
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and structure–activity relationships of mangostin analogs as cytotoxic agents†
Xiao-Qian Chiab,
Cheng-Ting Zi c,
Hong-Mei Lia,
Liu Yanga,
Yong-Feng Lvab,
Jin-Yu Liab,
Bo Houab,
Fu-Cai Renab,
Jiang-Miao Hu*a and
Jun Zhoua
c,
Hong-Mei Lia,
Liu Yanga,
Yong-Feng Lvab,
Jin-Yu Liab,
Bo Houab,
Fu-Cai Renab,
Jiang-Miao Hu*a and
Jun Zhoua
aState Key Laboratory of Phytochemistry and Plant Resources in West China, Yunnan Key Laboratory of Natural Medicinal Chemistry, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, People' s Republic of China. E-mail: hujiangmiao@mail.kib.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, People's Republic of China
cCollege of Science, Yunnan Agricultural University, Kunming 650201, People's Republic of China
First published on 12th December 2018
Abstract
In order to better understand the structure–activity relationship of mangostin, a series of xanthone derivatives based on α-mangostin were designed and synthesized. All the compounds were evaluated for their cytotoxicity against a panel of five human cancer cell lines (HL-60, SMMC-7721, A-549, MCF-7 and SW480) using MTT assays. Most of them showed cytotoxicity and most of all, compounds 1a and 2h showed the highest cytotoxic potency by HL-60 cancer cell lines with IC50 values of 5.96 μM and 6.90 μM respectively; compound 3e showed the highest cytotoxic potency against SMMC-7221 cancer cell line with IC50 values of 3.98 μM; compounds 2e and 2m showed lower cytotoxicity but higher selectivity than α-mangostin against HL-60 and SMMC-7221 cancer cell lines respectively. Structure–activity relationship analysis indicates that the maintenance of the isopentene group at C-8 is essential for the cytotoxic activity.
1. Introduction
α-Mangostin (1) (Fig. 1) is a kind of bioactive xanthone derivative, which can be isolated from the pericarps of the mangosteen fruit (Garcinia mangostana L.).1,2 This compound has shown increasing promise due to an abundance of therapeutic functions, including anti-tumor,3 anti-oxidant,4 anti-inflammatory,5,6 anti-bacterial,7–10 and inhibition of fatty acid synthase,11,12 neuraminidase,13 α-glycosidase14 and cholinesterase.15 The broad spectrum of mangostin against cancer cell lines has attracted considerable awareness; pharmacological16–19 and medicinal chemistry20 research of α-mangostin has been widely performed. Pharmacological studies have revealed that mangostin possesses potent antitumor activity both in vitro and in vivo. α-Mangostin has been found in the past few decades to exhibit anticancer properties on various cancer models in vivo. α-Mangostin can also be used in combination with other chemotherapeutic agents to increase therapeutic efficacy or reduce side effects.21–23 Despite its promising therapeutic values, there are no clinically approved drugs based on α-mangostin because of its high hydrophobicity, low selectivity24 and low bioavailability.25,26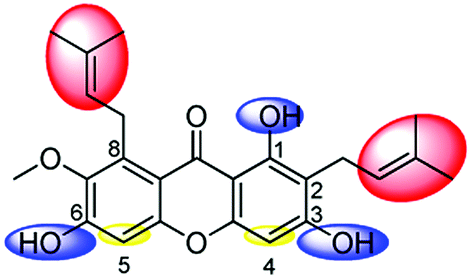 | ||
| Fig. 1 The structure of α-mangostin. | ||
To overcome these drawbacks, related α-mangostin derivatives have been synthesized through various modifications of the phenolic hydroxyl groups (C-1, C-3 and C-6 positions)10,27 and substitution reactions (C-4 and C-5 positions).28,29 Chemical isolation of oxidative cyclized isopentene groups (C-2 and C-8 positions) has been reported,30 and however, the modifications of the isopentene groups are limited. Thus, chemical modifications of the isopentene groups of mangostin were done herein to get more derivatives.
In this manuscript, a series of α-mangostin derivatives were synthesized and then all compounds were evaluated for cytotoxic activities against five human cancer cell lines, including HL-60 (leukemia), SMMC-7721 (hepatoma cells), A-549 (lung cancer), MCF-7 (breast cancer) and SW480 (colon cancer). To study the selectivity of tumor cells and normal cells, their growth inhibitory effect was evaluated against human normal pulmonary epithelial cells (BEAS-2B).
2. Results and discussion
2.1 Chemical synthesis
The synthesized compounds (1a–1l, 2a–2u, 3a–3e) can be divided into three groups according to the variety of functional groups: the isopentene groups (C-2 and C-8 positions), the phenolic hydroxyl groups (C-1, C-3 and C-6 positions) and the vacant sites of benzene ring (C-4 and C-5 positions).Treatment of α-mangostin with BrBn in the presence of K2CO3/acetone gave compound 1a; acetylation of α-mangostin with Ac2O in the addition of DMAP furnished a mixture of compounds 1b and 1c, which were separated by silica gel chromatography; the methylation compounds 1d and 1e were prepared by reaction of α-mangostin with (CH3)2SO4 in the presence of K2CO3, followed again by chromatographic separation; alkylation of α-mangostin to the desired compounds 1f and 1g was accomplished by reactions with BrCH2CHCH2 and K2CO3 in acetone at 65 °C, followed by partial catalytic hydrogenation to afford 78% and 75% yields respectively. For the prenylation compound 1h, α-mangostin was reacted with BrCH2CHC(CH3)2 at 65 °C for 12 h resulting in yields of 15%.31
To speak of, we also designed a series of α-mangostin derivatives by attaching different lengths (1–4) of carbon chains carboxylic ester groups to the free hydroxyl groups (at C-1, C-3 and C-6 positions) according to the references.32 Therefore compounds 1i–1l were synthesized in good yields (25–60%) by the reaction with various bromo-carboxylic acid esters and NaH (Scheme 1). Unfortunately, they showed complete absence of cytotoxicity.
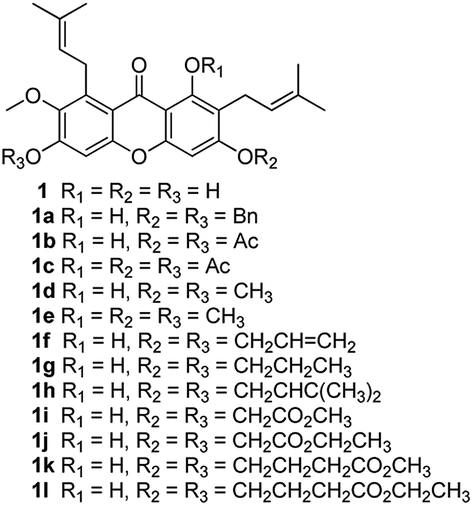 | ||
| Scheme 1 Reagents and conditions: BrBn, K2CO3, acetone, reflux, 80% for 1a; Ac2O, DMAP, DCM, cool temperature to room temperature, 80% for 1b and 1c; (CH3)2SO4, K2CO3, acetone, reflux, 80% for 1d and 1e; BrCH2CHCH2, K2CO3, acetone, reflux, 78% for 1f; Pd/C, H2, MeOH, rt, 75% for 1g; BrCH2CHC(CH3)2, K2CO3, acetone, reflux, 15% for 1h; BrCH2CO2CH3, NaH, acetone, reflux, 60% for 1i; BrCH2CO2CH2CH3, NaH, acetone, reflux, 60% for 1j; BrCH2CH2CH2CO2CH3, NaH, acetone, reflux, 25% for 1k; BrCH2CH2CH2CO2CH2CH3, NaH, acetone, reflux, 25% for 1l. | ||
In general, the alkylation of phenolic hydroxyl groups of mangostin can mainly cause the loss of cytotoxicity, so we focused on the structural modifications of isopentyl groups of mangostin.
 | ||
Scheme 2 Reagents and conditions: OsO4, NMO, acetone-H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), rt, 12–78%. :1), rt, 12–78%. | ||
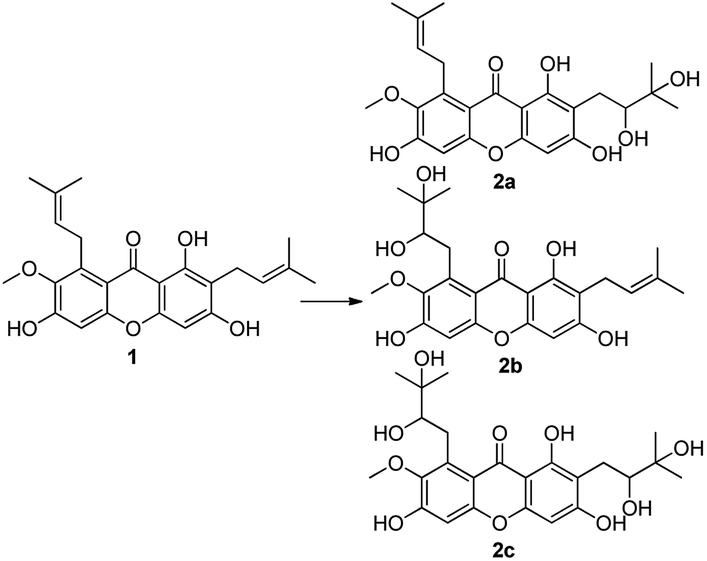
Firstly, α-mangostin derivatives based on the oxidation of isopentene group (at C-2 and C-8 positions) were synthesized. α-Mangostin was treated with OsO4 and NMO in mixed solvent (acetone:H2O = 1:1) to afford compounds 2a–2c, using a similar method as reported in the literature,33 with yields of 10%, 12% and 78% respectively. These products containing functional groups of O-diol act as key intermediates for the further synthesis of mangostin derivatives.
Compounds 2a and 2b have many identical partial structure similarities: one O-diol hydroxyl group and one isopentene group; the structural differences were substitution positions at C-2 or C-8 positions. As shown in Scheme 3, the isopentene group of compounds 2a and 2b was reduced to isopentyl group under H2, Pd/C to afford compounds 2d and 2e in high yields from 78% to 98%. Compounds 2r and 2t were separately prepared from α-mangostin (1) and γ-mangostin (2s) under the same conditions as for preparation of compounds 2d and 2e (Scheme 7). Compound 2d was prepared in 80% from 2a by catalytic hydrogenation, which served as starting material for the synthesis of oxidation product analogous. As shown in Scheme 4, compounds 2a, 2c and 2d, having the vicinal diol groups, were oxidized by NaIO4 in mixed solvent (THF:H2O = 2:1) to afford compounds 2f, 2g and 2h with good yields (40–60%) according to the relative reference.34
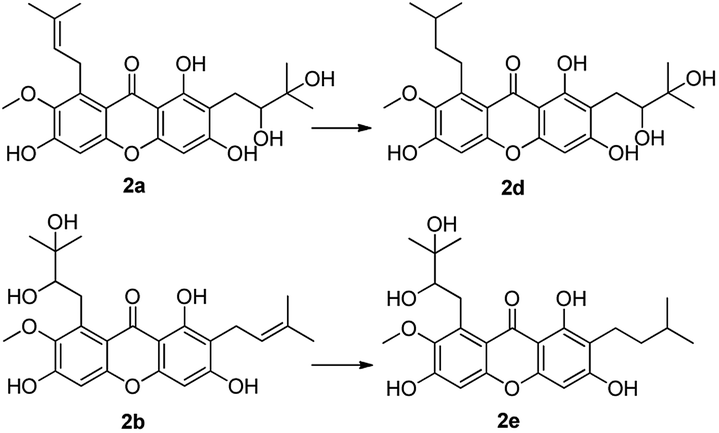 | ||
| Scheme 3 Reagents and conditions: H2, Pd/C, MeOH, rt, 78–98%. | ||
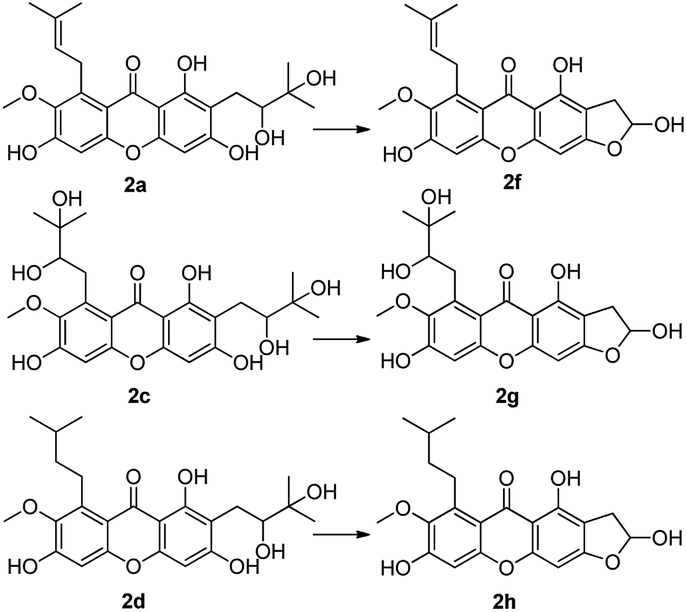 | ||
| Scheme 4 Reagents and conditions: NaIO4, THF–H2O (2:1), rt, 40–60%. | ||
With compounds 2a and 2d in hands, in order to increase their lipotropic properties, different contents of various alkylation products were obtained (Scheme 5). Methylation of compounds 2a and 2d with CH3I–NaH yielded the total methylation products 2i and 2j.
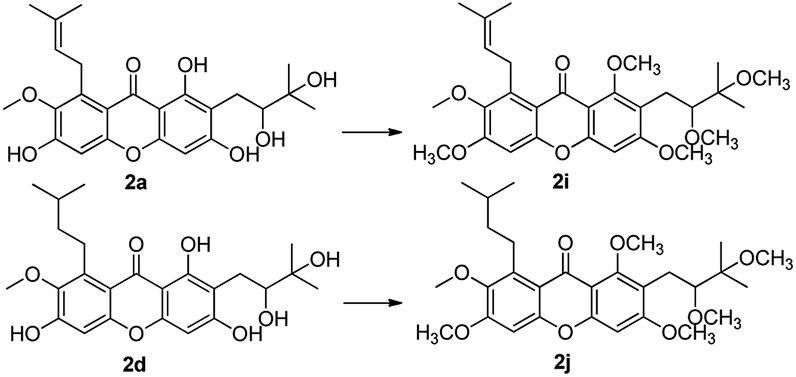 | ||
| Scheme 5 Reagents and conditions: NaH, CH3I, DMF, rt, 60%. | ||
In order to discuss the structure–activity relationship (SAR) more entirely, then we turned to synthesize the cyclization series of xanthone derivatives 2k–2p (Scheme 6) according to the relevant references.35,36 Initially, the cyclization of α-mangostin with DDQ afforded compound 2k with a yield of 40%; α-mangostin was oxidized with m-CPBA in the presence of NaHCO3 to yield compounds 2l and 2m followed again by chromatographic separation in 4% to 49% yields; compound 2n was obtained through an addition reaction in which α-mangostin was reacted with HCOOH with yield of 25%; the treatment of α-mangostin with p-TsOH produced compounds 2o, 2p and 2q, which were separated by silica gel chromatography with a relatively low yield of 12–23%; compound 2u was prepared by the cyclization of 2,4-dihydroxybenzoic acid and phloroglucin under the condition of Eaton's reagents at 60 °C.
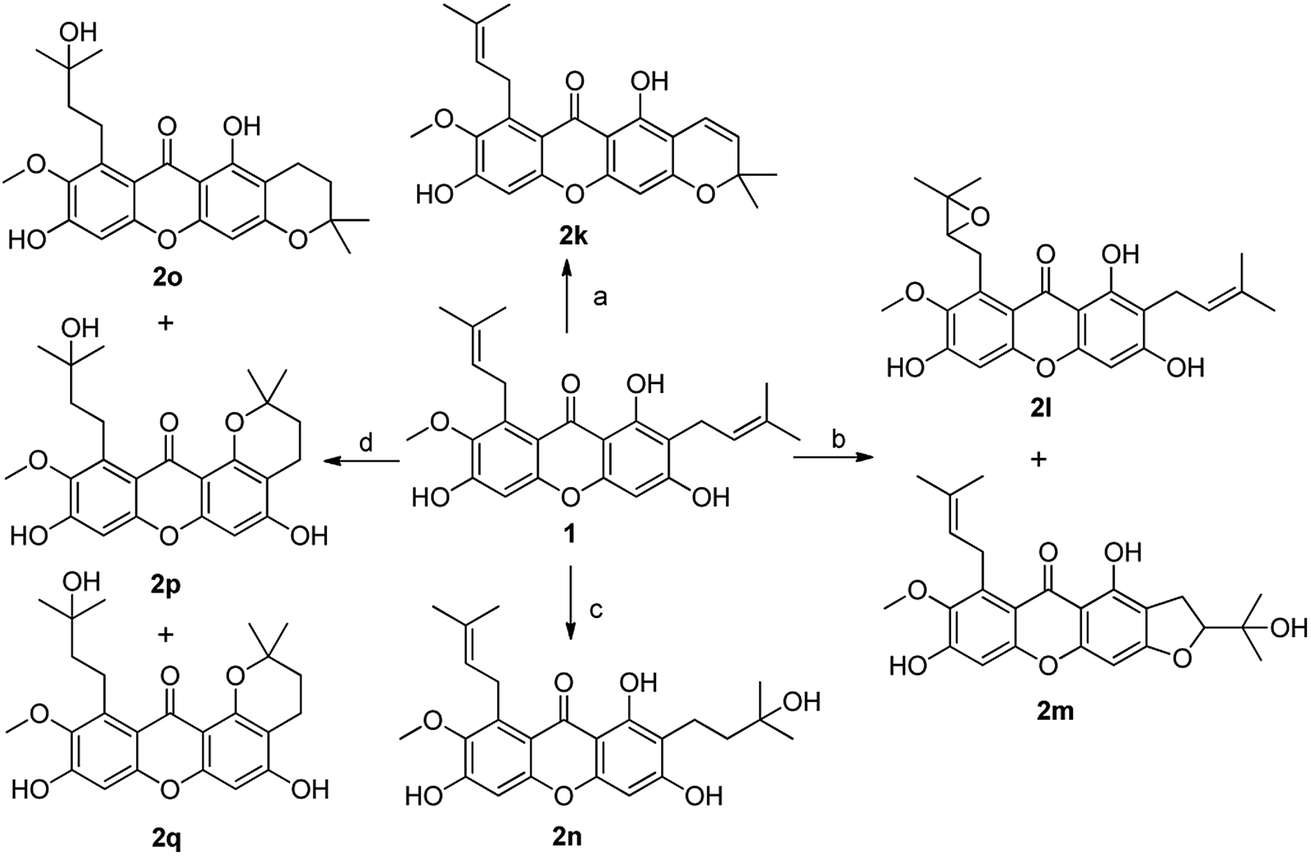 | ||
| Scheme 6 Reagents and conditions: (a) DDQ, toluene, reflux, 41%; (b) m-CPBA, NaHCO3, CH2Cl2, rt, 4–49%; (c) HCOOH, acetone, rt, 25%; (d) p-TsOH, toluene–CH2Cl2 (2:1), rt, 12–23%. | ||
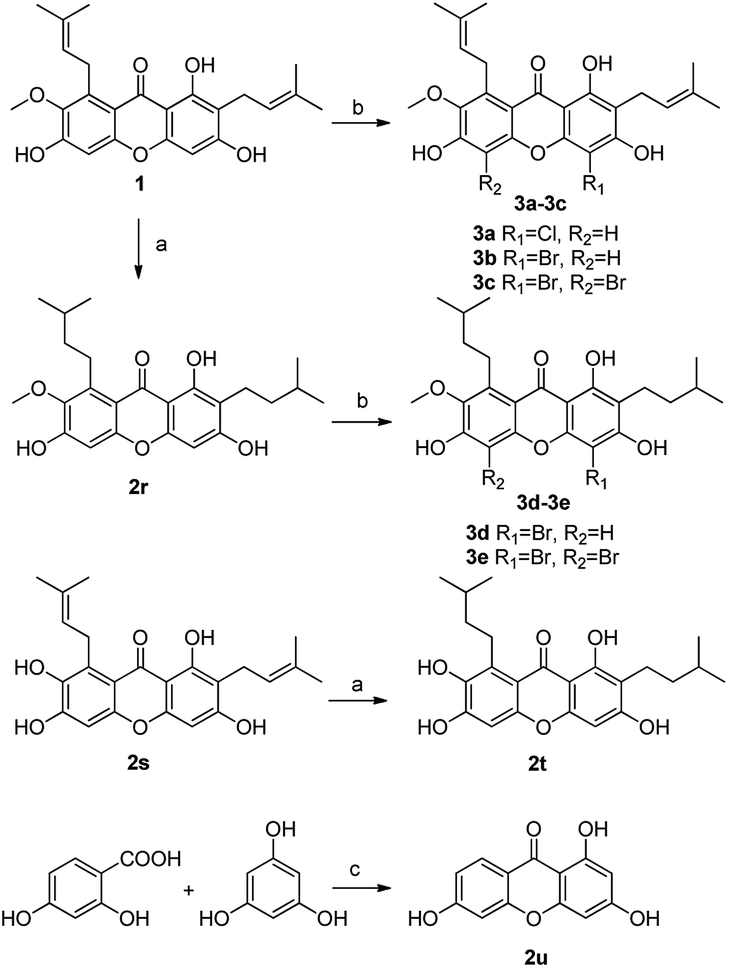 | ||
| Scheme 7 Reagents and conditions: (a) H2, Pd/C, MeOH, rt, 95%; (b) NCS/NBS, DCM or THF, rt, 12–60%; (c) Eaton's reagents, 60 °C, 60%. | ||
In conclusion, we had synthesized three series of mangostin derivatives. All reactions were described in the experimental section. All synthesized target compounds were purified by column chromatography (silica gel, 200–300 mesh, petroleum ether/ethyl acetate, 1:1 → 20:1) and their structures were elucidated by 1H NMR, 13C NMR, electrospray ionization mass spectrometry (ESI-MS) and high-resolution mass spectrometry (HR-ESIMS).
2.2 Evaluation of biological activity
The cytotoxicity of these derivatives was evaluated in vitro against five human cancer cell lines (HL-60, SMMC-7721, A-549, MCF-7 and SW480). Cisplatin (DDP) and Adriamycin (ADM) were taken as control drugs and their IC50 data were present in Tables 1, 2 and 3. The inhibitory of the tested compounds on cell viability was measured by the MTT colorimetric methods. A few of them showed higher potency than the parent compound and most of them displayed moderate cytotoxicity against all five cancer cell lines. In order to test the cytotoxicity of these derivatives with promising anticancer activity on normal cells, their growth inhibitory effect was evaluated against human normal pulmonary epithelial cells (BEAS-2B).| Compounds | IC50 ± SD (μM) | |||||
|---|---|---|---|---|---|---|
| HL-60 | SMMC-7721 | A-549 | MCF-7 | SW480 | BEAS-2B | |
| 1 | 15.04 ± 0.33 | 10.30 ± 0.48 | 13.82 ± 0.61 | 10.81 ± 1.12 | 14.45 ± 0.85 | 15.81 ± 1.02 |
| 1a | 5.96 ± 0.16 | 11.64 ± 0.61 | 10.27 ± 0.42 | 12.95 ± 0.44 | 15.85 ± 0.46 | NT |
| 1b | 11.92 ± 0.48 | 13.56 ± 0.32 | 11.60 ± 0.24 | 16.65 ± 1.32 | 16.17 ± 0.13 | NT |
| DDP | 3.19 ± 0.18 | 18.03 ± 0.49 | 13.75 ± 0.74 | 28.42 ± 3.71 | 14.77 ± 2.15 | >40 |
| ADM | 0.14 ± 0.00 | 0.90 ± 0.03 | 0.30 ± 0.01 | 0.90 ± 0.02 | 0.11 ± 0.01 | >40 |
| Compounds | IC50 ± SD (μM) | |||||
|---|---|---|---|---|---|---|
| HL-60 | SMMC-7721 | A-549 | MCF-7 | SW480 | BEAS-2B | |
| 1 | 15.04 ± 0.33 | 10.30 ± 0.48 | 13.82 ± 0.61 | 10.81 ± 1.12 | 14.45 ± 0.85 | 15.81 ± 1.02 |
| 2a | 18.22 ± 0.48 | 23.42 ± 1.29 | 19.82 ± 1.42 | 23.65 ± 0.64 | >40 | 18.31 ± 0.51 |
| 2b | >40 | >40 | >40 | >40 | >40 | NT |
| 2c | >40 | >40 | >40 | >40 | >40 | NT |
| 2d | 21.59 ± 2.41 | 26.87 ± 3.75 | 35.96 ± 0.99 | >40 | >40 | >40 |
| 2e | 18.65 ± 0.23 | >40 | >40 | >40 | >40 | >40 |
| 2f | 14.96 ± 0.80 | 11.93 ± 0.28 | 18.03 ± 1.49 | 19.75 ± 0.26 | 18.24 ± 0.79 | 27.20 ± 2.42 |
| 2g | >40 | >40 | >40 | >40 | >40 | NT |
| 2h | 6.90 ± 0.55 | 6.92 ± 0.55 | 11.77 ± 0.13 | 17.97 ± 0.23 | 15.86 ± 0.2 | 17.56 ± 0.60 |
| 2i | >40 | >40 | >40 | >40 | >40 | NT |
| 2j | >40 | >40 | >40 | >40 | >40 | NT |
| 2k | 13.72 ± 0.50 | 11.52 ± 0.17 | 11.47 ± 0.33 | 16.80 ± 1.04 | 17.35 ± 1.15 | 18.60 ± 0.70 |
| 2l | >40 | >40 | >40 | >40 | >40 | NT |
| 2m | >40 | 53.75 ± 0.26 | >40 | >40 | >40 | NT |
| 2n | 14.40 ± 0.05 | 26.31 ± 1.04 | 16.96 ± 0.77 | 24.62 ± 0.47 | 22.69 ± 2.35 | 29.34 ± 1.02 |
| 2o | >40 | >40 | >40 | >40 | >40 | NT |
| 2p | >40 | >40 | >40 | >40 | >40 | NT |
| 2q | NT | NT | NT | NT | NT | NT |
| 2r | 11.13 ± 0.15 | 15.68 ± 0.34 | 14.93 ± 0.15 | 8.29 ± 0.59 | 14.46 ± 0.15 | 14.35 ± 0.41 |
| 2s | 7.39 ± 0.33 | 6.57 ± 0.14 | 10.07 ± 0.59 | 5.33 ± 0.43 | 8.40 ± 0.67 | 7.43 ± 0.65 |
| 2t | 13.72 ± 0.06 | 6.51 ± 0.49 | 15.56 ± 0.17 | 12.32 ± 0.12 | 11.59 ± 0.52 | 6.48 ± 0.08 |
| 2u | >40 | >40 | >40 | >40 | >40 | NT |
| DDP | 3.19 ± 0.18 | 18.03 ± 0.49 | 13.75 ± 0.74 | 28.42 ± 3.71 | 14.77 ± 2.15 | >40 |
| ADM | 0.14 ± 0.00 | 0.90 ± 0.03 | 0.30 ± 0.01 | 0.90 ± 0.02 | 0.11 ± 0.01 | >40 |
| Compounds | IC50 ± SD (μM) | |||||
|---|---|---|---|---|---|---|
| HL-60 | SMMC-7721 | A-549 | MCF-7 | SW480 | BEAS-2B | |
| 1 | 15.04 ± 0.33 | 10.30 ± 0.48 | 13.82 ± 0.61 | 10.81 ± 1.12 | 14.45 ± 0.85 | 15.81 ± 1.02 |
| 3a | >40 | 34.67 ± 2.22 | >40 | >40 | >40 | >40 |
| 3b | >40 | 24.11 ± 0.16 | >40 | 22.18 ± 0.20 | >40 | >40 |
| 3c | 16.91 ± 0.19 | 8.07 ± 0.76 | 14.30 ± 0.85 | 16.01 ± 0.66 | 36.63 ± 0.74 | 21.83 ± 0.14 |
| 3d | 14.55 ± 0.40 | 10.76 ± 1.02 | 16.18 ± 0.13 | 21.92 ± 0.50 | 17.30 ± 0.49 | 16.05 ± 0.47 |
| 3e | 14.87 ± 0.33 | 3.98 ± 0.63 | 12.02 ± 0.19 | 19.66 ± 0.62 | 21.29 ± 0.44 | 20.74 ± 0.18 |
| DDP | 3.19 ± 0.18 | 18.03 ± 0.49 | 13.75 ± 0.74 | 28.42 ± 3.71 | 14.77 ± 2.15 | >40 |
| ADM | 0.14 ± 0.00 | 0.90 ± 0.03 | 0.30 ± 0.01 | 0.90 ± 0.02 | 0.11 ± 0.01 | >40 |
Overall, these synthesized compounds show a broad range of growth inhibitory effect against all five cancer cell lines tested. In general, the HL-60 cell line was most sensitive to these compounds. Noteworthy, compound 2h possesses remarkable anti-proliferation activity against all the tested cancer cell lines. Compound 2e, which solely possesses notable anti-proliferation activity against HL-60 cancer cells (IC50 = 18.65 ± 0.23 μM) is non-cytotoxic to BEAS-2B cell line. Moreover, several compounds (2a, 2f, 2k, 2n and 2r) with pretty anti-proliferation activity against all the tested cancer cell lines are observed to be less toxic to BEAS-2B cells compared with α-mangostin. The results suggest that these compounds are more sensitive to certain tested cancer cells than normal cells in vitro and hence possess good selectivity.
These data have allowed us to carry out a structure and activity relationship (SAR) study on the influence of the modifications of the isopentene group and halogen atoms in the cholinesterase inhibitory activities. The main conclusions can be summarized as follows:
(1) With respect to the SAR, the effects of the substitution reactions at phenolic hydroxyl groups were examined. Di-substitution at both C-3 and C-6 hydroxyl groups (1a–1l) causes totally decrease in the cytotoxicity of mangostin against the five tested cancer cell lines; while the acetylation form of mangostin (1b and 1c) can remain the cytotoxic activity, compound 1a possessed the most potent cytotoxicity against HL-60 cancer cell line with IC50 value of 5.96 ± 0.16 μM. In summary, the numbers of phenolic hydroxyl groups have certain effects on maintaining cytotoxicity.
(2) The oxidation of the isopentene group at C-8 causes drastically decreases in the cytotoxicity of mangostin against all the tested cancer cell lines. Compounds 2b, 2c, 2g, 2l and 2m displayed weak activity, all having IC50 > 40 μM, indicating that the isopentene group at C-8 was necessary for the cytotoxicity and the hydroxyl group at C-8 can cause totally loss of the cytotoxicity. However, one interesting exception is observed: compound 2e possesses notable anti-proliferation activity against HL-60 cancer cells with IC50 value of 18.65 ± 0.23 μM.
(3) Several structure features and their effects need to be pointed out. Oxidation of the isopentene group at C-2 (compounds 2a, 2d, 2f, 2k, 2m and 2n) generally resulted in slightly decreased activity or comparative activity with one exception. Compound 2h exhibited greater activity against HL-60 and SMMC-7221 cell lines with IC50 values of 6.90 ± 0.55 μM and 6.92 ± 0.55 μM respectively. These data indicate the number and position of hydroxyl group at C-2 have limited potency on the cytotoxicity and selectivity.
(4) By comparing compounds 1 and 2r, 2a and 2d, 2b and 2e, 2f and 2h, 2s and 2t, it is obvious to find that the reduction of the isopentene group to isopentyl group has no significant effect on the decrease or increase of cytotoxicity.
(5) The totally methylation of compounds 2a and 2d cause the disappearance of cytotoxicity of compound 2i and 2j. However, the cytotoxicity of compounds 2c and 2g with 5–7 hydroxyl groups also completely disappeared. Compound 2s (γ-mangostin) is more active than compound 1 (α-mangostin) indicating the importance of the presence of hydroxyl group at the C-7 position, whereby substituting it with methoxy group reduced the cytotoxicity. To summarize, a certain number of hydroxyl groups contribute to the maintenance of cytotoxic activity. The number and location of hydroxyl functional groups have different effects on cytotoxicity.
(6) A closer look at the data reveals that the effect of halogenation on the selective potency of these compounds is quite subtle. Some of the halogenated products showed better cytotoxicity, for example, compound 3e is up to three times more cytotoxic than the parent compound with IC50 value of 3.98 ± 0.63 μM for SMMC-7721 cell lines.
3. Experimental
3.1 Materials and methods
All reagents were purchased from Sigma-Aldrich or Aladdin or Innochem Co. Ltd. and were of commercial quality. They were used as received without further purification. Solvents were dried by standard methods prior to use. The other reagents were of analytical grade. Air and moisture sensitive reactions were performed under nitrogen atmosphere. All synthesized target compounds were purified by column chromatography (silica gel, petroleum ether/ethyl acetate, 1:1 ∼ 20:1) and their structures were elucidated by 1H NMR, 13C NMR, electrospray ionization mass spectrometry (ESI-MS) and high-resolution mass spectrometry (HR-ESIMS). Mass spectra were performed on an API QSTAR time-of-flight spectrometer (MDS Sciqaszex, Concord, Ontario, Canada) and LCMS-IT-TOF (Shimadzu, Kyoto, Japan) spectrometer. NMR spectra were recorded on Bruker AM-400 and DRX-500 instruments with TMS as the internal standard (Bruker, Bremerhaven, Germany). The chemical shifts were given in δ (ppm) with reference to the solvent signal. 1H NMR data were reported in the order of chemical shift, multiplicity (s, singlet; d, doublet; t, triplet; m, multiple resonances), number of protons, and coupling constant (J) in hertz (Hz). Column chromatography was performed on silica gel (200–300 and 300–400 mesh, Qingdao Marine Chemical Inc., Qingdao, China) with the indicated solvents. The fractions were monitored by TLC and the spots were visualized by UV light and sprayed with 10% H2SO4 in EtOH, followed by heating.
3.2 Synthetic procedures and crystallography
3,6-Di-O-benzyl-α-mangostin (1a). Yield 80%, 1H NMR (CDCl3, 500 MHz) δ 13.51 (s, 1H, 1-OH), 7.48–7.34 (m, 10H, Ar-H), 6.75 (s, 1H, H-5), 6.34 (s, 1H, H-4), 5.28 (br t, 2H, J = 7.0 Hz, H-12, H-17), 5.18, 5.15 (each s, each 2H, H-21, H-28), 4.15 (d, 2H, J = 6.5 Hz, H-16), 3.83 (s, 3H, 7-OCH3), 3.41 (d, 2H, J = 7.0 Hz, H-11), 1.86–1.59 (s, each 3H, H-14, H-15, H-19, H-20); 13C NMR (CDCl3, 125 MHz) δ 182.0 (C-9), 162.4 (C-3), 159.9 (C-1), 157.0 (C-6), 155.2 (C-10a), 155.0 (C-4a), 144.2 (C-7), 137.4 (C-8), 136.3, 135.7 (C-22, C-29), 131.8, 131.6 (C-13, C-18), 128.8, 128.6, 128.4, 128.1, 127.4, 127.2 (C-23, C-24, C-25, C-26, C-27, C-30, C-31, C-32, C-33, C-34), 123.3, 122.4 (C-12, C-17), 112.3 (C-8a), 111.8 (C-2), 104.1 (C-9a), 99.4 (C-5), 89.8 (C-4), 70.7, 70.2 (C-21, C-28), 61.0 (OCH3), 26.2 (C-16), 26.0, 25.9 (C-15, C-20), 21.6 (C-11), 18.2, 17.8 (C-14, C-19); positive ESIMS m/z 591 [M + H]+.
3,6-Di-O-acetyl-α-mangostin (1b). Yield 82%, 1H NMR (CDCl3, 500 MHz) δ 13.42 (s, 1H, 1-OH), 7.13 (s, 1H, H-5), 6.64 (s, 1H, H-4), 5.17 (br t, 2H, J = 7.0 Hz, H-12, H-17), 4.13 (d, 2H, J = 6.0 Hz, H-16), 3.77 (s, 3H, 7-OCH3), 3.32 (d, 2H, J = 7.0 Hz, H-11), 2.39, 2.34 (each s, each 3H, COCH3), 1.83, 1.78 (each s, each 3H, H-15, H-20), 1.68 (s, 6H, H-14, H-19); 13C NMR (CDCl3, 125 MHz) δ 182.9 (C-9), 168.5 (C-23), 168.1 (C-21), 161.0 (C-1), 154.9 (C-3), 154.1 (C-4a), 153.7 (C-10a), 149.4 (C-7), 146.7 (C-6), 139.2 (C-8), 132.4, 132.3 (C-13, C-18), 122.6, 121.3 (C-12, C-17), 116.9 (C-8a), 116.2 (C-2), 110.6 (C-5), 107.1 (C-9a), 100.3 (C-4), 61.7 (OCH3), 26.5 (C-16), 25.9, 25.7 (C-15, C-20), 22.3 (C-11), 21.0 (C-22), 20.9 (C-24), 18.2, 17.9 (C-14, C-19); positive ESIMS m/z 517 [M + Na]+.
1,3,6-Tri-O-acetyl-α-mangostin (1c). Yield 80%, 1H NMR (CDCl3, 500 MHz) δ 7.13 (s, 1H, H-5), 7.09 (s, 1H, H-4), 5.17 (br t, H, J = 7.0 Hz, H-12), 5.03 (br t, H, J = 7.0 Hz, H-17), 4.06 (d, 2H, H-16), 3.75 (s, 3H, 7-OCH3), 3.27 (d, 2H, H-11), 2.46, 2.37, 2.33 (each s, each 3H, COCH3), 1.82, 1.75, 1.67, 1.42 (each s, each 3H, H-14, H-15, H-19, H-20); 13C NMR (CDCl3, 125 MHz) δ 176.1 (C-9), 169.3 (C-21), 168.2 (C-22), 168.1 (C-23), 154.5 (C-3), 153.2 (C-1), 153.1 (C-10a), 148.8 (C-4a), 148.5 (C-7), 146.7 (C-6), 139.0 (C-8), 132.5, 131.8 (C-13, C-18), 123.6, 122.9 (C-12, C-17), 120.9 (C-8a), 118.9 (C-2), 113.6 (C-9a), 110.3 (C-5), 109.0 (C-4), 61.6 (OCH3), 26.2 (C-16), 25.8, 25.6 (C-15, C-20), 23.5 (C-11), 21.2, 21.0, 20.9 (C-21, C-23, C-25), 18.2, 17.9 (C-14, C-19); positive ESIMS m/z 537 [M + H]+.
3,6-Di-O-methyl-α-mangostin (1d). Yield 80%, 1H NMR (CDCl3, 500 MHz) δ 6.73 (s, 1H, H-5), 6.32 (s, 1H, H-4), 5.24 (br t, 2H, J = 7.3 Hz, H-12, H-17), 4.13 (d, 2H, J = 6.6 Hz, H-16), 3.95, 3.90 (each s, each 3H, 3-OCH3, 6-OCH3), 3.79 (s, 3H, 7-OCH3), 3.35 (d, 2H, J = 7.1 Hz, H-11), 1.82, 1.80 (each s, each 3H, H-14, H-19), 1.68 (s, 6H, H-15, H-20); 13C NMR (CDCl3, 125 MHz) δ 182.0 (C-9), 163.4 (C-3), 159.8 (C-1), 158.0 (C-10a), 155.4 (C-4a), 155.2 (C-7), 144.0 (C-6), 137.3 (C-8), 131.8, 131.7 (C-13, C-18), 123.2, 122.3 (C-12, C-17), 112.1 (C-8a), 111.5 (C-2), 104.0 (C-9a), 98.2 (C-5), 88.7 (C-4), 61.0 (7-OCH3), 56.0, 55.8 (3-OCH3, 6-OCH3), 26.2 (C-16), 25.9 (C-15, C-20), 21.4 (C-11), 18.2, 17.8 (C-14, C-19); positive ESIMS m/z 439 [M + H]+.
1,3,6-Tri-O-methyl-α-mangostin (1e). Yield 80%, 1H NMR (CDCl3, 500 MHz) δ 6.74 (s, 1H, H-5), 6.61 (s, 1H, H-4), 5.35 (br t, H, J = 6.5 Hz, H-12), 5.21 (br t, H, J = 7.0 Hz, H-17), 4.17 (d, 2H, J = 6.7 Hz, H-16), 3.98, 3.94, 3.89, 3.82 (each s, each 3H, 1-OCH3, 3-OCH3, 6-OCH3, 7-OCH3), 3.42 (d, 2H, J = 7.0 Hz, H-11), 1.82, 1.80 (each s, each 3H, H-15, H-20), 1.68 (s, 6H, H-14, H-19); 13C NMR (CDCl3, 125 MHz) δ 176.3 (C-9), 162.1 (C-3), 158.6 (C-1), 157.0 (C-10a), 156.6 (C-4a), 154.3 (C-6), 143.9 (C-7), 137.4 (C-8), 131.5, 131.3 (C-13, C-18), 123.8, 122.8 (C-12, C-17), 120.5 (C-2), 114.7 (C-9a), 110.9 (C-8a), 97.8 (C-5), 94.2 (C-4), 62.0 (1-OCH3), 60.9 (7-OCH3), 55.9, 55.9 (3-OCH3, 6-OCH3), 29.7 (C-16), 26.0, 25.9 (C-15, C-20), 22.4 (C-11), 18.2, 17.9 (C-14, C-19); positive ESIMS m/z 453 [M + H]+.
3,6-Bis(allyloxy)-1-hydroxy-7-methoxy-2,8-bis(3-methylbut-2-en-1-yl)-9H-xanthen-9-one (1f). Yield 78%, 1H NMR (CDCl3, 500 MHz) δ 13.49 (s, 1H, 1-OH), 6.69 (s, 1H, H-5), 6.27 (s, 1H, H-4), 6.08 (m, 2H, –CH
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) , H-22, H-25), 5.47, 5.34 (m, each 2H, CH2, H-23, H-26), 5.25 (br t, 2H, J = 6.0 Hz, H-12, H-17), 4.66, 4.60 (each d, each 2H, J = 5.0 Hz, –OCH2, H-21, H-24), 4.13 (d, 2H, J = 7.0 Hz, H-16), 3.81 (s, 3H, 7-OCH3), 3.38 (d, 2H, J = 7.0 Hz, H-11), 1.85, 1.79 (each s, each 3H, H-15, H-20), 1.68 (s, 6H, H-14, H-19); 13C NMR (CDCl3, 125 MHz) δ 182.0 (C-9), 162.2 (C-3), 159.9 (C-1), 156.9 (C-4a), 155.2 (C-10a), 155.0 (C-6), 144.1 (C-7), 137.4 (C-8), 132.5, 132.0 (C-22, C-25), 131.8, 131.6 (C-13, C-18), 123.3, 122.3 (C-12, C-17), 118.5, 111.7 (C-23, C-26), 112.1 (C-8a), 111.7 (C-2), 104.0 (C-9a), 99.2 (C-5), 89.6 (C-4), 69.4, 69.1 (C-21, C-24), 60.9 (7-OCH3), 26.2 (C-16), 26.0, 25.9 (C-15, C-20), 21.5 (C-11), 18.2, 17.9 (C-14, C-19); positive ESIMS m/z 491 [M + H]+.
, H-22, H-25), 5.47, 5.34 (m, each 2H, CH2, H-23, H-26), 5.25 (br t, 2H, J = 6.0 Hz, H-12, H-17), 4.66, 4.60 (each d, each 2H, J = 5.0 Hz, –OCH2, H-21, H-24), 4.13 (d, 2H, J = 7.0 Hz, H-16), 3.81 (s, 3H, 7-OCH3), 3.38 (d, 2H, J = 7.0 Hz, H-11), 1.85, 1.79 (each s, each 3H, H-15, H-20), 1.68 (s, 6H, H-14, H-19); 13C NMR (CDCl3, 125 MHz) δ 182.0 (C-9), 162.2 (C-3), 159.9 (C-1), 156.9 (C-4a), 155.2 (C-10a), 155.0 (C-6), 144.1 (C-7), 137.4 (C-8), 132.5, 132.0 (C-22, C-25), 131.8, 131.6 (C-13, C-18), 123.3, 122.3 (C-12, C-17), 118.5, 111.7 (C-23, C-26), 112.1 (C-8a), 111.7 (C-2), 104.0 (C-9a), 99.2 (C-5), 89.6 (C-4), 69.4, 69.1 (C-21, C-24), 60.9 (7-OCH3), 26.2 (C-16), 26.0, 25.9 (C-15, C-20), 21.5 (C-11), 18.2, 17.9 (C-14, C-19); positive ESIMS m/z 491 [M + H]+.
1-Hydroxy-7-methoxy-2,8-bis(3-methylbut-2-en-1-yl)-3,6-dipropoxy-9H-xanthen-9-one (1g). Yield 75%, 1H NMR (CDCl3, 500 MHz) δ 13.50 (s, 1H, 1-OH), 6.69 (s, 1H, H-5), 6.27 (s, 1H, H-4), 5.25 (br t, 2H, J = 7.0 Hz, H-12, H-17), 4.13 (d, 2H, J = 6.5 Hz, H-16), 4.03, 3.98 (each t, each 2H, J = 6.5 Hz, H-21, H-24), 3.81 (s, 3H, 7-OCH3), 3.36 (d, 2H, J = 7.0 Hz, H-11), 1.93–1.06 (m, 10H, –CH2, –CH3); 13C NMR (CDCl3, 125 MHz) δ 182.0 (C-9), 162.8 (C-3), 159.8 (C-1), 157.5 (C-4a), 155.3 (C-10a), 155.1 (C-6), 144.0 (C-7), 137.1 (C-8), 131.7, 131.3 (C-13, C-18), 123.3, 122.5 (C-12, C-17), 111.8 (C-2), 111.4 (C-8a), 103.8 (C-9a), 98.7 (C-5), 89.2 (C-4), 70.3, 70.0 (C-21, C-24), 60.8 (7-OCH3), 26.2 (C-16), 26.0, 25.9 (C-15, C-20), 22.5, 22.3 (C-22, C-25), 21.4 (C-11), 18.2, 17.8 (C-14, C-19), 10.7, 10.6 (C-23, C-26); positive ESIMS m/z 517 [M + Na]+.
3,6-Di-O-3,3-dimethylallyl-α-mangostin (1h). Yield 15%, 1H NMR (DMSO-d6, 500 MHz) δ 13.50 (s, 1H, 1-OH), 7.03 (d, 1H, J = 4.2 Hz, H-5), 6.55 (s, 1H, J = 4.3 Hz, H-4), 5.49, 5.46 (each t, each 1H, J = 6.6 Hz, H-22, H-27), 5.13 (br t, 2H, H-12, H-17), 4.71, 4.64 (each d, each 2H, J = 6.5 Hz, H-21, H-26), 3.99 (d, 2H, J = 6.0 Hz, H-16), 3.68 (s, 3H, 7-OCH3), 3.19 (d, 2H, J = 7.0 Hz, H-11), 1.77, 1.75, 1.75, 1.72, 1.69, 1.60, 1.59 (s, each 3H, H-14, H-15, H-19, H-20, H-24, H-25, H-29, H-30); 13C NMR (DMSO-d6, 125 MHz) δ 181.5 (C-9), 162.5 (C-3), 158.8 (C-1), 157.3 (C-4a), 154.8 (C-10a), 154.6 (C-6), 143.9 (C-7), 138.7, 138.2 (C-23, C-28), 135.7 (C-8), 130.7, 130.5 (C-13, C-18), 123.5 (C-12, C-17), 122.1 (C-22, C-27), 119.1 (C-8a), 118.8 (C-2), 102.8 (C-9a), 99.8 (C-5), 90.3 (C-4), 65.7, 65.4 (C-21, C-26), 60.3 (7-OCH3), 25.6 (C-16), 25.5 (C-15, C-20, C-25, C-30), 21.0 (C-11), 18.2, 18.1, 18.0, 17.6 (C-14, C-19, C-24, C-29); positive ESIMS m/z 547 [M + H]+.
Dimethyl-2,2′-((1-hydroxy-7-methoxy-2,8-bis(3-methylbut-2-en-1-yl)-9-oxo-9H-xanthene-3,6-diyl)bis(oxy))diacetate (1i). Yield 60%, 1H NMR (DMSO-d6, 500 MHz) δ 13.47 (s, 1H, 1-OH), 7.03 (s, 1H, H-5), 6.50 (s, 1H, H-4), 5.17 (m, 2H, H-12, H-17), 5.07, 4.98 (each s, each 2H, H-21, H-24), 4.01 (d, 2H, J = 6.3 Hz, H-16), 3.76, 3.73, 3.72 (each s, each 3H, 23-OCH3, 26-OCH3, 7-OCH3), 3.27 (d, 2H, J = 7.1 Hz, H-11), 1.77, 1.72, 1.61, 1.60 (each s, each 3H, H-14, H-15, H-19, H-20); 13C NMR (DMSO-d6, 125 MHz) δ 181.6 (C-9), 168.6, 168.5 (C-22, C-25), 161.5 (C-3), 159.0 (C-1), 156.4 (C-4a), 154.5 (C-10a), 154.4 (C-6), 143.7 (C-7), 136.2 (C-8), 130.9, 130.7 (C-13, C-18), 123.3, 121.9 (C-12, C-17), 111.4 (C-8a), 111.0 (C-2), 103.3 (C-9a), 99.9 (C-5), 90.2 (C-4), 65.2, 65.1 (C-21, C-24), 60.4 (7-OCH3), 52.1, 52.0 (C-23, C-26), 25.6 (C-16), 25.6, 25.5 (C-15, C-20), 21.0 (C-11), 18.0, 17.7 (C-14, C-19); negative ESIMS m/z 553 [M − H]−.
Diethyl-2,2′-((1-hydroxy-7-methoxy-2,8-bis(3-methylbut-2-en-1-yl)-9-oxo-9H-xanthene-3,6-diyl)bis(oxy))diacetate (1j). Yield 60%, 1H NMR (DMSO-d6, 500 MHz) δ 13.49 (s, 1H, 1-OH), 7.04 (s, 1H, H-5), 6.51 (s, 1H, H-4), 5.20, 5.15 (each t, each 1H, J = 7.0 Hz, H-12, H-17), 5.06, 4.97 (each s, each 2H, H-21, H-25), 4.19 (m, 4H, H-23, H-27), 4.02 (d, 2H, J = 7.0 Hz, H-16), 3.76 (s, 3H, 7-OCH3), 3.28 (d, 2H, J = 7.0 Hz, H-11), 1.77, 1.72, 1.61, 1.60 (each s, each 3H, H-14, H-15, H-19, H-20), 1.22 (m, 6H, H-24, H-28); 13C NMR (DMSO-d6, 125 MHz) δ 181.6 (C-9), 168.1, 168.0 (C-22, C-26), 161.5 (C-3), 159.0 (C-1), 156.5 (C-4a), 154.6 (C-10a), 154.5 (C-6), 143.8 (C-7), 136.2 (C-8), 130.9, 130.8 (C-13, C-18), 123.3, 121.9 (C-12, C-17), 111.4 (C-8a), 111.0 (C-2), 103.3 (C-9a), 99.9 (C-5), 90.3 (C-4), 65.3, 65.2 (C-21, C-25), 61.0, 60.9 (C-23, C-27), 60.4 (7-OCH3), 25.6 (C-16), 25.6, 25.5 (C-15, C-20), 21.0 (C-11), 18.0, 17.7 (C-14, C-19), 14.1 (C-24, C-28); positive ESIMS m/z 605 [M + Na]+.
Dimethyl-4,4′-((1-hydroxy-7-methoxy-2,8-bis(3-methylbut-2-en-1-yl)-9-oxo-9H-xanthene-3,6-diyl)bis(oxy))dibutanoate (1k). Yield 25%, 1H NMR (DMSO-d6, 500 MHz) δ 13.44 (s, 1H, 1-OH), 6.87 (s, 1H, H-5), 6.38 (s, 1H, H-4), 5.10 (br t, 2H, J = 7.0 Hz, H-12, H-17), 4.10, 4.05 (each t, each 2H, H-21, H-26), 3.93 (d, 2H, J = 6.5 Hz, H-16), 3.67 (s, 3H, 7-OCH3), 3.61, 3.60 (each s, each 3H, H-25, H-30), 3.14 (d, 2H, J = 7.0 Hz, H-11), 2.50 (m, 4H, H-23, H-28), 2.02 (m, 4H, H-22, H-27), 1.74, 1.68, 1.60, 1.59 (each s, each 3H, H-14, H-15, H-19, H-20); 13C NMR (DMSO-d6, 125 MHz) δ 181.3 (C-9), 172.9, 172.9 (C-24, C-29), 162.3 (C-3), 158.8 (C-1), 157.2 (C-4a), 154.7 (C-10a), 154.6 (C-6), 143.6 (C-7), 135.7 (C-8), 130.5 (C-13, C-18), 123.4, 122.3 (C-12, C-17), 110.7 (C-8a), 110.4 (C-2), 102.8 (C-9a), 99.3 (C-5), 89.7 (C-4), 67.8, 67.3 (C-21, C-26), 60.3 (7-OCH3), 51.4 (C-25, C-30), 29.9, 29.8 (C-23, C-28), 25.5 (C-16), 25.4, 24.0, 23.9 (C-15, C-20, C-22, C-27), 20.9 (C-11), 17.9, 17.5 (C-14, C-19); positive ESIMS m/z 633 [M + Na]+.
Diethyl-4,4′-((1-hydroxy-7-methoxy-2,8-bis(3-methylbut-2-en-1-yl)-9-oxo-9H-xanthene-3,6-diyl)bis(oxy))dibutanoate (1l). Yield 25%, 1H NMR (CDCl3, 500 MHz) δ 13.49 (s, 1H, 1-OH), 6.72 (s, 1H, H-5), 6.29 (s, 1H, H-4), 5.23 (br t, 2H, J = 7.0 Hz, H-12, H-17), 4.15 (m, 8H, H-21, H-25, H-27, H-31), 4.09 (t, 2H, J = 6.0 Hz, H-16), 3.79 (s, 3H, 7-OCH3), 3.35 (d, 2H, J = 7.1 Hz, H-11), 2.56 (dt, 4H, J = 7.2, 12.5 Hz, H-23, H-29), 2.20 (m, 4H, H-22, H-28), 1.85, 1.79 (each s, each 3H, H-15, H-20), 1.68 (s, 6H, H-14, H-19), 1.27 (t, 6H, H-26, H-32); 13C NMR (CDCl3, 125 MHz) δ 182.0 (C-9), 173.1, 172.9 (C-24, C-30), 162.5 (C-3), 159.9 (C-1), 157.2 (C-4a), 155.3 (C-10a), 155.1 (C-6), 144.0 (C-7), 137.3(C-8), 131.8, 131.5 (C-13, C-18), 123.2, 122.5 (C-12, C-17), 112.1 (C-8a), 111.5 (C-2), 104.0 (C-9a), 98.8 (C-5), 89.3 (C-4), 67.6, 67.2 (C-21, C-27), 60.9, 60.6 (C-25, C-31), 60.5 (7-OCH3), 30.6 (C-23, C-29), 26.2 (C-16), 25.9, 25.8, 24.4, 24.3 (C-15, C-20, C-22, C-28), 21.4 (C-11), 18.2, 17.8 (C-14, C-19), 14.2 (C-26, C-32); positive ESIMS m/z 661 [M + Na]+.
:1) to afford 2a (4.4 mg, 10%), 2b (5 mg, 12%) and 2c (37 mg, 78%).
1,3,6-Trihydroxy-2-(2,3-dihydroxy-3-methylbutyl)-7-methoxy-8-(3-methyl-2-butenyl)xanthone (2a). Yield 10%, 1H NMR (CD3OD, 500 MHz) δ 6.70 (s, 1H, H-5), 6.27 (s, 1H, H-4), 5.21 (br t, 1H, J = 6.5 Hz, H-2′′), 4.07 (d, 2H, J = 6.5 Hz, H-1′′), 3.75 (s, 3H, 7-OCH3), 3.61 (dd, 1H, J = 2.5, 10.0 Hz, H-2′), 3.04 (dd, 1H, J = 2.5, 14.0 Hz, H-1′), 2.68 (dd, 1H, J = 10.0, 14.0 Hz, H-1′), 1.82, 1.66 (each s, each 3H, H-4′′, H-5′′), 1.26 (s, 6H, H-4′, H-5′); 13C NMR (CD3OD, 125 MHz) δ 183.2 (C-9), 164.4 (C-3), 162.1 (C-1), 158.1 (C-6), 156.8 (C-4a), 156.6 (C-10a), 144.9 (C-7), 138.5 (C-8), 131.9 (C-3′), 125.1 (C-2′′), 112.2 (C-8a), 109.8 (C-2), 103.8 (C-9a), 102.8 (C-5), 93.9 (C-2′), 80.0 (C-4), 74.0 (C-3′), 61.3 (OCH3), 27.1 (C-1′), 26.0 (C-1′′), 25.8, 25.6, 25.2, 18.3; negative ESIMS m/z 443 [M − H]−.
1,3,6-Trihydroxy-2-(3-methyl-2-butenyl)-7-methoxy-8-(2,3-dihydroxy-3-methylbutyl)xanthone (2b). Yield 12%, 1H NMR (CD3OD, 500 MHz) δ 6.77 (s, 1H, H-5), 6.28 (s, 1H, H-4), 5.22 (m, 1H, H-2′), 3.84 (s, 3H, 7-OCH3), 3.66 (dd, 1H, J = 2.8, 10.2 Hz, H-2′′), 3.54 (dd, 2H, J = 2.8, 12.3 Hz, H-1′′), 3.33 (m, 2H, H-1′), 1.77, 1.65 (each s, each 3H, H-4′′, H-5′′), 1.34, 1.33 (each s, each 3H, H-4′, H-5′); 13C NMR (CD3OD, 125 MHz) δ 184.0 (C-9), 164.2 (C-3), 161.5 (C-1), 158.3 (C-4a), 156.7 (C-10a), 156.4 (C-6), 145.9 (C-7), 136.5 (C-8), 131.8 (C-3′), 123.7 (C-2′), 112.9 (C-8a), 111.8 (C-2), 103.7 (C-9a), 103.2 (C-5), 93.3 (C-4), 80.7 (C-2′′), 74.3 (C-3′′), 60.9 (OCH3), 29.6 (C-1′′), 26.0, 25.8, 25.4, 22.2 (C-1′), 17.9; negative ESIMS m/z 443 [M − H]−.
1,3,6-Trihydroxy-7-methoxy-2,8-bis(2,3-dihydroxy-3-methylbutyl)-9H-xanthen-9-one (2c). Yield 78%, 1H NMR (CD3OD, 500 MHz) δ 6.74 (s, 1H, H-5), 6.29 (s, 1H, H-4), 3.85 (s, 3H, 7-OCH3), 3.65 (m, 1H, H-2′′), 3.61 (m, 1H, H-2′), 3.56 (m, 2H, H-1′′), 3.02 (dd, 1H, J = 2.4, 14.0 Hz, H-1′), 2.68 (dd, 1H, J = 10.2, 14.0 Hz, H-1′), 1.33, 1.32, 1.26, 1.25 (each s, each 3H, H-4′, H-5′, H-4′′, H-5′′); 13C NMR (CD3OD, 125 MHz) δ 183.9 (C-9), 164.9 (C-3), 161.9 (C-1), 158.3 (C-4a), 156.7 (C-10a), 156.7 (C-6), 145.9 (C-7), 136.5 (C-8), 112.8 (C-8a), 110.0 (C-2), 103.7 (C-9a), 103.3 (C-5), 94.0 (C-4), 80.6 (C-2′′), 79.8 (C-2′), 74.3 (C-3′′), 74.0 (C-3′), 61.0 (OCH3), 29.6 (C-1′′), 25.9, 25.8 (C-1′), 25.6, 25.4, 25.2; negative ESIMS m/z 477 [M − H]−; HRESIMS m/z 477.1766 [M − H]− (calcd for C24H29O10, 477.1766).
:1 ∼ 2:1) to afford 2d, 2e or 2r respectively.
1,3,6-Trihydroxy-2-(2,3-dihydroxy-3-methylbutyl)-7-methoxy-8-isopentyl-9H-xanthen-9-one (2d). Yield 78%, 1H NMR (CD3OD, 500 MHz) δ 6.62 (s, 1H, H-5), 6.21 (s, 1H, H-4), 3.78 (s, 3H, 7-OCH3), 3.60 (dd, 1H, J = 2.4, 10.1 Hz, H-2′), 3.26 (m, 2H, H-1′′), 3.01 (dd, 1H, J = 2.4, 14.1 Hz, H-1′), 2.65 (dd, 1H, J = 10.1, 14.1 Hz, H-1′), 1.71 (m, 1H, H-3′′), 1.41 (m, 2H, H-2′′), 1.26 (s, 6H, H-4′′, H-5′′), 1.00, 0.98 (each s, each 3H, H-4′, H-5′); 13C NMR (CD3OD, 125 MHz) δ 183.1 (C-9), 164.2 (C-1), 162.1 (C-3), 157.8 (C-4a), 156.7 (C-10a), 156.5 (C-6), 144.6 (C-7), 140.4 (C-8), 112.1 (C-8a), 109.6 (C-2), 103.8 (C-9a), 102.6 (C-5), 93.9 (C-4), 80.0 (C-2′′), 74.0 (C-3′′), 61.5 (OCH3), 41.4 (C-2′), 30.1 (C-3′), 26.3 (C-1′′), 25.8 (C-1′), 25.7, 25.1, 23.0; negative ESIMS m/z 445 [M − H]−; HRESIMS m/z 445.1870 [M − H]− (calcd for C24H29O8, 445.1868).
1,3,6-Trihydroxy-2-isopentyl-7-methoxy-8-(2,3-dihydroxy-3-methylbutyl)-9H-xanthen-9-one (2e). Yield 78%, 1H NMR (CD3OD, 500 MHz) δ 6.66 (d, 1H, J = 1.8 Hz, H-5), 6.18 (d, 1H, J = 1.6 Hz, H-4), 3.84 (s, 3H, 7-OCH3), 3.63 (dd, 1H, J = 2.7, 10.5 Hz, H-2′′), 3.48 (m, 2H, H-1′′), 2.56 (m, 2H, H-1′), 1.55 (m, 1H, H-3′), 1.37 (m, 2H, H-2′), 1.32 (s, 6H, H-4′′, H-5′′), 0.95, 0.93 (each s, each 3H, H-4′, H-5′); 13C NMR (CD3OD, 125 MHz) δ 183.8 (C-9), 164.2 (C-1), 161.5 (C-3), 158.0 (C-4a), 156.6 (C-10a), 156.1 (C-6), 145.7 (C-7), 136.3 (C-8), 112.8 (C-8a), 112.8 (C-2), 103.6 (C-9a), 103.2 (C-5), 93.3 (C-4), 80.8 (C-2′), 74.3 (C-3′), 60.9 (OCH3), 39.1 (C-2′′), 29.6 (C-1′), 29.5 (C-3′′), 25.9, 25.3, 23.1, 21.2 (C-1′′); negative ESIMS m/z 445 [M − H]−; HRESIMS m/z 445.1867 [M − H]− (calcd for C24H29O8, 445.1868).
Tetrahydro-α-mangostin (2r). Yield 95%, 1H NMR (CD3OD, 500 MHz) δ 6.60 (s, 1H, H-5), 6.17 (s, 1H, H-4), 3.77 (s, 3H, 7-OCH3), 3.24 (m, 2H, H-1′′), 2.57 (m, 2H, H-1′), 1.70 (m, 1H, J = 6.6, 13.1 Hz, H-3′), 1.56 (m, 1H, J = 6.6, 13.1 Hz, H-3′′), 1.39 (m, 4H, H-2′, H-2′′), 0.99, 0.97, 0.95, 0.94 (each s, each 3H, H-4′, H-5′, H-4′′, H-5′′); 13C NMR (CD3OD, 125 MHz) δ 183.1 (C-9), 163.6 (C-1), 161.7 (C-3), 157.6 (C-4a), 156.7 (C-6), 156.0 (C-10a), 144.5 (C-7), 140.4 (C-8), 112.5 (C-8a), 112.2 (C-2), 103.7 (C-9a), 102.5 (C-5), 93.0 (C-4), 61.5 (7-OCH3), 41.5 (C-2′), 39.1 (C-2′′), 30.1 (C-3′), 29.5 (C-3′′), 26.2 (C-1′′), 23.1 (C-4′, C-5′), 23.0 (C-4′′, C-5′′), 21.2 (C-1′); negative ESIMS m/z 413 [M − H]−.
:H2O = 2:1) was added NaIO4 (26 mg, 0.12 mmol) at cool temperature. After the addition was completed, the reaction solution was allowed to warm to room temperature. After stirring for 4 h, the reaction mixture was diluted with water, extracted with ethyl acetate (3 × 10 mL). The organic phase solvent was washed with brine, dried over anhydrous sodium sulfate, and then concentrated in vacuo to give a yellow solid. The crude product was purified on column chromatograph using petroleum ether/ethyl acetate (2:1 ∼ 4:1) to afford 2f, 2g or 2h.
1,3,6-Trihydroxy-7-methoxy-8-(3-methylbut-2-en-1-yl)-2H-furo[3,2-b]xanthen-5(3H)-one (2f). Yield 60%, 1H NMR (CD3OD, 500 MHz) δ 6.64 (s, 1H, H-5), 6.21 (s, 1H, H-4), 5.19 (m, 1H, H-2′′), 4.83 (t, 1H, J = 5.7 Hz, H-2′), 4.02 (t, 2H, J = 6.2 Hz, H-1′′), 3.74 (s, 3H, 7-OCH3), 2.91 (m, 2H, H-1′), 1.81, 1.66 (each s, each 3H, H-4′′, H-5′′); 13C NMR (CD3OD, 125 MHz) δ 183.1 (C-9), 164.2 (C-3), 162.4 (C-1), 158.0 (C-6), 156.7 (C-4, C-10a), 144.8 (C-7), 138.5 (C-8), 131.8 (C-3′′), 125.1 (C-2′′), 112.1 (C-8a), 107.0 (C-2), 103.7 (C-9a), 102.8 (C-5), 99.0 (C-2′), 93.5 (C-4), 61.3 (OCH3), 30.9 (C-1′), 27.1 (C-1′′), 26.0, 18.3; negative ESIMS m/z 383 [M − H]−; HRESIMS m/z 383.1134 [M − H]− (calcd for C21H19O7, 383.1136).
1,3,6-Trihydroxy-7-methoxy-8-(2,3-dihydroxy-3-methylbutyl)-2H-furo[3,2-b]xanthen-5(3H)-one (2g). Yield 50%, 1H NMR (CD3OD, 500 MHz) δ 6.67 (s, 1H, H-5), 6.21 (s, 1H, H-4), 4.83 (t, 1H, J = 5.7 Hz, H-2′), 3.84 (s, 3H, 7-OCH3), 3.63 (m, 1H, H-2′′), 3.48 (m, 2H, H-1′′), 2.91 (dd, 2H, J = 5.7, 14.4 Hz, H-1′), 1.32, 1.31 (each s, each 3H, H-4′′, H-5′′); 13C NMR (CD3OD, 125 MHz) δ 183.8 (C-9), 164.7 (C-3), 162.3 (C-1), 158.2 (C-6), 156.7 (C-4a), 156.6 (C-10a), 145.9 (C-7), 136.4 (C-8), 112.7 (C-8a), 107.3 (C-2), 103.6 (C-9a), 103.3 (C-5), 98.9 (C-2′), 93.7 (C-4), 80.6 (C-2′′), 74.3 (C-3′′), 60.9 (OCH3), 30.9 (C-1′), 29.6 (C-1′′), 25.9, 25.4; negative ESIMS m/z 417 [M − H]−; HRESIMS m/z 417.1187 [M − H]− (calcd for C21H21O9, 417.1191).
1,3,6-Trihydroxy-7-methoxy-8-isopentyl-2H-furo[3,2-b]xanthone-5(3H)-one (2h). Yield 60%, 1H NMR (CD3OD, 500 MHz) δ 6.65 (s, 1H, H-5), 6.23 (s, 1H, H-4), 4.84 (t, 1H, J = 5.7 Hz, H-2′), 3.79 (s, 3H, 7-OCH3), 3.29 (m, 2H, H-1′′), 2.94 (m, 2H, H-1′), 1.72 (m, 1H, H-3′′), 1.42 (m, 2H, H-2′′), 1.00, 0.99 (each s, each 3H, H-4′′, H-5′′); 13C NMR (CD3OD, 125 MHz) δ 183.1 (C-9), 164.2 (C-3), 162.5 (C-1), 157.9 (C-6), 156.7 (C-4a), 156.7 (C-10a), 144.7 (C-7), 140.4 (C-8), 112.1 (C-8a), 107.0 (C-2), 103.8 (C-9a), 102.6 (C-5), 99.0 (C-2′), 93.5 (C-4), 61.3 (OCH3), 41.5 (C-2′′), 30.9 (C-1′), 30.1 (C-3′′), 26.2 (C-1′′), 23.0; negative ESIMS m/z 385 [M − H]−; HRESIMS m/z 385.1295 [M − H]− (calcd for C21H21O7, 385.1293).
:1) to afford 2i or 2j.
2-(2,3-Dimethoxy-3-methylbutyl)-1,3,6,7-tetramethoxy-8-(3-methylbut-2-en-1-yl)-9H-xanthen-9-one (2i). Yield 60%, 1H NMR (DMSO-d6, 500 MHz) δ 6.98 (s, 1H, H-5), 6.82 (s, 1H, H-4), 5.15 (br t, 1H, J = 7.0 Hz, H-17), 3.99 (dd, 2H, J = 6.6, 15.6 Hz, H-16), 3.92, 3.77, 3.68 (each s, each 3H, 1-OCH3, 3-OCH3, 6-OCH3, 7-OCH3), 3.39 (dd, 1H, J = 3.0, 10.0 Hz, H-11), 3.15 (s, 3H, 12-OCH3), 2.93 (s, 3H, 13-OCH3), 2.85 (dd, 1H, J = 10.0, 13.4 Hz, H-12), 2.66 (dd, 1H, J = 3.0, 13.4 Hz, H-11), 1.76, 1.58, 1.15, 1.11 (each s, each 3H, C-14, C-15, C-19, C-20); 13C NMR (DMSO-d6, 125 MHz) δ 175.2 (C-9), 162.5 (C-3), 158.5 (C-1), 157.0 (C-10a), 156.2 (C-4a), 153.7 (C-6), 143.6 (C-7), 135.6 (C-8), 130.3 (C-18), 123.9 (C-17), 118.4 (C-2), 113.7 (C-8a), 110.0 (C-9a), 98.5 (C-5), 94.5 (C-12), 84.6 (C-4), 77.2 (C-13), 61.3 (1-OCH3), 60.4 (7-OCH3), 60.1 (12-OCH3), 56.3 (3-OCH3, 6-OCH3), 48.9 (13-OCH3), 25.6, 25.3 (C-16), 23.8 (C-11), 22.1, 20.6, 18.0; positive ESIMS m/z 537 [M + Na]+; HRESIMS m/z 537.2472 [M + Na]+ (calcd for C29H38NaO8, 537.2464).
2-(2,3-Dimethoxy-3-methylbutyl)-8-isopentyl-1,3,6,7-tetramethoxy-9H-xanthen-9-one (2j). Yield 60%, 1H NMR (DMSO-d6, 500 MHz) δ 6.93 (s, 1H, H-5), 6.78 (s, 1H, H-4), 3.92, 3.91, 3.76, 3.70 (each s, each 3H, 1-OCH3, 3-OCH3, 6-OCH3, 7-OCH3), 3.37 (dd, 1H, J = 3.0, 10.0 Hz, H-12), 3.24 (m, 2H, H-16), 3.14, 2.92 (each s, each 3H, 12-OCH3, 13-OCH3), 2.84 (m, 1H, H-11), 2.64 (dd, 1H, J = 3.0, 13.0 Hz, H-11), 1.14, 1.10, 0.95, 0.94 (each s, each 3H, C-14, C-15, C-19, C-20); 13C NMR (DMSO-d6, 125 MHz) δ 175.1 (C-9), 162.4 (C-3), 158.6 (C-1), 156.8 (C-4a), 156.2 (C-10a), 153.7 (C-6), 143.4 (C-7), 137.5 (C-8), 118.3 (C-2), 113.7 (C-8a), 110.0 (C-9a), 98.3 (C-12), 94.4 (C-5), 84.6 (C-13), 77.2 (C-4), 61.2, 60.6, 60.1, 56.3, 56.2 (1-OCH3, 3-OCH3, 6-OCH3, 12-OCH3, 13-OCH3), 48.9 (C-17), 28.3 (C-18), 24.2 (C-16), 23.7 (C-11), 22.5, 22.5, 22.1, 20.6; positive ESIMS m/z 539 [M + Na]+; HRESIMS m/z 539.2625 [M + Na]+ (calcd for C29H40NaO8, 539.2621).
A solution of mangostin (0.1 mmol) and DDQ (27 mg, 0.12 mM) in toluene (2 mL) was placed in a flask with round bottom (10 mL), after slowly heating up to 120 °C. After stirring for 4 h, the reaction mixture was concentrated in vacuo to give a yellow solid. The crude product was purified on column chromatograph using petroleum ether/ethyl acetate (9:1) to afford 2k.
9-Hydroxycalabaxanthone (2k). Yield 40%, 1H NMR (CD3OD, 500 MHz) δ 6.66 (s, 1H, H-5), 6.62 (d, 1H, J = 10.0 Hz, H-1′), 6.13 (s, 1H, H-4), 5.61 (d, 1H, J = 10.0 Hz, H-2′), 5.20 (br t, 1H, J = 6.5 Hz, H-2′′), 4.03 (d, 2H, J = 6.5 Hz, H-1′′), 3.75 (s, 3H, 7-OCH3), 1.81, 1.66, 1.43 (each s, each 3H, H-4′, H-5′, H-4′′, H-5′′); 13C NMR (CD3OD, 125 MHz) δ 183.2 (C-9), 161.0 (C-1), 158.9 (C-3), 158.2 (C-4a), 157.5 (C-10a), 156.7 (C-6), 145.0 (C-7), 138.5 (C-8), 131.9 (C-3′′), 128.3 (C-2′), 125.0 (C-2′′), 116.4 (C-1′), 112.1 (C-8a), 105.3 (C-2), 104.4 (C-9a), 102.9 (C-5), 94.9 (C-4), 79.0 (C-3′), 61.3 (OCH3), 28.6, 27.1 (C-1′′), 26.0, 18.3; negative ESIMS m/z 407 [M − H]−.
A solution of mangostin (0.1 mmol) in DCM (2 mL) was placed under an atmosphere of nitrogen, a m-CPBA solution in DCM (2 mL) was then added drop wise over 10 min. The reaction mixture was stirred at 0 °C for 12 h, washed with 10% sodium hydrogen sulfite solution (10 mL), saturated hydrogen carbonate solution (10 mL), saturated sodium chloride (10 mL) and dried with anhydrous sodium sulfate. The reaction mixture was concentrated in vacuo to give a yellow solid and the crude product was purified on column chromatograph using petroleum ether/ethyl acetate (9:1) to afford 2l and 2m respectively.
1-((3,3-Dimethyloxiran-2-yl)methyl)-3,6,8-trihydroxy-2-methoxy-7-(3-methylbut-2-en-1-yl)-9H-xanthen-9-one (2l). Yield 4%, 1H NMR (DMSO-d6, 600 MHz) δ 13.82 (s, 1H, 1-OH), 6.79 (s, 1H, H-5), 6.30 (s, 1H, H-4), 5.28 (d, 1H, J = 4.8 Hz, H-2′′), 5.15 (t, 1H, J = 6.6 Hz, H-2′), 4.02 (t, 2H, J = 6.0 Hz, H-1′′), 3.71 (d, H, J = 6.3 Hz, H-1′), 3.70 (s, 3H, 7-OCH3), 2.79 (dd, H, J = 16.8 Hz, H-1′), 1.77, 1.62, 1.30, 1.24 (each s, each 3H, H-4′, H-5′, H-4′′, H-5′′); 13C NMR (DMSO-d6, 150 MHz) δ 181.4 (C-9), 161.2 (C-3), 159.6 (C-1), 157.3 (C-4a), 154.9 (C-10a), 154.2 (C-6), 143.4 (C-7), 136.5 (C-8), 130.5 (C-3′), 123.6 (C-2′), 109.8 (C-8a), 102.8 (C-2), 102.1 (C-10a), 101.8 (C-5), 93.4 (C-4), 79.0 (C-2′′), 66.9 (C-3′′), 60.2 (OCH3), 25.8 (C-1′′), 25.6 (C-5′), 25.3, 25.0, 21.2 (C-1′, C-4′′, C-5′′), 18.1(C-4′); negative ESIMS m/z 425 [M − H]−; HRESIMS m/z 425.1600 [M − H]− (calcd for C24H25O7, 425.1600).
Mangostanin (2m). Yield 49%, 1H NMR (DMSO-d6, 500 MHz) δ 13.63 (s, 1H, 1-OH), 6.78 (s, 1H, H-5), 6.37 (s, 1H, H-4), 5.15 (br t, 1H, J = 6.5 Hz, H-2′′), 4.73 (m, 1H, H-2′), 3.99 (d, 2H, J = 6.6 Hz, H-1′′), 3.69 (s, 3H, 7-OCH3), 3.03 (d, 2H, J = 8.6 Hz, H-1′), 1.75, 1.61, 1.14, 1.12 (each s, each 3H, H-4′, H-5′, H-4′′, H-5′′); 13C NMR (DMSO-d6, 125 MHz) δ 181.5 (C-9), 166.6 (C-1), 157.2 (C-6), 157.1 (C-10a), 156.4 (C-4a), 154.7 (C-3), 143.6 (C-7), 136.3 (C-8), 130.4 (C-3′′), 123.6 (C-2′′), 109.8 (C-8a), 107.8 (C-2), 102.9 (C-9a), 101.7 (C-5), 91.6 (C-2′), 88.0 (C-4), 70.0 (C-3′), 60.2 (OCH3), 26.0 (C-1′), 25.8, 25.7, 25.6 (C-1′′), 25.0, 18.0; negative ESIMS m/z 425 [M − H]−.
1,3,6-Trihydroxy-2-(3-hydroxy-3-methylbutyl)-7-methoxy-8-(3-methylbut-2-en-1-yl)-9H-xanthen-9-one (2n). Yield 25%, 1H NMR (CD3OD, 500 MHz) δ 6.70 (s, 1H, H-5), 6.24 (s, 1H, H-4), 5.22 (t, 1H, J = 6.5 Hz, H-2′′), 3.82 (s, 3H, 7-OCH3), 3.34–3.40 (m, 4H, H-1′, H-1′′), 1.77 (s, 3H, H-4′), 1.73 (m, 2H, H-2′), 1.65 (s, 3H, H-5′′), 1.33 (s, 6H, H-4′, H-5′); 13C NMR (CD3OD, 125 MHz) δ 183.2 (C-9), 163.7 (C-3), 161.6 (C-1), 160.0 (C-6), 156.8 (C-10a), 156.2 (C-4a), 144.7 (C-7), 139.9 (C-8), 131.7 (C-3′′), 123.8 (C-2′′), 112.2 (C-8a), 111.5 (C-2), 103.8 (C-9a), 102.7 (C-5), 93.1 (C-4), 71.9 (C-3′), 61.5 (OCH3), 45.6 (C-2′), 29.0, 26.0, 23.6 (C-1′′), 22.2 (C-1′), 17.9; negative ESIMS m/z 427 [M − H]−.
3-Isomangostin hydrate (2o). Yield 23%, 1H NMR (CD3OD, 500 MHz) δ 6.65 (s, 1H, H-5), 6.12 (s, 1H, H-4), 3.82 (s, 3H, 7-OCH3), 3.36 (m, 2H, H-1′′), 2.65 (m, 2H, H-1′), 1.83 (t, 2H, J = 6.8 Hz, H-2′), 1.72 (m, 2H, H-2′′), 1.34, 1.32 (s, 12H, H-4′, H-5′, H-4′′, H-5′′); 13C NMR (CD3OD, 125 MHz) δ 183.2 (C-9), 161.9 (C-1), 161.6 (C-3), 158.8 (C-4a), 157.0 (C-10a), 156.0 (C-6), 144.9 (C-7), 139.7 (C-8), 111.8 (C-8a), 104.7 (C-2), 103.6 (C-9a), 102.9 (C-5), 94.8 (C-4), 77.1 (C-3′), 71.9 (C-3′′), 61.5 (OCH3), 45.5 (C-2′′), 32.8 (C-2′), 29.0, 27.0, 23.6 (C-1′′), 17.0 (C-1′); negative ESIMS m/z 427 [M − H]−.
1-Isomangostin hydrate (2p). Yield 12%, 1H NMR (CD3OD, 500 MHz) δ 6.66 (s, 1H, H-5), 6.29 (s, 1H, H-4), 3.80 (s, 3H, 7-OCH3), 3.37 (m, 2H, H-1′′), 2.66 (m, 2H, H-1′), 1.82 (t, 2H, J = 6.9 Hz, H-2′), 1.75 (m, 2H, H-2′′), 1.40 (s, 6H, H-4′, H-4′′), 1.32 (s, 6H, H-5′, H-5′′); 13C NMR (CD3OD, 125 MHz) δ 179.0 (C-9), 161.9 (C-1), 161.6 (C-2), 158.2 (C-4a), 157.1 (C-10a), 155.8 (C-6), 144.8 (C-7), 139.5 (C-8), 114.7 (C-8a), 107.6 (C-9a), 106.2 (C-2), 102.2 (C-5), 93.9 (C-4), 76.7 (C-3′), 71.9 (C-3′′), 61.4 (OCH3), 45.5 (C-2′′), 32.6 (C-2′), 30.8, 29.2, 26.8 (C-1′′), 24.0, 23.2, 18.1 (C-1′); negative ESIMS m/z 427 [M − H]−.
1-Isomangostin (2q). Yield 20%, 1H NMR (CD3OD, 500 MHz) δ 6.61 (s, 1H, H-5), 6.05 (s, 1H, H-4), 5.21 (br t, H, J = 6.5 Hz, H-2′′), 4.02 (d, 2H, J = 6.5 Hz, H-1′′), 3.75 (s, 3H, 7-OCH3), 2.62 (t, 2H, J = 6.8 Hz, H-1′), 1.81 (m, 2H, H-2′), 1.81, 1.67, 1.33 (each s, each 3H, H-4′, H-5′, H-4′′, H-5′′); 13C NMR (CD3OD, 125 MHz) δ 183.1 (C-9), 161.8 (C-3), 161.5 (C-1), 158.3 (C-4a), 156.8 (C-10a), 155.9 (C-6), 144.8 (C-7), 138.4 (C-8), 131.6 (C-3′′), 125.2 (C-2′′), 111.9 (C-8a), 104.6 (C-9a), 103.5 (C-2), 102.9 (C-5), 94.8 (C-4), 77.0 (C-3′), 61.3 (7-OCH3), 32.8 (C-2′), 27.1, 27.0 (C-4′, C-4′′), 26.0 (C-2′′), 18.3 (C-5′, C-5′′), 17.0 (C-1′′); negative ESIMS m/z 409 [M − H]−.
γ-Mangostin (2s). 1H NMR (CD3OD, 500 MHz) δ 6.66 (s, 1H, H-5), 6.22 (s, 1H, H-4), 5.25 (br t, 2H, J = 6.5 Hz, H-2′, H-2′′), 4.11 (d, 2H, J = 6.7 Hz, H-1′′), 3.34 (m, 2H, H-1′), 1.83, 1.78 (each s, each 3H, H-4′, H-4′′), 1.65 (s, 6H, H-5′, H-5′′); 13C NMR (CD3OD, 125 MHz) δ 183.6 (C-9), 163.4 (C-3), 161.5 (C-1), 156.3 (C-4a), 154.0 (C-10a), 153.2 (C-6), 142.0 (C-7), 131.7, 131.6 (C-3′, C-3′′), 129.5 (C-8), 124.8, 124.0 (C-2′, C-2′′), 112.2 (C-8a), 111.1 (C-2), 103.9 (C-9a), 100.9 (C-5), 92.9 (C-4), 26.6 (C-1′′), 26.1, 26.0 (C-4′, C-4′′), 22.2 (C-1′), 18.3, 17.9 (C-5′, C-5′′); negative ESIMS m/z 395 [M − H]−.
Tetrahydro-γ-mangostin (2t). Yield 96%, 1H NMR (CD3OD, 500 MHz) δ 6.64 (s, 1H, H-5), 6.21 (s, 1H, H-4), 3.35 (m, 2H, H-1′′), 2.60 (m, 2H, H-1′), 1.70, 1.57 (dt, each 1H, J = 6.6, 13.2 Hz, H-3′, H-3′′), 1.42 (m, 4H, H-2′, H-2′′), 1.00, 0.99, 0.95, 0.94 (each s, each 3H, H-4′, H-5′, H-4′′, H-5′′); 13C NMR (CD3OD, 125 MHz) δ 183.6 (C-9), 163.4 (C-1), 161.7 (C-3), 156.1 (C-4a), 154.1 (C-10a), 153.0 (C-6), 141.8 (C-7), 131.6 (C-8), 112.3 (C-8a), 112.2 (C-2), 103.9 (C-9a), 100.8 (C-5), 92.9 (C-4), 40.4, 39.2 (C-2′, C-2′′), 30.0 (C-3′), 29.5 (C-3′′), 25.8 (C-1′′), 23.1 (C-4′, C-5′, C-4′′, C-5′′), 21.2 (C-1′); negative ESIMS m/z 399 [M − H]−.
1,3,6-Trihydroxy-9H-xanthen-9-one (2u). Yield 60%, 1H NMR (CD3OD, 500 MHz) δ 7.97 (d, 1H, H-8), 6.82 (dd, 1H, J = 2.2, 8.8 Hz, H-7), 6.73 (d, 1H, J = 2.2 Hz, H-2), 6.27 (d, 1H, J = 2.2 Hz, H-2), 6.13 (d, 1H, J = 2.2 Hz, H-2); 13C NMR (CD3OD, 125 MHz) δ 181.2 (C-9), 166.7 (C-1), 165.7 (C-3), 164.6 (C-7), 159.4 (C-4a), 159.4 (C-10a), 128.3 (C-8), 114.7 (C-8a), 114.2 (C-7), 103.1 (C-9a, C-2), 99.0 (C-5), 95.0 (C-4); negative ESIMS m/z 243 [M − H]−.
:1) to afford 3a–3e.
4-Chloro-α-mangostin (3a). Yield 30%, 1H NMR (CD3OD, 500 MHz) δ 6.77 (s, 1H, H-5), 5.21 (br t, 2H, H-2′, H-2′′), 4.05 (d, 2H, J = 6.5 Hz, H-1′′), 3.76 (s, 3H, 7-OCH3), 3.34 (m, 2H, H-1′), 1.82, 1.78, 1.66, 1.65 (each s, each 3H, H-4′, H-5′, H-4′′, H-5′′); 13C NMR (CD3OD, 125 MHz) δ 182.7 (C-9), 160.5 (C-1), 159.6 (C-3), 158.8 (C-10a), 156.5 (C-6), 151.2 (C-4a), 145.4 (C-7), 138.5 (C-8), 132.1, 131.9 (C-3′, C-3′′), 125.0, 123.5 (C-2′, C-2′′), 112.8 (C-8a), 111.6 (C-2), 103.8 (C-9a), 103.0 (C-5), 98.5 (C-4), 61.3 (7-OCH3), 27.1 (C-1′′), 26.0, 26.0 (C-4′, C-4′′), 22.9 (C-1′), 18.3, 18.0 (C-1′, C-1′′); negative ESIMS m/z 443 [M − H]−.
4-Bromo-α-mangostin (3b). Yield 12%, 1H NMR (CDCl3, 500 MHz) δ 13.69 (s, 1H, 1-OH), 6.97 (s, 1H, H-5), 5.25 (br t, 2H, H-2′, H-2′′), 4.08 (d, 2H, J = 6.5 Hz, H-1′′), 3.82 (s, 3H, 7-OCH3), 3.46 (m, 2H, H-1′), 1.83, 1.82, 1.71, 1.69 (each s, each 3H, H-4′, H-5′, H-4′′, H-5′′); 13C NMR (CDCl3, 125 MHz) δ 181.8 (C-9), 160.0 (C-1), 156.8 (C-3), 155.5 (C-10a), 155.0 (C-6), 150.7 (C-4a), 143.0 (C-7), 137.2 (C-8), 133.4, 132.4 (C-3′, C-3′′), 122.9, 121.4 (C-2′, C-2′′), 111.9 (C-8a), 110.6 (C-2), 104.3 (C-9a), 101.9 (C-5), 86.4 (C-4), 62.1 (7-OCH3), 26.6 (C-1′′), 25.9, 25.8 (C-4′, C-4′′), 22.3 (C-1′), 18.3, 17.9 (C-1′, C-1′′); negative ESIMS m/z 488 [M − H]−.
4,5-Dibromo-α-mangostin (3c). Yield 50%, 1H NMR (CD3OD, 500 MHz) δ 5.20 (br t, 2H, H-2′, H-2′′), 4.03 (d, 2H, J = 6.5 Hz, H-1′′), 3.75 (s, 3H, 7-OCH3), 3.36 (d, 2H, J = 7.0 Hz, H-1′), 1.82, 1.79, 1.66, 1.66 (each s, each 3H, H-4′, H-5′, H-4′′, H-5′′); 13C NMR (CD3OD, 125 MHz) δ 182.6 (C-9), 160.4 (C-1), 160.1 (C-3), 156.1 (C-10a), 153.4 (C-6), 152.1 (C-4a), 145.2 (C-7), 137.3 (C-8), 132.5, 132.4 (C-3′, C-3′′), 124.5, 123.1 (C-2′, C-2′′), 113.2 (C-8a), 112.4 (C-2), 104.5 (C-9a), 97.6 (C-5), 87.8 (C-4), 62.0 (7-OCH3), 27.1 (C-1′′), 26.0, 26.0 (C-4′, C-4′′), 23.1 (C-1′), 18.4, 18.0 (C-1′, C-1′′); negative ESIMS m/z 567 [M − H]−; HRESIMS m/z 564.9863 [M − H]− (calcd for C24H23Br2O6, 564.9867).
4-Bromo-tetrahydro-α-mangostin (3d). Yield 12%, 1H NMR (DMSO-d6, 500 MHz) δ 13.86 (s, 1H, 1-OH), 6.79 (s, 1H, H-5), 3.74 (s, 3H, 7-OCH3), 3.20 (m, 2H, H-1′′), 2.61 (m, 2H, H-1′), 1.65 (dt, H, J = 6.5, 13.0 Hz, H-3′′), 1.53 (dt, H, J = 6.5, 13.0 Hz, H-3′), 1.33 (dt, 4H, J = 16.5, 7.0 Hz, H-2′, H-2′′), 0.95, 0.93, 0.91, 0.89 (each s, each 3H, H-4′, H-5′, H-4′′, H-5′′); 13C NMR (DMSO-d6, 125 MHz) δ 181.2 (C-9), 159.0 (C-1), 158.5 (C-3), 157.2 (C-10a), 154.5 (C-6), 150.2 (C-4a), 143.6 (C-7), 138.5 (C-8), 112.4 (C-8a), 109.6 (C-2), 102.9 (C-9a), 101.6 (C-5), 86.7 (C-4), 60.4 (7-OCH3), 39.0, 37.5 (C-2′, C-2′′), 28.3, 27.7 (C-3′, C-3′′), 24.6 (C-1′′), 22.5, 22.4 (C-4′, C-4′′, C-5′, C-5′′), 20.6 (C-1′); negative ESIMS m/z 491 [M − H]−; HRESIMS m/z 491.1072 [M − H]− (calcd for C24H28BrO6, 491.1069).
4,5-Dibromo-tetrahydro-α-mangostin (3e). Yield 60%, 1H NMR (DMSO-d6, 500 MHz) δ 13.54 (s, 1H, 1-OH), 3.70 (s, 3H, 7-OCH3), 3.16 (m, 2H, H-1′′), 2.59 (m, 2H, H-1′), 1.63 (dt, H, J = 6.5, 13.0 Hz, H-3′′), 1.53 (dt, H, J = 6.5, 13.0 Hz, H-3′), 1.32 (m, 4H, H-2′, H-2′′), 0.93, 0.92, 0.90, 0.89 (each s, each 3H, H-4′, H-5′, H-4′′, H-5′′); 13C NMR (DMSO-d6, 125 MHz) δ 182.7 (C-9), 160.7 (C-1), 160.6 (C-3), 156.3 (C-10a), 153.4 (C-6), 152.0 (C-4a), 145.4 (C-7), 139.2 (C-8), 114.6 (C-8a), 112.3 (C-2), 104.6 (C-9a), 98.1 (C-5), 88.8 (C-4), 63.3 (7-OCH3), 41.0, 39.3 (C-2′, C-2′′), 30.1, 29.5 (C-3′, C-3′′), 26.4 (C-1′′), 24.3, 24.2 (C-4′, C-4′′, C-5′, C-5′′), 22.4 (C-1′); negative ESIMS m/z 571 [M − H]−; HRESIMS m/z 569.0170 [M − H]− (calcd for C24H27Br2O6, 569.0174).
3.3 Biological assays
The following human cancer cell lines were used: HL-60, SMMC-7721, A-549, MCF-7 and SW-480. These cells were obtained from ATCC (Manassas, VA, USA). All the cells were cultured in RPMI-1640 or DMEM medium (Hyclone, Logan, UT, USA), supplemented with 10% fetal bovine serum at 37 °C in a humidified atmosphere with 5% CO2. Cell viability was assessed by conducting colorimetric measurements of the amount of insoluble formazan formed in living cells based on the reduction of MTS (Sigma, St. Louis, MO, USA). Briefly, 100 μM of adherent cells were seeded into each well of a 96-well cell culture plate and allowed to adhere for 12 h before drug addition, while suspended cells were seeded just before drug addition, both with an initial density of 1 × 105 cells per mL in 100 μM medium, Each tumour cell line was exposed to the test compound at various concentrations in triplicate for 48 h, with cisplatin and paclitaxel (Sigma) as positive controls. After the incubation, MTS (100 μg) was added to each well and the incubation continued for 4 h at 37 °C. The cells were lysed with 100 μM of 20% SDS–50% DMF after removal of 100 μM medium. The optical density of the lysate was measured at 490 nm in a 96-well microtiter plate reader (Bio-Rad 680). The IC50 value of each compound was calculated by Reed and Muench's method.4. Conclusions
In order to enrich the types of mangostin derivatives and improve the structure–activity relationship, in this investigation, we report the synthesis of a series of α-mangostin derivatives based on three kinds of different functional groups. They have been assessed for their cytotoxicity against a panel of human cancer cell lines, including HL-60, SMMC-7721, A-549, MCF-7 and SW480. Most of them exhibited good cytotoxicity against all five cancer cell lines evaluated and several of them were even better than α-mangostin. Structure–activity relationship (SAR) analysis reveals that the isopentene group at C-8 is critical for retaining the exceptional cytotoxicity of α-mangostin; the oxidation form of isopentene group at C-8 causes the loss of the cytotoxicity. Based on the SAR studies, further study is under progress. Some active compounds were obtained in this study and our results suggest that some of these compounds have potential for further development as anticancer agents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by grants of National Key Research and Development Program of China (2017YFD0201402), the Yunnan Provincial Science and Technology Department (2017ZF003-04 and 2015HB093). The authors thank the staff of analytical group and the staff of natural drug activity screening centre of the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, for measurements of all spectra and the activity screening of all compounds.Notes and references
- J. M. Hu, J. J. Chen, Y. X. Zhao, R. R. Wang, Y. T. Zheng and J. Zhou, Acta Bot. Yunnanica, 2006, 28, 319–322 CAS.
- W. Jinsart, B. Ternai, D. Buddhasukh and G. M. Polya, Phytochemistry, 1992, 31, 3711–3713 CrossRef CAS PubMed.
- K. J. Zhang, Q. L. Gu, K. Yang, X. J. Ming and J. X. Wang, Planta Med., 2017, 83, 188–202 CAS.
- H. A. Jung, B. N. Su, W. J. Keller, R. G. Mehta and A. D. Kinghorn, J. Agric. Food Chem., 2006, 54, 2077–2082 CrossRef CAS PubMed.
- L.-G. Chen, L.-L. Yang and C.-C. Wang, Food Chem. Toxicol., 2008, 46, 688–693 CrossRef CAS PubMed.
- S. Tewtrakul, C. Wattanapiromsakul and W. Mahabusarakam, J. Ethnopharmacol., 2009, 121, 379–382 CrossRef CAS PubMed.
- S. Narasimhan, S. Maheshwaran, I. A. Abu-Yousef, A. F. Majdalawieh, J. Rethavathi, P. E. Das and P. Poltronieri, Molecules, 2017, 22, 275–288 CrossRef PubMed.
- S. M. Lin, J. J. Koh, T. T. Aung, F. H. Lim, J. G. Li, H. X. Zou, L. Wang, R. Lakshminarayanan, C. Verma, Y. J. Wang, D. T. H. Tan, D. R. Cao, R. W. Beuerman, L. Ren and S. P. Liu, J. Med. Chem., 2017, 60, 1362–1378 CrossRef CAS PubMed.
- G. Gopalakrishnan, B. Banumathi and G. Suresh, J. Nat. Prod., 1997, 60, 519–524 CrossRef CAS PubMed.
- W. Mahabusarakam, K. Kuaha, P. Wilairat and W. C. Taylor, Planta Med., 2006, 72, 912–916 CrossRef CAS PubMed.
- H. Z. Jiang, X. F. Quan, W. X. Tian, J. M. Hu, P. C. Wang, S. Z. Huang, Z. Q. Cheng, W. J. Liang, J. Zhou, X. F. Ma and Y. X. Zhao, Bioorg. Med. Chem. Lett., 2010, 20, 6045–6047 CrossRef CAS PubMed.
- P. Li, W. X. Tian and X. F. Ma, Mol. Cancer, 2014, 13, 138–149 CrossRef PubMed.
- H. W. Ryu, M. J. Curtis-Long, S. Jung, Y. M. Jin, J. K. Cho, Y. B. Ryu, W. S. Lee and K. H. Park, Bioorg. Med. Chem., 2010, 18, 6258–6264 CrossRef CAS PubMed.
- H. W. Ryu, J. K. Cho, M. J. Curtis-Long, H. J. Yuk, Y. S. Kim, S. Jung, Y. S. Kim, B. W. Lee and K. H. Park, Phytochemistry, 2011, 72, 2148–2154 CrossRef CAS PubMed.
- K. Y. Khaw, S. B. Choi, S. C. Tan, H. A. Wahab, K. L. Chan and V. Murugaiyah, Phytomedicine, 2014, 21, 1303–1309 CrossRef CAS PubMed.
- C. H. Lee, T. H. Ying, H. L. Chiou, S. C. Hsieh, S. H. Wen, R. H. Chou and Y. H. Hsieh, Oncotarget, 2017, 8, 47425–47439 Search PubMed.
- N. Cai, S. J. Xie, D. B. Qiu, C. C. Jia, C. Du, W. Liu, J. J. Chen and Q. Zhang, J. Funct. Foods, 2016, 26, 309–318 CrossRef CAS.
- B. Bin Hafeez, A. Mustafa, J. W. Fischer, A. Singh, W. X. Zhong, M. O. Shekhani, L. Meske, T. Havighurst, K. Kim and A. K. Verma, Antioxid. Redox Signaling, 2014, 21, 682–699 CrossRef PubMed.
- J. H. Yoo, K. Kang, E. H. Jho, Y. W. Chin, J. Kim and C. W. Nho, Food Chem., 2011, 129, 1559–1566 CrossRef CAS.
- M.-H. Wang, K.-J. Zhang, Q.-L. Gu, X.-L. Bi and J.-X. Wang, Chin. J. Nat. Med., 2017, 15, 81–93 Search PubMed.
- A. F. A. Aisha, K. M. Abu-Salah, Z. Ismail and A. Majid, Molecules, 2012, 17, 2939–2954 CrossRef CAS PubMed.
- Y. Nakagawa, M. Iinuma, T. Naoe, Y. Nozawa and Y. Akao, Bioorg. Med. Chem., 2007, 15, 5620–5628 CrossRef CAS PubMed.
- Y. Xia, Y. Li, K. D. Westover, J. M. Sun, H. X. Chen, J. M. Zhang and D. E. Fisher, PLoS One, 2016, 11, e0155217 CrossRef PubMed.
- R. Benjakul, L. Kongkaneramit, N. Sarisuta, P. Moongkarndi and C. C. Muller-Goymann, Anticancer Drugs, 2015, 26, 824–834 CrossRef CAS PubMed.
- R. K. Verma, W. Yu, A. Shrivastava, S. Shankar and R. K. Srivastava, Sci. Rep., 2016, 6, 32743 CrossRef CAS PubMed.
- Y. Zhao, G. S. Tang, Q. Tang, J. Zhang, Y. Y. Hou, E. B. Cai, S. L. Liu, D. H. Lei, L. X. Zhang and S. J. Wang, Eur. J. Drug Metab. Pharmacokinet., 2016, 41, 605–613 CrossRef CAS PubMed.
- X. J. Zhang, X. Li, S. F. Ye, Y. Zhang, L. Tao, Y. Gao, D. D. Gong, M. Y. Xi, H. Y. Meng, M. Q. Zhang, W. L. Gao, X. L. Xu, Q. L. Guo and Q. D. You, Med. Chem., 2012, 8, 1012–1025 CAS.
- X. Fei, M. Jo, B. Lee, S. B. Han, K. Lee, J. K. Jung, S. Y. Seo and Y. S. Kwak, Bioorg. Med. Chem. Lett., 2014, 24, 2062–2065 CrossRef CAS PubMed.
- E. V. Buravlev, O. G. Shevchenko and A. V. Kutchin, Bioorg. Med. Chem. Lett., 2015, 25, 826–829 CrossRef CAS PubMed.
- Y. W. Chin, H. A. Jung, H. Chai, W. J. Keller and A. D. Kinghorn, Phytochemistry, 2008, 69(3), 754–758 CrossRef CAS PubMed.
- L. D. Ha, P. E. Hansen, O. Vang, F. Duus, H. D. Pham and L. H. D. Nguyen, Chem. Pharm. Bull., 2009, 57, 830–834 CrossRef CAS PubMed.
- C. Wang, P. Wu, J. F. Shi, Z. H. Jiang and X. Y. Wei, Eur. J. Med. Chem., 2015, 100, 139–150 CrossRef CAS PubMed.
- X. Huo, X. F. Ren, Y. F. Xu, X. Y. Li, X. G. She and X. F. Pan, Tetrahedron: Asymmetry, 2008, 19, 343–347 CrossRef CAS.
- K. Mori and C. Y. Yang, Tetrahedron, 2016, 72, 4593–4607 CrossRef CAS.
- C. F. Morelli, M. Biagiotti, V. M. Pappalardo, M. Rabuffetti and G. Speranza, Nat. Prod. Res., 2015, 29, 750–755 CrossRef CAS PubMed.
- Y. L. Ren, S. Matthew, D. D. Lantvit, T. N. Ninh, H. B. Chai, J. R. Fuchs, D. D. Soejarto, E. J. C. de Blanco, S. M. Swanson and A. D. Kinghorn, J. Nat. Prod., 2011, 74, 1117–1125 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Abbreviation for the manuscript, NMR and MS spectrum of synthesized compounds. See DOI: 10.1039/c8ra08409b |
| This journal is © The Royal Society of Chemistry 2018 |