
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective synthesis of E-vinylsilanes and E,E-divinylsilanes via platinum-catalyzed hydrosilylation of alkynes with secondary silanes†
P. Żak *,
M. Bołt and
C. Pietraszuk
*,
M. Bołt and
C. Pietraszuk
Adam Mickiewicz University in Poznań, Faculty of Chemistry, Umultowska 89b, 61-614 Poznań, Poland. E-mail: pkw@amu.edu.pl
First published on 30th November 2018
Abstract
Platinum-N-heterocyclic carbene complex with the formula [Pt(IPr*OMe)(dvtms)] (where IPr*OMe = 1,3-bis{2,6-bis(diphenylmethyl)-4-methoxyphenyl}imidazol-2-ylidene, dvtms = divinyltetramethyldisiloxane) exhibits a high catalytic activity towards the β-E-selective hydrosilylation of terminal acetylenes with many secondary silanes (46 examples). Depending on the ratio of the reagent concentrations, the products of mono- or disubstitution are selectively obtained. Moreover, the one-pot sequential hydrosilylation of two different alkynes with secondary silane was also achieved over the same platinum catalyst.
Introduction
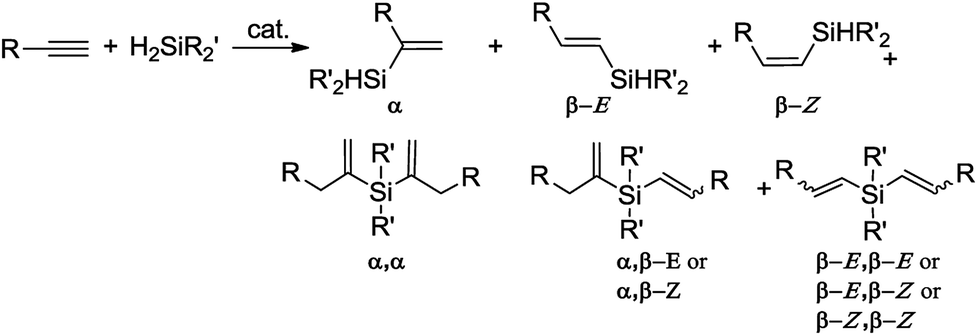
Hydrosilylation of terminal alkynes is one of the most powerful methods for the preparation of alkenyl functional organosilicon compounds,1 which are useful intermediates in several synthetic transformations.2 An important issue hampering the widespread use of this method is the need to control the regio- and stereochemistry, because the hydrosilylation of a terminal alkyne might generate β-Z, β-E and α-vinylsilanes (Scheme 1). Moreover, the reaction is often accompanied by side-processes, such as dehydrogenative silylation, isomerization and hydrogenation of olefins, olefin oligomerization and redistribution of silanes.1 | ||
| Scheme 1 Hydrosilylation of terminal alkynes with trisubstituted silanes. | ||
An additional problem appears in the reactions with secondary silanes, where the presence of two Si–H bonds permits formation of mono- and disubstituted products (Scheme 2).
 | ||
| Scheme 2 Hydrosilylation of terminal alkynes with disubstituted silanes. | ||
Although the hydrosilylation of olefins has been investigated using various transition metal complexes as catalysts,1,3 only a few reports describe the selective hydrosilylation of terminal alkynes with secondary silanes. In 2014, Deng et al. reported that hydrosilylation of alkynes with H2SiPh2 in the presence of the three-coordinate cobalt(I) complex led to the formation of respective monoalkenylsubstituted silanes.4 The ruthenium complex [Ru(t-BuPNP)(H)2(H2)] has been also applied in the hydrosilylation of alkynes with H2SiPh2 to form mainly β-Z-vinylsilanes (in all cases mixtures of stereoisomers were obtained).5 In 2016, Lu's and Huang's groups independently demonstrated that cobalt complexes showed Markovnikov-selectivity in the hydrosilylation of terminal alkynes with H2SiPh2, by employing iminopyridine oxazoline6 and pyridine bis(oxazoline)7 ligands, respectively. In 2018, Huang and co-workers described co-catalyzed asymmetric synthesis of Si-stereogenic vinylhydrosilanes via hydrosilylation of terminal alkynes with H2SiPhAr (where Ar ≠ Ph).8 Recently, we have demonstrated that platinum complexes bearing bulky N-heterocyclic carbene ligands can be used as selective catalysts for the hydrosilylation of a range of compounds containing unsaturated carbon–carbon bonds.9 Our work was focused on hydrosilylation of alkenes and alkynes with trisubstituted silanes and only three example of hydrosilylation of acetylene with disubstituted silane was reported.
As presented above, there is only a limited number of catalytic systems enabling selective formation of single isomer in hydrosilylation of terminal alkynes with disubstituted silanes (Scheme 2). Moreover, these catalytic systems lead to β-Z- or α-adducts and the scope of silanes is limited to two examples. To our surprise, literature peruse revealed a lack of reliable procedure for selective synthesis of the β-E-vinylsilanes via hydrosilylation of terminal alkynes with secondary silanes. According to our knowledge, there are also no literature reports on one-pot sequential hydrosilylation of two different terminal alkynes with secondary silane. We found only one example described the multicomponent silylation of alkynes and alcohols with dihydrosilane, resulted in high stereoselectivity as well as high yield construction of functional E-vinylsilyl ethers.10 It is thus of great importance to broaden the hydrosilylation of 1-alkynes to multicomponent silylation, as it would open up new strategy to design structurally diverse organosilicon compounds including chiral organosilanes.
Inspired by the encouraging preliminary results showing β-E-selective hydrosilylation of terminal alkynes with secondary silanes in the presence of platinum catalysts bearing bulky N-heterocyclic carbene ligand,9 we decided to expand the scope of tested 1-alkynes and disubstituted silanes.
Herein, we report a highly efficient and selective hydrosilylation of terminal alkynes with secondary silanes, occurring in the presence of platinum complex of the formula [Pt(IPr*OMe)(dvtms)] I (where IPr*OMe = 1,3-bis{2,6-bis-(diphenylmethyl)-4-methoxy-phenyl}imidazol-2-ylidene, dvtms = divinyltetramethyldi-siloxane). We report procedures that enable stereoselective synthesis of β-E-isomers of mono- or dialkenylsubstituted silanes, depending on the molar ratio of reagents used.
Results and discussion
Mono-hydrosilylation of alkynes with secondary silanes
On the basis of our previous work,9 we attempted to develop a synthetic protocol for the formation of Si–C bond via hydrosilylation of terminal alkynes with disubstituted silanes. Our examination started with optimization of the reaction, including the selection of a suitable amount and type of solvent, catalyst concentration, reaction time and temperature. H2SiMePh (1a) and 1-heptyne (2a) were used as model substrates. The platinum(0) complex I was synthesized according to a recently published procedure.11 The addition of 5 × 10−2 mol% of I to a 1.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of H2SiMePh (1a) and 1-heptyne (2a) in toluene at 40 °C led to highly preferred formation of β-E-isomer of monoalkenylsubstituted product in 58% yield, as revealed by GC/MS analysis. 1H NMR spectroscopy showed that the coupling constants between olefinic protons for the major product took the value JH–H = 19.2 Hz, that indicates the formation of β-E-isomer. In optimized conditions β-E-isomer was obtained as an exclusive product of the reaction (Scheme 3).
:1 mixture of H2SiMePh (1a) and 1-heptyne (2a) in toluene at 40 °C led to highly preferred formation of β-E-isomer of monoalkenylsubstituted product in 58% yield, as revealed by GC/MS analysis. 1H NMR spectroscopy showed that the coupling constants between olefinic protons for the major product took the value JH–H = 19.2 Hz, that indicates the formation of β-E-isomer. In optimized conditions β-E-isomer was obtained as an exclusive product of the reaction (Scheme 3).
 | ||
| Scheme 3 Hydrosilylation of 1-heptyne with H2SiMePh catalyzed by I. | ||
To evaluate the effect of temperature and catalyst concentration on the model reaction, we performed a series of catalytic tests using different solvents. The results are collected in Table 1.
| Entry | Solvent/T[°C] | [I] [mol%] | Time [h] | Conv.b [%] | β-E:β-Z:αc |
|---|---|---|---|---|---|
| a Reaction condition: argon; [1a]:[2a] = 1.1:1.b Determined by GC analysis.c Determined by GC analysis and confirmed by 1H NMR spectroscopy of the crude reaction mixture.d Without solvent.e β-E,β-E product was detected (up to 20% for entry 1–4, 27% for entry 7). |
|||||
| 1 | Toluene/100 | 1 × 10−3 | 24 | 100 | 70:10:20e |
| 2 | Toluene/100 | 5 × 10−2 | 1 | 100 | 78:10:12e |
| 3 | Benzene/60 | 5 × 10−2 | 12 | 98 | 87:5:8e |
| 4 | Toluene/60 | 5 × 10−2 | 6 | 100 | 95:3:2e |
| 5 | Toluene/40 | 5 × 10−2 | 24 | 58 | 98:1:1 |
| 6 | Toluene/40 | 1 × 10−1 | 24 | 100 | 97:1:2 |
| 7 | —/40d | 1 × 10−1 | 24 | 100 | 96:1:3e |
| 8 | THF/40 | 1 × 10−1 | 24 | 85 | 95:2:3 |
| 9 | Toluene/35 | 1 × 10−1 | 24 | 100 | 100:0:0 |
| 10 | Toluene/35 | 1 × 10−2 | 24 | 70 | 100:0:0 |
| 11 | Toluene/RT | 1 × 10−1 | 24 | 85 | 100:0:0 |
| 12 | Toluene/RT | 2 × 10−1 | 24 | 100 | 100:0:0 |
As shown in Table 1, the best results were achieved when conducting the reaction in toluene at 35 °C in the presence of 10−1 mol% of complex I (entry 9). When the catalyst loading was lower than 10−1 mol%, complete conversion of 1-heptyne was not achieved (entry 10). The appropriate choice of solvent had a significant impact on the course of the reaction and from among all the solvents tested, toluene was found to be the best one in terms of both activity and selectivity to the anti-Markovnikov hydrosilylation. An effective transformation of substrates was also observed when the reaction was performed in THF or benzene, but then a mixture of three isomers was obtained (entry 3 and 8). Optimization tests indicated also that the concentration of acetylene have influence on the reaction effectiveness and selectivity. When the process was carried out in solvent-free conditions, 27% of dialkenylsubstituted silane was detected (entry 7). The same problem was observed for the reaction conducted at the temperature above 40 °C (entry 1–4). Moreover, when the reaction was carried out at 100 °C, it was necessary to isolate the expected products immediately after full conversion of the substrates as otherwise the product 3 disappeared and polymer products were formed. The tests performed have shown that the process can be carried out at room temperature but then catalyst I had to be used in a slightly higher concentration to achieve quantitative yields (entry 12). Conditions of entry 9 were found optimal as they allowed quantitative conversion by using a relatively low catalyst loading.
The results are presented in Table 2.
| Entry | H2SiR1R2 1 | Alkyne 2 | Product | Isolated yield [%] |
|---|---|---|---|---|
| a Reaction conditions: toluene, 35 °C, argon, [1]:[2] = 1.1:1, [I] = 10−1 mol%.b [I] = 5 × 10−1 mol%. |
||||
| 1 | 1a | 2a | 3 | 92 |
| 2 | 1b | 2a | 4 | 93 |
| 3 | 1c | 2a | 5 | 90 |
| 4 | 1d | 2i | 6 | 92 |
| 5 | 1b | 2b | 7 | 89 |
| 6 | 1a | 2b | 8 | 92 |
| 7 | 1c | 2g | 9 | 92 |
| 8 | 1b | 2c | 10 | 93 |
| 9 | 1b | 2f | 11 | 89 |
| 10 | 1b | 2f | 12 | 87 |
| 11 | 1a | 2e | 13 | 89b |
| 12 | 1a | 2d | 14 | 87b |
| 13 | 1b | 2d | 15 | 89b |
| 14 | 1a | 2h | 16 | 94 |
| 15 | 1b | 2h | 17 | 92 |
| 16 | 1a | 2i | 18 | 91 |
| 17 | 1b | 2i | 19 | 90 |
| 18 | 1c | 2i | 20 | 90 |
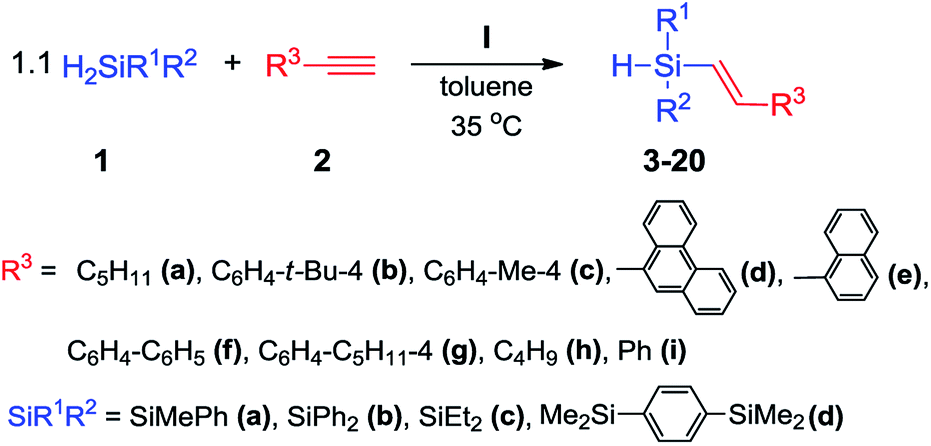
Having an active and selective catalyst in hand, the range of substrates was extended to determine versatility of the method (Scheme 4).
 | ||
| Scheme 4 Hydrosilylation of alkynes (1 equiv.) with secondary silanes (1.1 equiv.) catalyzed by I. | ||
We tested four different secondary silanes containing alkyl (1c), aryl (1b) and a mixture of alkyl and aryl substituents (1a, 1d) located at silicon atom. Alkynes bearing alkyl, phenyl or substituted phenyl groups were the source of multiple carbon–carbon bonds. For all of the substrates tested, nearly quantitative yields and exclusive formation of β-E product was achieved. For the variety of substrates that were used, we did not observe meaningful difference in the reaction course. Only in the case of alkynes containing alkyl substituent (2a, 2h) we were able to shorten the reaction time to 2 h. In all cases, the temperature rise caused significant acceleration of the reaction rate, however lowering of the selectivity was also observed. Upon using sterically crowded substrates such as 9-ethynylphenanthrene (2d) and 1-ethynylnaphthalene (2e), a slightly higher loading of catalyst I had to be used to achieve high yields. We did not observe the formation of dialkenylsubstituted silanes and other products of competitive reactions for any of the reactions studied. Within the research course, 18 compounds were isolated and spectroscopically characterized (3–20) (see ESI† for detailed analytical data).
Symmetrical bis-hydrosilylation of alkynes with secondary silanes
Encouraged by the high activity of catalyst I in hydrosilylation of terminal acetylenes with secondary silanes used in practical nearly equimolar ratio, we examined the reactions with 2 equiv. of alkynes. The performed tests revealed that under such conditions the reactions selectively produced β-E,β-E dialkenylsubstituted silanes in high yields (Scheme 5). | ||
| Scheme 5 Hydrosilylation of terminal acetylenes (2 equiv.) with secondary silanes (1 equiv.) catalyzed by complex I. | ||
The results are presented in Table 3.
| Entry | H2SiR1R2 1 | Alkyne 2 | Product | Isolated yield [%] |
|---|---|---|---|---|
| a Reaction conditions: toluene, 35 °C, argon, [1]:[2] = 1:2, [I] = 2 × 10−1 mol%, 24 h. |
||||
| 1 | 1a | 2a | 21 | 92 |
| 2 | 1b | 2a | 22 | 90 |
| 3 | 1c | 2a | 23 | 92 |
| 4 | 1d | 2a | 24 | 94 |
| 5 | 1d | 2j | 25 | 90 |
| 6 | 1b | 2c | 26 | 90 |
| 7 | 1a | 2k | 27 | 89 |
| 8 | 1b | 2b | 28 | 91 |
| 9 | 1a | 2b | 29 | 92 |
| 10 | 1b | 2f | 30 | 89 |
| 11 | 1a | 2f | 31 | 87 |
| 12 | 1c | 2f | 32 | 88 |
| 13 | 1a | 2h | 33 | 91 |
| 14 | 1b | 2h | 34 | 90 |
| 15 | 1c | 2h | 35 | 89 |
| 16 | 1a | 2i | 36 | 90 |
| 17 | 1b | 2i | 37 | 92 |
| 18 | 1c | 2i | 38 | 92 |
| 19 | 1d | 2i | 39 | 92 |
The results presented in Table 3 show that irrespective of the type of acetylenes and silanes used, all reactions are stereoselective and lead exclusively to the product of E geometry of the resulting substituted vinylene bonds. All reactions were performed at 35 °C, as temperature rise caused lowering of their selectivities in each case. When the reactions were carried out at temperatures higher than 80 °C, the desired β-E,β-E-product was found to be accompanied by 15% β-Z,β-Z-isomer similarly to mono-hydrosilylation, the reactions were performed in toluene, as a change of solvent to THF or benzene resulted in the formation of a mixture of geometric isomers of desired products. Satisfyingly, in the optimized conditions a range of functional groups were compatible, including t-butyl, methyl, ethyl, phenyl, n-hexyl, n-heptyl, methoxy to afford the 19 products (21–39) in excellent yields (up to 94% isolated yield).
Unsymmetrical bis-hydrosilylation of alkynes with secondary silanes
The successful catalytic hydrosilylation between 2 equiv. of terminal acetylenes and 1 equiv. of secondary silanes prompted us to carry out two successive reactions with separate alkynes to probe the feasibility of a one-pot procedure leading to a unsymmetrical dialkenyl functionalized organosilicon derivatives (Scheme 6).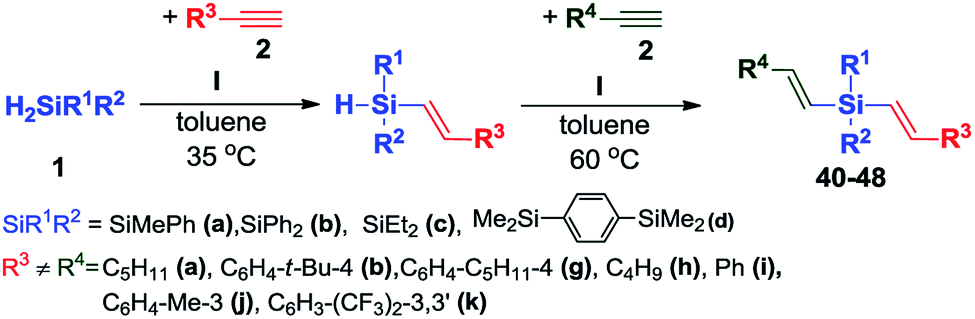 | ||
| Scheme 6 Unsymmetrical bis-hydrosilylation. | ||
Hence, we performed a series of reactions between selected secondary silanes and two different acetylenes containing aryl or alkyl groups. In this procedure, we treated equimolar amounts of silane (1) and alkyne (2) with platinum complex I and the reaction mixture was heated until full conversion of substrates was detected by GC. Then the second type of acetylene (2) was added and the catalytic system was left for the night. The second step was performed at 60 °C, which allowed getting the quantitative conversion of both reagents without decrease in reaction selectivity. The second step of the reaction can also be carried out at 40 °C, but then a longer reaction time is needed. After solvent evaporation, the remaining mixture were purified by column chromatography. Further spectroscopic characterization of the pure products confirmed their identity and purity (see the ESI† for detailed analytical data). In all cases, we observed the complete functionalization of the two hydrogen atoms of silanes and the formation of unsymmetrical products 40–48 with exclusive E-stereochemistry around the newly formed Si–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH double bonds (Table 4):
CH double bonds (Table 4):
| Entry | H2SiR1R2 1 | Alkyne 2 | Alkyne 2 | Product | Isolated yield [%] |
|---|---|---|---|---|---|
| a Reaction conditions: toluene, 35 °C/60 °C, [1]:[2]:[2] = 1:1:1, [I] = 2 × 10−1 mol%, 24 h, argon. |
|||||
| 1 | 1a | 2i | 2a | 40 | 90 |
| 2 | 1a | 2i | 2b | 41 | 90 |
| 3 | 1a | 2k | 2a | 42 | 88 |
| 4 | 1b | 2g | 2a | 43 | 92 |
| 5 | 1a | 2a | 2b | 44 | 90 |
| 6 | 1b | 2k | 2i | 45 | 86 |
| 7 | 1a | 2k | 2i | 46 | 88 |
| 8 | 1a | 2g | 2i | 47 | 90 |
| 9 | 1b | 2g | 2j | 48 | 92 |
It is worth mentioning that all obtained products (46 examples) are air-stable and soluble in many popular solvents (i.e. dichloromethane, THF, toluene and chloroform), which makes them easy to purify via column chromatography. In all cases, the yields of isolated compounds are given. In all experiments performed under specified conditions no competitive reactions were observed. All adducts obtained were characterized by spectroscopic methods. 1H NMR spectra indicated that the only olefinic protons observed are characterized by the coupling constant JH–H of ca. 19 Hz, which suggests that E-isomers were obtained exclusively (see ESI† for detailed analytical data).
Experimental section
General methods and reagents
Unless otherwise indicated, all operations were carried out under dry argon, using standard Schlenk techniques. All syntheses and catalytic tests were performed in an open system. 1H NMR and 13C NMR spectra were recorded in CDCl3 on a Varian 400 operating at 402.6 and 101.2 MHz, respectively. 29Si NMR were recorded on a Brucker Ascend 400 Nanobay operating at 79.50 MHz. GC analyses were carried out on an Agilent 7890B instrument. GC/MS analyses were performed on a Varian Saturn 2100 T equipped with DB-5, 30 m capillary column and ion trap detector. Elementar analyses were performed using Vario EL III apparatus (Elementar). Thin layer chromatography (TLC) was conducted on plates coated with a 250 mm thick silica gel layer and column chromatography was performed on silica gel 60 (70–230 mesh) using hexane/dichloromethane.Reagents were purchased from commercial sources and, unless otherwise indicated, were used without further purification. [Pt(IPr*OMe)(dvtms)] was prepared according to the literature procedure.10 Toluene was dried prior to use over CaH2 and stored under argon.
General procedures for hydrosilylation of terminal alkynes with disubstituted silanes
See Table 2, entry 1–18: A 5 mL glass reactor was charged with toluene (3 mL), disubstituted silane (0.23 mmol, 1.1 equiv.), terminal alkyne (0.21 mmol, 1 equiv.) and internal standard (decane or dodecane, 20 μL). Then platinum catalyst I was added in the amount of 2.1 × 10−7 mol or 1.05 × 10−6 mol for reaction with 1-ethynylnaphthalene and 9-ethynylphenanthrene. The reaction mixture was stirred and heated in an oil bath at 35 °C until full conversion of Si–H was detected. Conversion of the substrates was monitored by gas chromatography. Reaction yields and selectivities were calculated on the basis of the GC/MS and 1H NMR spectra of the reaction mixture.See Table 3, entry 1–19: A 5 mL glass reactor was charged with toluene (3 mL), disubstituted silane (0.23 mmol, 1 equiv.), terminal alkyne (0.46 mmol, 2 equiv.) and internal standard (decane or dodecane, 20 μL). Then 4.6 × 10−7 mol platinum catalyst I was added. The reaction mixture was stirred and heated in an oil bath at 35 °C until full conversion of Si–H was detected. Conversion of the substrates was monitored by gas chromatography. Reaction yields and selectivities were calculated on the basis of the GC/MS and 1H NMR spectra of the reaction mixture.
See Table 4, entry 1–9: A 5 mL glass reactor was charged with toluene (3 mL), silane (0.19 mmol, 1 equiv.), terminal alkyne (0.19 mmol, 1 equiv.) and internal standard (decane or dodecane, 20 μL). Then 3.8 × 10−7 mol platinum catalyst I was added. The reaction mixture was stirred and heated in an oil bath at 35 °C until full conversion of Si–H was detected (GC analysis). Then the second type of terminal alkyne (0.19 mmol, 1 equiv.) was added and the catalytic system was left for the night at 60 °C. Conversion of the substrates was monitored by gas chromatography. Reaction yields and selectivities were calculated on the basis of the GC/MS and 1H NMR spectra of the reaction mixture.
General procedures for the synthesis of hydrosilylation products
Products 3–20 (Table 2): A 25 mL glass reactor equipped with a reflux condenser and connected to gas and vacuum line was charged under argon with toluene (10 mL), disubstituted silane (1.1 mmol) and terminal alkyne (1 mmol). The mixture was heated to 35 °C in an oil bath and platinum catalyst I was added in the amount of 10−6 mol or 5 × 10−6 mol for reaction with 1-ethynylnaphthalene and 9-ethynylphenanthrene. The reaction mixture was stirred and heated in an oil bath at 35 °C until full conversion of Si–H was detected (GC analysis). Then the solvent was evaporated under vacuum and the resulting product was purified by column chromatography (silica gel 60, n-hexane or n-hexane/DCM = 10/1). Evaporation of the solvent gave an analytically pure sample.Products 21–39 (Table 3): A 25 mL glass reactor equipped with a reflux condenser and connected to gas and vacuum line was charged under argon with toluene (10 mL), disubstituted silane (1.1 mmol) and terminal alkyne (2.2 mmol). The mixture was heated to 35 °C in an oil bath and platinum catalyst I was added in the amount of 2.2 × 10−6 mol. The reaction mixture was stirred and heated in an oil bath at 35 °C until full conversion of Si–H was detected (GC analysis). Then the solvent was evaporated under vacuum and the resulting product was purified by column chromatography (silica gel 60, n-hexane or n-hexane/DCM = 10/1). Evaporation of the solvent gave an analytically pure sample.
Products 40–48 (Table 4): A 25 mL glass reactor equipped with a reflux condenser and connected to gas and vacuum line was charged under argon with toluene (10 mL), disubstituted silane (1.1 mmol) and terminal alkyne (1.1 mmol). The mixture was heated to 35 °C in an oil bath and platinum catalyst I was added in the amount of 2.2 × 10−6 mol. The reaction mixture was stirred and heated in an oil bath at 35 °C until full conversion of Si–H was detected (GC analysis). Then the second type of terminal alkyne (1.1 mmol) was added and the catalytic system was left for night at 60 °C. Then the solvent was evaporated under vacuum and the resulting product was purified by column chromatography (silica gel 60, n-hexane or n-hexane/DCM = 10/1). Evaporation of the solvent gave an analytically pure sample.
Conclusions
We found that a platinum(0) complex I bearing bulky N-heterocyclic carbene ligand effects hydrosilylation of alkynes with secondary silanes to produce β-E-alkenylsilanes with high yield and excellent stereoselectivity. A broad range of terminal acetylenes and secondary silanes undergo this reaction to afford a lot of symmetrical and unsymmetrical organosilicon compounds which can be used as liquid crystals,12 solid polymeric electrolytes,13 coupling agents14 or monomers for UV induced polymerization.15 We established the conditions for fully selective synthesis of mono- as well as dialkenylsubstituted β-E-adducts. Additionally, sequential hydrosilylation of two different terminal alkynes with disubstituted silane was also achieved in the presence of the same catalyst. This one-pot method is atom economical and allows construction of valuable organosilicon compounds from relatively simple and readily available starting materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the National Science Centre (Poland) (SONATA Project No. UMO-2016/23/D/ST5/00417).Notes and references
- (a) B. Marciniec, J. Gulinski, W. Urbaniak and Z. W. Kornetka, Comprehensive Handbook on Hydrosilylation, ed. B. Marciniec, Pergamon Press, Oxford, 1992 Search PubMed; (b) B. M. Trost and Z. T. Ball, Synthesis, 2005, 115, 853–887 CrossRef; (c) B. Marciniec, H. Maciejewski, C. Pietraszuk and P. Pawluć, Hydrosilylation: A Comprehensive Review on Recent Advances, ed. B. Marciniec, Springer, 2009 CrossRef; (d) D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef CAS; (e) D. Lim and E. Anderson, Synthesis, 2012, 7, 983–1010 Search PubMed; (f) Y. Nakajima and S. Shimada, RSC Adv., 2015, 5, 20603–20616 RSC; (g) C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946–8975 CrossRef CAS PubMed; (h) M. Zaranek, B. Marciniec and P. Pawluć, Org. Chem. Front., 2016, 3, 1337–1344 RSC; (i) J. Sun and L. Deng, ACS Catal., 2016, 6, 290–300 CrossRef CAS.
- (a) A. Hosomi, M. Endo and H. Sakurai, Chem. Lett., 1975, 5, 941–942 CrossRef; (b) W. P. Weber, Silicon Reagents for Organic Synthesis, Springer, Berlin, 1983 CrossRef; (c) E. W. Colvin, Silicon Reagents in Organic Synthesis, Academic Press, London, 1988 Search PubMed; (d) E. Langkopf and D. Schinzer, Chem. Rev., 1995, 95, 1375–1408 CrossRef CAS; (e) M. A. Brook, Silicon in Organic, Organometallic, and Polymer Chemistry, Wiley, 2000 Search PubMed; (f) T. Bunlaksananusorn, A. L. Rodriguez and P. Knochel, Chem. Commun., 2001, 745–746 RSC; (g) K. Miura and A. Hosomi, Silicon in Organic Synthesis, in Main Group Metals in Organic Synthesis, ed. H. Yamamoto, K. Oshima, Wiley-VCH, 2004, Vol. 2, pp. 409–592 Search PubMed; (h) S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763–2793 CrossRef CAS PubMed; (i) S. Bracegirdle and E. A. Anderson, Chem. Soc. Rev., 2010, 39, 4114–4129 RSC; (j) S. E. Denmark and J. H.-C. Liu, Angew. Chem., 2010, 122, 3040–3049 CrossRef; (k) Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893–4901 RSC; (l) S. E. Denmark and R. F. Sweis, Organosilicon Compounds in Cross-Coupling Reactions in Metal-Catalyzed Cross-Coupling Reactions and More, ed. A. de Meijere, S. Brase, M. Oestreich, Wiley-VCH, 2014, Vol. 2, pp. 475–532 Search PubMed.
- (a) M. D. Greenhalgh, D. J. Frank and S. P. Thomas, Adv. Synth. Catal., 2014, 356, 584–590 CrossRef CAS; (b) M. D. Greenhalgh, A. S. Jones and S. P. Thomas, ChemCatChem, 2015, 7, 190–222 CrossRef CAS; (c) T. Takahashi, F. Bao, G. Gao and M. Ogasawara, Org. Lett., 2003, 5, 3479–3481 CrossRef CAS PubMed.
- Z. Mo, J. Xiao and L. Deng, J. Am. Chem. Soc., 2014, 136, 17414–17417 CrossRef CAS PubMed.
- C. Conifer, C. Gunanathan, T. Rinesch, M. Hölscher and W. Leitner, Eur. J. Inorg. Chem., 2015, 333–339 CrossRef CAS.
- J. Guo and Z. Lu, Angew. Chem., Int. Ed., 2016, 55, 1–5 CrossRef.
- Z. Zuo, J. Yang and Z. Huang, Angew. Chem., Int. Ed., 2016, 128, 10997–11001 CrossRef.
- H. Wen, X. Wan and Z. Huang, Angew. Chem., Int. Ed., 2018, 21, 6319–6323 CrossRef PubMed.
- P. Żak, M. Bołt, M. Kubicki and C. Pietraszuk, Dalton Trans., 2018, 47, 1903–1910 RSC.
- J.-X. Xu, M.-Y. Chen, Z.-J. Zheng, J. Cao, Z. Xu, Y.-M. Cui and L.-W. Xu, ChemCatChem, 2017, 9, 3111–3116 CrossRef CAS.
- P. Żak, M. Bołt, J. Lorkowski, M. Kubicki and C. Pietraszuk, ChemCatChem, 2017, 9, 3627–3631 CrossRef.
- (a) G. Shanker, M. Prehm and C. Tschierske, J. Mater. Chem., 2012, 22, 168–174 RSC; (b) R. Alvarez and G. H. Mehl, Mol. Cryst. Liq. Cryst., 2005, 439, 258–267 Search PubMed.
- X. –G. Sun, J. Hou and J. B. Kerr, Electrochim. Acta, 2005, 50, 1139–1147 CrossRef CAS.
- R. Januszewski, I. Kownacki, H. Maciejewski, B. Marciniec and A. Szymanska, Eur. J. Inorg. Chem., 2017, 4, 851–856 CrossRef.
- (a) R. A. Ortiz, M. Sangermano, R. Bongiovanni, A. E. G. Valdes, L. B. Duarte, I. P. Saucedo and A. Priola, Prog. Org. Coat., 2006, 57, 159–164 CrossRef; (b) R. Acosta Ortiz, M. de Lourdes Guillen Cisneros and G. A. Garcia, Polymer, 2005, 46, 10663–10671 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08640k |
| This journal is © The Royal Society of Chemistry 2018 |