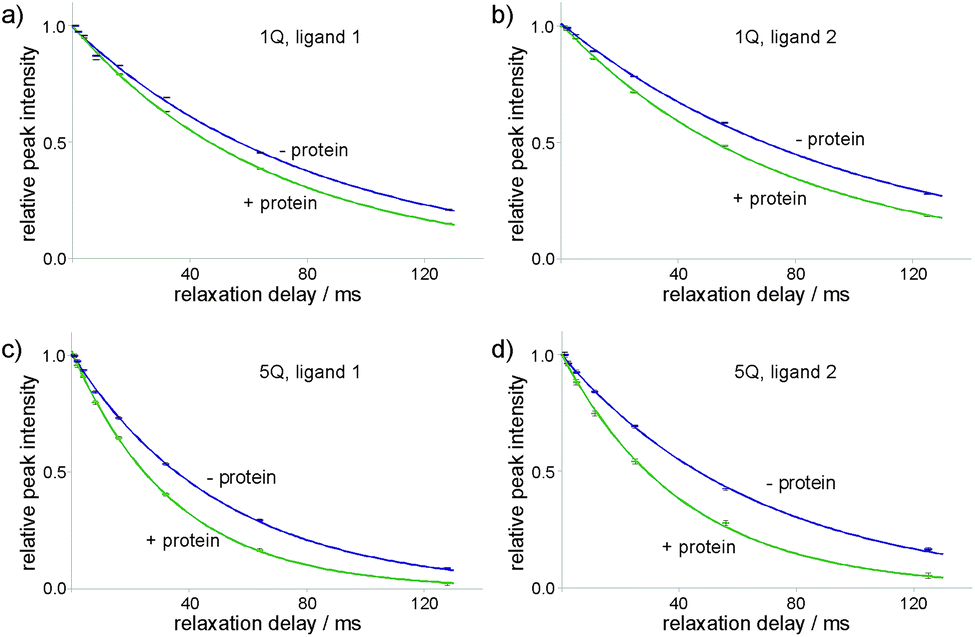 | ||
| Fig. 4 Decay of the merged F4 group peak intensity with increasing relaxation delay Δ, together with the exponential decay trends. Data are shown for ligand 1 (panels (a) and (c)) and ligand 2 (panels (b) and (d)). Relative peak intensities of 1Q (panels (a) and (b)) and 5Q (panels (c) and (d)) coherence peaks are shown. Purple markers and lines correspond to the data for the ligands alone, green ones correspond to the ligands with added protein. The protein concentration for ligand 1 was 8 μM and for ligand 2: 3 μM. | ||
Differential relaxation of 5Q and 1Q was analysed as a function of protein concentration and is illustrated in Fig. 5, which shows the relative change of the relaxation rates normalized to the values of the apo-state (ΔR/Rlig) and its dependence on the protein concentration. It is evident from the figure that increasing protein concentration leads to a sizeable difference of 5Q and 1Q relaxation; ΔR/Rlig is typically about two times larger for 5Q than for 1Q. At low protein concentration relative relaxation changes are comparable for 5Q and 1Q coherences. In contrast, at higher protein concentration significant differences are found. Clearly, 5Q coherences are more sensitive to protein binding than 1Q coherences. In other words, 5Q coherences exhibit the same relative changes in the T2 value as the 1Q coherence but already at significantly smaller protein concentrations. Additionally, closer inspection of Fig. 5a shows that NGAL/ligand 1 displays a linear dependence of ΔR/Rlig vs. protein concentration indicating that the measured relaxation rate is a population average of contributions from the apo and bound state, respectively. Conversely, the ΔR/Rlig analysis for β-catenin/ligand 2 clearly reveals non-linearity (Fig. 5b). This could be due to exchange contributions (between free and bound state) and which is typically found for weakly binding ligands. In this case the exchange contribution is given by pLpLPkexΔω2/kex, and pLpLP does not scale linearly with increasing protein concentration. However, given the low affinity (KD is high μM) of the β-catenin/ligand 2 complex the population of the bound state is rather small and at low protein concentrations the ligand exists predominantly in the free state (pL ≈ 1.0) and thus the product of the populations pLpLP ≈ pLP. The expected non-linearity (due to pLpLP) occurs only at substantially higher protein concentrations (simulations are given in the ESI†).
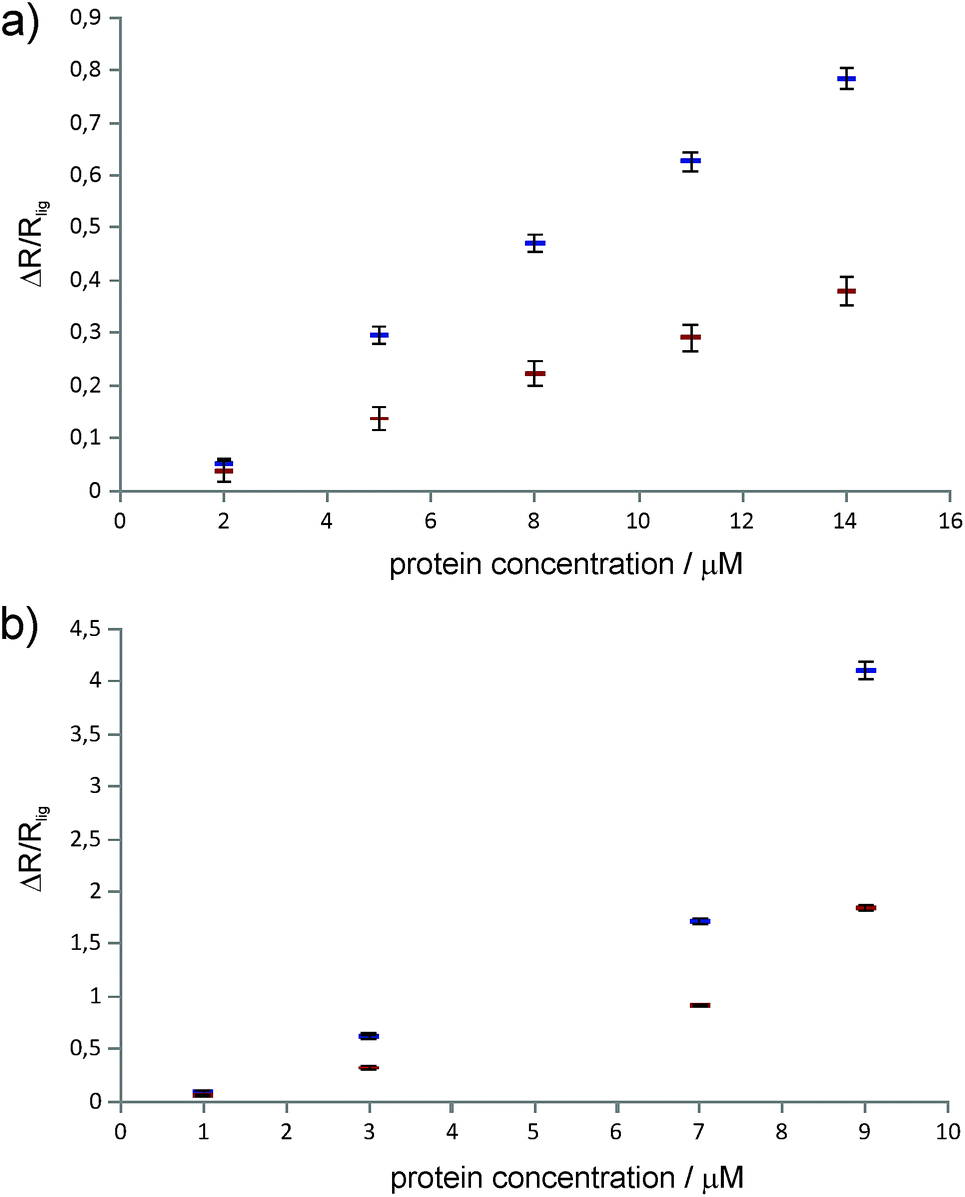 | ||
| Fig. 5 Relative change of 1Q (red markers) and 5Q (blue markers) relaxation rates of the ligand (with respect to the relaxation rate of the ligand alone) at different protein concentrations. (a) Ligand 1 with NGAL protein, (b) ligand 2 with β-catenin protein. | ||
Therefore we concluded that the observed non-linear concentration dependence of β-catenin is due to protein oligomerization (aggregation) leading to a substantial increase in the relaxation rate of the bound state. This is also in agreement with the empirical observation that β-catenin displays low solubility and tends to aggregate in solution. The superior sensitivity of the proposed methodology to probe protein binding even at low protein concentrations might thus become important for future NMR-supported programs aiming at β-catenin inhibitors as anti cancer drugs.
The 5Q experiment is of course less sensitive (in terms of signal-to-noise ratio) than the 1Q experiment. The estimated 5Q/1Q sensitivity ratio is about 10%. Therefore the number of scans needed for data acquisition should be increased to obtain sufficient sensitivity. In our case the number of scans employed for 5Q experiment for ligand 1 (whose concentration was 1 mM) was 40 which corresponds to ca. 14 minutes of the total measurement time to record the data for 8 different relaxation delays (from 1 ms up to 128 ms). For ligand 2 (0.5 mM concentration) the number of scans was increased to 160, which corresponds to ca. 49 minutes of the total measurement time (for 7 different relaxation delays, from 1 ms to 125 ms). The issue of sensitivity is being constantly alleviated with the development of NMR equipment. Despite lower signal-to-noise ratio, 5Q experiment is very powerful in terms of possibility of detection of binding event. In this case the difference between relaxation rates of a free ligand and ligand in a presence of protein is much larger than in the case of 1Q experiment.
Of course, the proposed 5Q experiment requires presence of the SF5 group in the ligand. Therefore we expect that the main application would be competition experiments. The SF5-containing molecule can be used as a spy to report binding events of other ligands present in the solution. High specificity of the method allows screening large sets of ligands at a time, including also those containing fluorine atom(s). The observed spectrum would always contain only one signal, which makes the analysis straightforward.
Experimental
NMR samples
The NMR samples used in our measurements were 450 μL in volume and contained 5% DMSO-D6 to ensure lock signal and proper solubility of the ligands. The samples were prepared using 2–14 μM NGAL with 1 mM ligand 1 and 1–9 μM beta-catenin with 500 μM ligand 2.NMR experiments
All NMR experiments were performed on a Bruker Avance III HD+600 MHz spectrometer equipped with an X(CPF) 1H-decoupled gradient (QP) probe and Bruker Avance III HDX 700 MHz spectrometer equipped with Cryo-QCI-F probe, operating at 19F frequencies of 564 MHz and 659 MHz, respectively. The acquisition parameters were gathered in the Table 1.| Ligand | Coherence order | Number of scans | Relaxation delays (Δ), ms | Spectral width, Hz | Number of time-domain points |
|---|---|---|---|---|---|
| Ligand 1 | 1Q | 4 | 1, 2, 4, 8, 16, 32, 64, 128 | 65789 |
32768 |
| 5Q | 40 | 1, 2, 4, 8, 16, 32, 64, 128 | 65789 |
32768 |
|
| Ligand 2 | 1Q | 16 | 1, 2.2, 5, 11.2, 25, 55.9, 125 | 65789 |
32768 |
| 5Q | 160 | 1, 2.2, 5, 11.2, 25, 55.9, 125 | 65789 |
32768 |
Protein expression and purification
Recombinant human beta-catenin (bCat) was produced as a H6-MBP-3C-bCat141–781 construct from a petM44 plasmid in E. coli BL21(DE3) in LB media. After reaching an OD600 = 0.7–0.9, the cultures were put for 5 min on ice before induction with 0.4 mM IPTG (isopropyl-β-D-1-thiogalactopyranoside) at 18 °C. After 20 hours of expression, the cells were collected by centrifugation and resuspended in 40 mL buffer containing 20 mM Tris, 150 mM NaCl, 5 mM β-mercaptoethanol, 1 mM benzamidine and protease inhibitor (Roche cOmplete Mini, EDTA free). After sonication and centrifugation, proteins were purified by Ni2+ affinity chromatography (HisTrap Chelating HP, 5 mL, GE Healthcare). The obtained protein (∼60 μM) was diluted to a concentration of 5 μM and cleaved overnight at 4 °C with 3C protease (20 mM Tris, 150 mM NaCl, 1% glycerol, 2 mM β-mercaptoethanol). The solution was again loaded onto a Ni2+ column. The cleaved tag, the His6-tagged 3C protease and the bCat141–781 was eluted with a gradient (0–200 mM imidazole) of 20 CV. Appropriate fractions were loaded onto a gel filtration column equilibrated in 20 mM Tris, 150 mM NaCl, 0.5 mM DTT, pH = 7.45 (HiLoad 26/60 Superdex 200pg, GE Healthcare). Beta-catenin containing fractions were diluted to 5 μM concentration and stored at −20 °C.Recombinant neutrophil gelatinase-associated lipocalin (NGAL) was produced as a H6-TEV-NGAL construct from a petM11 plasmid in BL21pLysS cells in LB media. After reaching an OD600 = 0.8, protein expression was induced with 0.8 mM IPTG (isopropyl-β-D-1-thiogalactopyranoside) at 30 °C. After overnight expression, the cells were collected by centrifugation and resuspended in 40 mL PBS buffer. After sonication and centrifugation, proteins were purified by Ni2+ affinity chromatography (HisTrap Chelating HP, 5 mL, GE Healthcare). The obtained protein was dialyzed overnight to Tris buffer (20 mM Tris, 50 mM NaCl, pH = 7.4). The protein was cleaved with TEV-protease overnight in Tris buffer (1 mM DTT, 0.5 mM EDTA), and loaded on a gel filtration column (HiLoad 16/60 Superdex 75pg, GE Healthcare). NGAL-containing fractions were pooled and concentrated to 0.5 mL, and the protein was denatured with 0.5 g guanidine hydrochloride. After 20 minutes incubation at 70 °C, the protein was loaded on a desalting column (PD10, Sephadex™ G-25 M, GE Healthcare) equilibrated with 6 M guanidine hydrochloride. The denatured protein was dialyzed to Tris buffer. After two days, the precipitates were centrifuged and the protein was stored at −20 °C.
Conclusions
In summary, we have presented a 19F 5Q relaxation experiment for probing of ligand binding to protein targets. The experiment offers superior sensitivity compared to conventional 19F 1Q methodologies and thus allows the identification and characterization of weak binders typically found in early stages of drug discovery programs. Exchange line broadening due to reversible protein binding can be a significant contribution to transverse relaxation and is thus exploited in NMR ligand screening. The potentially beneficial effect of exchange contributions to NMR probing of ligand screening has been extensively discussed and reviewed by Dalvit and co-workers.18 As the chemical exchange contribution to the transverse relaxation scales with the square of the coherence order, this term in the proposed 5Q experiment can be as much as twenty-five times larger than the corresponding term in the typically performed 1Q experiment. The significant increase in sensitivity will be advantageous in drug screening programs as ligand binding events can be detected at lower populations of the protein bound state. This substantially decreases the required amount of protein material, which is still a limiting factor in large scale screening initiatives. Consequently, we are proposing to use this 19F 5Q methodology for searching SF5 reporter molecules as potential spy compounds for the screening of chemical libraries. Furthermore, versatile synthetic approaches exist to SF5-containing chemical scaffolds offering access to diversity-oriented fragment screening libraries. As such, this novel 19F 5Q approach provides a rich avenue in drug discovery programs where protein availability and low concentration of protein ligand complexes are limiting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Initial Training Network, FLUOR21, funded by the FP7 Marie Curie Actions of the European Commission (FP7-PEOPLE-2013-ITN-607787) and the Austrian Science Fund Grant (W1258), DK: “Integrative Structural Biology”. A. Z.-K. was supported by an FWF Lise-Meitner Postdoctoral Fellowship (M 2084). This work was financially supported by the Czech Academy of Sciences (RVO: 61388963). The authors thank prof. Andrzej Ejchart of Institute of Biochemistry and Biophysics of the Polish Academy of Sciences, for his valuable suggestions.References
- J. Tsai, J. T. Lee, W. Wang, J. Zhang, H. Cho, S. Mamo, R. Bremer, S. Gillette, J. Kong, N. K. Haass, K. Sproesser, L. Li, K. S. M. Smalley, D. Fong, Y.-L. Zhu, A. Marimuthu, H. Nguyen, B. Lam, J. Liu, I. Cheung, J. Rice, Y. Suzuki, C. Luu, C. Settachatgul, R. Shellooe, J. Cantwell, S.-H. Kim, J. Schlessinger, K. Y. J. Zhang, B. L. West, B. Powell, G. Habets, C. Zhang, P. N. Ibrahim, P. Hirth, D. R. Artis, M. Herlyn and G. Bollag, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3041–3046 CrossRef CAS PubMed.
- D. A. Erlanson, S. W. Fesik, R. E. Hubbard, W. Jahnke and H. Jhoti, Nat. Rev. Drug Discovery, 2016, 15, 605–619 CrossRef CAS PubMed.
- G. Bollag, J. Tsai, J. Zhang, C. Zhang, P. Ibrahim, K. Nolop and P. Hirth, Nat. Rev. Drug Discovery, 2012, 11, 873–886 CrossRef CAS PubMed.
- A. J. Souers, J. D. Leverson, E. R. Boghaert, S. L. Ackler, N. D. Catron, J. Chen, B. D. Dayton, H. Ding, S. H. Enschede, W. J. Fairbrother, D. C. S. Huang, S. G. Hymowitz, S. Jin, S. L. Khaw, P. J. Kovar, L. T. Lam, J. Lee, H. L. Maecker, K. C. Marsh, K. D. Mason, M. J. Mitten, P. M. Nimmer, A. Oleksijew, C. H. Park, C.-M. Park, D. C. Phillips, A. W. Roberts, D. Sampath, J. F. Seymour, M. L. Smith, G. M. Sullivan, S. K. Tahir, C. Tse, M. D. Wendt, Y. Xiao, J. C. Xue, H. Zhang, R. A. Humerickhouse, S. H. Rosenberg and S. W. Elmore, Nat. Med., 2013, 19, 202–208 CrossRef CAS PubMed.
- P. J. Hajduk and J. Greer, Nat. Rev. Drug Discovery, 2007, 6, 211–219 CrossRef CAS PubMed.
- P. J. Hajduk, J. C. Mack, E. T. Olejniczak, C. Park, P. J. Dandliker and B. A. Beutel, J. Am. Chem. Soc., 2004, 126, 2390–2398 CrossRef CAS PubMed.
- C. Dalvit, P. E. Fagerness, D. T. A. Hadden, R. W. Sarver and B. J. Stockman, J. Am. Chem. Soc., 2003, 125, 7696–7703 CrossRef CAS PubMed.
- B. Meyer and T. Peters, Angew. Chem., Int. Ed., 2003, 42, 864–890 CrossRef CAS PubMed.
- M. Mayer and B. Meyer, Angew. Chem., Int. Ed., 1999, 38, 1784–1788 CrossRef CAS PubMed.
- C. Dalvit, P. Pevarello, M. Tato, M. Veronesi, A. Vulpetti and M. Sundström, J. Biomol. NMR, 2000, 18, 65–68 CrossRef CAS.
- P. J. Hajduk, E. T. Olejniczak and S. W. Fesik, J. Am. Chem. Soc., 1997, 119, 12257–12261 CrossRef CAS.
- T. Moschen, S. Grutsch, M. A. Juen, C. H. Wunderlich, C. Kreutz and M. Tollinger, J. Med. Chem., 2016, 59, 10788–10793 CrossRef CAS PubMed.
- C. Dalvit, Prog. Nucl. Magn. Reson. Spectrosc., 2007, 51, 243–271 CrossRef CAS.
- C. Dalvit, N. Mongelli, G. Papeo, P. Giordano, M. Veronesi, D. Moskau and R. Kümmerle, J. Am. Chem. Soc., 2005, 127, 13380–13385 CrossRef CAS PubMed.
- C. Dalvit, A. D. Gossert, J. Coutant and M. Piotto, Magn. Reson. Chem., 2011, 49, 199–202 CrossRef CAS PubMed.
- C. Dalvit, E. Ardini, M. Flocco, G. P. Fogliatto, N. Mongelli and M. Veronesit, J. Am. Chem. Soc., 2003, 125, 14620–14625 CrossRef CAS PubMed.
- C. Dalvit, M. Flocco, S. Knapp, M. Mostardini, R. Perego, B. J. Stockman, M. Veronesi and M. Varasi, J. Am. Chem. Soc., 2002, 124, 7702–7709 CrossRef CAS PubMed.
- C. Dalvit and M. Piotto, Magn. Reson. Chem., 2017, 55, 106–114 CrossRef CAS PubMed.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- H.-J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637–643 CrossRef PubMed.
- W. A. Sheppard, J. Am. Chem. Soc., 1960, 82, 4751–4752 CrossRef CAS.
- W. A. Sheppard, J. Am. Chem. Soc., 1962, 84, 3072–3076 CrossRef CAS.
- R. W. Taft, S. Ehrenson, I. C. Lewis and R. E. Click, J. Am. Chem. Soc., 1959, 81, 5352–5361 CrossRef CAS.
- L. J. Sæthre, N. Berrah, J. D. Bozek, K. J. Børve, T. X. Carroll, E. Kukk, G. L. Gard, R. Winter and T. D. Thomas, J. Am. Chem. Soc., 2001, 123, 10729–10737 CrossRef.
- J. E. True, T. D. Thomas, R. W. Winter and G. L. Gard, Inorg. Chem., 2003, 42, 4437–4441 CrossRef CAS PubMed.
- P. R. Savoie and J. T. Welch, Chem. Rev., 2015, 115, 1130–1190 CrossRef CAS PubMed.
- C. Dalvit and A. Vulpetti, Chem. - Eur. J., 2016, 22, 7592–7601 CrossRef CAS PubMed.
- T. Yuwen, P. Vallurupalli and L. E. Kay, Angew. Chem., Int. Ed., 2016, 55, 11490–11494 CrossRef CAS PubMed.
- C. Dalvit and A. Vulpetti, Magn. Reson. Chem., 2012, 50, 592–597 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information on 5Q coherence generation, doublet components merging procedure, relaxation simulation, chemical synthesis of ligand 1. See DOI: 10.1039/c8ra09296f |
| This journal is © The Royal Society of Chemistry 2018 |