DOI:
10.1039/C8RA08285E
(Paper)
RSC Adv., 2018,
8, 39352-39361
The efficiency and mechanism of dibutyl phthalate removal by copper-based metal organic frameworks coupled with persulfate†
Received
7th October 2018
, Accepted 19th November 2018
First published on 26th November 2018
Abstract
Copper-based metal organic framework (Cu-BTC) was prepared and used to remove dibutyl phthalate (DBP) in the presence of persulfate (PS). The surface characteristics, textural properties, and stability of activated Cu-BTC (denoted as Cu-BTC-A) were evaluated by scanning electron microscope (SEM), X-ray diffraction (XRD), Fourier transform infrared (FTIR), X-ray photoelectron spectroscopy (XPS), Raman spectroscopy, N2 physical adsorption–desorption, electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV). The effects of parameters such as initial pH, PS concentration, catalyst dosage, and free-radical quenchers have been investigated. The results showed that DBP could be removed in a wide pH range by Cu-BTC-A via mechanisms of adsorption and heterogeneous catalytic reaction. Unfortunately, the DBP removal was not completed because of radical scavenging reactions in Cu-BTC-A cages where PS can enter freely but DBP is blocked outside. Another explanation was that Cu-BTC-A showed a low adsorption capacity for DBP because the molecular size of DBP (15.84 × 11.00 × 7.56 Å) is larger than microporous cages (approximately 9 × 9 Å in diameter) of Cu-BTC-A.
1. Introduction
Metal–Organic Frameworks (MOFs), a class of crystalline materials having infinite network structures built with multitopic organic ligands and metal clusters, have evoked significant research interest in recent years.1,2 MOFs have been widely used because of their attractive features such as high crystallinity, open porous structure, high surface area, and a high volume fraction of metal ions.3–6 Indeed, MOFs first received tremendous attention as adsorbents for gas (hydrogen or methane) storage.7,8 With the research and development of MOFs, it is well realized that they are a kind of potential heterogeneous catalysts for water purification.9,10 Although more and more new methods are developed for organic wastewater treatment,9–11 advanced oxidation processes (AOPs) are considered to be superior to others in view of the creation of highly reactive radicals, such as hydroxyl radical (·OH), sulfate radical (SO4˙−), and superoxide radical (O2˙−).12,13 Sulfate radical-based AOP (SR-AOP) has been the focus of recent research on the removal of refractory organic pollutants from water due to its a variety of advantages, including high redox potential of SO4˙−, a wide range of application pH, easy operation, and low harmful secondary products.2,11,14 However, the generation of SO4˙− by decomposition of persulfate (PS) or peroxymonosulfate (PMS) relies on extra energy and/or catalysts (eqn (1)–(3)).13,15| | |
Photochemical activation: S2O82− + hv → 2SO4˙−
| (1) |
| | |
Thermal activation: S2O82− + heat → 2SO4˙−
| (2) |
| | |
Chemical activation: S2O82− + Mn+ → SO4˙− + M(n+1)+ + SO42−
| (3) |
Among these activation methods, chemical activation using ferrous ions as catalysts appears to be an inexpensive and practical way, but the formation of a great deal of iron sludge limits its widespread application.16 To avoid iron sludge formation, a worldwide effort has been exhausted to search alternative catalysts with excellent catalytic activity and long useful lives.10,17,18 Many studies indicated that heterogeneous AOPs exhibited better efficiency than homogeneous reactions through enhancing generation of free radicals and expanding effective pH range. As aforementioned, the central metals and coordinative unsaturated sites make MOFs possible to be excellent heterogeneous catalysts. Recently, several attempts have been made to judge the feasibility of MOFs as heterogeneous catalysts.2,10,19 For example, MIL-53(Fe) was employed to activate hydrogen peroxide (H2O2) for degradation of methylene blue dye under visible light.19 However, this process showed weak Fenton activity because the chemical state of iron in MIL-53(Fe) structure is saturated with Fe(III). Other Fe-based metal–organic frameworks including MIL-101(Fe), MIL-100(Fe), and MIL-88B(Fe) were also examined as catalysts of PS to remove acid orange 7 in aqueous solution,10 with low degradation efficiency due to the weak reaction activity between Fe(III) and PS. Therefore, synthesis of MOFs containing Fe(II) or other unsaturated metal sites is necessary to enhance the catalytic performance of MOFs coupled with PS or H2O2. Nevertheless, most Fe-based MOFs usually contain Fe(III) rather than Fe(II) because of the high reducibility of Fe(II) that would be oxidized during the synthesis process.
It is well known that copper metal–organic framework Cu3(BTC)2(H2O)3 (Cu-BTC, BTC = 1,3,5-benzene-tricarboxylate) is a commonly used porous coordination polymer containing framework Cu(II) species. In addition, the chemical state of copper would not change and the crystalline nature of Cu-BTC still keep well after removing the coordinated water (see Fig. S2†).20 Moreover, Cu(II) species had been reported to exhibit catalytic activity on PS and generate SO4˙−.18 Hence, Cu-BTC has been speculated to be an efficient catalyst of H2O2 for oxidation of aromatic substrates.20 The biodegradability of wastewater was significantly improved while Cu-BTC was used as a heterogeneous catalyst of H2O2 for treatment of olive oil mill wastewater.21 Huang et al. (2015) also employed Cu-MOFs as heterogeneous catalysts of H2O2 for oxidation of simulated phenol wastewater, which obtained satisfactory results with a phenol degradation level of 99% and a maximum COD removal efficiency of 93% under optimum conditions.22 Thus, it is reasonable to expect that Cu-BTC would be an effective catalyst of PS for degradation of persistent pollutants with production of reactive radicals. Hence, the objective of this work is to evaluate the feasibility and mechanism of Cu-BTC as catalyst of PS for removal of dibutyl phthalate (DBP).
Very recently, we have reported the potential application of Co-BTC for oxidation of DBP2 and herein, Cu-BTC was synthesized and its catalytic activity as a heterogeneous catalyst of PS was evaluated. The possible removal mechanism of DBP was investigated, as well as the catalytic mechanism. DBP, a kind of commonly used phthalic acid esters, was selected as the target pollutant to evaluate the catalytic performance due to its toxicity to living organisms and wide use in various industries.23,24 The results of this study will present a new extension of MOFs and also make a contribution to the development of AOPs.
2. Materials and methods
2.1 Materials
Unless otherwise specified, all chemicals and reagents were of reagent grade and used without further purification. Copper(II) nitrate trihydrate (Cu(NO3)2·3H2O) was obtained from Sinopharm Chemical Reagent Co., Ltd (Beijing, China). DBP (purity > 99.5% (GC)), 1,3,5-benzenetricarboxylic acid (BTC) were purchased from Aladdin chemistry Co., Ltd (Shanghai, China). Sodium persulfate (Na2S2O8, 99.0%) and all other chemicals such as dimethyl formamide (DMF), sodium hydroxide (NaOH) and sulfuric acid (H2SO4) were provided by Sinopharm Chemical Reagent Co., Ltd (Beijing, China).
2.2 Preparation of Cu-BTC
Cu-BTC was prepared via a method similar to that reported in the literature with certain modification.1,20 Briefly, copper nitrate (1.94 g) was dissolved in 30 mL of deionized water and benzene-1,3,5-tricarboxylic acid (BTC) (0.84 g) was dissolved in 30 mL of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ethanol-DMF solvent mixture, then these two solutions were mixed together and stirred for 15 min under vigorous magnetic stirring at room temperature, followed by transferring into a Teflon-lined stainless steel autoclave with a volume of 100 mL and heated up to 100 °C for 24 h. In order to get the optimal synthesis condition, the temperature of 120, 140, 160, 180 °C and the reaction time of 12, 18, 36, 48 h were also used to prepare Cu-BTC. After the heat treatment, the autoclave was allowed to cool naturally to room temperature, and the collected products were then refined through centrifugation at 6000 rpm for 2 min. To remove the excess solvent, the obtained blue powder was extensively rinsed with deionized water (3 × 20 mL) along with alternation of ethanol (3 × 20 mL), and then dried in full vacuum at 60 °C for 24 h.
:1 ethanol-DMF solvent mixture, then these two solutions were mixed together and stirred for 15 min under vigorous magnetic stirring at room temperature, followed by transferring into a Teflon-lined stainless steel autoclave with a volume of 100 mL and heated up to 100 °C for 24 h. In order to get the optimal synthesis condition, the temperature of 120, 140, 160, 180 °C and the reaction time of 12, 18, 36, 48 h were also used to prepare Cu-BTC. After the heat treatment, the autoclave was allowed to cool naturally to room temperature, and the collected products were then refined through centrifugation at 6000 rpm for 2 min. To remove the excess solvent, the obtained blue powder was extensively rinsed with deionized water (3 × 20 mL) along with alternation of ethanol (3 × 20 mL), and then dried in full vacuum at 60 °C for 24 h.
2.3 Catalytic potential of Cu-BTC
A shake-flask test was used to evaluate the catalytic and adsorptive performance of Cu-BTC for removal of DBP in the presence of PS. Typically, the experiments were performed in 250 mL serum bottles containing 100 mL of 5.0 mg L−1 DBP solution. The reaction was initialed by adding PS and Cu-BTC, immediately, the serum bottles were put in the rotary shaker (ZHWY-20102C, Shanghai, China) with operating parameters of 180 rpm and 25 ± 0.2 °C. At each predetermined time intervals, 1.0 mL aliquot of reaction solution was taken into a 10 mL serum bottle where 1.0 mL of ethanol was added previously for quenching the reaction. The sample was filtered through a 0.45 μm nylon membrane filter and analyzed for DBP concentration. To assess the effect of PS dosage, Cu-BTC dosage, initial pH, reaction temperature, and free-radical quenchers on DBP removal, a series of similar experiments were carried out. The initial pH was adjusted by 1 M sodium hydroxide (NaOH) or sulfuric acid (H2SO4) and monitored by pH meter (Shanghai LeiCi PHS-25) equipped with a pH electrode.
2.4 Characterization
Details of characterization of Cu-BTC-A with scanning electron microscope (SEM), X-ray diffraction (XRD), Raman spectra, Fourier transform infrared (FTIR), N2 physical adsorption–desorption, X-ray photoelectron spectroscopy (XPS), electrochemical impedance spectra and (EIS) and cyclic voltammograms (CV) are displayed in the ESI (Text S1).†
2.5 Analytical methods
Details of the HPLC analysis method used to detect the concentration of DBP are presented in the ESI (Text S2).†
3. Results and discussion
3.1 Structural and morphological characterization
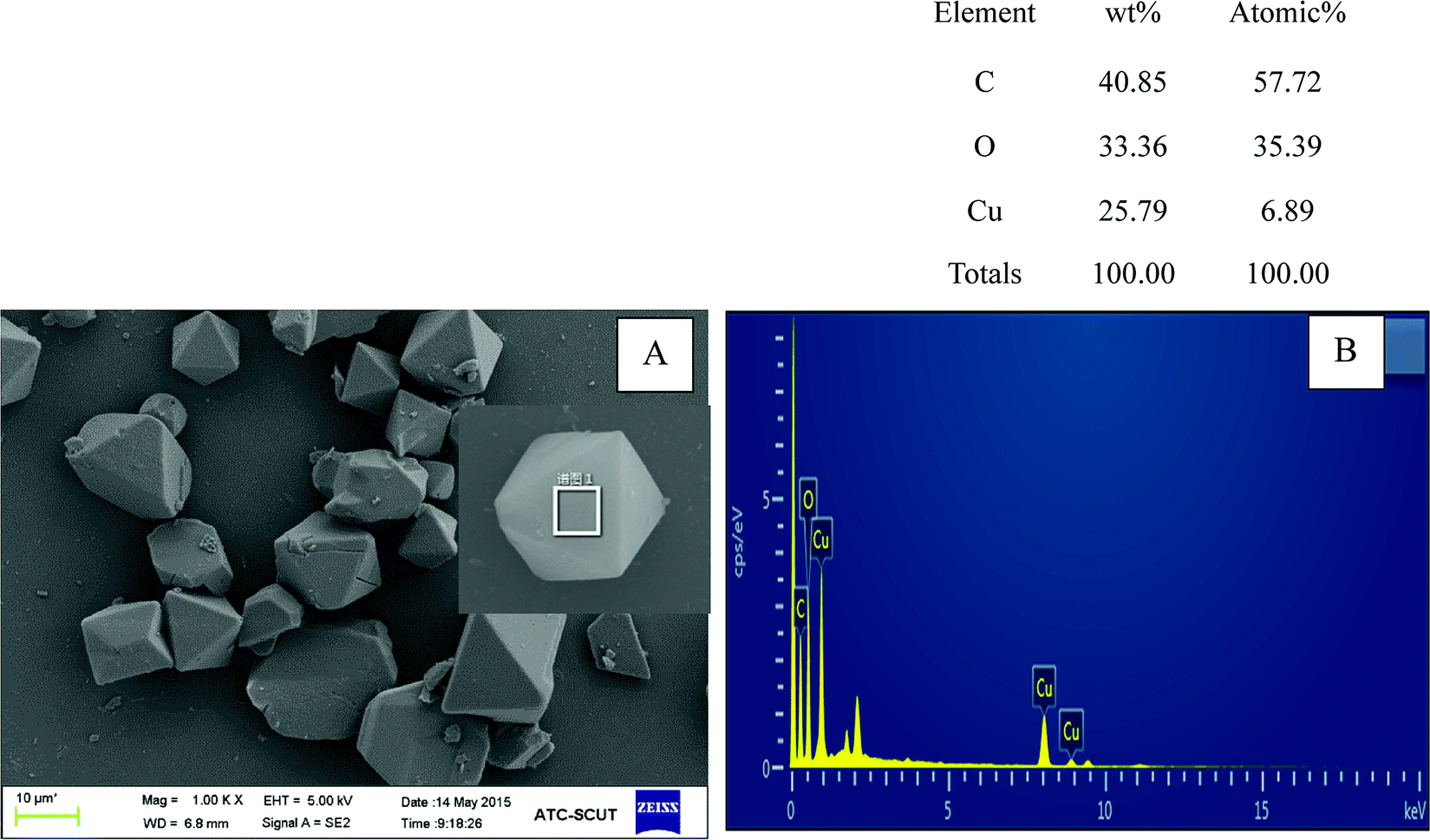
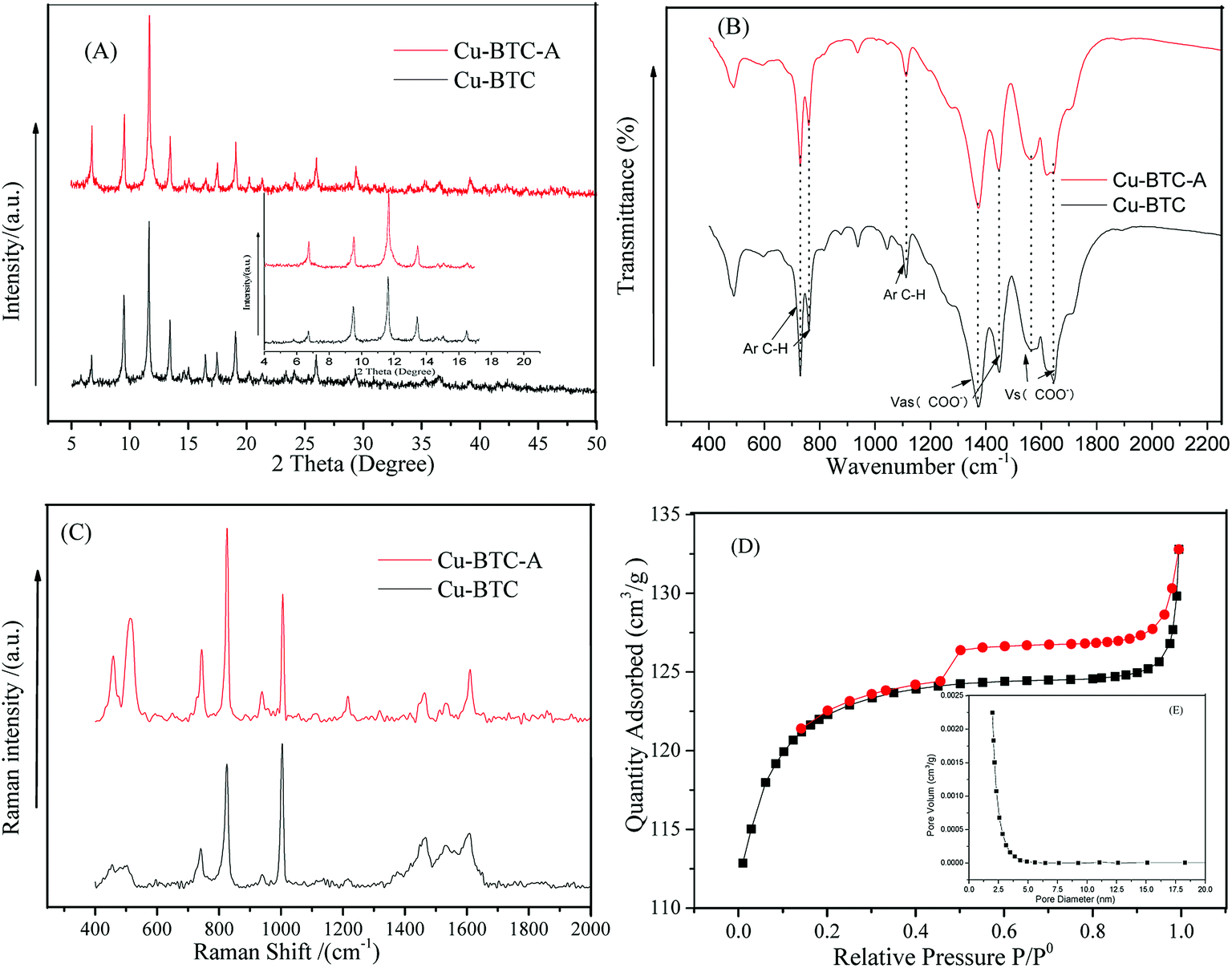
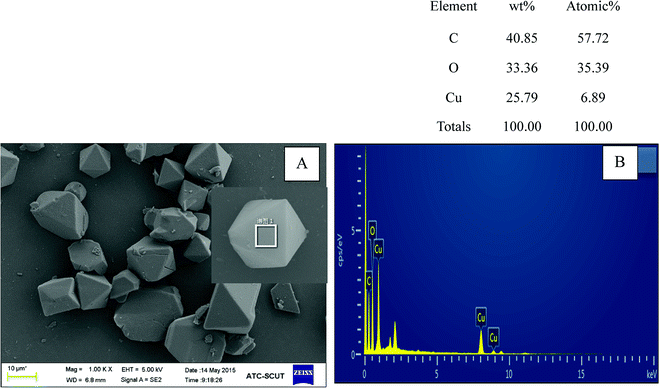
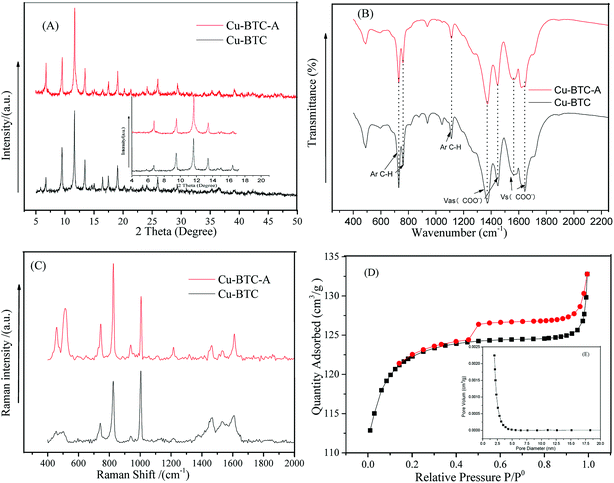
Fig. 1A depicted the scanning electron microscopy (SEM) image of as-synthesized Cu-BTC. Apparently, the image showed a octahedron-shaped structure which possessed an average size of ca. 10 μm. The elements of Cu, C, and O were main compositions of the as-synthesized Cu-BTC, which can be confirmed by energy-dispersive X-ray spectroscopy (EDS) (Fig. 1B). The crystal phases of as-synthesized Cu-BTC were investigated by XRD analysis. As shown in Fig. 2, diffraction peaks located at 7.2, 9.8, 12, 13.8, 14.2, 14.8, 18, 21, 26.7, and 29.5° were indexed to the indices (200), (220), (222), (400), (331), (420), (511), (440), (553) and (555) of Cu-BTC.20,25 Moreover, the XRD pattern was essentially identical to that of the stimulated XRD pattern of Cu-BTC powder,26 verifying the successful synthesis of Cu-BTC. However, weak peaks were also found at 37.5° and 42.5° assigned to Cu2O as a by-product of Cu-BTC.27 The FT-IR spectroscopy and Raman spectra were conducted to identify characteristic functional groups associated with as-synthesized Cu-BTC. As seen in Fig. 2B, the FT-IR spectra of Cu-BTC exhibited characteristic absorption peaks at 1665, 1589, 1396, 1442, 1112, 763, and 735 cm−1. The strong bands at 1396 cm−1 and 1442 cm−1 were due to the presence of asymmetric (vas(COO−)) while bands at 1589 cm−1 and 1665 cm−1 were both attributed to the symmetric (vs(COO−)) vibrations of carboxyl groups. Meanwhile, the absorption peak at 1112 cm−1 denoted the C–H bending modes of the benzene ring in the plane. These bands together with the out of plane C–H bending vibration of benzene ring located at 763 and 735 cm−1 reflected the presence of dicarboxylate.28 The Raman spectra exhibited a ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) modes of the benzene ring at 1606 and 1002 cm−1, and the peaks at 816 and 720 cm−1 were attributed to the (C–H) bending vibrations and ring bending of out-of-plane, respectively. Meanwhile, the doublet at 1521 and 1458 cm−1 corresponded to the vibrational modes of vas(COO−) and vs(COO−).29 The observed bands at the low-frequency region of 500 and 442 cm−1 were assigned to the presence of Cu(II) species.29 These results indicated the formation of “nodes” and “bridges” with the respective Cu(II) and BTC. The permanent porosity of Cu-BTC was verified by N2 sorption at 77 K via Brunauer–Emmett–Teller (BET) analysis, as shown in Fig. 2D. The BET surface areas and pore volume were 410 m2 g−1 and 0.196 cm3 g−1, respectively, these results fully agree with those of previous study.30 It was interesting to note that a hysteresis loop beginning at ∼P/P0 = 0.45 and ending at P/P0 = 1.0 was indicative of the presence of mesoporosity,25 which could be the explanation for the low surface areas. But the difference between the adsorption and desorption was less than 10 cm3 g−1, revealing a small amount of mesopores in the structure. As shown in the inset of Fig. 2D, the BJH pore size calculated from the desorption branch was smaller than 5 nm and the pore volume was lower than 0.0025 cm3 g−1, further confirmed that most of pores in Cu-BTC were micropores. According to these characterization results, we can conclude that Cu-BTC material was successfully synthesized.
C) modes of the benzene ring at 1606 and 1002 cm−1, and the peaks at 816 and 720 cm−1 were attributed to the (C–H) bending vibrations and ring bending of out-of-plane, respectively. Meanwhile, the doublet at 1521 and 1458 cm−1 corresponded to the vibrational modes of vas(COO−) and vs(COO−).29 The observed bands at the low-frequency region of 500 and 442 cm−1 were assigned to the presence of Cu(II) species.29 These results indicated the formation of “nodes” and “bridges” with the respective Cu(II) and BTC. The permanent porosity of Cu-BTC was verified by N2 sorption at 77 K via Brunauer–Emmett–Teller (BET) analysis, as shown in Fig. 2D. The BET surface areas and pore volume were 410 m2 g−1 and 0.196 cm3 g−1, respectively, these results fully agree with those of previous study.30 It was interesting to note that a hysteresis loop beginning at ∼P/P0 = 0.45 and ending at P/P0 = 1.0 was indicative of the presence of mesoporosity,25 which could be the explanation for the low surface areas. But the difference between the adsorption and desorption was less than 10 cm3 g−1, revealing a small amount of mesopores in the structure. As shown in the inset of Fig. 2D, the BJH pore size calculated from the desorption branch was smaller than 5 nm and the pore volume was lower than 0.0025 cm3 g−1, further confirmed that most of pores in Cu-BTC were micropores. According to these characterization results, we can conclude that Cu-BTC material was successfully synthesized.
 |
| | Fig. 1 SEM image (A) and EDS (B) of as-synthesized Cu-BTC; synthesis condition: T (temperature) = 100 °C; t (time) = 24 h. | |
 |
| | Fig. 2 XRD pattern (A), FT-IR spectra (B), and Raman spectra (C) of Cu-BTC before and after activation; N2 physical adsorption–desorption of as-synthesized Cu-BTC (D) (inset: pore size distribution of mesopores). Synthesis condition: T (temperature) = 100 °C; t (time) = 24 h. | |
3.2 Effect of thermo-activation and catalytic activity of Cu-BTC
It has previously been reported that Cu(II) complexes show excellent catalytic activity for decomposition of H2O2 only if the metal coordination sphere appears in unsaturated conditions.31 Hence, in order to get coordinative unsaturated Cu(II) metal sites, the as-synthesized Cu-BTC was activated under 230 °C for 2 h with the medium of nitrogen in a horizontal quartz tube, the obtained product was denoted as Cu-BTC-A. The temperature of 230 °C was chosen based on the TGA data of Cu-BTC from previous studies.32 The sky blue crystal powder turned to an evident dark blue after heating, which showed good agreement with previous research.26 These results indicated coordinated water removal and Cu-BTC-A was able to decompose PS in theory because of the open unsaturated Cu(II) metal sites. The XRD, FT-IR, and Raman spectra were also investigated after thermo-activation (see Fig. 2). There was no obvious change in the XRD pattern and FT-IR spectrum before and after activation, implying that the crystal structure powder of Cu-BTC was not damaged during the thermal activation. Some small differences were observed in two samples of XRD pattern, whereas the intensity ratio of I200/I220 significantly increased while the I331/I420 decreased for Cu-BTC-A sample. These results were in good accordance with the calculated patterns of dehydrated Cu-BTC in previous research.26 Additionally, it was evident that peaks in the 800–400 cm−1 range of Raman spectra involving Cu(II) species became dramatically stronger. All these features can be explained in terms of a change in the first coordination sphere of Cu(II) species upon removal of water,31 which demonstrated activating of Cu-BTC and leaving unsaturated Cu(II) sites available for catalyzing PS.
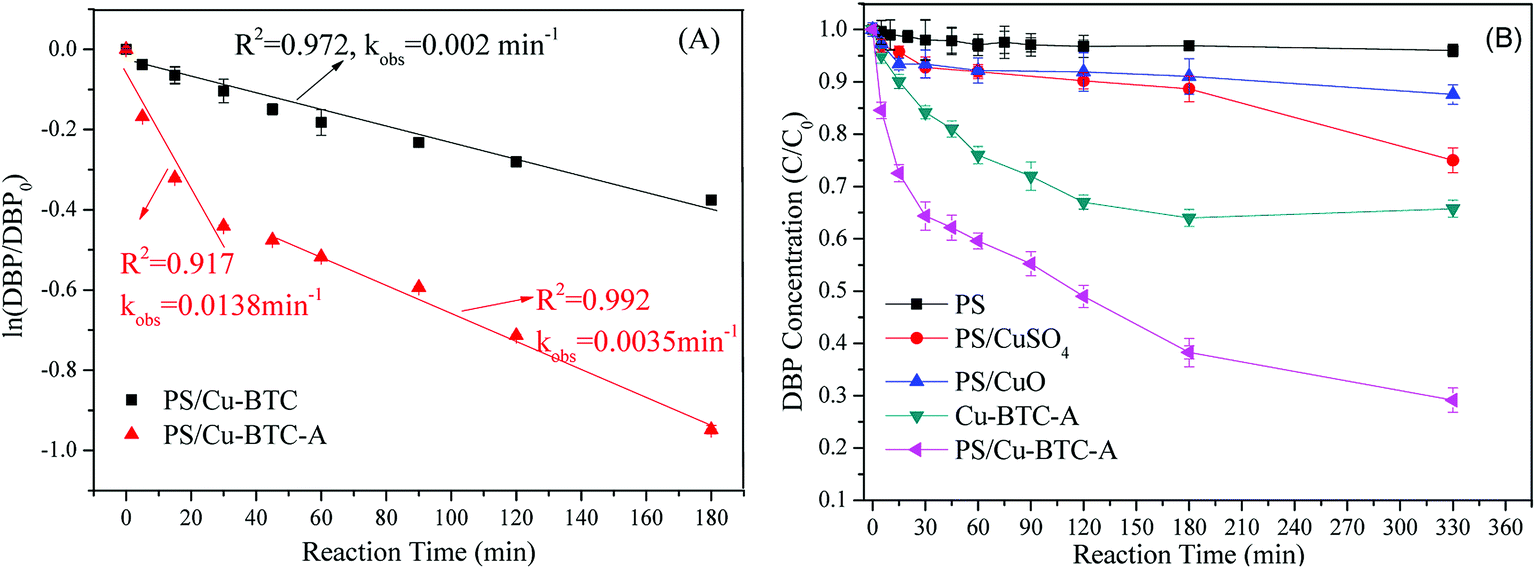
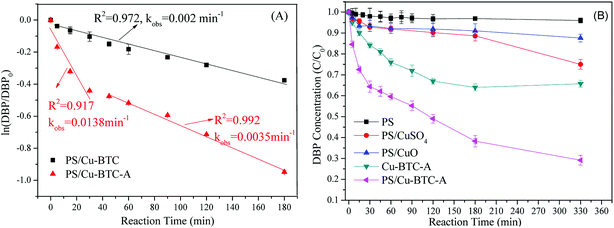
The catalytic performance of Cu-BTC before and after activation was conducted. As displayed in Fig. 3A, the pseudo-first-order rate constant of DBP removal was 0.002 min−1 in Cu-BTC/PS system. However, the DBP degradation behavior in the Cu-BTC-A/PS system was markedly different from that of Cu-BTC/PS system, where DBP removal showed two-stage first-order degradation kinetic. The two-stage kinetic was composed of an initial rapid removal stage (first-stage) and a followed slow removal stage (second-stage). The high kobs of first stage could be mostly ascribed to the both adsorption and degradation reactions occur on the Cu-BTC-A surface simultaneously. The reason of lower kobs of second stage may be explained by desorption of DBP from surface or active sites of Cu-BTC-A and scavenge reactions in the cages of Cu-BTC-A and resulted in the slow removal of DBP. Meanwhile, the two-stage kinetic also does a good explaining for the phenomenon that a turning point appeared on the curve of the DBP removal in the PS/Cu-BTC-A system (Fig. 3B). Similar result had previously been observed in other heterogeneous catalytic material in the literature.33 In Fig. 3B, it was worth to note that about 25% of DBP removal was observed in the system with only Cu-BTC-A, confirming that Cu-BTC-A showed weak adsorption capacity for DBP. However, an obvious improvement of DBP removal was noticed when both PS and Cu-BTC-A were added into the DBP solutions, where 70.9% of DBP was removed. This fact further proved that Cu-BTC after activation showed catalytic performance for decomposition of PS and generating free radicals. Though there is H2O that could re-coordinate Cu(II) sites of Cu-BTC-A during the catalytic reaction, Cu(II) will coordinate to PS firstly due to the much more reactive nucleophile of PS than that of H2O.34 Hence, the subsequent experiments were conducted using Cu-BTC-A.
 |
| | Fig. 3 (A) Effect of thermo-activation on pseudo-constant of DBP removal; (B) DBP removal in different systems. Experiment condition: DBP = 0.018 mM; PS = 1.08 mM; catalyst = 1.0 g L−1; T = 25 °C; no pH adjustment. | |
To achieve a better understanding of the role of synthesis process parameters on the catalytic performance, Cu-BTC-A was synthesized under different temperature for different reaction time. As illustrated in Fig. S5 and S6,† Cu-BTC-A synthesized at 100 °C for 24 h showed the highest removal efficiency of DBP. Consequently, the subsequent experiments were conducted using Cu-BTC-A synthesized at 100 °C for 24 h.
3.3 Effect of initial pH and scavengers on DBP degradation
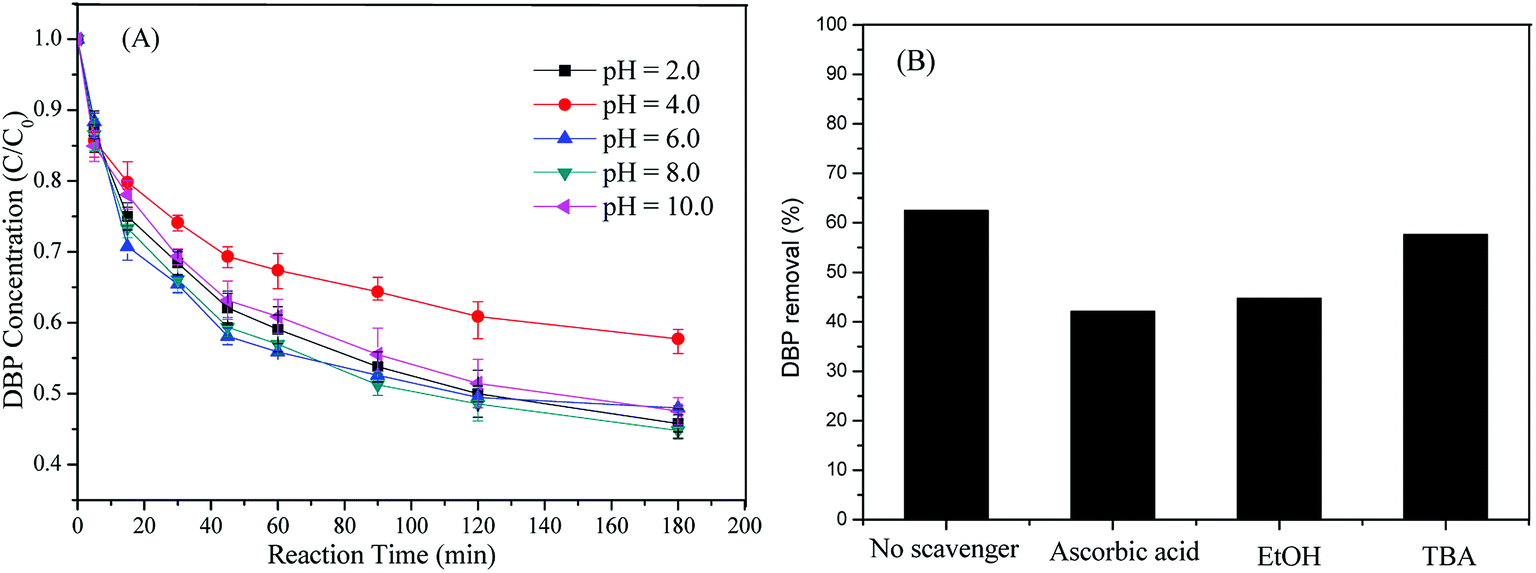
As shown in Fig. 4A, heterogeneous catalysts of Cu-BTC-A worked effectively over a broad pH range from 2.0 to 10.0, compared with the conventional catalysts including Fe2+ (pH < 3), zero-valent iron, and Fe3O4.13,16,35 However, DBP removal at pH 4.0 was a little lower than those of other pH values. This result was largely different from the homogeneous system16 where the acidic condition was more favorable to organic pollutants degradation. In addition, it seemed that there was no evident difference in DBP removal among different initial pH values except pH 4.0. But this heterogeneous process exhibited better efficient at neutral solutions (pH 6.0 and pH 8.0), attaining a maximum value of DBP removal at a pH of 6.0. This fact reveals that this catalyst is a good option for treatment of wastewaters without pre-adjusting their pH due to the pH of industrial organic contaminated wastewaters is often around 7.0.33
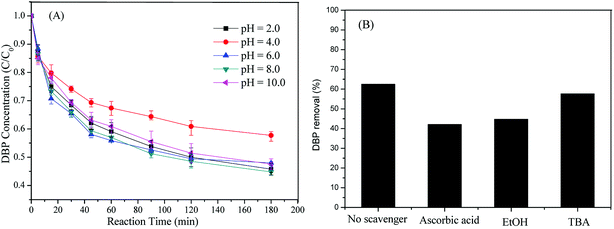 |
| | Fig. 4 (A) Effect of initial pH on DBP removal; (B) effect of radical scavengers on DBP removal. Experiment condition: DBP = 0.018 mM; PS = 1.08 mM; Cu-BTC-A = 1.0 g L−1; T = 25 °C; no pH adjustment. | |
DBP can be removed by adsorption, via a catalytic degradation, or by both processes. In order to gain deep insight into DBP removal mechanism, scavenging experiments were investigated. Ascorbic acid, tertiary butanol (TBA), and ethanol (EtOH) are well-known scavengers for quenching free radicals (SO4˙− and ·OH) because of their priority reaction with radicals with high second-order rate constants.36 Fig. 4B displayed the DBP removal in the presence of scavengers with a concentration of 18 mM. Evidently, DBP decomposition was declined from 62.5% to 42.1% and 44.7% after the addition of ascorbic acid and EtOH, respectively, indicating the existence of SO4˙− or ·OH. However, it was noted that the inhibition on DBP with TBA was smaller than those in the presence of ascorbic acid and EtOH under exactly the same conditions. This result may be explained that both SO4˙− and ·OH are responsible for DBP degradation due to that TBA is a perfect scavenger for ·OH rather than SO4˙− while both ascorbic acid and EtOH are good candidates for quenching SO4˙− and ·OH. In addition, our previous studies35,36 found that most of DBP removal was inhibited with the presence of radical scavengers in ZVI/PS and Fe0@Fe3O4/PS systems. The inhibition on DBP removal by the same scavengers in this study was much weaker than those two systems. This was most likely that partial DBP removal was attributed to the absorption by Cu-BTC-A.
3.4 Effect of PS and Cu-BTC-A dosages on DBP removal
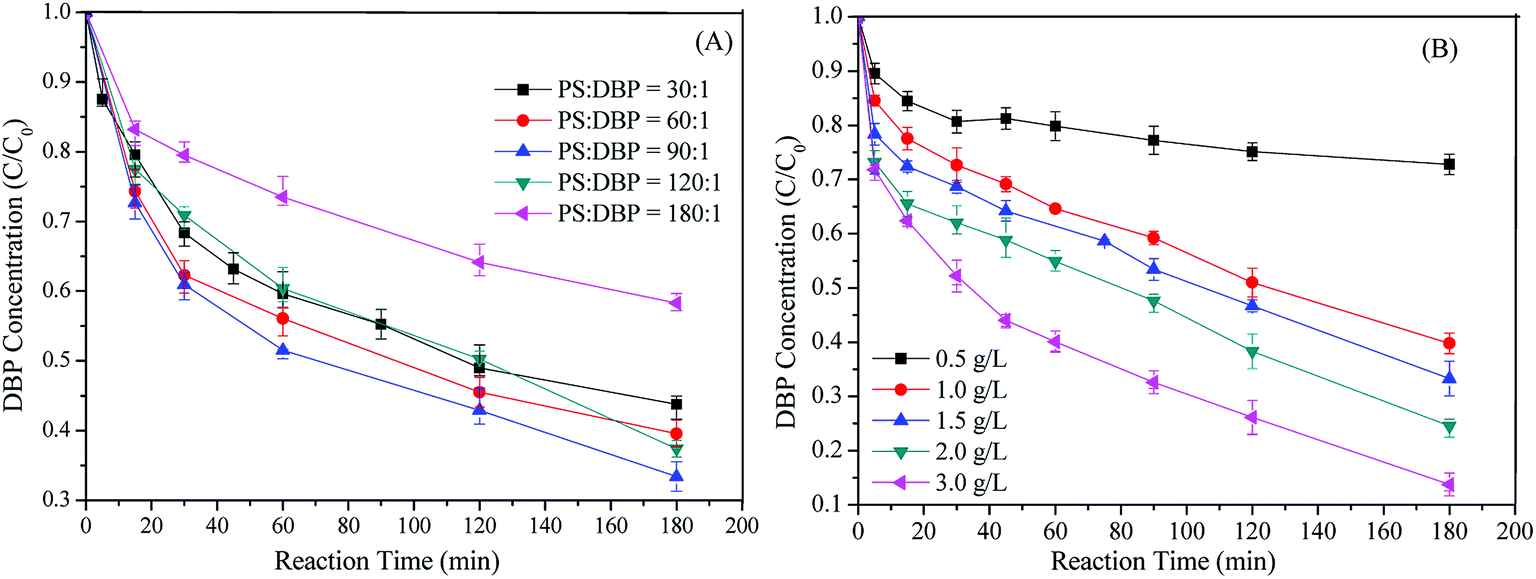
As illustrated in Fig. 5A, DBP removal efficiency slowly increased as the molar ratio of [PS]/[DBP] increasing from 30:1 to 90:1. This indicated the existence of the adsorption mechanism for the DBP removal. But the conclusion that Cu-BTC-A could catalyze PS to generate SO4˙− also confirmed by the enhancement on DBP removal. Hence, it is reasonable to increase PS dosage for enhancing DBP removal. Nevertheless, a further increase of PS concentration resulted in lower degradation efficiency when [PS]/[DBP] exceeds 90/1. This could be elucidated by the well known scavenging effect of SO4˙− (eqn (4) and (5)), which leaded to competitive reactions between pollutants and SO4˙−. Therefore, [PS]/[DBP] of 90/1 was perfect for this system, as the use of excess oxidant not only was wasteful and costly but also decreased the DBP removal efficiency. Furthermore, it should be noted that the highest DBP removal was only 68.9% within 180 min. The possible explanation was due to the lack of catalytic sites of Cu(II) comparing to PS/Cu2+ system, as the amount of Cu(II) in the Cu-BTC was only 4.67% of atomic concentration based on XPS analysis in this study. In order to confirm this speculation, the effect of Cu-BTC-A dosages on DBP removal was also conducted.| | |
SO4˙− + SO4˙− → 2SO42−
| (4) |
| | |
S2O82− + SO4˙− → SO42− + S2O8˙−
| (5) |
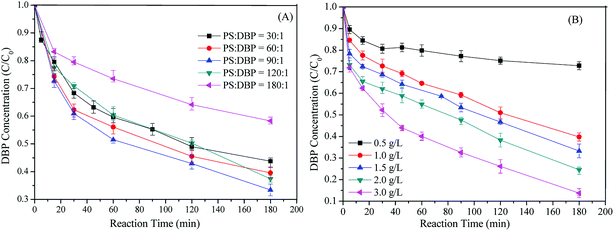 |
| | Fig. 5 (A) Effect of PS concentration on DBP removal; (B) effect of catalyst dosages on DBP removal. Experiment condition: DBP = 0.018 mM; PS = 1.08 mM (for B); Cu-BTC-A = 1.0 g L−1 (for A); T = 25 °C; no pH adjustment. | |
Fig. 5B showed the relationship between DBP removal and Cu-BTC-A dosages. It was found that DBP removal was considerable accelerating with increasing Cu-BTC-A dosages. When Cu-BTC-A dosage increased from 0.5, 1.0, 1.5, 2.0 to 3.0 g L−1, DBP removal efficiency increased from 25.5%, 52.4%, 63.6%, 72.5% to 88.7%, respectively. This behavior was related to the increased catalytic sites for PS contacting and resulted in more free radicals, implying that the density of metal ions play an important role in its catalytic performance.
3.5 Effect of temperature on DBP removal
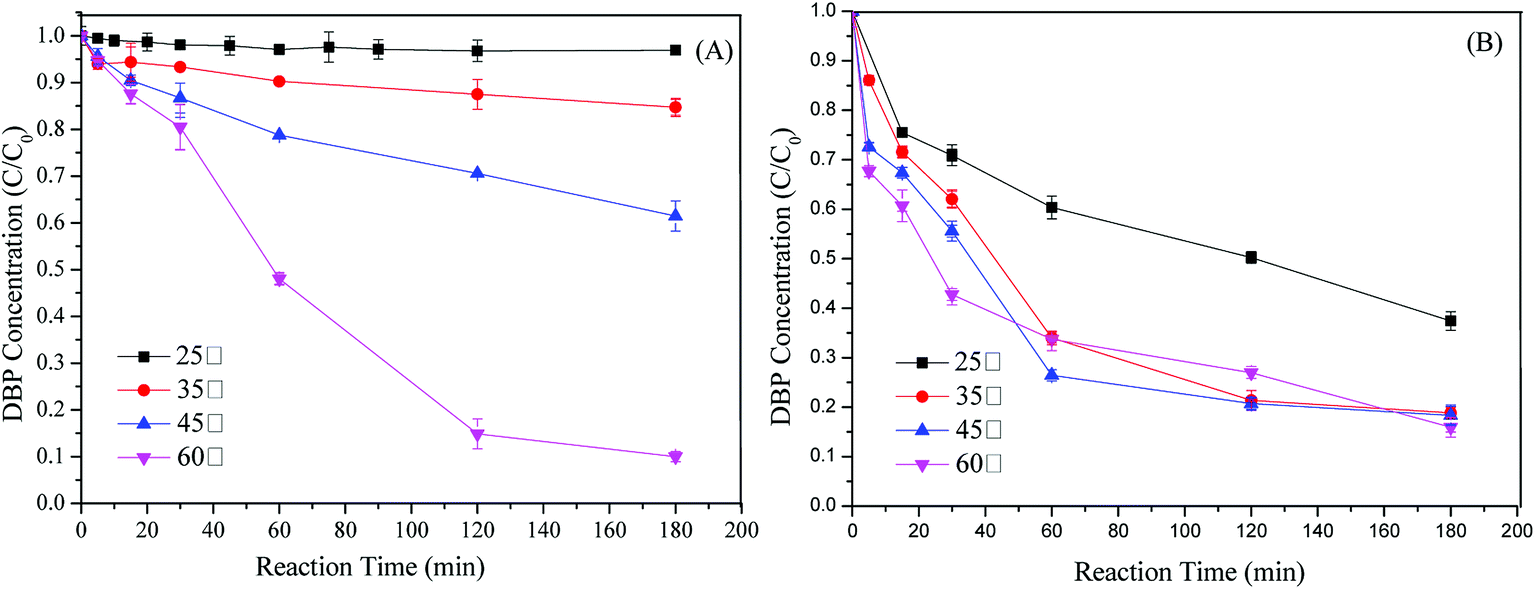
Thermocatalytic decomposition of PS shows high efficiency for pollutants removal although it usually requires high costs for equipment and operation.15 Effect of temperature on DBP removal was conducted to enhance understanding the mechanism between the catalytic and adsorptive performance of Cu-BTC-A. As illustrated in Fig. 6A, increasing temperature can significantly promote DBP degradation without Cu-BTC-A. When the temperature was increased from 45 °C to 60 °C, a great degree of enhancement on the conversion of DBP was observed from the original value of 41.6% to 96.4% in 180 min. However, DBP degradation efficiency declined when both Cu-BTC-A and PS were added into the solutions of 60 °C, where DBP removal rate was 82.3%. This was probably due to the scavenging reactions between a large amount of reactive radicals, which generated via the dual catalysis of thermal and Cu-BTC-A. If Cu-BTC-A only shows adsorption ability on DBP and no catalytic performance on PS, DBP removal rate in the PS/Cu-BTC-A system should be higher than that of only PS system. Hence, the opposite results confirmed the speculation that Cu-BTC-A shows catalytic performance on PS and generating free radicals. In addition, it was also remarkable that the presence of Cu-BTC-A significantly accelerated the removal rate of DBP at temperatures of 25, 35, 45 °C. This fact revealed that high DBP degradation was mainly due to the heterogeneous catalytic process in low temperatures. Furthermore, the study of electrochemical performance (Fig. 7) also confirmed electron transfer undergoing on the interface of Cu-BTC-A and PS.
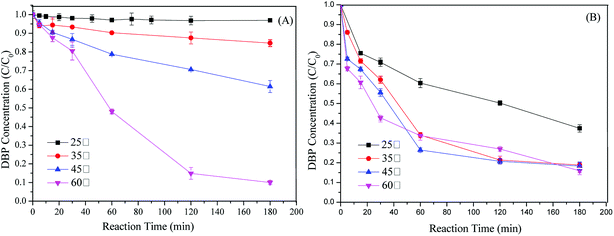 |
| | Fig. 6 (A) Effect of temperature on DBP removal in the system with only PS; (B) effect of temperature on DBP degradation in PS/Cu-BTC system. Experiment condition: DBP = 0.018 mM; PS = 2.16 mM; Cu-BTC-A = 1.0 g L−1; T = 25 °C; no pH adjustment. | |
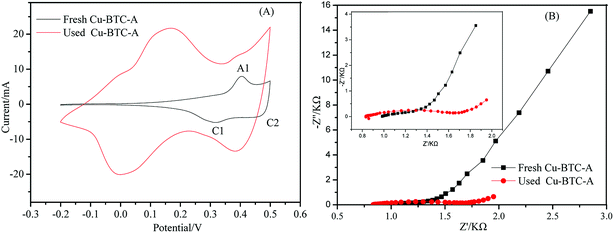 |
| | Fig. 7 (A) Cyclic voltammogram curves in 1.0 M KOH solution recorded at a rate of 0.1 mV s−1; and (B) Nyquist plots of electrochemical impedance spectra for Cu-BTC-A. | |
3.6 Electrochemical performance and stability of Cu-BTC-A
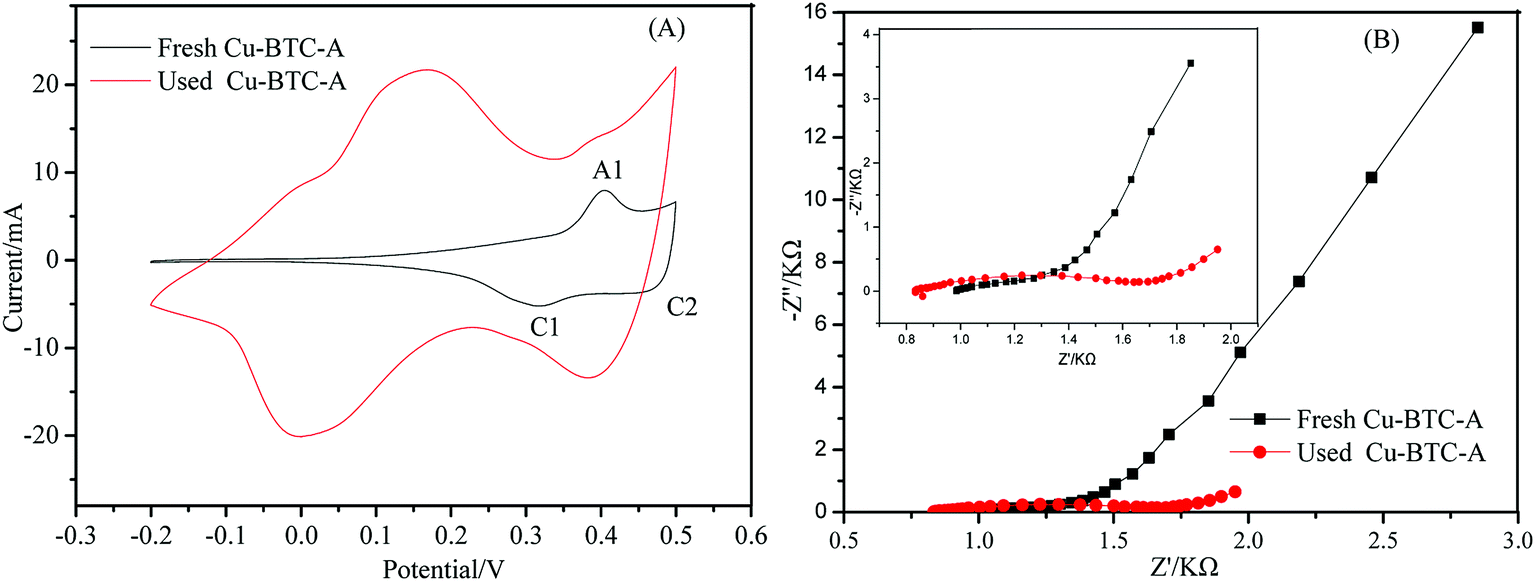
The redox potential which concerns electron translation is a crucial factor for catalysts during the catalysis process. The electrochemical performance of Cu-BTC-A was evaluated by cyclic voltammograms (CV) and electrochemical impedance spectra (EIS). As seen in Fig. 7, the appearance of peak A1 at 0.41 V was ascribed to the anodic oxidation of Cu(II) to Cu(III) while the peak C2 located at 0.48 V was related to the cathodic reduction of Cu(III) to Cu(II).37 Another broad peak (C1) at about 0.3 V may be associated with two possible reduction reactions: the subsequent conversion of Cu(II) to Cu(I) and the reduction directly from Cu(II) to Cu(0).37 This meant that most of copper in Cu-BTC-A was in Cu(II) valence state which would show good catalytic performance.38,39 One of the difference between the used and fresh Cu-BTC-A is that the two cathodic peaks attributed to the reduction of Cu(III) thus forming Cu(II) and Cu(0) in the used Cu-BTC-A was more evident than that of fresh Cu-BTC-A. Another difference was their peaks potentials shifted to more negative values in the curve of used Cu-BTC-A, which indicated that the electrochemical processes of Cu occur in Cu-BTC-A during the direct sweep. According to the literature,40 this phenomenon was due to the strong complexation of Cu(II). Moreover, the peak-to-peak separation between the reduction and oxidation peaks were significantly increased. This result can be attributed to the fact that the electron-transfer was gradually obstructed in the used Cu-BTC-A. Similar results were also observed in other MOFs.2 The Nyquist plots of the fresh and used Cu-BTC-A were studied with the aim to investigate charge transfer mechanisms, as displayed in Fig. 6B. Both of them showed a semicircle that corresponds to the interface electron transfer resistance (Ret) in the high-frequency region and the linear region in the low.41 However, an increase of interfacial Ret value was observed at the used Cu-BTC-A (left inset). This might be attributed to the formation of Cu(III) in Cu-BTC-A and reduced electron transfer efficiency. The phenomena could also be explained by the fact that DBP was absorbed on Cu-BTC-A surface and blocked electron transfer. It looked like the result of impedance was consistent with the behavior observed in CV curves discussed above. Hence, a conclusion could be deduced that electron transfer underwent on the interface of fresh Cu-BTC-A and involved in heterogeneous catalytic reaction.
The stability of catalyst is a key factor for its application, and it is necessary to evaluate the stability of Cu-BTC-A from the view of economic and environmental points. Thus, Cu-BTC-A was tested by XRD patterns and IR spectra after reactions. As depicted in Fig. 8A, the XRD pattern of used Cu-BTC-A was similar to that of fresh Cu-BTC-A, suggesting that the structure of Cu-BTC-A was not damaged after reactions. This also can be confirmed by the FT-IR spectra of used Cu-BTC-A (Fig. 8B). In other words, the organic ligands which act as the “bridges” keep well and make Cu-BTC-A present a good stability during the catalytic reaction process.
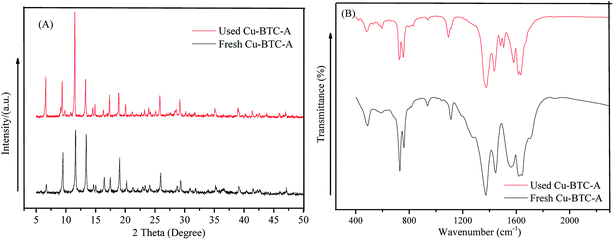 |
| | Fig. 8 (A) XRD patterns of the fresh and used Cu-BTC-A; (B) FT-IR spectra of the fresh and used Cu-BTC-A. | |
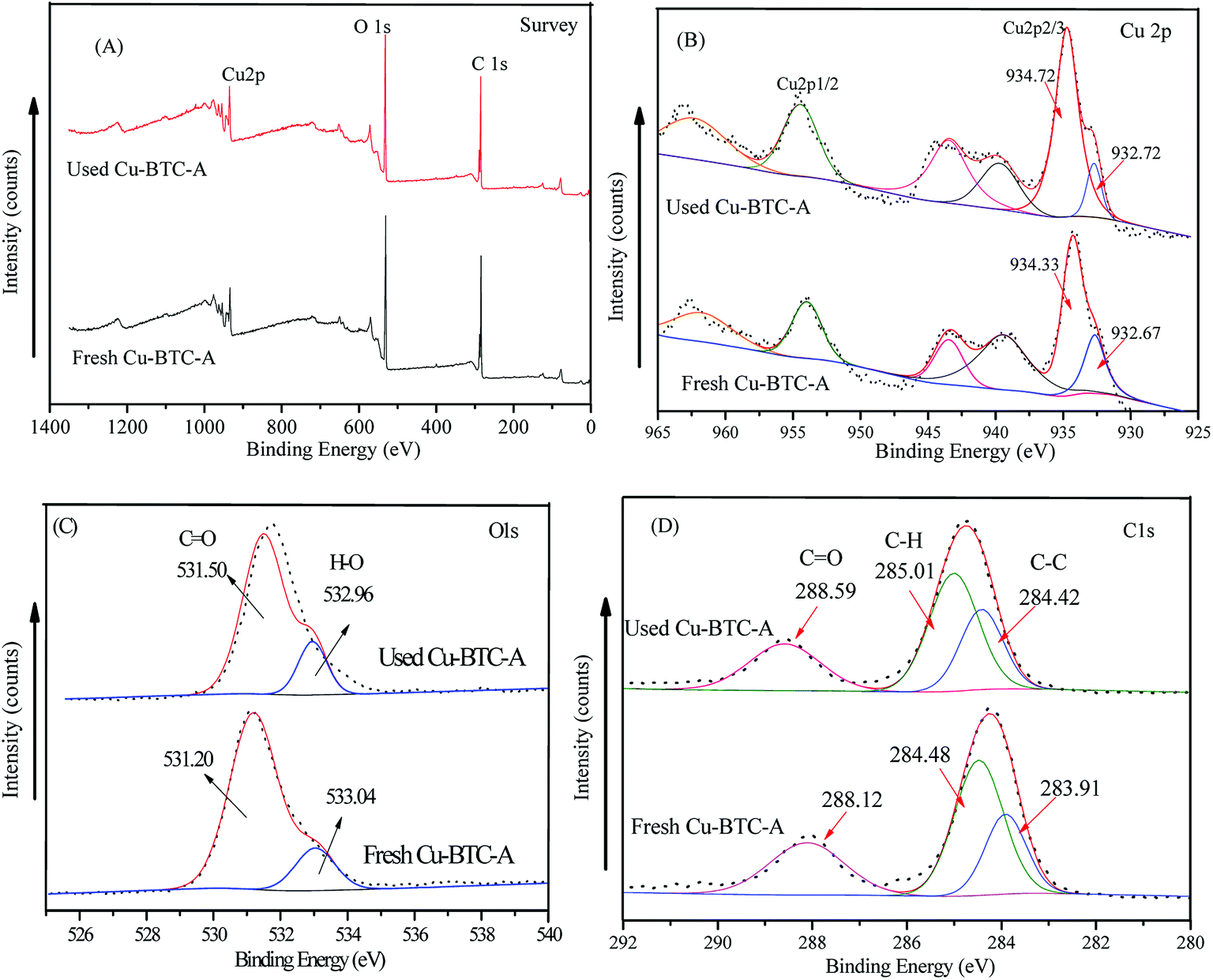
X-ray photoelectron spectroscopy (XPS) analyses were tested to better understand the chemical states of elemental composition on the surface of Cu-BTC-A before and after oxidation process. For Cu 2p (Fig. 9B), the peaks located at about 932.7 eV and 934.5 eV were assigned to Cu(I) and Cu(II), respectively in Cu 2p2/3, while the peak presented at 954.8 eV was contributed to Cu(II) species. Meanwhile, the peaks at 938–946 eV and 961–965 eV belonged to the shake-up satellite peaks. The appearance of a small portion of Cu(I) species was due to the impurity Cu2O, the data of XRD also showed the spectra of Cu2O. However, it was very interesting to note that the intensity of peaks at 932.7 eV for Cu(I) decreased after reaction, as shown in the used Cu-BTC-A sample. A possible reason for this result might be attributed to electron transfer from PS to Cu(I) during the oxidation process. The spectra of O 1s could be deconvoluted into two peaks centered at 533.04 and 531.2 eV (Fig. 9C), which was attributed to the oxygen in water and BTC. As seen in the spectra of C 1s, the peak located at around 284 eV and 285 eV corresponded to C–C and C–H while the peak at binding energy of 288 eV may be assigned to the presence of CO, indicating the presence of BTC.42 It was worth to note that there was no significant difference between the used and fresh Cu-BTC-A. These results indicated that Cu-BTC-A showed good stability.
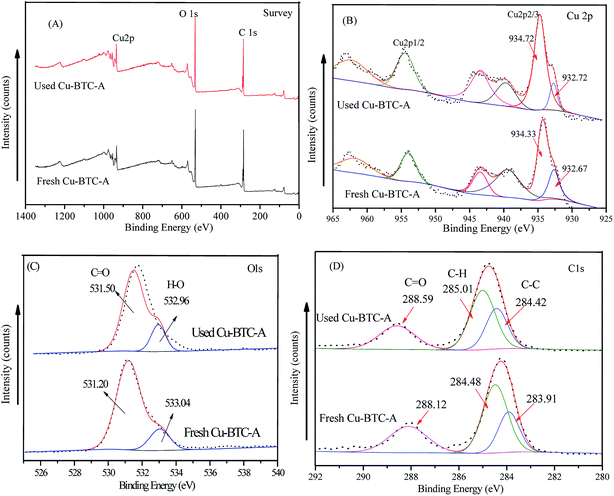 |
| | Fig. 9 The XPS spectra of the fresh and used Cu-BTC-A: (A) survey; (B) Cu 2p; (C) O 1s; (D) C 1s. | |
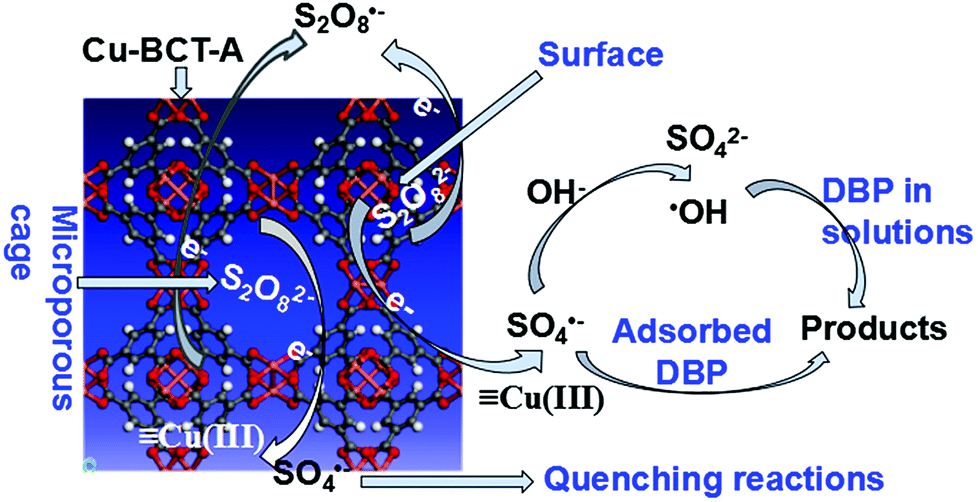
3.7 The mechanism for removal of DBP by Cu-BTC-A
It is well known that catalytic reaction mainly occurs on the surface of catalyst in a heterogeneous catalytic reaction system. Hence, catalytic activity of Cu-BTC-A is largely dependent on its chemical composition, elemental valence, crystal structure and surface state.43,44 In this work, significant enhancement in removal of DBP can be attributed to SO4˙− and ·OH oxidation, which was confirmed by the scavenging experiment. However, the drop of DBP removal rate in presence of EtOH was higher than that of TBA meant that SO4˙− was one of the oxidizing species in DBP removal. In addition to radicals oxidizing, Cu-BTC-A absorption also made a contribution to DBP removal, as discussed previously. Hence, the mechanism of DBP removal could be inferred, as in Scheme 1. At the initial stage, DBP was absorbed on the surface of Cu-BTC-A. Afterwards, PS was decomposed by metal sites of Cu(II) via electron transfer with generating SO4˙− (eqn (6))18,38,39 that would attack DBP absorbed on the surface of Cu-BTC-A, as well as in solutions. On the other hand, SO4˙− would directly convert into ·OH via the reaction with hydroxyl ion (eqn (7)) and water (eqn (8)).| |
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Cu(II) + S2O82− → Cu(III) + SO4˙− + SO42− Cu(II) + S2O82− → Cu(III) + SO4˙− + SO42−
| (6) |
| | |
SO4˙− + OH− → SO42− + ·OH
| (7) |
| | |
SO4˙− + H2O → SO42− + ·OH + H+
| (8) |
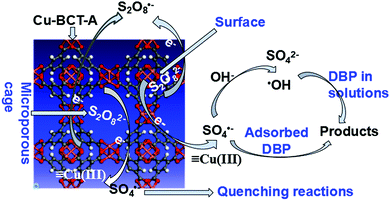 |
| | Scheme 1 Proposed mechanism for removal of DBP by Cu-BTC-A. | |
Then the formation of Cu(III) would reduce by PS (eqn (9)), and the oxidized Cu(II) will be recovered. Meanwhile, Cu(III) also reacted with the small amounts of impurity Cu2O (eqn (10)), which made the amount of Cu(I) reduce after reaction, as presented in XPS analysis.
| | |
Cu(III) + S2O82− → Cu(II) + S2O8˙−
| (9) |
| | |
Cu(III) + Cu(I) → 2Cu(II)
| (10) |
The structure of Cu-BTC-A contains mesoporous cages approximately 9 × 9 Å in diameter.1 In order to confirm whether or not catalytic oxidation reaction takes place in the cage of Cu-BTC-A, molecular size of DBP and PS were calculated based on the Bondi's method of van der Waals Volumes.45 The calculation results indicated that dimensions of PS and DBP molecular size were 8.25 × 5.21 × 5.05 Å and 15.84 × 11.00 × 7.56 Å, respectively. This revealed that the cage of Cu-BTC-A prevented DBP molecule from entering while PS molecule could freely pass through. As a result, SO4˙− generated in the cage triggered a series of quenching reactions, as displayed in eqn (11)–(15), which made DBP degradation decline. This data also made a good explanation for the fact that PS concentration made a less effect on DBP removal.
| | |
SO4˙− + SO4˙− → 2SO42−
| (11) |
| | |
SO4˙− + OH− → SO42− + ·OH
| (12) |
| | |
SO4˙− + H2O → SO42− + ·OH + H+
| (13) |
| | |
SO4˙− + Cu(II) → Cu(III) + SO42−
| (14) |
| | |
SO4˙− + S2O82− → SO42− + S2O8˙−
| (15) |
Hence, an effort to enlarge cages of Cu-BTC-A for control the entrance of DBP and improving pollutants degradation is needed in further research. The homogeneous reaction made a very small contribution to DBP removal due to its low activity on DBP removal (see Fig. 2B), which could be neglected compared to heterogeneous oxidation and adsorption mechanisms. Based on the results analysis, it can be concluded that Cu-BTC-A could behave as a stable and efficient heterogeneous catalyst of PS for removal of organic compounds that their molecular size is smaller than 9 × 9 Å by a combination of adsorption and degradation.
4. Conclusions
This study reported the synthesis of an effective and relatively stable copper-based metal organic framework and its novel application as a good heterogeneous catalyst upon the removal of DBP via adsorption and heterogeneous oxidation process. Parameters influencing the DBP removal efficiency were investigated and the optimal removal conditions were obtained. The electrochemical performance and stability of Cu-BTC-A were also conducted, indicating that Cu-BTC-A with good stability exhibited catalytic performance via electron transfer. However, DBP removal was not complete due to radical scavenging reactions in the cage of Cu-BTC-A where PS can enter freely while DBP was blocked outside due to its large molecular size. Considering the degradation efficiency of practical application, enlarging the size of Cu-BTC-A cage is significant for catalytic degradation of large molecular organic compounds. Hence, further work is devoted to modifying this material and improved free radicals utilization efficiency. Our findings might extend the applications of Cu-MOFs as an efficient heterogeneous catalyst of PS for water purification.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 51808177). One of the authors, Huanxuan Li, would like to acknowledge financial support from the Chinese Scholarship Council (CSC).
References
- S. S. Y. Chui, S. M. F. Lo, P. H. C. Jonathan, A. G. Orpen and D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- H. X. Li, J. Q. Wan, Y. W. Ma, Y. Wang, Y. M. Chen and X. Chen, J. Hazard. Mater., 2016, 318, 154–163 CrossRef CAS PubMed.
- X. Li, H. L. Liu, X. S. Ji, G. Y. Li, T. C. An and Y. P. Gao, Sci. Total Environ., 2017, 621, 1533–1541 CrossRef PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- M. Dinca and J. R. Long, J. Am. Chem. Soc., 2005, 127, 9376–9377 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- S. Lin, Z. L. Song, G. B. Che, A. Ren, P. Li, C. B. Liu and J. S. Zhang, Microporous Mesoporous Mater., 2014, 193, 27–34 CrossRef CAS.
- M. J. Pu, Y. W. Ma, J. Q. Wan, Y. Wang, J. M. Wang and M. L. Brusseau, Catal. Sci. Technol., 2017, 7(5), 1129–1140 RSC.
- X. Qin, S. W. Fang, L. Zhao, P. H. Shi, J. C. Fan, Y. L. Min, Q. J. Xu and W. F. Yao, Sci. Total Environ., 2017, 601–602, 99–108 CrossRef CAS PubMed.
- H. Tekin, O. Bilkay, S. S. Ataberk, T. H. Balta, I. H. Ceribasi, F. D. Sanin, F. B. Dilek and U. Yetis, J. Hazard. Mater., 2006, 136, 258–265 CrossRef CAS PubMed.
- H. X. Li, J. Q. Wan, Y. W. Ma, Y. Wang and Y. M. Chen, Chem. Eng. J., 2014, 237, 487–496 CrossRef CAS.
- Y. Xu, H. Lin, Y. K. Li and H. Zhang, Sci. Total Environ., 2017, 609, 644–654 CrossRef CAS PubMed.
- R. H. Waldemer, P. G. Tratnyek, R. L. Johnson and J. T. Nurmi, Environ. Sci. Technol., 2007, 41, 1010–1015 CrossRef CAS PubMed.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS PubMed.
- G. S. Liu, H. S. Lv, H. Y. Sun and X. Z. Zhou, Ind. Eng. Chem. Res., 2018, 57, 2396–2403 CrossRef CAS.
- H. Y. Liang, Y. Q. Zhang, S. B. Huang and I. Hussain, Chem. Eng. J., 2013, 218, 384–391 CrossRef CAS.
- J. J. Du, Y. P. Yuan, J. X. Sun, F. M. Peng, X. Jiang, L. G. Qiu, A. J. Xie, Y. H. Shen and J. F. Zhu, J. Hazard. Mater., 2011, 190, 945–951 CrossRef CAS PubMed.
- S. Marx, W. Kleist and A. Baiker, J. Catal., 2011, 281, 76–87 CrossRef CAS.
- S. De Rosa, G. Giordano, T. Granato, A. Katovic, A. Siciliano and F. Tripicchio, J. Agric. Food Chem., 2005, 53, 8306–8309 CrossRef CAS PubMed.
- K. Huang, Y. Xu, L. G. Wang and D. F. Wu, RSC Adv., 2015, 5, 32795–32803 RSC.
- Z. Huang, Y. W. Ma, Y. Wang, J. Q. Wan and H. P. Zhang, Bioresour. Technol., 2010, 101, 7767–7772 CrossRef PubMed.
- M. Ghisari and E. C. Bonefeld-Jorgensen, Toxicol. Lett., 2009, 189, 67–77 CrossRef CAS PubMed.
- G. W. Zhan and H. C. Zeng, Chem. Commun., 2016, 52, 8432–8435 RSC.
- K. Schlichte, T. Kratzke and S. Kaskel, Microporous Mesoporous Mater., 2004, 73, 81–88 CrossRef CAS.
- S. Loera-Serna, M. A. Oliver-Tolentino, M. L. López-Núñez, A. Santana-Cruz, A. Guzmán-Vargas, R. Cabrera-Sierra, H. I. Beltrán and J. Flores, J. Alloys Compd., 2012, 540, 113–120 CrossRef CAS.
- S. Maiti, A. Pramanik, U. Manju and S. Mahanty, Microporous Mesoporous Mater., 2016, 226, 353–359 CrossRef CAS.
- C. Prestipino, L. Regli, J. G. Vitillo, F. Bonino, A. Damin, C. Lamberti, A. Zecchina, P. L. Solari, K. O. Kongshaug and S. Bordiga, Chem. Mater., 2006, 18, 1337–1346 CrossRef CAS.
- A. R. Abbasia and M. Rizvandi, Ultrason. Sonochem., 2018, 40, 465–471 CrossRef PubMed.
- A. N. Pham, G. W. Xing, C. J Miller and T. D Waite, J. Catal., 2013, 301, 54–64 CrossRef CAS.
- Q. X. Luo, X. D. Song, M. Jia, S. E. Park, C. Hao and Y. Q. Li, Appl. Catal., A, 2014, 478, 81–90 CrossRef CAS.
- A. C. K. Yip, F. L. Y. Lam and X. Hu, Ind. Eng. Chem. Res., 2005, 44, 7983–7990 CrossRef CAS.
- Y. Rebeca, G. Salvador, S. Persi, S. Manuel, H. G. Juan, G. Z. Eduardo, A. I. Ilich and A. P. Julia, New J. Chem., 2015, 39, 5112–5115 RSC.
- H. X. Li, J. Q. Wan, Y. W. Ma and Y. Wang, Chem. Eng. J., 2016, 301, 315–324 CrossRef CAS.
- H. X. Li, J. Q. Wan, Y. W. Ma and Y. Wang, Sci. Total Environ., 2016, 562, 889–897 CrossRef CAS PubMed.
- Y. Wan, X. M. Wang, H. Sun, Y. B. Li, K. Zhang and Y. H. Wu, Int. J. Electrochem. Sci., 2012, 7, 7902–7914 CAS.
- C. S. Liu, K. Shih, C. X. Sun and F. Wang, Sci. Total Environ., 2012, 416, 507–512 CrossRef CAS PubMed.
- X. L. Zhang, M. B. Feng, R. J. Qu, H. Liu, L. S. Wang and Z. Y. Wang, Chem. Eng. J., 2016, 301, 1–11 CrossRef CAS.
- M. A. Oliver-Tolentino, A. Guzmán-Vargas, E. M. Arce-Estrada and A. ManzoRobledo, ECS Trans., 2010, 29, 399–408 CAS.
- X. Yang, Y. B. Tang, X. Huang, H. T. Xue, W. P. Kang, W. Y. Li, T. W. Ng and C. S. Lee, J. Power Sources, 2015, 284, 109–114 CrossRef CAS.
- H. Y. Niu, S. L. Liu, Y. Q. Cai, F. C. Wu and X. L. Zhao, Microporous Mesoporous Mater., 2016, 219, 48–53 CrossRef CAS.
- A. D. Bokare and W. Choi, J. Hazard. Mater., 2014, 275, 121–135 CrossRef CAS PubMed.
- A. Y. Zhang, T. Lin, Y. Y. He and Y. X. Mou, J. Hazard. Mater., 2016, 311, 81–90 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08285e |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ac,
Jialing Qina,
Yayun Zhang
*ac,
Jialing Qina,
Yayun Zhang