
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComparison of two water oxidation electrocatalysts by copper or zinc supermolecule complexes based on porphyrin ligand†
Zhaodi Huanga,
Meixi Zhanga,
Huan Lina,
Shuo Dinga,
Bin Donga,
Di Liu c,
Hong Wang*b,
Fangna Dai*a and
Daofeng Suna
c,
Hong Wang*b,
Fangna Dai*a and
Daofeng Suna
aSchool of Materials Science and Engineering, College of Science, China University of Petroleum (East China), Qingdao, Shandong 266580, People's Republic of China
bSchool of Materials Science and Engineering, Beijing University of Chemical Technology, Beijing, 100029, People's Republic of China
cSchool of Chemical and Environmental Engineering, Shandong University of Science and Technology, Qingdao, Shandong 266590, People's Republic of China. E-mail: fndai@upc.edu.cn; wanghong@mail.buct.edu.cn
First published on 30th November 2018
Abstract
Based on the 5,10,15,20-tetra(cyanophenyl)porphyrin (CNTCPP) ligand, two supermolecule complexes formulated as [Cu(CNTCPP)]·2DMF (Cu-CNTCPP) and [Zn(CNTCPP)] (Zn-CNTCPP) have been synthesized and structurally characterized by single-crystal X-ray diffraction. Cu-CNTCPP features a 0 dimensional (0D) supramoleculer structure, whereas Zn-CNTCPP is a 2D structure. With coordination unsaturated points, Cu-CNTCPP exhibits electrocatalytic activity toward oxygen evolution reaction (OER) with low potential in 1.0 M KOH, generating a current density of 10 mA cm−2 at an potential of 1.66 V vs. RHE, which means it an efficient electrocatalytic material for OER.
Introduction
With increasing energy consumption and the greenhouse effect due to carbon dioxide emission, seeking renewable and non-carbon energy alternatives is an urgent and important issue.1,2 The water splitting reaction provides a convenient chemical method for renewable energy storage.3,4 As the oxidative half reaction of water splitting, the oxygen evolution reaction (OER) is kinetically sluggish and leads to significant overpotential and energy loss.5 Developing highly efficient electrocatalysts for OER has emerged as one of the most research hot spots in clean energy generation and storage.6,7 The most reported precious-metal electrocatalysts such as IrO2 and RuO2 are the state-of-art OER catalysts, but they are limited by low abundance and high cost for large-scale application.8,9 Therefore, there is tremendous interest in developing inexpensive and earth-abundant OER catalysts.10 Keeping the advantages of low cost, high chemical stability, and electrocatalytic ability, the most reported catalysts for OER are 3d transition metal (e.g. Fe, Cu, Co, Ni and Mn) derived oxides or hydroxides, phosphates, perovskites, chalcogenides and so on,11–15 but there are scarce reports of supermolecule complexes used as direct electrocatalyst for OER.16On the other hand, supermolecule complexes are a class of interesting materials which have been intensively investigated in diverse fields, such as luminescence, gas storage, sensing, magnetics, catalysis, and so on.17–21 The supermolecule complex structures and properties are mainly dependent on functional ligands and metal nodes.22–24 As we know, porphyrins are valuable building ligands in supermolecule complex systems; their extended square planar ligand favors the formation of ordered frameworks.25–27 Besides that, porphyrins can be metalated in their center, and could function as catalysis active centers,28–30 so porphyrins or metallporphyrin molecules have been used as active constituents for constructing a variety of supermolecule complexes.31–33 For example, reports on lithium-ion battery electrodes have been recorded based on iron-porphyrin materials.34–36 Co-based porphyrin and graphitic materials was predicted to be the OER catalyst by density functional theory (DFT) calculation.37 Analysis research of porphyrines as catalysts for electrochemical reduction of O2 and oxidation of H2O have also been reported.38 However, only a few supermolecule complexes show sufficient electrocatalytic OER activity at the high electric potential experienced during electrolysis (1.6–2.0 V vs. RHE).
Inspired by the above reports, we choose the 5,10,15,20-tetra(cyanophenyl)porphyrin (CNTCPP) ligand as organic linker for building supermolecule complexes based on the following reasons: (i) the ligand possesses delocalized π-electron system, which can provide an intense electrical conductivity;39 (ii) four cyanide bridged coordination groups and the rigid phenyl in the ligand can afford more coordination opportunities, which will leading to diverse structures;40 (iii) the electron withdrawing group of cyanide and high symmetry framework are favor for electron transferring in the ordered supermolecule structures; (iv) the central flat surface structure would help to increase the active surface area, which can ensure efficient contact between the electrolyte and active catalysts.41,42 Herein, we demonstrate two supermolecule complexes [Cu(CNTCPP)]·2DMF (Cu-CNTCPP) and [Zn(CNTCPP)] (Zn-CNTCPP) by assembling the designed CNTCPP ligand with cupper (II) or zinc(II) ions, respectively. Cu-CNTCPP are zero-dimensional (0D) complex with cupper (II) metalated in the porphyrin center, Zn-CNTCPP is a 2D structure. Although Zn-CNTCPP has been reported by Jiang and coworkers, only its structure was focused upon in their work.43 The OER properties of Cu-CNTCPP and Zn-CNTCPP have been investigated. Particularly, due to the different coordination situations, the Cu-CNTCPP materials afforded better performance than Zn-CNTCPP in 1.0 M KOH, indicating it a promising new class of non-noble catalyst for OER.
Materials and general methods
All of the chemicals were obtained from commercial sources and were used without further purification, except CNTCPP ligand was synthesized through the condensation of 4-formylbenzonitrile with pyrrole, catalyzed by BF3·OEt2, followed by oxidation with p-chloranil (Fig. S1†).44 C, H, N of elemental analysis were measured on an EA 1110 elemental analyzer. The powder diffraction data were collected on a Cu-Kα radiation using an X-Pert PRO MPD diffractometer. Thermogravimetric analyses were performed in the temperature ranged from 40 to 800 °C with 10 °C min−1 rate of heat using a Mettler Toledo Thermogravimetric analysis (TGA) instrument. The purity of Cu-CNTCPP and Zn-CNTCPP were confirmed by comparison of their simulated and experimental powder X-ray diffraction (Fig. S2 and S3†). TGA measurement shows that Cu-CNTCPP and Zn-CNTCPP lose uncoordinated solvents below 200 °C and can be stable up to 580 and 560 °C, respectively. (Fig. S4 and S5†). Method for electrochemical measurements are displayed in ESI.†The X-ray datum of complexes were obtained on an Agilent Super nova with Cu-Kα radiation (λ = 1.54178) and MoKα (λ = 0.71073) at room temperature, respectively. The absorption corrections were decided by employing the SADABS program. The structures and hydrogen atoms were refined to utilize the SHELX-97 program 33–34 through the full-matrix least-squares by fitting on F2 and anisotropic thermal parameters, respectively. There are many disordered solvent molecules existed in the cavity of Cu-CNTCPP, which can not be achieved through the reasonable modeling. Hence, the diffuse electron was removed by the PLATON/SQUEEZE routine 35. The summary of the structure data is displayed in Tables SI1 and SI2.† Crystallographic data have been deposited at the Cambridge Crystallographic Data Center (CCDC: 1450485 for Cu-CNTCPP, 1449406 for Zn-CNTCPP), which can be obtained from the Cambridge Crystallographic Data Center, 12, Union Road, Cambridge CB21EZ, U.K.
Results and discussion
Crystal Structure
Complex of Cu-CNTCPP crystallizes in the monoclinic space group P2/c, and the asymmetric unit contains two Cu2+ (Cu1 and Cu2) ions and two CNTCPP ligands. As displayed in Fig. 1a, Cu1 is surrounded by four N atoms from one CNTCPP ligands' central porphyrin ring with the average Cu–N bond length of 2.006 Å. Cu2 adopt similar coordination mode with the average Cu–N bond length of 2.003 Å. The non-coordinated sites of Cu1 and Cu2 in the axial direction mean the possible presence of large number of bridging and terminal hydroxyl or water ligands, which could provide high concentration of potential active sites. The angles between the side benzene rings and the central Cu1-porphyrin ring are 85.828°, 69.302°, 73.563°, and 68.479°, respectively, the angles between the side benzene rings and the central Cu2-porphyrin ring are 84.213°, 67.916°, 67.704°, and 68.709°, respectively (Fig. 1b). The CNTCPP porphyrin ring are metallic by Cu2+ ion, and all the four cyanide groups in the CNTCPP ligands are non-coordinated. The distance between Cu1 and Cu2 in the two-porphyrin ring is 8.599 Å. There are weak C–H⋯N (3.695 Å and 3.743 Å) interactions between the 0D Cu-porphyrin molecules to connect it into 3D structure (Fig. 1c and d). There is microspore view along c axis and the uncoordinated DMF molecules blocked the pores. | ||
| Fig. 1 (a and b) The angles between the side benzene rings and the central Cu1 and Cu2-porphyrin ring. (c and d) 3D framework viewed from c axis. | ||
X-ray single-crystal diffraction reveals complex of Zn-CNTCPP crystallizes in the monoclinic space group P21/n, and the asymmetric unit contains one Zn2+ ion and one CNTCPP ligand. As displayed in Fig. 2a, Zn2+ is surrounded by four N atoms from one CNTCPP ligands' central porphyrin ring with the average Zn–N bond length of 2.046 Å and there are weak interactions between the two cyanide groups from two different CNTCPP ligands with the Zn2+ ions (Zn–N bond length of 2.645 Å). The CNTCPP porphyrin ring central are metallic by zinc ion to form 0D molecule, and the weak Zn–N bond between the two of cyanide groups in the CNTCPP ligands connect another two zinc(II) ions from the metallic porphyrin ring to form 2D supramolecular structure. The angles between the side benzene rings and the central Zn-porphyrin ring are 86.849°, 64.595°, 86.849°, and 64.595°, respectively (Fig. 2b). Each ligand connects three Zn(II) ions, and each Zn(II) attaches three different CNTCPP ligands, generating a 2D network (Fig. 2c). There are weak C1–H⋯N8 (3.259 Å) supramolecular hydrogen bonds between the 2D layers to connect it into 3D structures. View along b axis, there is micro-pores with dimension of 4.5 × 8.2 nm (Fig. 2d). The crystallographic data of the two complexes are summarized in Tables S1 and S2.†
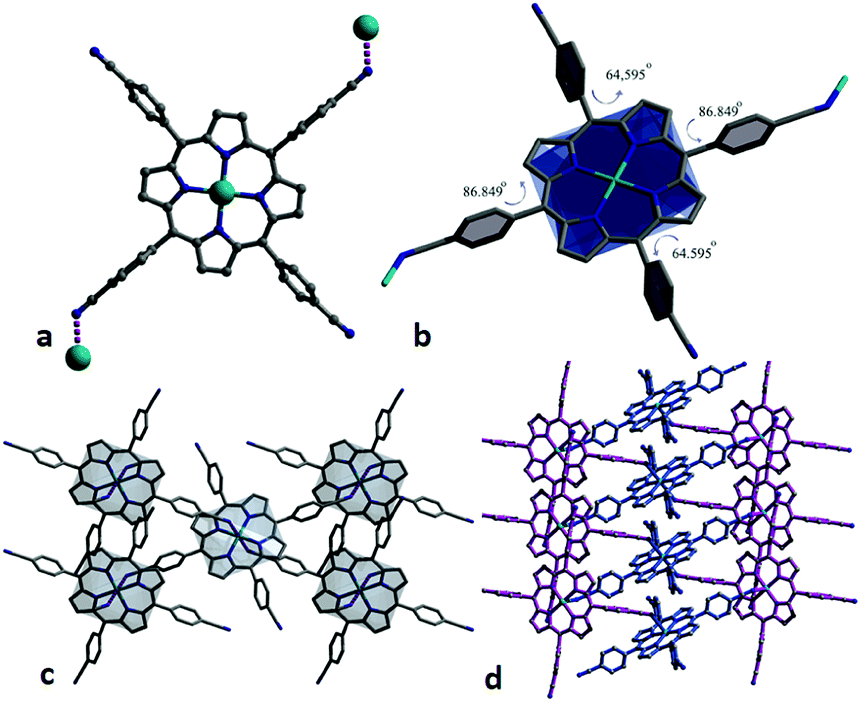 | ||
| Fig. 2 (a) Coordination environment of the ligand and Zn(II) in complex. (b) Angles between the side benzene rings and the central Zn-porphyrin ring. (c) 2D layer viewed from b axis. (d) 3D space filling representation viewed from a axis. | ||
Electrocatalytic activity
Many researchs show promising electrocatalytic properties for a myriad of electrochemical reactions such as OER. Different studies reported various optimal compositions of copper, to achieve maximum OER performance. But slightly literatures reported the positive result got by supramolecular with copper structures. As the Cu2+ ions are unsaturated coordinated in Cu-CNTCPP, there are enriched active sites, we therefore investigate the electrocatalytic activity of the Cu-CNTCPP. It is necessary to understand the origination of the OER activity of the as-prepared catalyst, for comparison, we have also measured the electrocatalytic OER activity of the comparative catalysts under the same conditions except Cu-CNTCPP: CNTCPP ligand, mixture of CuCl2 and CNTCPP ligand by manual grinding (named mixture of CNTCPP and Cu2+), mixture of CuCl2 and CNTCPP ligand by reflux in hot DMF solution (named amorphous Cu-CNTCPP), and Cu(OH)2 by conventional precipitin reaction (named Cu(OH)2). Fig. 3a shows the polarization curves of Cu-CNTCPP and the comparative catalysts. The potential required for a current density of 10 mA cm−2, which is a metric associated with solar fuel synthesis.45 As shown, the Cu-CNTCPP delivers a current density of 10 mA cm−2 at a potential of 1.66 V, whereas a potential as high as 1.74 V, 1.84 V and 1.78 V are required for CNTCPP ligand, mixture of CNTCPP and Cu2+ and amorphous Cu-CNTCPP to achieve the same current density, respectively. And for Cu(OH)2, it fails to reach 10 mA cm−2 in 1.0 M KOH. The OER kinetics of these electrodes is estimated by their corresponding Tafel plots. As shown in Fig. 3b, the Tafel slope of the Cu-CNTCPP is 83.9 mV dec−1, compared to 90.1 mV dec−1, 165.4 mV dec−1, 120.9 mV dec−1 and 267.4 mV dec−1 for CNTCPP, mixture of CNTCPP and Cu2+, amorphous Cu-CNTCPP and Cu(OH)2, respectively.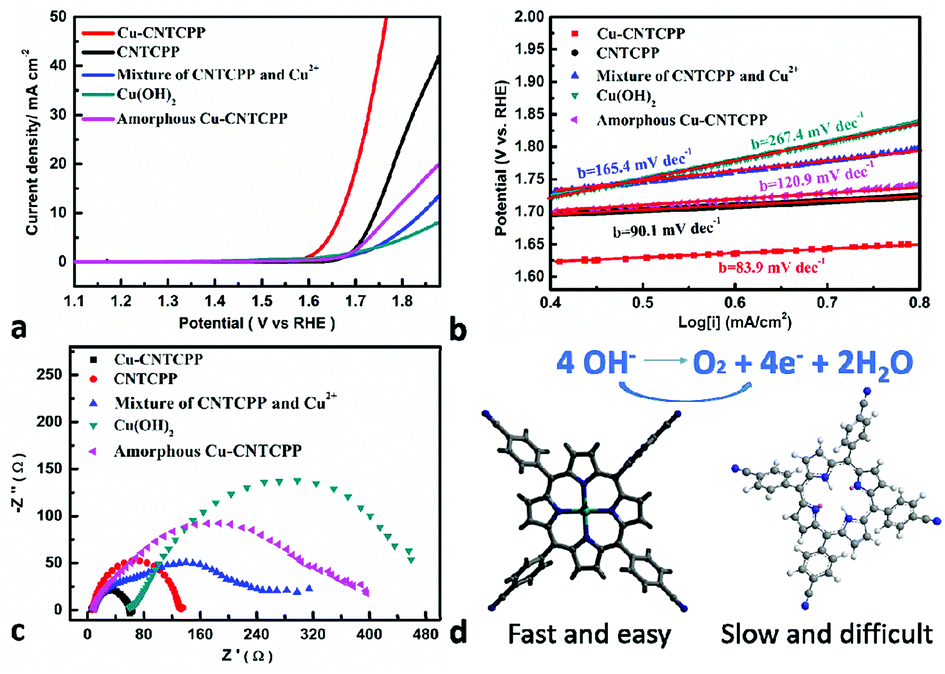 | ||
| Fig. 3 Electrocatalytic activity of the Cu-CNTCPP for OER. (a) Polarization curves. (b) Tafel plots in 1.0 M KOH solution (5 mV s−1). (c) Nyquist plots examined at 0.5 V (vs. SCE). (d) Comparison of coordinated and non-coordinated CNTCPP. | ||
To get further insight into the activity of as-synthesized Cu-CNTCPP modified electrodes toward OER, electrochemical impedance spectroscopy (EIS) analysis was also performed. Fig. 3c describe the obtained Nyquist plots of Cu-CNTCPP and the comparative catalysts, the charge-transfer resistance (Rct) at the surface of the catalysts is determined from the diameter of a semicircle at high frequencies in the Nyquist plot. Generally, Rct value varies inversely with the electrocatalytic activity. That is, smaller diameter corresponds to faster OER kinetics. The Rct values of Cu-CNTCPP (60 Ω) are much lower than the other catalysts. Thus, such a low Rct value of Cu-CNTCPP indicates that its high electrocatalytic activity for OER could be ascribed to the highly conductive Cu2+ hybrid CNTCPP ligands improving the charge transfer characteristics of Cu-CNTCPP. All the results show that the proprieties of Cu-CNTCPP are better than the comparative catalysts. As we know the electrocatalytic reaction is primarily a surface phenomenon, the axial positions of coordinated Cu2+ ions are naked, so it could offer sufficient active sites for OER. In the long-range ordered crystals of Cu-CNTCPP, CNTCPP is supposed to promote electron transport at heterogeneous surface due to its excellent electrical conductive property, which act as an ideal platform for loading Cu2+, so it showed higher catalytic activity than pure CNTCPP ligands, mixture of CNTCPP and Cu2+ and amorphous Cu-CNTCPP.
From the crystal structure of Zn-CNTCPP, there are weak interactions between the two cyanide groups from two different CNTCPP ligands with the Zn2+ ions (Zn–N bond length of 2.645 Å), so it will partially block the catalytic active sites of Zn ions, and the OER performance of it also verified our speculation. Similar with Cu-CNTCPP, we have also measured the electrocatalytic OER activity of Zn-CNTCPP and the comparative catalysts under the same conditions for comparison: ZnCl2 and CNTCPP ligand mixture by manual grinding (named mixture of CNTCPP and Zn2+), ZnCl2 and CNTCPP ligand mixture by reflux in hot DMF solution (named amorphous Zn-CNTCPP), and Zn(OH)2 by conventional precipitin reaction (named Zn(OH)2). Fig. 4a shows the polarization curves of Zn-CNTCPP and the comparative catalysts. As shown, the Zn-CNTCPP deliver a current density of 10 mA cm−2 at a potential of 1.71 V, whereas a potential as high as 1.74 V are required for CNTCPP ligand to achieve the same current density. And for mixture of CNTCPP and Zn2+, amorphous Zn-CNTCPP and Zn(OH)2, it fails to reach 10 mA cm−2 in 1.0 M KOH., the Tafel slope of the Zn-CNTCPP is 87.5 mV dec−1, compared to 272.1 mV dec−1 and 348.2 mV dec−1 for mixture of CNTCPP and Zn2+, amorphous Zn-CNTCPP, respectively (Fig. 4b). The Rct values of Zn-CNTCPP are 140 Ω (Fig. S6†). All the results show the OER properties of Zn-CNTCPP are inferior to Cu-CNTCPP, that coincides with our speculation, that is the weak interactions of Zn–N bond of central Zn(II) ions partially block the catalytic active sites of Zn ions, but the axial positions of coordinated Cu2+ ions are naked, so it offer sufficient active sites for OER and afford more convenient electrocatalytic reaction.
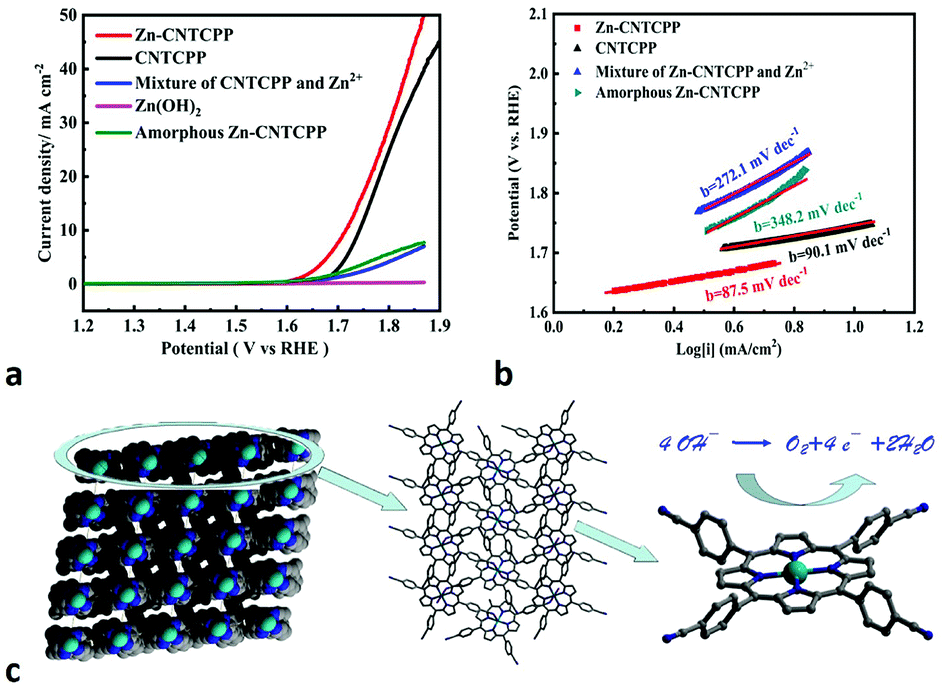 | ||
| Fig. 4 Electrocatalytic activity of the Zn-CNTCPP for OER. (a) Polarization curves. (b) Tafel plots in 1.0 M KOH solution (5 mV s−1). (c) Comparison of coordinated and non-coordinated CNTCPP. | ||
The electrochemical double-layer capacitance (Cdl) was investigated under non-faradaic condition to estimate the electrochemical surface area (ECSA). The cyclic voltammograms at various scan rates of the Cu-CNTCPP, Zn-CNTCPP and CNTCPP were shown in Fig. 5a–c, according to which the Cdl was calculated. As shown in Fig. 5d, the Cdl of the Cu-CNTCPP, Zn-CNTCPP and CNTCPP are 0.531, 0.254 and 0.104 mF cm−2, suggesting that the Cu-CNTCPP may have an advantage in active surface area and active sites over others for oxygen production. Catalytic activity and stability towards OER are two valid parameters for oxygen production. In order to investigate the stability in alkaline solution of the prepared Cu-CNTCPP and Zn-CNTCPP, continuous CV was performed within the potential range of 1.45 to 1.7 V (vs. RHE) for 2000 cycles at scan rate of 50 mV s−1. As shown in Fig. 6a and c, clearly, Cu-CNTCPP exhibits slight loss compared with Zn-CNTCPP. Furthermore, the continuous electrolysis at different current density were monitored over 10 h. Moreover, taking further insight from the i–t curves obtained at 10 mA cm−2, it can be seen that the Cu-CNTCPP keeps approximately 83% compared with the Zn-CNTCPP of 54%. Notably, Cu-CNTCPP exhibits a slight variation over the entire time, indicating the outstanding stability. (Fig. 6b and d). In a word, Cu-CNTCPP with lower potential, Tafel slope and higher estimated Cdl, compared to others as depicted in Table 1, has the potential to be developed to be superior catalytic oxygen production from electrolyzed water.
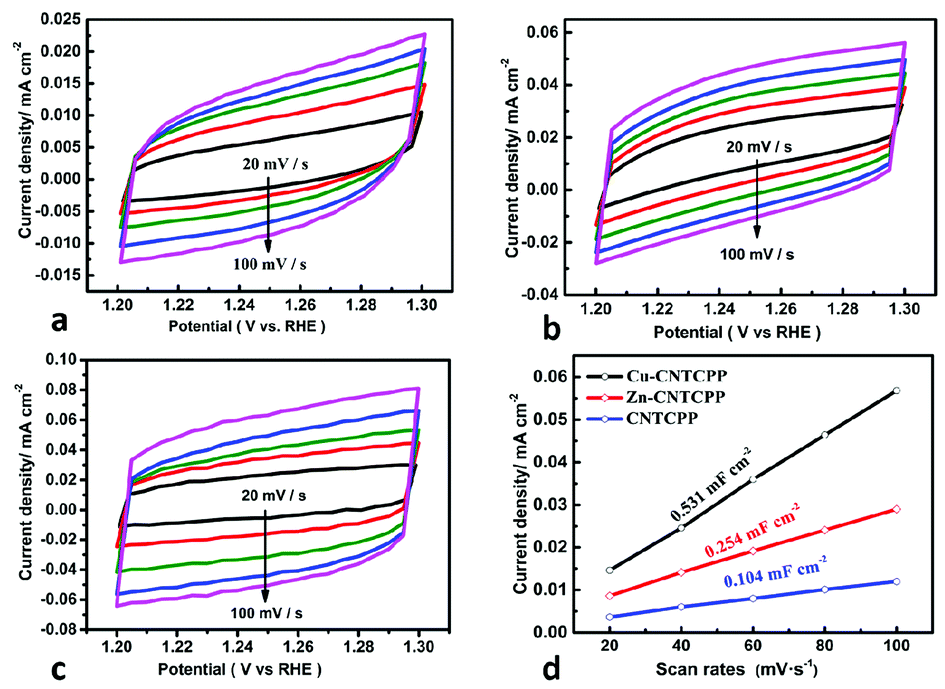 | ||
| Fig. 5 Cyclic voltammograms (CV) of (a) Cu-CNTCPP. (b) Zn-CNTCPP. (c) CNTCPP. (d) Estimated Cdl. | ||
 | ||
| Fig. 6 (a) Polarization curves of Cu-CNTCPP, for initially and after 2000 potential sweeps. (b) Long-term stability test carried out at constant current densities of 10, 20, 30 mA cm−2 for Cu-CNTCPP (inset: time dependent current density curves at current densities of 10 mA cm−2). (c) Polarization curves of Zn-CNTCPP, for initially and after 2000 potential sweeps. (d) Time dependent current density curves for Zn-CNTCPP. | ||
| Electrocatalyst | Electrolytes | η10 (mV) | Tafel slope [mV dec−1] | References |
|---|---|---|---|---|
| Cu-CNTCPP | 1 M KOH | 430 | 83.9 | This work |
| Zn-CNTCPP | 1 M KOH | 480 | 87.5 | This work |
| CNTCPP | 1 M KOH | 510 | 90.1 | This work |
| Mn3O4/CoSe2 | 1 M KOH | 450 | 49 | 1 (ref. 46) |
| Zn-Co-LDH | 0.1 M KOH | 460 | 101 | 2 (ref. 47) |
| Fe doped Co3O4 | 1 M KOH | 370 | 60 | 3 (ref. 48) |
| Fe3O4@Co9S8/rGO-2 | 1 M KOH | 400 | 54.5 | 4 (ref. 49) |
In a word, the OER performance of Zn-CNTCPP with coordination saturation is inferior to the Cu-CNTCPP with coordination unsaturation. The main reason for the difference between Cu-CNTCPP and Zn-CNTCPP in OER performance is due to the presence of weak Zn–N bonds of Zn-CNTCPP, which will partially block the catalytic active sites of Zn ions. Based on the structures and its outstanding OER performance of Cu-CNTCPP, we supposed its reaction mechanism in the OER process, as the axial direction of Cu2+ ions are unsaturated-coordinated, which provides mainly catalytically active sites used for fast dissipation of the electrons generated during OER (Scheme 1): a Cu atom with an oxidation state of 2+ serves as the center, it could be connected by solvent –OH and H2O ligands in 1.0 M KOH solution. As H2O adsorption on the surface is favorable thermodynamically, the OER directly splits H2O on a Cu2+ site to produce an adsorbed –OH. –OH then loses a proton to form an O atom, so a Cu–O− group would form (steps I and II). After the nucleophilic attack of a solvent molecule on the Cu–O− group, a hydroperoxo CuOOH group was yielded (step III). Then a superoxo (step IV) and molecular O2 (step V) could formed. Finally, O2 is evolved, and the catalyst is recovered when a H2O molecule connects to the Cu site (step VI).
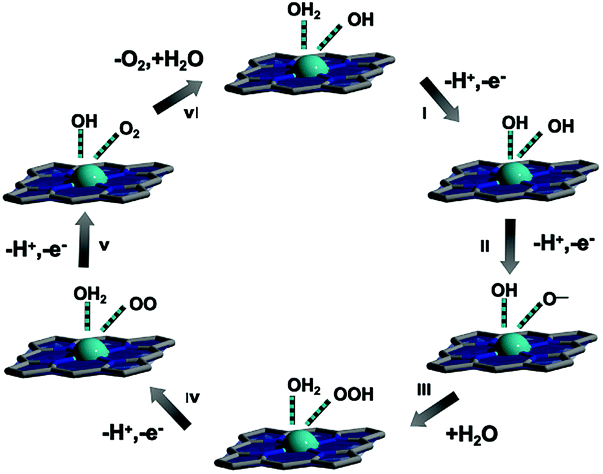 | ||
| Scheme 1 Proposed reaction mechanism of Cu-CNTCPP for OER. | ||
In summary, complexes of Cu-CNTCPP and Zn-CNTCPP have been successfully synthesized and used as OER catalyst by utilizing of conductive CNTCPP porphyrin ligands. As an advanced OER catalyst, Cu-CNTCPP and Zn-CNTCPP catalyst exhibit a low potential of 1.66 V and 1.71 V. This designed 0D Cu-CNTCPP supramoleculer structure possesses plenty of catalytic active sites and high electrical conductivity, which can lead to better OER activity. Compared with those non-noble-metal electrocatalytic materials reported recently, Cu-CNTCPP electrode presents a quite large exchange current density and a relatively small Tafel slope, suggesting it is a promising low-cost and earth-abundant metallic electrocatalyst for OER. We anticipate that the design of the OER electrocatalyst will lead improved strategies and broad the scope for future exploration in MOFs fields.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSF of China (Grant No. 21771191), Taishan Scholar Foundation (ts201511019), the Shandong Natural Science Fund (ZR2017QB012), the Fundamental Research Funds for the Central Universities (14CX02213A, 16CX05015A), the Foundation of State Key Laboratory of Structural Chemistry (20160006), and the Applied Basic Research Projects of Qingdao (16-5-1-95-jch).References
- H. Shin, H. Xiao and W. A. Goddard, J. Am. Chem. Soc., 2018, 140, 6745–6748 CrossRef CAS PubMed.
- Z. D. Huang, J. H. Liu, Z. Y. Xiao, H. Fu, W. D. Fan, B. Xu, B. Dong, D. Liu, F. N. Dai and D. F. Sun, Nanoscale, 2018 10.1039/C8NR06877A.
- M. R. Gao, X. Cao, Q. Gao, Y. F. Xu, Y. R. Zheng, J. Jiang and S. H. Yu, ACS Nano, 2014, 8, 3970–3978 CrossRef CAS PubMed.
- C. Tang, N. Y. Cheng, Z. H. Pu, W. Xing and X. P. Sun, Angew. Chem., Int. Ed., 2018, 54, 9351–9355 CrossRef PubMed.
- F. Song, L. C. Bai, A. moysiadou, S. Lee, C. Hu, L. Liardet and X. L. Hu, J. Am. Chem. Soc., 2018, 140, 7748–7759 CrossRef CAS PubMed.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925–13931 CrossRef CAS PubMed.
- H. Y. Zou, B. W. He, P. Y. Kuang, J. G. Yu and K. Fan, ACS Appl. Mater. Interfaces, 2018, 10, 22311–22319 CrossRef CAS PubMed.
- X. H. Ren, J. Zhou, X. Qi, Y. D. Liu, Z. Y. Huang, Z. J. Li, Y. Q. Ge, S. C. Dhanabalan, J. S. Ponraj, S. Y. Wang, J. X. Zhong and H. Zhang, Adv. Energy Mater., 2017, 7, 1700396 CrossRef.
- J. Li, Y. C. Wang, T. Zhou, H. Zhang, X. H. Sun, J. Tang, L. J. Zhang, A. M. Al-Enizi, Z. Q. Yang and G. F. Zheng, J. Am. Chem. Soc., 2015, 137, 14305–14312 CrossRef CAS PubMed.
- F. Song and X. L. Hu, J. Am. Chem. Soc., 2014, 136, 16481–16484 CrossRef CAS PubMed.
- C. Jin, F. L. Lu, X. C. Cao, Z. R. Yang and R. Z. Yang, J. Mater. Chem. A, 2013, 1, 12170–12177 RSC.
- M. R. Gao, Y. F. Xu, J. Jiang, Y. R. Zheng and S. H. Yu, J. Am. Chem. Soc., 2012, 134, 2930–2933 CrossRef CAS PubMed.
- L. T. Yan, L. Cao, P. C. Dai, X. Gu, D. D. Liu, L. J. Li, Y. Wang and X. B. Zhao, Adv. Funct. Mater., 2017, 27, 1703455 CrossRef.
- D. D. Wang, X. Chen, D. G. Evans and W. S. Yang, Nanoscale, 2013, 5, 5312–5315 RSC.
- X. Long, J. K. Li, S. Xiao, K. Y. Yan, Z. L. Wang, H. N. Chen and S. H. Yang, Angew. Chem., 2014, 126, 7714–7718 CrossRef.
- H. J. Liang, W. Shuang, Y. T. Zhang, S. J. Chao, H. J. Han, X. B. Wang, H. Zhang and L. Yang, ChemElectroChem, 2017, 5, DOI:10.1002/celc.201701074.
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- S. L. Zhao, Y. Wang, J. C. Dong, C. T. He, H. J. Yin, P. F. An, K. Zhao, X. F. Zhang, C. Gao, L. J. Zhang, J. W. Lv, J. X. Wang, J. Q. Zhang, A. M. Khattak, N. A. Khan, Z. X. Wei, J. Zhang, S. Q. Liu, H. J. Zhao and Z. Y. Tang, Nat. Energy, 2016, 1, 16184–16819 CrossRef CAS.
- X. F. Lu, P. Q. Liao, J. W. Wang, J. X. Wu, X. W. Chen, C. T. He, J. P. Zhang, G. R. Li and X. M. Chen, J. Am. Chem. Soc., 2016, 138, 8336–8339 CrossRef CAS PubMed.
- G. Lu and J. T. Hupp, J. Am. Chem. Soc., 2010, 132, 7832–7833 CrossRef CAS PubMed.
- C. Zou and C. D. Wu, Dalton Trans., 2012, 41, 3879–3888 RSC.
- Y. X. Guo, X. Feng, T. Y. Han, S. Wang, Z. G. Lin, Y. P. Dong and B. Wang, J. Am. Chem. Soc., 2014, 136, 15485–15488 CrossRef CAS PubMed.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- C. Dey, T. Kundu, B. P. Biswal, A. Mallick and R. Banerje, Acta Crystallogr., 2014, 70, 3–10 CrossRef CAS PubMed.
- C. Wang, D. M. Liu and W. B. Lin, J. Am. Chem. Soc., 2013, 135, 13222–13234 CrossRef CAS PubMed.
- D. Y. Du, J. S. Qin, S. L. Li, Z. M. Su and Y. Q. Lan, Chem. Soc. Rev., 2014, 43, 4615–4632 RSC.
- A. Fateeva, J. Clarisse, G. Pilet, J. M. Grenèche, F. Nouar, B. K. Abeykoon and M. J. Rosseinsky, Cryst. Growth Des., 2015, 15, 1819–1826 CrossRef CAS.
- M. C. Das, Q. Guo, Y. He, J. Kim, C. G. Zhao, K. Hong and B. Chen, J. Am. Chem. Soc., 2012, 134, 8703–8710 CrossRef CAS PubMed.
- O. K. Farha, A. M. Shultz, A. A. Sarjeant, S. T. Nguyen and J. T. Hupp, J. Am. Chem. Soc., 2011, 133, 5652–5655 CrossRef CAS PubMed.
- J. Yang, Z. Wang, Y. S. Li, Q. X. Zhuang, W. R. Zhao and J. L. Gu, RSC Adv., 2016, 6, 69807–69814 RSC.
- T. Rhauderwiek, N. Heidenreich, H. Reinsch, S. Øien, K. Lomachenko, U. Ruett, A. Soldatov, K. P. Lillerud and N. Stock, Cryst. Growth Des., 2017, 17, 3462–3474 CrossRef CAS.
- H. Shahroosvand, S. Zakavi, A. Sousaraeia, E. Mohajeranic and M. Mahmoudi, Dalton Trans., 2015, 44, 8364–8368 RSC.
- C. N. He, S. Wu, N. Q. Zhao, C. S. Shi, E. Z. Liuc and J. J. Li, ACS Nano, 2013, 7, 4459–4469 CrossRef CAS PubMed.
- M. M. Thackeray, C. Wolvertonb and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854–7863 RSC.
- U. I. Koslowski, I. A. Wurmbach, S. Fiechter and P. Bogdanoff, J. Phys. Chem. C, 2008, 112, 15356–15366 CrossRef CAS.
- S. Kattel, P. Atanassov and B. Kiefer, Phys. Chem. Chem. Phys., 2013, 15, 148–153 RSC.
- J. D. Baran, H. Grönbeck and A. Hellman, J. Am. Chem. Soc., 2014, 136, 1320–1326 CrossRef CAS PubMed.
- F. N. Dai, W. D. Fan, J. H. Bi, P. Jiang, D. D. Liu, X. R. Zhang, H. Lin, C. F. Gong, R. M. Wang, L. L. Zhang and D. F. Sun, Dalton Trans., 2015, 45, 61–65 RSC.
- K. Artyushkova, B. Kiefer, B. Halevi, A. K. Gericke, R. Schloglc and P. Atanassov, Chem. Commun., 2013, 49, 2539–2541 RSC.
- S. M. Vinko, O. Ciricosta and J. S. Wark, Nat. Commun., 2014, 5, 3533 CrossRef CAS PubMed.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- J. M. Thomsen, S. W. Sheeha, S. M. Hashmi, J. Campos, U. Hintermair, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2014, 136, 13826–13834 CrossRef CAS PubMed.
- S. Dong and J. Z. Jiang, Acta Crystallogr., 2011, 67, 431, DOI:10.1107/S160053681100849X.
- J. A. Hedo, M. Kasuga, E. V. Obberghen, J. Roth and C. R. Kahn, J. Org. Chem., 1981, 46, 4792–4795 CrossRef.
- D. A. Smith, D. N. Reynolds and L. K. Woo, J. Am. Chem. Soc., 1993, 115, 2511–2513 CrossRef CAS.
- M. R. Gao, Y. F. Xu, J. Jiang, Y. R. Zheng and S. H. Yu, J. Am. Chem. Soc., 2012, 134, 2930–2933 CrossRef CAS PubMed.
- C. Qiao, Y. Zhang, Y. Zhu, C. Cao, X. Bao and J. Xu, J. Mater. Chem. A, 2015, 3, 6878–6883 RSC.
- C. Xiao, X. Lu and C. Zhao, Chem. Commun., 2014, 50, 10122–10125 RSC.
- J. Yang, G. Zhu, Y. Liu, J. Xia, Z. Ji, X. Shen and S. Wu, Adv. Funct. Mater., 2016, 26, 4712–4721 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, PXRD, TGA curves, crystal data and structure refinement for complexes, method for electrochemical measurements. CCDC 1450485 and 1449406. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra08338j |
| This journal is © The Royal Society of Chemistry 2018 |