
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA facile synthesis methodology for preparation of Ag–Ni-reduced graphene oxide: a magnetically separable versatile nanocatalyst for multiple organic reactions and density functional study of its electronic structures†
Madhurya Chandel,
Priyanka Makkar,
Barun Kumar Ghosh,
Debabrata Moitra and
Narendra Nath Ghosh *
*
Nano-materials Lab, Department of Chemistry, Birla Institute of Technology and Science, Pilani K. K. Birla Goa Campus, Goa-403726, India. E-mail: naren70@yahoo.com; Fax: +91 832 2557033; Tel: +91 832 2580318
First published on 12th November 2018
Abstract
Here, we report a simple ‘in situ’ co-precipitation reduction synthesis method for the preparation of nanocatalysts composed of Ag, Ni nanoparticles, and reduced graphene oxide (RGO). First-principles calculations based on Density Functional Theory (DFT) were performed to obtain the electronic structures and properties of Ag–Ni-graphene superlattice and to understand the interfacial interactions which exist at the interface between Ag, Ni, and graphene. The catalytic performance of the synthesized catalysts (AgxNi(1−x))yRGO(100−y) were evaluated for four reactions (i) reduction of 4-nitrophenol (4-NP) in the presence of excess NaBH4 in aqueous medium, (ii) A3 coupling reaction for the synthesis of propargylamines, (iii) epoxidation of styrene, and (iv) ‘Click reaction’ for the synthesis of 1,2,3-triazole derivatives. For all of these reactions the catalyst composed of Ag, Ni, and RGO, exhibited significantly higher catalytic activity than that of pure Ag, Ni, and RGO. Moreover, an easy magnetic recovery of this catalyst from the reaction mixture after completion of the catalytic reactions and the good reusability of the recovered catalyst is also reported here. To the best of our knowledge, this is the first time the demonstration of the versatile catalytic activity of (AgxNi(1−x))yRGO(100−y) towards multiple reactions, and the DFT study of its electronic structure have been reported.
1. Introduction
For the last couple of decades, the applications of graphene and graphene-based materials have been explored extensively in myriad fields including electronics, sensors, solar cells, structural materials, catalysis, microwave absorbing materials, supercapacitors, etc.1–5 Graphene has proved its capability to become a material of paramount importance by exhibiting several excellent properties (such as, electrical conductivity, mechanical strength, chemical stability, lightweight, etc.).1,2,6,7 Several researchers have reported the application of graphene or Reduced Graphene Oxide (RGO) as the support to construct heterogeneous catalysts, where catalytically active sites are immobilized on the surface of graphene or RGO sheets.6–11 However, to the best of our knowledge, to date, the development of graphene-based catalysts, which can exhibit versatile high catalytic activity towards multiple organic reactions as well as easy magnetic recovery from reaction mixtures, is limited.10,12To develop an efficient, but versatile and magnetically separable catalyst, we have synthesized the catalysts which are composed of varying amounts of Ag, Ni nanoparticles, and RGO. We have investigated the catalytic activity of the synthesized catalysts towards four important reactions: (i) reduction of 4-nitrophenol (4-NP) in the presence of NaBH4, (ii) A3 coupling reaction for the synthesis of propargylamines, (iii) epoxidation of styrene, and (iv) ‘Click reaction’ for the synthesis of 1,2,3-triazole derivatives in aqueous medium. We have chosen to develop a versatile catalyst for these reactions because of the following reasons:
(i) Reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP): nitrophenols and their derivatives are widely used, or produced in many industries, such as dye-based industries, textiles, leather, pesticides, pharmaceuticals, iron and steel manufacturing, foundries, refinery, etc.8,11,13 Effluents of these industries contain a significant amount of 4-NP and other nitrophenols. These effluents, when coming in contact with natural water bodies, cause water pollution, because these nitrophenols are harmful to the kidney, liver, central nervous systems, etc.11,14,15 Therefore, to address this environmental pollution issue removal or destruction of 4-NP (or similar nitrophenols) from the polluted aquatic system is very crucial. On the other hand, reduction of 4-NP produces 4-AP, which is an important chemical for many pharmaceutical industries for the production of antipyretic and analgesic drugs, etc.10,11,14,16 Moreover, several researchers have investigated the reduction of 4-NP to 4-AP as a model reaction to evaluate the catalytic activity of variety of nanostructured catalysts because this reaction is easy to perform, convincing, and trustworthy for this purpose.8,9,11,13,14,16
(ii) A3 coupling reaction: this reaction can be used to synthesize propargylamines through a one-pot reaction using the combination of three starting materials (e.g. aldehyde, amine, and alkyne). Propargylamines are an important class of compound for the preparation of several biologically active nitrogen compounds, and also crucial intermediates for many pharmaceuticals and natural products.17,18 Traditional methods for propargylamines synthesis (such as the nucleophilic addition of Grignard reagents or lithium acetylide to imines, etc.) involve multistep synthesis or purification.19–21 Use of moisture sensitive reagents in a regulated reaction condition is one of the limitations associated with these traditional methods which limits the large-scale reaction.18,21 To avoid these problems, the coupling of aldehydes, amines, and alkynes via A3 coupling reaction over the heterogeneous catalysts has emerged as a promising approach to synthesize propargylamines.17,18,22,23
(iii) Epoxidation of styrene: epoxidation is an important reaction for the chemical industries because of the wide range applications of epoxy resins, including synthesis of many commodities, useful intermediates for the synthesis of many pharmaceutical products, and fine chemicals.24,25 Conventional methodologies for the epoxidation of styrene (such as the non-catalytic process using chlorine, co-epoxidation process, organic peroxide and peracid-based catalytic process, etc.) suffer from some disadvantages, such as the production of the large amount of undesirable by-products, harsh reaction conditions, cost-effectiveness, separation problem associated with homogeneous catalysts, etc.10,26,27 Therefore, till date, several researchers are engaged in developing novel catalysts for chlorine-free epoxidation reaction.27–30
(iv) Click reaction: synthesis of triazoles by ‘Click reaction’ has gained immense attention due to the use of triazoles as biologically active chemicals, agrochemicals, pharmaceuticals, drug molecules with significant anti-HIV activity, antimicrobial activity against Gram-positive bacteria, etc.31–34 Several researchers have developed various types of heterogeneous catalysts for the synthesis of triazole derivatives.23,32,34,35 However, in some cases the limitations associated with these procedures are the requirement of elevated reaction temperature, the formation of undesired products, low yields, use of homogeneous catalysts which are difficult to separate from products, etc.31,35,36
Though several nanostructured catalysts exhibit high catalytic activity due to their high surface area, their tendency to form agglomerates and difficulty in separation by easy filtration process are major limitations for large-scale applications. To overcome these limitations immobilization of nanocatalysts on high surface area support and developing magnetically separable catalyst are attractive strategies.10,14,18,34,37,38 Therefore, to develop a catalyst which will exhibit catalytic activity towards all of the above mentioned organic reactions along with easy magnetic separation, we have designed the catalyst where Ag and Ni nanoparticles are intimately coexisting, and these nanoparticles are immobilized on the surface of nanometer thin sheets of RGO. The reasons for choosing Ni and Ag nanoparticles as catalytically active components of this catalyst is that Ni and Ag nanoparticles can participate in these reactions by stabilizing intermediates through the formation of coordination complexes and thus help the reactions to proceed forward.22,23,39–41 Moreover, as Ni nanoparticles are magnetic in nature, the presence of Ni introduces a magnetic character in the catalyst and makes the catalyst magnetically separable. RGO provides a high surface area, which helps the reactant molecules to get adsorbed on the surface of the catalyst and to come in contact with catalytically active sites (here Ag and Ni). It is also expected that the high conducting nature of RGO will help in the electron transfer process of these catalysis reactions.1,3,6
Herein, we are reporting an ‘in situ’ co-precipitation reduction technique for the preparation of a versatile catalyst, ((AgxNi(1−x))yRGO(100−y)), for (i) reduction reaction of 4-nitrophenol (4-NP) in presence of NaBH4, (ii) A3 coupling reaction for the synthesis of propargylamines, (iii) epoxidation reaction of styrene, and (iv) ‘Click reaction’ for the synthesis of 1,2,3-triazole derivatives in aqueous medium and magnetic recovery and reusability of the catalyst. We have also performed the first-principles calculations based on Density Functional Theory (DFT) to understand how the interfacial interactions between Ag, Ni, and RGO influence their electronic structures, due to which the synergistic effect originates and the synthesized catalyst exhibits superior catalytic activity. To the best of our knowledge, this is the first time the catalyst, containing Ag, Ni nanoparticles and RGO, has been reported which can exhibit catalytic property for these four reactions.
2. Experimental
2.1. Materials
Acetone, hydrazine hydrated (N2H4·H2O), polyvinylpyrrolidone (PVP), sodium hydroxide (NaOH), sodium nitrate, sulphuric acid (H2SO4), hydrochloric acid (HCl), sodium nitrate (NaNO3), 4-nitrophenol (4-NP), and nickel(II)nitrate hexahydrate (Ni(NO3)2·6H2O) were purchased from Fischer Scientific. Silver nitrate (AgNO3), potassium permanganate (KMnO4), sodium azide (NaN3), 30% H2O2 solution, ethylene glycol, acetonitrile, methanol, dichloromethane, toluene, chloroform, diethylamine, and aniline were purchased from Merck, India. Sodium borohydride (NaBH4), aldehydes, piperidine, phenylacetylene, styrene, styrene oxide, cyclohexene oxide, epichlorohydrin, tert-butyl hydroperoxide (TBHP in 5–6 M decane), and graphite powder (mean particle size of <20 mm) were purchased from Sigma-Aldrich. All the chemicals were used without further purification. Distilled water was used throughout the experiment.2.2. Synthesis of ((AgxNi(1−x))yRGO(100−y))
We have synthesized the nanocatalysts composed of the different amount of Ag nanoparticle, Ni nanoparticle, and RGO, in two steps. In the first step, graphene oxide (GO) was prepared by using the modified Hummers method (method of GO synthesis is provided in ESI†).42 In the next step, a mixture of ethylene glycol (EG) and PVP (molar ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was prepared, and the calculated amount of GO sheets were dispersed in this mixture. In this dispersion AgNO3 and Ni(NO3)·6H2O were added according to the compositions of the nanocatalyst (i.e. (AgxNi(1−x))yRGO(100−y), where x = 0–1, and y = 0–100). After the addition of metal nitrate salts, the mixture was stirred until the metal salts were dissolved. In this mixture, NaOH pellets were added with stirring till the pH of the mixture became ∼10, and then N2H4 was added dropwise keeping the metal ion: N2H4 molar ratio 1:40. After complete addition of N2H4, this reaction mixture was refluxed at 85 °C for 15 min and then cooled to room temperature. The precipitate thus formed was magnetically separated from the reaction mixture by using a magnet externally. The collected precipitate was washed with distilled water until the pH of the washing reached ∼7. Finally, the precipitate was washed with acetone and dried at 60 °C for 10 h. As a representative, the detail of the synthesis procedure for (Ag0.27Ni0.73)37RGO63 preparation has been described in ESI.† To perform the control experiments, we have also prepared pure Ag, and pure Ni nanoparticles using the aforesaid method, without adding RGO. Pure RGO was also synthesized by reducing GO with N2H4. Several compositions of catalysts, containing only Ag and Ni nanoparticles (i.e. AgxNi(1−x)), were also prepared using the same method. The code of the catalysts and the corresponding compositions are listed in Table S1 (ESI†).
:1) was prepared, and the calculated amount of GO sheets were dispersed in this mixture. In this dispersion AgNO3 and Ni(NO3)·6H2O were added according to the compositions of the nanocatalyst (i.e. (AgxNi(1−x))yRGO(100−y), where x = 0–1, and y = 0–100). After the addition of metal nitrate salts, the mixture was stirred until the metal salts were dissolved. In this mixture, NaOH pellets were added with stirring till the pH of the mixture became ∼10, and then N2H4 was added dropwise keeping the metal ion: N2H4 molar ratio 1:40. After complete addition of N2H4, this reaction mixture was refluxed at 85 °C for 15 min and then cooled to room temperature. The precipitate thus formed was magnetically separated from the reaction mixture by using a magnet externally. The collected precipitate was washed with distilled water until the pH of the washing reached ∼7. Finally, the precipitate was washed with acetone and dried at 60 °C for 10 h. As a representative, the detail of the synthesis procedure for (Ag0.27Ni0.73)37RGO63 preparation has been described in ESI.† To perform the control experiments, we have also prepared pure Ag, and pure Ni nanoparticles using the aforesaid method, without adding RGO. Pure RGO was also synthesized by reducing GO with N2H4. Several compositions of catalysts, containing only Ag and Ni nanoparticles (i.e. AgxNi(1−x)), were also prepared using the same method. The code of the catalysts and the corresponding compositions are listed in Table S1 (ESI†).
2.3. Catalytic activity tests
| dCt/dt = −kappCt | (1) |
| ln(Ct/Co) = ln(At/Ao) = −kapp t | (2) |
The value of kapp was calculated from the slope of the ln(At/Ao) vs. time plot.
:2) as eluent. The catalysis reaction was performed at different reaction conditions and it was observed that the highest yield was obtained when the reaction temperature was 100 °C, reaction time was 12 h and acetonitrile as the solvent. After optimizing the reaction conditions, the A3 coupling reaction was also performed by using different types of aldehydes and amines to demonstrate the effectiveness of the synthesized catalyst.
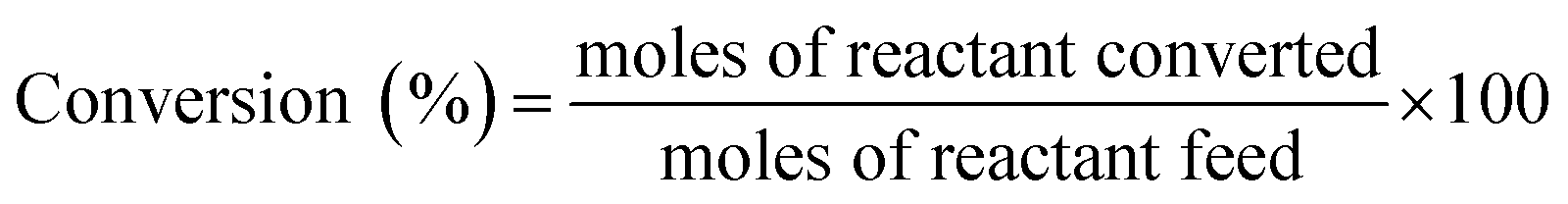 | (3) |
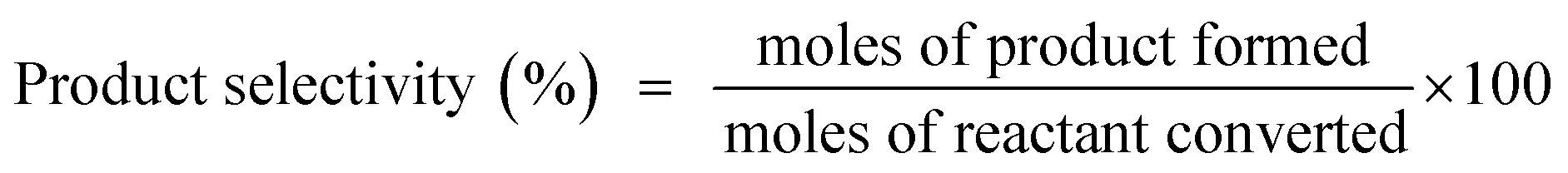 | (4) |
The reaction was conducted under different reaction conditions. It was observed that the highest amount of styrene conversion was achieved when the reaction was performed at 100 °C for 10 h with 1:2 styrene:TBHP ratio. Therefore, this reaction condition was used when the reaction was performed with the catalysts having different compositions.
2.4. Characterization
The synthesized pure Ni, pure Ag, AgxNi(1−x), and (AgxNi(1−x))yRGO(100−y) catalysts were characterized by X-ray diffraction (XRD), thermogravimetric analysis (TGA), Fourier Transform Infrared spectra (FT-IR), Raman, Field Emission Scanning Electron Microscope (FESEM), energy dispersive X-ray analysis (EDX), and Vibrating Sample Magnetometer (VSM). The products which were obtained from catalysis reactions were characterized by Liquid Chromatography-Mass Spectrometer (LC-MS), Proton Nuclear Magnetic Resonance (1H NMR), Gas Chromatography (GC), and Differential Scanning Calorimetric (DSC) (details of the characterization are discussed in ESI†).2.5. First-principles calculations
We have performed the first-principles quantum mechanical calculations based on DFT to obtain the electronic structures of Ni, Ag, Ag–Ni interface, and Ag–Ni-graphene superlattice. Here, we have used Quantum ESPRESSO computational package43 to calculate the ground state structures, binding energy, Density of States (DOS), and Projected Density of states (PDOS) using a plane wave set and pseudopotentials. DFT was used with Generalized Gradient Approximation (GGA)44 and parameterized by Perdew, Burke, and Ernzerhof (PBE).45 Kohn–Sham orbitals were expanded in a plane-wave basis set up to the kinetic energy cutoff 25 Ry (340 eV). The convergence criterion for the self-consistence calculation was 10−7 Ry per Bohr (0.0257 eV Å−1). For calculating the final electronic properties of the structures, and empirical dispersion-corrected density functional theory (DFT-D2) approach, which was proposed by Grimme46,47 was employed. To account for the intermolecular interactions and van der Waals (vdW) interactions, the Grimme-D2 correction was used.12In the present study five systems were investigated (i) superlattice of pure Ag with face-centered cubic structure (space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), (ii) pure Ni with face-centered cubic structure (space group Fmm), (iii) graphene superlattice, (iv) Ag–Ni interface, and (v) Ag–Ni-graphene superlattice. Ultrasoft pseudopotential for these systems were constructed by using 17, 16, and 4 electrons for Ag (4p64d105s1), Ni (3p63d84s2), and C (2s22p2), respectively. From the Quantum ESPRESSO website, the pseudopotentials of Ag, Ni, and C were chosen.48 To select the k-points mesh, the Monkhorst–Pack approach was used49 and the details of k points for each system are provided in the computational details section in the ESI.† The Brillouin zone integration of these systems was performed with Methfessel–Paxton smearing technique50 for Ag (111) slab, and Ni (111) slab, Marzari–Vanderbilt smearing technique51 for graphene, Ag–Ni interface, and Ag–Ni-graphene superlattice. Here, the smearing parameter was 0.005 Ry. The Binding energy of Ag–Ni-graphene superlattice was calculated from the difference between the total energy of the Ag–Ni-graphene superlattice and the sum of each system alone (Ag–Ni interface and graphene). The sizes of the unit cells of the simulated systems are listed in Table S2 (ESI†). The details of the sample input files for the geometric optimization of Ag, Ni, graphene, Ag–Ni, and Ag–Ni-graphene superlattice are provided in the computational details section in ESI.†
m), (ii) pure Ni with face-centered cubic structure (space group Fmm), (iii) graphene superlattice, (iv) Ag–Ni interface, and (v) Ag–Ni-graphene superlattice. Ultrasoft pseudopotential for these systems were constructed by using 17, 16, and 4 electrons for Ag (4p64d105s1), Ni (3p63d84s2), and C (2s22p2), respectively. From the Quantum ESPRESSO website, the pseudopotentials of Ag, Ni, and C were chosen.48 To select the k-points mesh, the Monkhorst–Pack approach was used49 and the details of k points for each system are provided in the computational details section in the ESI.† The Brillouin zone integration of these systems was performed with Methfessel–Paxton smearing technique50 for Ag (111) slab, and Ni (111) slab, Marzari–Vanderbilt smearing technique51 for graphene, Ag–Ni interface, and Ag–Ni-graphene superlattice. Here, the smearing parameter was 0.005 Ry. The Binding energy of Ag–Ni-graphene superlattice was calculated from the difference between the total energy of the Ag–Ni-graphene superlattice and the sum of each system alone (Ag–Ni interface and graphene). The sizes of the unit cells of the simulated systems are listed in Table S2 (ESI†). The details of the sample input files for the geometric optimization of Ag, Ni, graphene, Ag–Ni, and Ag–Ni-graphene superlattice are provided in the computational details section in ESI.†
3. Results and discussion
3.1. Structure and morphology of the synthesized (AgxNi(1−x))yRGO(100−y) catalyst
The room temperature wide-angle XRD was used to identify the crystalline phases which were present in the synthesized catalysts. As a representative, XRD patterns of GO, pure Ag, pure Ni, Ag0.50Ni0.50, Ni30RGO70, and (Ag0.27Ni0.73)37RGO63 are shown in Fig. 1. The XRD patterns of other compositions of the catalysts are shown in Fig. S1–S2 (ESI†). In case of pure Ag nanoparticles, diffraction peaks at 2θ = 38.2°, 44.3°, 64.6°, and 77.3°, which correspond to the (111), (200), (220), and (222) planes of FCC phase of Ag (ICDD no. 04–0783), were observed. XRD pattern of pure Ni sample showed the peaks at 2θ = 44.3°, 51.7° and 76.4° corresponding to (111), (200), and (220) planes of FCC phase of Ni (ICDD no. 04–0850). In the XRD patterns of the AgxNi(1−x) catalysts, the characteristic diffraction peaks of the only Ag and Ni were present (Fig. S1, ESI†). In case of pure GO sample, an intense peak at 2θ = 9.76° and a small peak at 2θ = 42.14° were observed which represented the (001) and (101) planes of GO, respectively.3,4,10 In the XRD pattern of (AgxNi(1−x))yRGO(100−y) samples, all the peaks which are characteristic diffraction peaks of Ag and Ni were present indicating the presence of pure Ni and Ag nanoparticles in these samples (Fig. S2, ESI†). The crystallite size of Ag and Ni particle was calculated by Scherrer's equation using the diffraction peak corresponding to (111) plane of Ag and (111) plane of Ni. The crystallite sizes of Ni and Ag nanoparticles were ∼11 nm and ∼15 nm, respectively. It was also observed that in the XRD patterns of (AgxNi(1−x))yRGO(100−y) samples no diffraction peaks of GO were present. Therefore, it was concluded that during the synthesis of these nanocatalysts by the ‘in situ’ co-precipitation reduction technique GO was converted to RGO. TGA, FT-IR, and Raman spectroscopy of these samples also indicated this conversion of GO to RGO.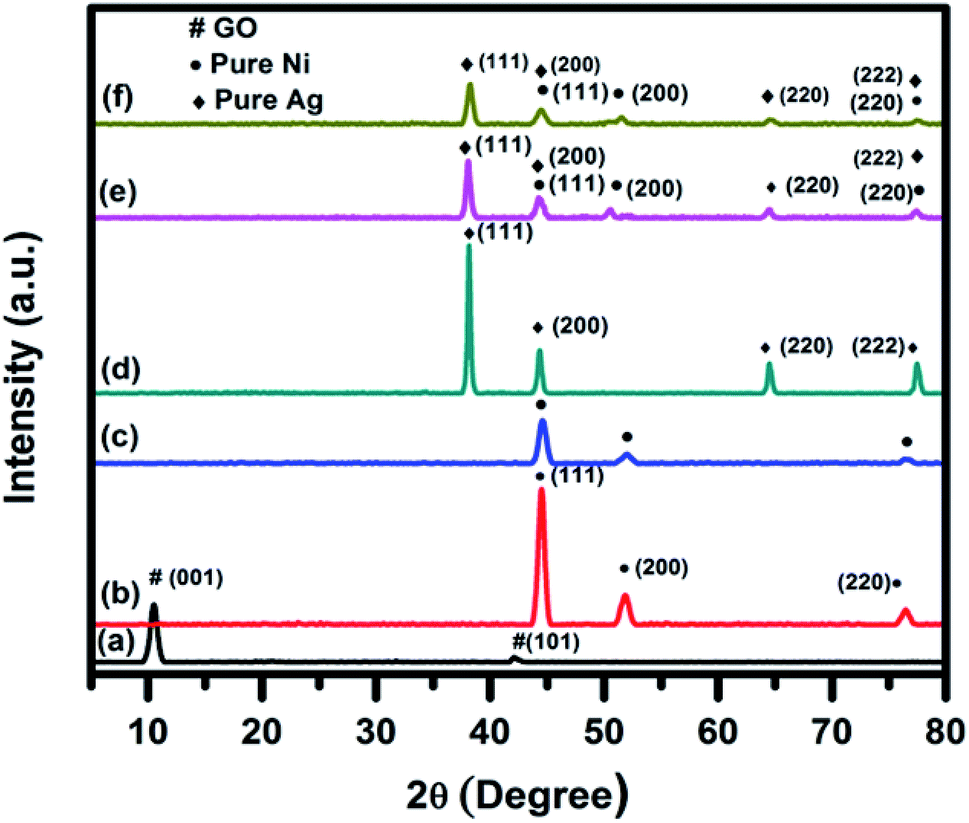 | ||
| Fig. 1 Room temperature wide angle powder XRD pattern of (a) GO, (b) pure Ni, (c) Ni30RGO70, (d) pure Ag, (e) Ag0.50Ni0.50, and (f) (Ag0.27Ni0.73)37RGO63. | ||
TGA and DTA of the synthesized catalysts were performed to understand the effect of temperature on the catalysts and also to determine the amount of RGO present in the catalyst (Fig. 2). In the TGA-DTA thermogram of pure GO following points were observed (i) ∼11% weight loss occurred in the temperature range of 30–110 °C, which might be due to the loss of surface adsorbed water, (ii) in the temperature range of 110–290 °C, ∼31% weight loss in TGA and a sharp exothermic peak at 240 °C in DTA indicated the removal of oxygen-containing functional groups (e.g. epoxy, carbonyl, carboxylic group) from GO in this temperature range, (iii) the complete decomposition of GO occurred in the temperature range of 300–625 °C. The exothermic nature of this decomposition was indicated by the presence of an exothermic peak at 460 °C in the DTA. The TGA thermogram of pure RGO showed (i) ∼5% weight loss in 30–110 °C temperature range due to the removal of adsorbed water, and (ii) a continuous weight loss till the complete decomposition of RGO in the temperature range of 110–625 °C. DTA of RGO also showed an exothermic peak at 500 °C. From TGA-DTA thermograms of the synthesized catalysts two major points were observed (i) similar to GO and RGO, the loss of surface adsorbed water (∼5 wt%) occurred in the temperature range of 30–110 °C, (ii) in the temperature range of 110–550 °C, the continuous weight loss due to the decomposition of RGO from the catalyst. This major weight loss step is similar to pure RGO. Here, the important points were noted (i) for the (AgxNi(1−x))yRGO(100−y) samples, no distinguishable weight loss step in the temperature range of 110–290 °C was observed, which was noticed in case of pure GO. This fact indicated that (AgxNi(1−x))yRGO(100−y) samples contain only a few numbers of oxygen-containing functional groups, and GO was converted to RGO via the partial reduction of these groups during the synthesis of the catalysts. (ii) the decomposition of RGO occurred in the catalysts at a relatively lower temperature (∼550 °C) than that RGO (∼625 °C). This could be due to the presence of Ag and Ni particles in the catalyst, which facilitated the decomposition of carbon, (iii) in case of the catalysts ((AgxNi(1−x))yRGO(100−y)), after complete decomposition of RGO, the remaining amount of the undecomposed catalyst matched well with the expected amount of Ag and Ni in the catalyst. This is an indication that, the ‘in situ’ co-precipitation reduction method, which we have adopted to prepare the catalysts, is capable of producing nanocatalysts with the desired composition of Ag, Ni, and RGO.
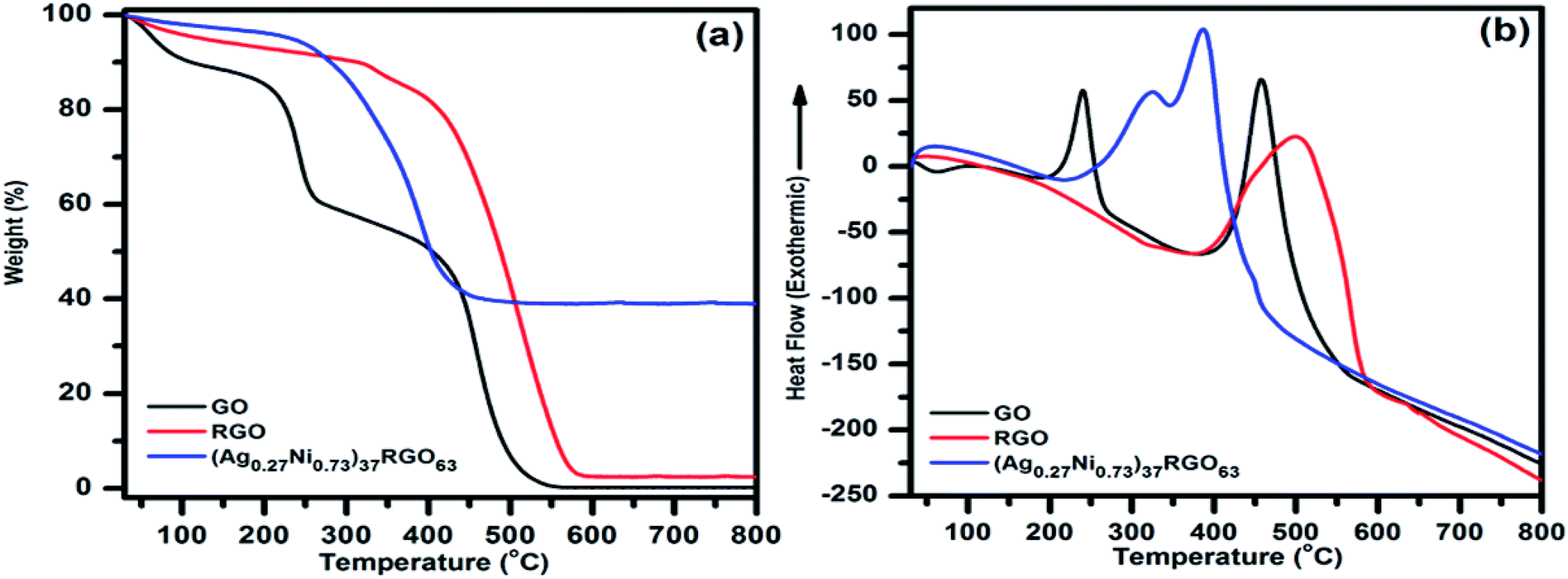 | ||
| Fig. 2 (a) TGA and (b) DTA curve of GO, RGO, and (Ag0.27Ni0.73)37RGO63. | ||
The FT-IR spectra of GO, pure RGO, and the catalyst (Ag0.27Ni0.73)37RGO63 are shown in Fig. S3 (ESI†). In case of GO the peaks at (i) 1384 cm−1 for the C–O stretching vibration of –CO2H group, (ii) 1234 cm−1 for the C–O stretching vibration of an epoxy group, (iii) 1053 cm−1 for C–O stretching vibration were observed, which indicated the presence of carboxyl, epoxy, and carbonyl groups on the surface of GO sheets.3,52 Moreover, the peak at 1620 cm−1 represented the contribution of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C skeletal vibration of the graphitic domain of GO.3,52 In the FT-IR spectra of RGO and the catalyst, the disappearance of the peaks at 1730 and 1234 cm−1, and decrease in the intensity of the peak at 1053 cm−1 indicated the reduction of the functional groups of GO during the preparation of RGO and the catalysts. It was also noted that the band at 1620 cm−1 (in GO sample) has been red shifted to 1545 cm−1 (for RGO) and 1580 cm−1 (for catalyst). The red shifting of this band, which corresponds to the CC skeletal vibration, can be interpreted as the partial restoration of π–π conjugation of graphene sheet in the pure RGO as well as the catalyst.3,52 These results clearly indicated the formation of RGO from GO during the synthesis of the catalysts.
C skeletal vibration of the graphitic domain of GO.3,52 In the FT-IR spectra of RGO and the catalyst, the disappearance of the peaks at 1730 and 1234 cm−1, and decrease in the intensity of the peak at 1053 cm−1 indicated the reduction of the functional groups of GO during the preparation of RGO and the catalysts. It was also noted that the band at 1620 cm−1 (in GO sample) has been red shifted to 1545 cm−1 (for RGO) and 1580 cm−1 (for catalyst). The red shifting of this band, which corresponds to the CC skeletal vibration, can be interpreted as the partial restoration of π–π conjugation of graphene sheet in the pure RGO as well as the catalyst.3,52 These results clearly indicated the formation of RGO from GO during the synthesis of the catalysts.
Fig. S4 (ESI†) presents the Raman spectra of the pure GO, pure RGO, and (Ag0.27Ni0.73)37RGO63 catalyst. In case of GO, two characteristic peaks at 1337 and 1589 cm−1 were observed which correspond to the D and G bands, respectively. In the case of RGO, these bands were shifted to 1328 and 1580 cm−1. Raman spectra of the catalyst showed the further shifting of these D and G bands to the lower wave numbers i.e. at 1315 and 1563 cm−1, respectively. The shifting of the Raman shift values of D and G bands to the lower bands could be attributed to the formation of RGO in the catalyst due to the reduction of GO.4,10 The ID/IG ratio of GO, RGO, and the catalyst were 0.98, 1.02, and 1.15, respectively. This increase of ID/IG for RGO and the catalyst compared to GO might be due to the decrease in the average size of sp2 domains upon the reduction of GO during the formation of the catalyst.4,10
FESEM micrographs of the synthesized pure RGO, Ag nanoparticles, Ni nanoparticle, Ag0.50Ni0.50, and (Ag0.27Ni0.73)37RGO63 nanocomposite are shown in Fig. 3. Nanometer thin sheets were clearly visible in the micrograph of RGO. The micrographs of pure Ni and Ag showed that in both the cases almost spherical nanoparticles, having the particle size of 30–50 nm range were attached closely to each other. In case of Ag0.50Ni0.50 sample, Ag and Ni nanoparticles were randomly attached to each other. However, the large agglomerations of only Ni or only Ag particles were not observed. The micrograph of (Ag0.27Ni0.73)37RGO63 revealed that closely attached Ag and Ni particles were immobilized on the surface of RGO sheets. Color mapping of Ag0.50Ni0.50 and (Ag0.27Ni0.73)37RGO63 also showed the intimate attachment of Ag and Ni particles (Fig. S5–S7, ESI†). EDS analysis of the samples also confirmed the composition of the composites (Fig. S8 and S9, ESI†).
 | ||
| Fig. 3 FESEM images of (a) pure RGO, (b) pure Ni, (c) pure Ag, (d) Ag0.50Ni0.50, (e) (Ag0.27Ni0.73)37RGO63. | ||
3.2. Formation mechanism of (AgxNi(1−x))yRGO(100−y) nanocomposites
In this ‘in situ’ co-precipitation reduction method the formation of Ni and Ag nanoparticles might occur due to the following reactions:53–55 When Ni(NO3)2 was dissolved in ethylene glycol, Ni2+ ion formed the coordination complex with glycol (eqn (5)).| Ni2+ + 3EG → [Ni(EG)3]2+ | (5) |
| [Ni(EG)3]2+ + mN2H4 → [Ni(N2H4)m]2+ + 3EG | (6) |
| [Ni(N2H4)m]2+ + 2OH− → Ni(OH)2 + mN2H4 | (7) |
| 2Ni(OH)2 + N2H4 → 2Ni0 + N2 + 4H2O | (8) |
When this complex reacted with N2H4, the ligand (i.e. EG) was replaced by N2H4 and formed another complex [Ni(N2H4)m]2+ (eqn (6)). Ni(OH)2 was formed when [Ni(N2H4)m]2+ was reacted with OH− (eqn (7)). The formation of nickel hydroxide from hydrazine complex occurred because Ni(OH)2 is more stable than Ni[(N2H4)m]2+, so this transformation is thermodynamically feasible.53 Then, the formation of Ni nanoparticles occurred via the reduction of nickel hydroxide by hydrazine (eqn (8)).
Ag nanoparticles were produced via the reduction of Ag+ ions by ethylene glycol and N2H4. Ethylene glycol was converted to acetaldehyde after losing one molecule of H2O (eqn (9)). Then, Ag+ ions were reduced by acetaldehyde and formed Ag nanoparticles (eqn (10)). Simultaneously, N2H4 also reduced Ag+ and produced Ag nanoparticles (eqn (11)).56–58
| HO–(CH2)2–OH → CH3CHO + H2O | (9) |
| 2Ag+ + 2CH3CHO → 2Ag0 + 2H+ + CH3COCOCH3 | (10) |
| 4Ag+ + N2H4 → 4Ag0 + N2 + 4H+ | (11) |
In this ‘in situ’ co-precipitation reduction technique to synthesize (AgxNi(1−x))yRGO(100−y), hydrazine also reduced GO (particularly its epoxy groups (eqn (12))1 and converted GO to RGO. Generally, this reduction process initiated from the edges of GO flakes and then moved towards the basal planes.1 Gao et al. have proposed the plausible reaction mechanism for the reduction of the epoxy groups of GO by N2H4. The general reaction for this reduction reaction can be presented as eqn (12).1
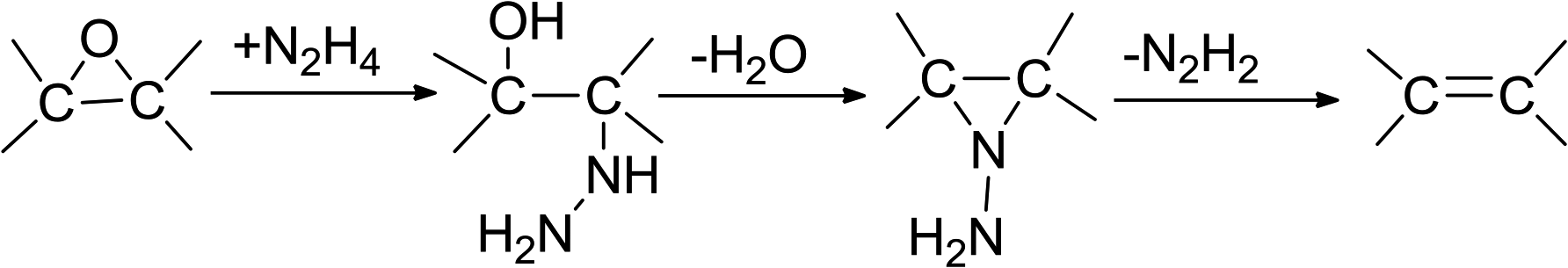 | (12) |
In this synthesis route, PVP played the role of a capping agent, which prevented the agglomeration of Ag and Ni nanoparticles and restricted their growth. Here, the formation of Ag and Ni nanoparticles and conversion of GO to RGO occurred simultaneously. The unreduced or partially reduced oxygen-containing functional groups of RGO helped to immobilize the Ag and Ni nanoparticles on the surface of nanometer thin RGO sheets. These metal nanoparticles also acted as a spacer between the RGO layers and prevented the agglomeration of RGO sheets.59,60
3.3. First-principles calculations of electronic structure
To understand the interfacial interactions between Ag, Ni, and graphene in the synthesized catalysts, we have performed the first-principles calculations based on DFT. The superlattice structures of Ag, Ni, graphene, Ag–Ni interface, and Ag–Ni-graphene before and after full relaxation are presented in Fig. S10 and S11 (ESI†). The lattice parameters obtained for the unit cell of Ag and Ni were 4.08 and 3.52 Å, respectively. These values matched well with the reported values. E. Schiavo et al. and F. Cinquini et al. have reported the values of lattice parameter of Ag and Ni 4.09 Å (ref. 61 and 62) and 3.52 Å,63 respectively. In the Ag–Ni superlattice, the binding energy between Ag and Ni was −5.68 eV, which indicated the existence of the interaction between Ag and Ni in the interface. In Ag–Ni-graphene superlattice, the binding energy between Ag–Ni interface and graphene was −3.32 eV, which also indicated the existence of strong interaction between them. The equilibrium interlayer distance between Ag–Ni interface and graphene was 3.29 Å.The electronic total charge density for the interface between Ag and Ni is shown in Fig. 4d, which clearly shows the orbital overlap between Ag and Ni in the interface. From this plot, it was observed that due to this interaction electron deficient centres were formed on/near the Ni atoms. The higher value of electronegativity of Ag (1.93) than that of Ni (1.91) might be responsible for this. The difference charge density plot of Ag–Ni (Fig. 4f), where the blue color represents the charge depletion and the red color represents the charge accumulation, also indicated the formation of electron deficient center on/near Ni atoms. The charge density distribution plot (Fig. 4e) and the difference charge density plot (Fig. 4g) of Ag–Ni-graphene clearly exhibited the interaction between Ag–Ni, C–Ag, and C–Ni in the interface.
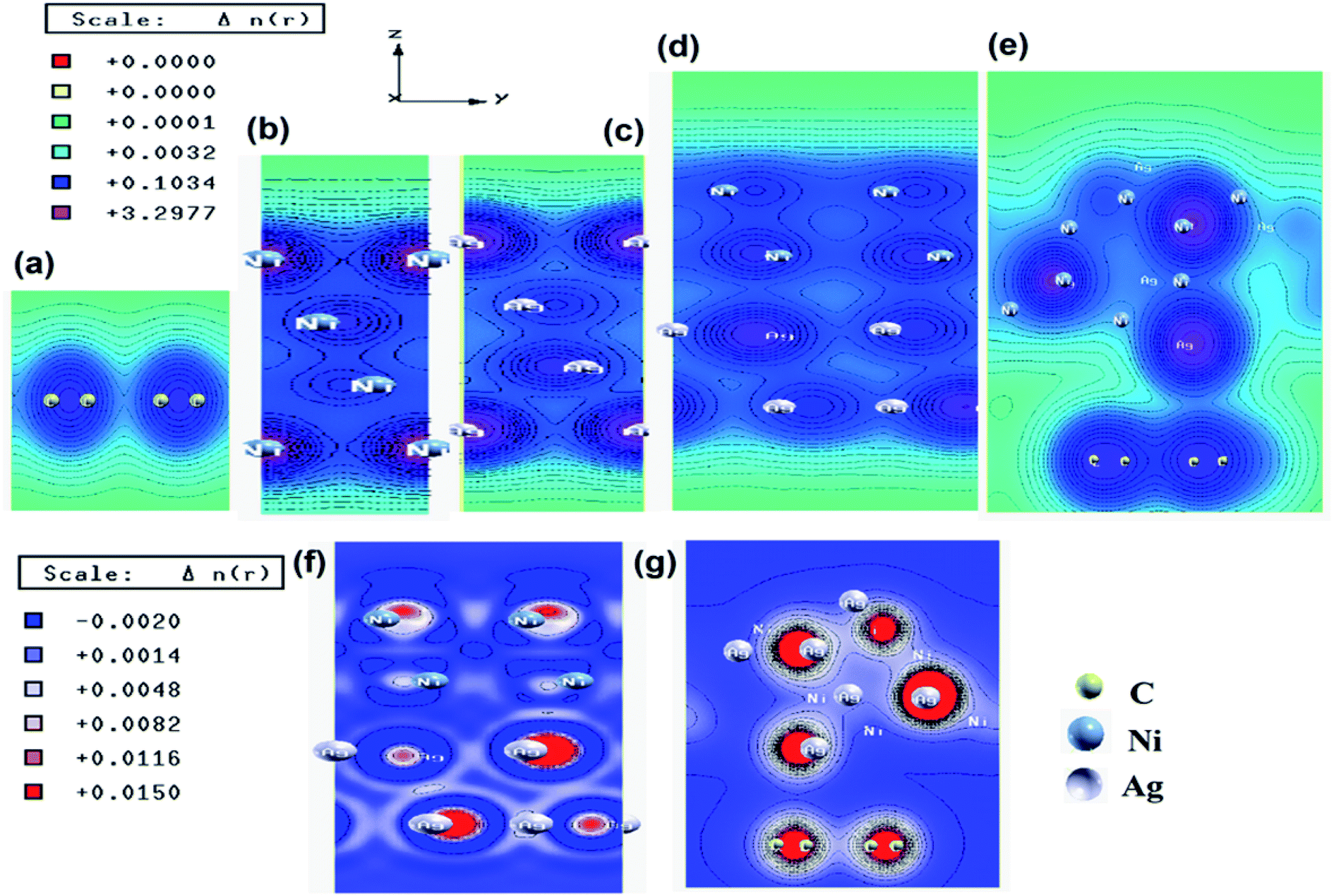 | ||
| Fig. 4 Electronic total charge density plot of (a) graphene, (b) Ni slab, (c) Ag slab, (d) Ag–Ni interface, (e) Ag–Ni-graphene superlattice, difference charge density plots of (f) Ag–Ni interface, and (g) Ag–Ni-graphene superlattice (where red color represents charge accumulation and blue color represents charge depletion in difference charge density plot). | ||
For in-depth understanding of the interactions which exist in the interfaces between Ag–Ni, and Ag–Ni-graphene, their Density of States (DOS), Projected Density of States (PDOS), and band structures were studied (Fig. S12–19, ESI†). Zero band gap of graphene was clearly shown by the band structure of graphene (Fig. S17, ESI†). This plot also showed that the valence bands and the conduction bands are either separated by a gap or overlapped with each other which intersect in two equivalent points, known as Dirac points in the first Brillouin zone.64,65 The band structures of Ag, Ni, and Ag–Ni-graphene also showed their metallic character (Fig. S12–19, ESI†). In the band structure of Ag–Ni interface the appearance of new bands near the Fermi level compared to Ag and Ni indicated the effect of Ag on the electronic property of Ni. PDOS of Ag–Ni interface (Fig. S15–S16, ESI†) also indicated the strong hybridization between Ni 3d, Ni 4s, Ag 4d and Ag 5s. The PDOS on the d band of Ni, which is positioned between −4 eV to −5 eV below the Fermi level, was found to be enhanced, and the width of d-band was broadened, indicating the hybridization of Ni 3d with Ag 4d states.66 PDOS of Ag–Ni-graphene (Fig. S18–S19, ESI†) indicated the strong hybridization between C 2p states of graphene and Ni 3d and Ag 4d at valance and conduction band. Due to this hybridization, we can predict that the conductivity of the composite will be increased when graphene will be incorporated in the system. The EIS measurements (Nyquist plot, Fig. S20, ESI†) of the synthesized catalysts supported this by showing the charge transfer resistance (Rct) of (Ag0.27Ni0.73)37RGO63 (0.144Ω) is much lower than that of RGO (2.073Ω), Ag0.50Ni0.50 (0.275Ω), Ni(0.85Ω), and Ag (3.11Ω), indicating the enhanced conductivity of (Ag0.27Ni0.73)37RGO63. The DFT calculations showed that the strong interfacial interaction and orbital level hybridization between Ag, Ni, and graphene in Ag–Ni-graphene superlattice originate the formation of electron deficient Ni centres and enhancement of electrical conductivity in Ag–Ni-graphene. These factors might play important roles in the intermediate formation, and faster charge transfer process during the catalysis reactions. Therefore, we can predict that the synthesized catalyst (AgxNi(1−x))yRGO(100−y) will show better catalytic activity than that of pure Ag, or pure Ni, or Ag–Ni. This prediction agrees well with our experimental results which have been discussed in details in Sections 3.4.
3.4. Catalytic activity of (AgxNi(1−x))yRGO(100−y)
| Composites | Initiation time (min) | Completion time (min) | Apparent rate constant (kapp) s−1 |
|---|---|---|---|
| Pure Ni | 8 | 30 | 2.46 × 10−3 |
| Ni40RGO60 | 4 | 18 | 6.66 × 10−3 |
| Pure Ag | 3 | 8 | 10.32 × 10−3 |
| Ag0.05Ni0.95 | 4 | 22 | 1.50 × 10−3 |
| Ag0.15Ni0.85 | 2 | 14 | 4.75 × 10−3 |
| Ag0.25Ni0.75 | 0 | 7 | 10.03 × 10−3 |
| Ag0.27Ni0.73 | 5 | 15 | 5.76 × 10−3 |
| (Ag0.05Ni0.95)90RGO10 | 2 | 15 | 4.83 × 10−3 |
| (Ag0.05Ni0.95)70RGO30 | 1 | 7 | 13.87 × 10−3 |
| (Ag0.05Ni0.95)60RGO40 | 0 | 4 | 19.60 × 10−3 |
| (Ag0.05Ni0.95)50RGO50 | 0 | 8 | 9.10 × 10−3 |
| (Ag0.05Ni0.95)40RGO60 | 2 | 10 | 8.39 × 10−3 |
| (Ag0.15Ni0.85)33.5RGO66.5 | 1 | 10 | 7.86 × 10−3 |
| (Ag0.27Ni0.73)37RGO63 | 0 | 6 | 10.83 × 10−3 |
| (Ag0.37Ni0.63)40.5RGO59.5 | 1 | 9 | 5.10 × 10−3 |
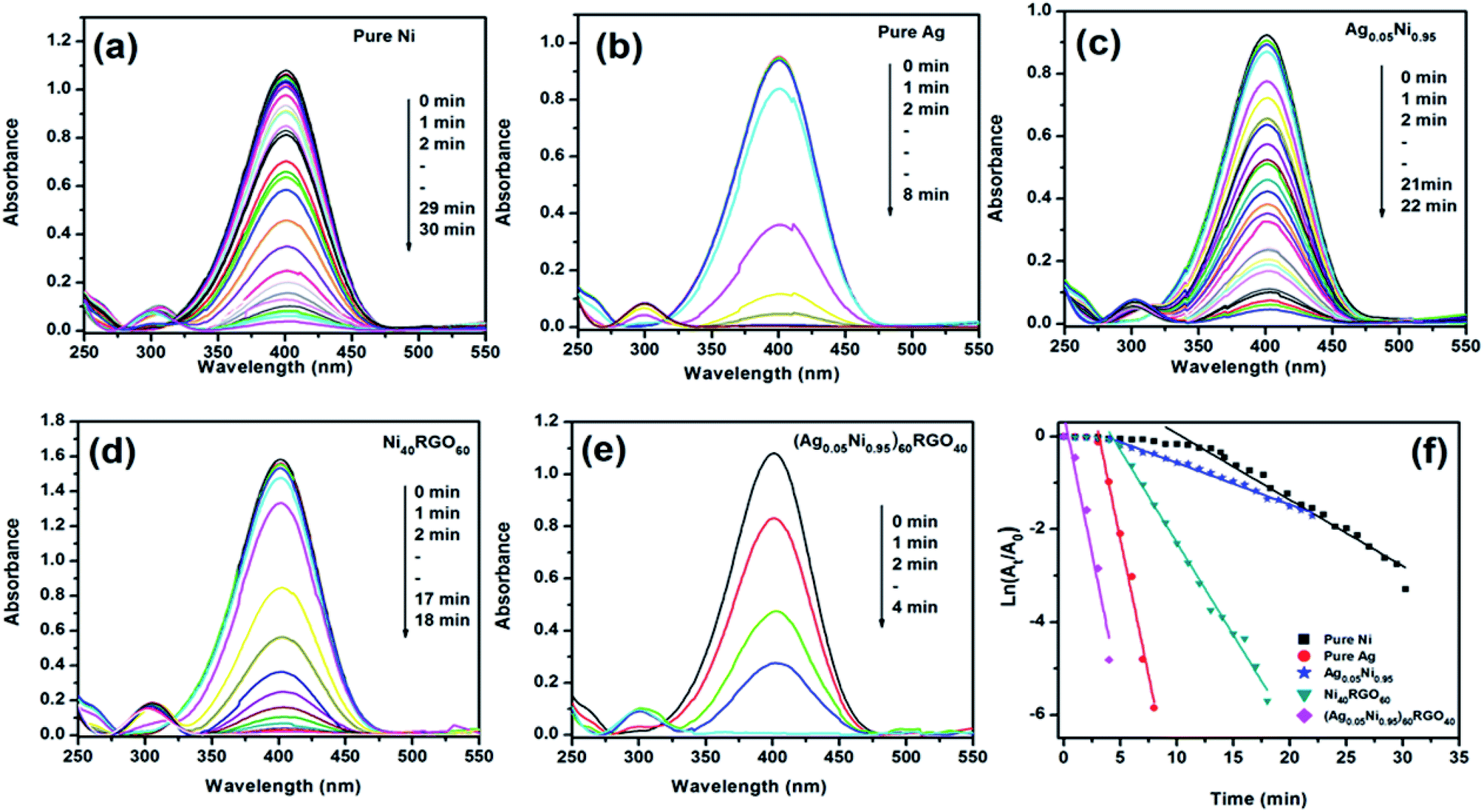 | ||
| Fig. 5 Time dependent UV-Vis spectral changes of the reaction mixture of 4-NP catalyzed by (a) pure Ni, (b) pure Ag, (c) Ag0.05Ni0.95, (d) Ni40RGO60, (e) (Ag0.05Ni0.95)60RGO40, and (f) pseudo first order kinetic plot of 4-NP reduction reaction with pure Ni, Pure Ag, Ag0.05Ni0.95, Ni40RGO60, and (Ag0.05Ni0.95)60RGO40. | ||
The conversion of 4-NP to 4-AP is a six electron transfer reduction reaction. When this reaction is carried out in the presence of metal nanoparticle catalyst and excess NaBH4, it proceeds via electron transfer mechanism where relaying of electrons occur from BH4− (donor) to 4-NP (acceptor). In the aqueous medium generation of hydrogen from BH4− occurs via the first adsorption of BH4− on the surface of catalyst (metal nanoparticles), followed by electron transfer (ET) to the metal nanoparticles.11,14,15 According to Ding et al. the metal nanoparticles play the role of storing electrons after ET from BH4−.41 The H atoms thus generated attack 4-NP and reduce it.11
Several factors are responsible for the high catalytic performance of (Ag0.05Ni0.95)60RGO40 towards this reaction, such as (i) nanosize of the Ag and Ni particles helps to adsorb BH4− ions on their surface, (ii) from the DFT study we have observed that, presence of Ag atoms creates electron deficient sites on/near Ni atoms. These electron deficient Ni atoms become more effective to collect electrons from BH4− via electron transfer process and to generate hydrogen efficiently. Thus the presence of Ag makes the catalysts more efficient towards the reduction of 4-NP in presence of NaBH4, (iii) presence of RGO in the catalyst also helps to enhance its catalytic activity by providing larger surface area. RGO exhibits high adsorption capacity of 4-NP due to π–π stacking interaction.3,10 Rout et al. have reported that due to the strong adsorption of 4-NP onto the surface of RGO, the NO bond of –NO2 group becomes stretched out (from 1.23 Å to 1.28 Å) and this helps to activate the –NO2 group for the reduction reaction and convert to –NH2 group.9 (iv) DFT study also showed the existence of hybridization in the orbital level between Ag, Ni, and graphene. Due to these interfacial interactions, the electrical conductivity of the catalyst enhances. (EIS measurements also showed the increase of the conductivity of catalyst due to the presence of RGO Fig. S20, ESI†). This increase of conductivity, as well as the highly conjugated π bonding RGO system facilitates the charge transfer between the catalytically active sites and the reactant molecules (i.e. 4-NP). These factors improve the efficiency of the catalyst ((Ag0.05Ni0.95)60RGO40).
 | (13) |
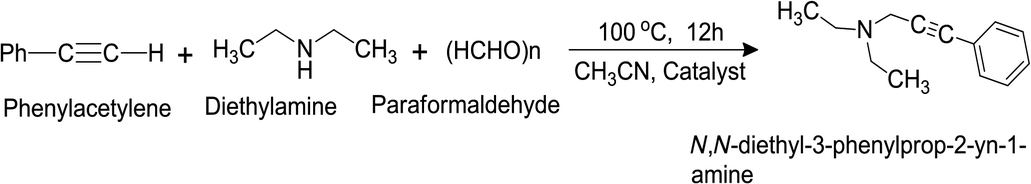
This reaction was performed in presence of the catalysts with varying compositions. When the reactions were conducted without any catalyst or with pure RGO no product was formed. When pure Ni particle was used as a catalyst, ∼71% yield was obtained. Incorporation of Ag nanoparticle in the catalyst resulted in the enhancement of the yield. For example, the yield was increased to ∼80% when Ag0.50Ni0.50 was the catalyst. To investigate the effect of RGO in the catalyst, the catalysts were prepared by immobilizing Ni nanoparticles on the surface of RGO (e.g. Ni10RGO90, Ni20RGO80, and Ni30RGO70 where the amount of RGO was 90, 80 and 70 wt%, respectively). Ni30RGO70 showed better catalytic performance (yield ∼80%) than that of pure Ni (yield ∼71%). As it has been observed that the presence of Ag and RGO with Ni enhanced the yield, now the catalysts were prepared where both Ag and Ni particles were immobilized on the surface of RGO and found to be beneficial in terms of catalytic activity. The % of yield of the product obtained by the catalysts having different compositions are listed in Table S4 (ESI†). It was observed that the catalyst (Ag0.27Ni0.73)37RGO63 generated the highest yield of ∼95%. The % of yields of the product which were obtained at different reaction conditions are listed in Table S5 (ESI†). To demonstrate the applicability of this catalyst for the A3 reaction, several other propargylamines were also synthesized by using different amines and aldehydes. It was observed that reasonably good yield (∼80–87%) was obtained and listed in Table 2. The LC-MS, 1H NMR, and FT-IR data of the products are provided in ESI (Spectral data section, Fig. S27–29, ESI†).
| Entry | Alkyne | Amine | Aldehyde | Product | Yield (%) |
|---|---|---|---|---|---|
| a Reaction condition: aldehyde (2 mmol), amine (2.4 mmol), phenylacetylene (3 mmol) catalyst (Ag0.27Ni0.73)37RGO63 50 mg, acetonitrile 10 ml, reaction temperature 100 °C, reaction time 12 h. | |||||
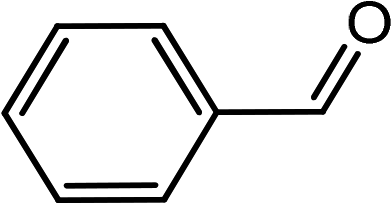
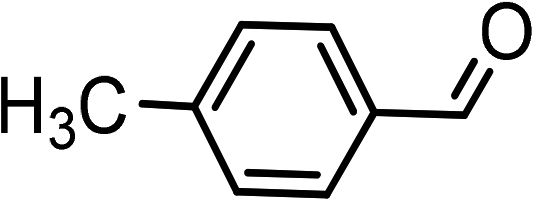
| 1 | Ph–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H C–H |
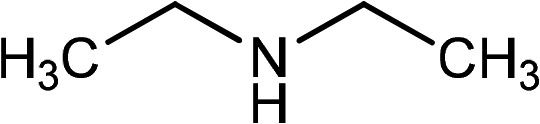 |
(HCHO)n | 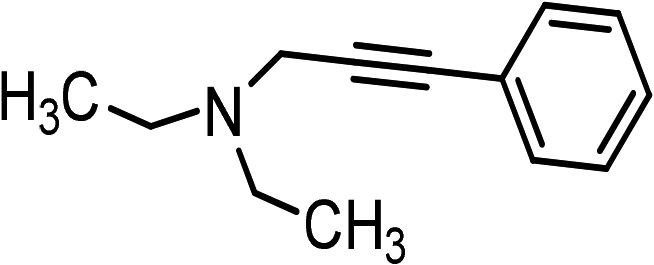 |
95 |
| 2 | Ph–CC–H |
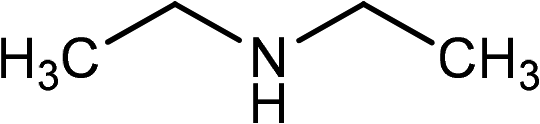 |
 |
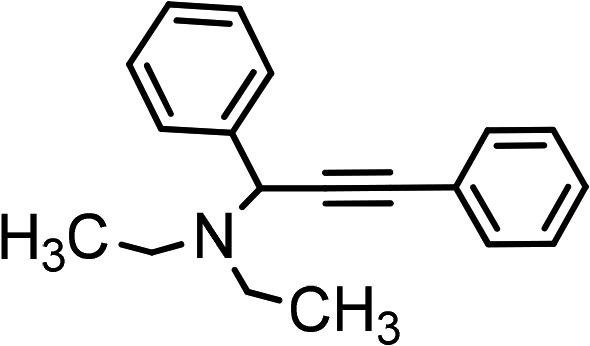 |
84 |
| 3 | Ph–CC–H |
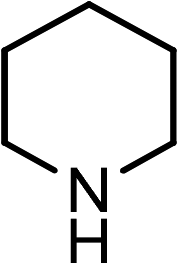 |
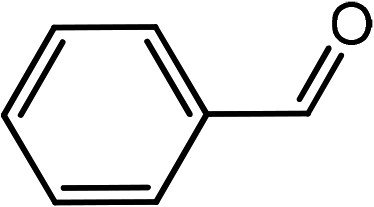 |
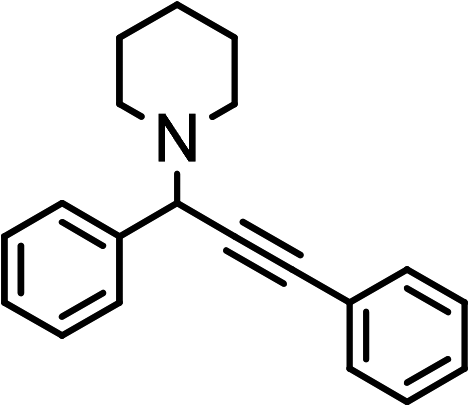 |
88 |
| 4 | Ph–CC–H |
 |
 |
 |
86 |
| 5 | Ph–CC–H |
 |
 |
 |
87 |
| 6 | Ph–CC–H |
 |
 |
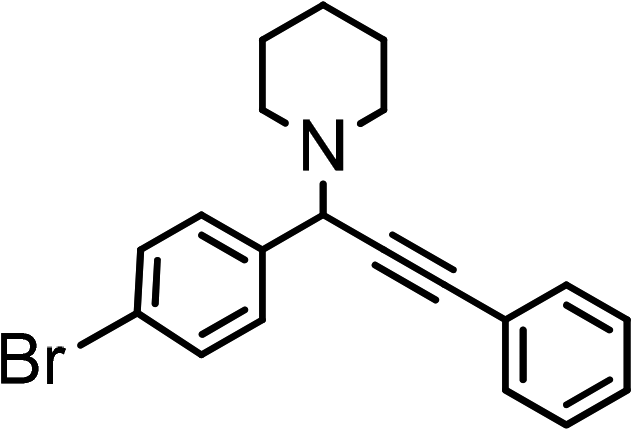 |
80 |
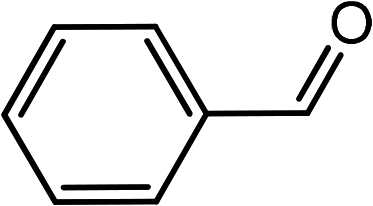
| 7 | Ph–CC–H |
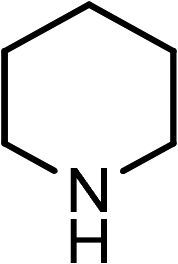 |
(HCHO)n | 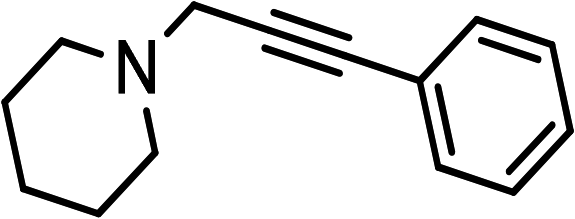 |
80 |
| 8 | Ph–CC–H |
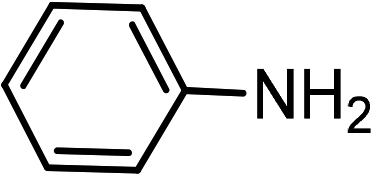 |
 |
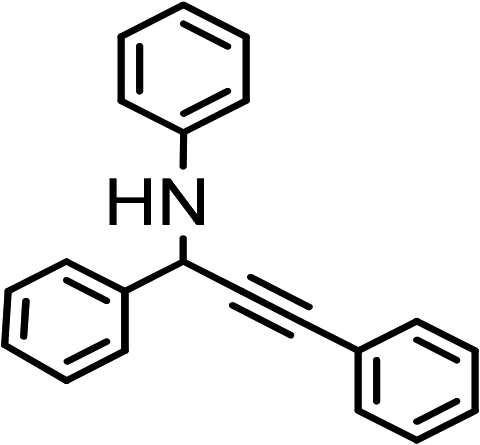 |
87 |
| 9 | Ph–CC–H |
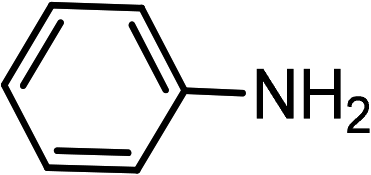 |
 |
 |
83 |
| 10 | Ph–CC–H |
 |
 |
 |
78 |
| 11 | Ph–CC–H |
 |
 |
 |
80 |
The comparison of catalytic activity in terms of % yield of N,N-diethyl-3-phenylprop-2-yn-1-amine of different reported catalysts with that of (Ag0.27Ni0.73)37RGO63 is listed in Table S6 (ESI†) and it was observed that the catalytic activity of the synthesized catalyst is comparable with the reported catalysts.17,18,23,67
The reaction mechanisms for nanoparticles catalysed A3 coupling reaction, have been proposed by different researchers.17,68,69 The reaction generally proceeds via the activation of C– H bond of the alkyne in the first step by the formation of a metal acetylide intermediate, which is generated by the reaction of alkyne and the metal nanoparticles. In the second step, this metal acetylide reacts with iminium ion (generated ‘in situ’ from the reaction of aldehyde and amine) and forms the corresponding propargylamine. The tentative reaction mechanism is shown in Scheme 1.
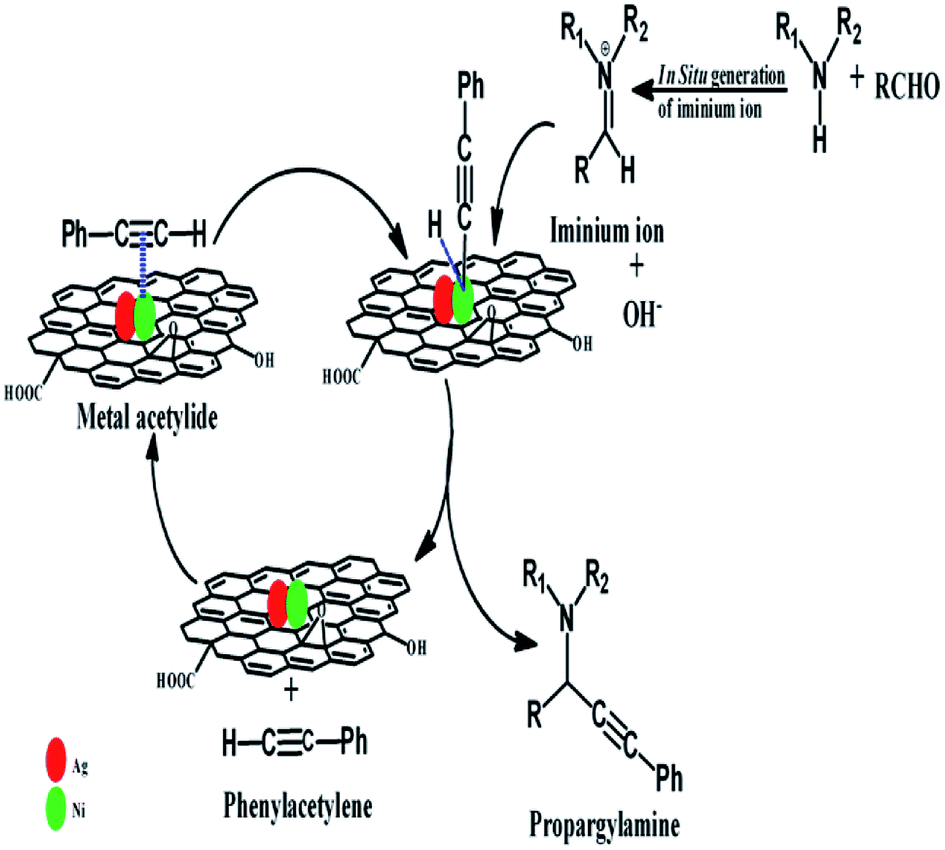 | ||
| Scheme 1 The tentative reaction mechanism for synthesis of propargylamines via A3 coupling reaction. | ||
In this reaction, the electron deficient centres on the Ni, which are generated due to the presence of Ag in the catalyst, helps to coordinate –CC– of phenylacetylene with Ni and subsequently formation of Ni-acetylide intermediate more efficiently. Here also, RGO helps to adsorb the reactant molecules on the surface of the catalyst by providing larger surface area and π–π stacking interaction. The enhancement of the conductivity of the catalyst due to the presence of RGO facilitates the charge transfer between catalytically active sites (i.e. metal nanoparticles) and reactant molecules. Therefore, the catalysts which contain Ag, Ni, and RGO show superior catalytic activity than pure Ag, or pure Ni, or Ni–Ag, or Ni-RGO.
 | (14) |
The reaction was performed in acetonitrile medium by varying the reaction conditions (e.g., temperature, reaction time, styrene:TBHP ratio) and catalyst compositions. When the reaction was performed without catalyst or with pure RGO no desirable product was formed. Table S7 (ESI†) summarizes the effects of catalyst composition on the % of styrene conversion and % of styrene oxide formation. It was observed that the presence of Ag nanoparticle along with Ni particle in the catalyst composite was beneficial for obtaining better styrene conversion and selectivity of styrene oxide formation (Fig. S22, ESI†). The catalytic performance of the catalyst was further improved when Ag and Ni nanoparticles were dispersed on the surface of RGO. When the reaction was performed in the presence of (Ag0.27Ni0.73)37RGO63 catalyst the highest amount of styrene conversion (96%) with 89% styrene oxide formation selectivity were achieved. The catalytic performance of (Ag0.27Ni0.73)37RGO63 at different reaction conditions are listed in Table S8 (ESI) (Fig. S23 and S24, ESI†). The catalytic performance of (Ag0.27Ni0.73)37RGO63 towards the epoxidation reaction of styrene was found to be comparable and in some cases better than that of reported catalysts (Table S9, ESI†).
Several researchers have addressed the epoxidation of styrene on the various catalyst (e.g. Ag, Au, Au–BaTiO3, etc.).24,25,30,39,40 The plausible reaction mechanism for the oxidation of styrene to styrene oxide is presented in Scheme 2. In the first step of the reaction, a metal alkyl peroxy intermediate (Intermediate-I) is formed via interaction between catalytically active metal nanoparticles and TBHP. When styrene reacts with intermediate I, two type of intermediates (Intermediate IIa and Intermediate IIb) are formed which are isomers of oxametallacyclic species. From Intermediate IIa, epoxide (i.e. styrene oxide) is formed via sharpless mechanism where the transfer of oxygen occurs to the olefinic bond. On the other hand benzaldehyde forms as a byproduct from Intermediate IIb via breakage of the C–C bond.24
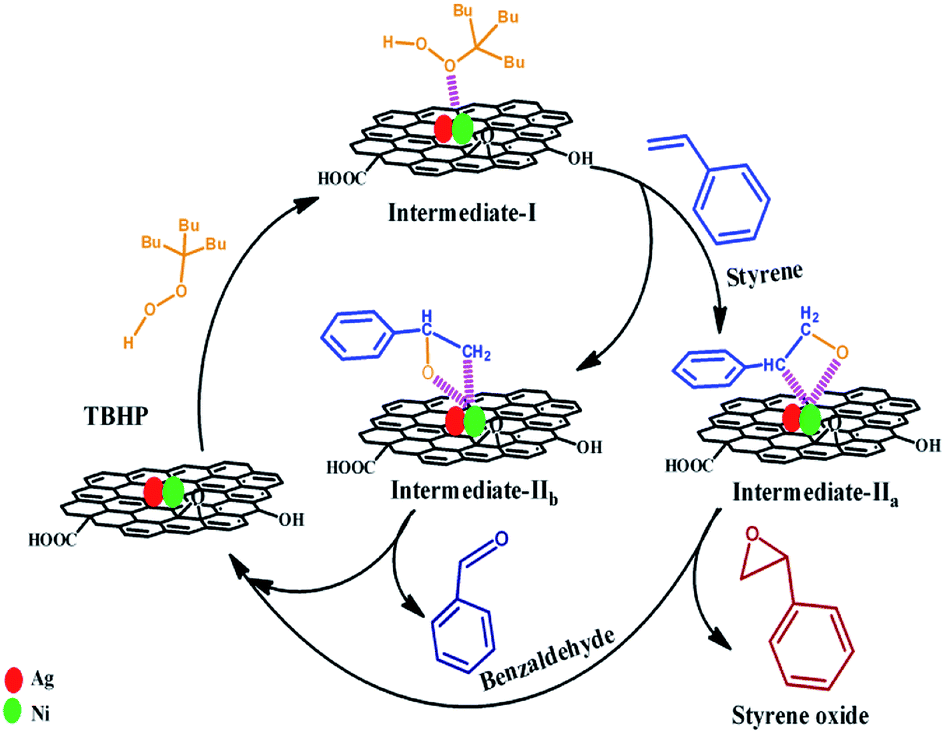 | ||
| Scheme 2 Plausible reaction mechanism of formation of styrene oxide and benzaldehyde from styrene in presence of TBHP catalyzed by (Ag0.27Ni0.73)37RGO63. | ||
In the synthesized catalyst (AgxNi(1−x))yRGO(100−y) presence of electron deficient Ni centres helps in the formation of Intermediates IIa and IIb from Intermediate I via formation of coordination complexes with metal nanoparticles. Moreover, the presence of RGO helps to adsorb styrene molecule on the surface of the catalyst by π–π interaction. Due to these factors, the catalyst containing Ag, Ni, and RGO showed better catalytic activity than pure Ni, pure Ag, and Ag0.50Ni0.50.
 | (15) |
First, the reaction was performed without any catalyst or with pure RGO and no product was obtained. Then the reactions were carried out in presence of the synthesized catalysts, which are composed of varying amount of Ni, Ag, and RGO. When pure Ni and pure Ag nanoparticles were used as a catalyst, the yield was ∼50% and ∼48%, respectively. When Ag0.50Ni0.50 was used as catalyst the product yield was increased to ∼62%, which clearly indicated that the presence of Ag positively influences the catalytic activity property of the catalyst. It has also been observed that, when Ni nanoparticle was dispersed on the surface of RGO, the efficiency of the catalyst was also increased. For example, when Ni30RGO70 was used as catalyst the yield was ∼66% which is higher than that of pure Ni-catalyzed reaction. This enhancement of the catalytic activity might be due to the following reasons (i) dispersion of Ni particles on the RGO surface prevents the agglomeration of nanoparticles. Therefore, the reactant molecules get more access to the catalytically active sites, (ii) the reactant molecules (e.g. styrene oxide, phenylacetylene) are adsorbed more on the surface of the RGO containing catalysts, because of the π–π interaction between RGO sheets and phenyl group of the reactant molecules, (iii) the high conductivity of RGO facilitates the electron transfer process during the reaction.
As it has been observed that the presence of Ag and RGO helped in the enhancement of the catalytic activity of Ni, we have tested the catalytic activity of the catalyst, containing varying amount of Ag, Ni, and RGO. The change in % of product yield with the variation of the composition of the synthesized catalyst is listed in Table 3. It has been observed that the catalyst containing 10 wt% Ag, 27% Ni, and 63 wt% RGO (i.e. (Ag0.27Ni0.73)37RGO63) showed the highest catalytic activity with ∼91% yield. The performance of this catalyst for this reaction was compared with the reported catalysts (Table S10, ESI†) and found to be comparable and in some cases superior to some of the reported results. We have also performed the Click reaction by replacing styrene oxide with cyclohexane oxide (and epichlorohydrine) and found the % of the yield of the corresponding products are ∼88% and ∼67%.
| Catalyst | Yield (%) |
|---|---|
| a Reaction condition: styrene oxide (1 mmol 0.115 ml), phenylacetylene (1 mmol 0.110 ml) sodium azide (1.5 mmol 97.5 mg), water (3 ml), reaction time 12h, reaction temperature = 100 °C, catalyst dose = 25 mg. | |
| Pure Ni | 55 |
| Pure Ag | 53 |
| Ni10RGO90 | 26 |
| Ni20RGO80 | 66 |
| Ni30RGO70 | 66 |
| Ag0.50Ni0.50 | 62 |
| (Ag0.15Ni0.85)33.5RGO66.5 | 89 |
| (Ag0.27Ni0.73)37RGO63 | 91 |
| (Ag0.37Ni0.63)40.5RGO59.5 | 90 |
| (Ag0.45Ni0.55)44RGO56 | 92 |
The tentative reaction mechanism for this Click reaction was proposed by several researchers10,32,33,35 and shown in Scheme 3. In this reaction, the metal nanoparticles play the role of a catalytically active site and act as a bifunctional catalyst which combines one pot ring opening of epoxy and 1,3-dipolar cyclo addition.10,32 Initially, catalytically active metal azide is formed which helps to activate epoxide and facilitates the delivery of azide ion during the ring opening of epoxide. Thus, the organoazide intermediate (Intermediate-I) is formed from epoxy and azide. During the same time, another intermediate, metal-acetylide complex (Intermediate-II) forms via π complexation between the metal nanoparticle and phenylacetylene. Then coordination of organoazide (Intermediate-I) with the metal centre of metal acetylide (Intermediate II) occurs which enhances the nucleophilicity of –CC–. Then 1,3 dipolar cyclo addition reaction results in the formation of the new C–N bond between the nucleophilic β-carbon atom of the acetylide and the terminal electrophilic nitrogen atom of the azide (Intermediate-III).10,32 After protonation, intermediate-III yields the β-hydroxy-1,2,3-triazole. In this reaction pathway formation of metal azide via interaction of metal nanoparticles and N3−, and formation of the metal-acetylide complex (which forms via π complexation between the metal nanoparticle and phenylacetylene) play critical roles. As the DFT study showed that the presence of Ag causes the formation of electron deficient centres near/on Ni atoms, the formation of metal azide and metal-acetylide occurs efficiently with these electron deficient Ni centres. Moreover, the presence of RGO in the synthesized catalyst also helps to absorb the reactant materials (e.g. phenylacetylene, styrene oxide) on the surface of the catalyst. These factors are responsible for the superior catalytic activity of (AgxNi(1−x))RGO(100−x) than pure Ag, Ni, or Ag0.50Ni0.50.
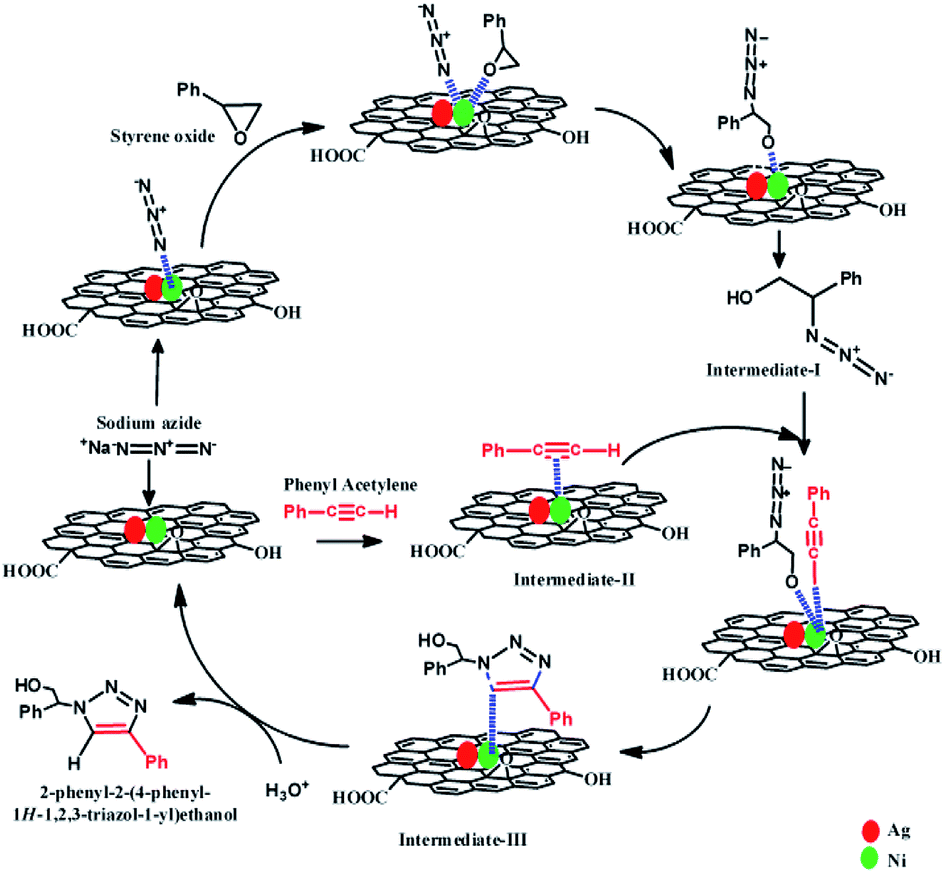 | ||
| Scheme 3 The tentative reaction mechanism involved for synthesis of 2-phenyl-2-(4-phenyl-1H-1,2,3-triazol-1-yl)ethanol via Click reaction catalyzed by (Ag0.27Ni0.73)37RGO63 catalyst. | ||
The details of the spectral analysis of products, which were formed in the A3 coupling reaction, epoxidation reaction of styrene and Click reaction, are provided in ESI (Spectra data section, Fig. S25–S29, ESI†).
3.5. Reusability
The presence of Ni nanoparticle introduced the magnetic character in (AgxNi(1−x))yRGO(100−y) nanocatalyst. Using VSM the saturation magnetization (Ms) and coercivity (Hc) values of the catalysts were determined. For example, Ms and Hc values of (Ag0.27Ni0.73)37RGO63, were 9.81 emu g−1 and 146.5 Oe, respectively (Fig. S30, ESI†). The magnetic character of this catalyst allowed its easy magnetic separation from the reaction mixture. After completion of all the reactions, the catalyst was separated from the reaction mixture by applying a magnet externally (Fig. S31, ESI†) and then washed with distilled water for several times and dried in a vacuum oven at 60 °C for 12 h. The recovered catalyst was used in the next cycle of the catalysis reactions. No significant loss of activity was observed up to 6th reuse for all four reactions and shown in Fig. 6. XRD patterns and FESEM micrograph of the reused catalyst also confirmed that the crystal structure and morphology of the catalyst remained same after 6th catalysis cycle (Fig. S32, ESI†). These results thus clearly demonstrated the structural robustness of the catalyst.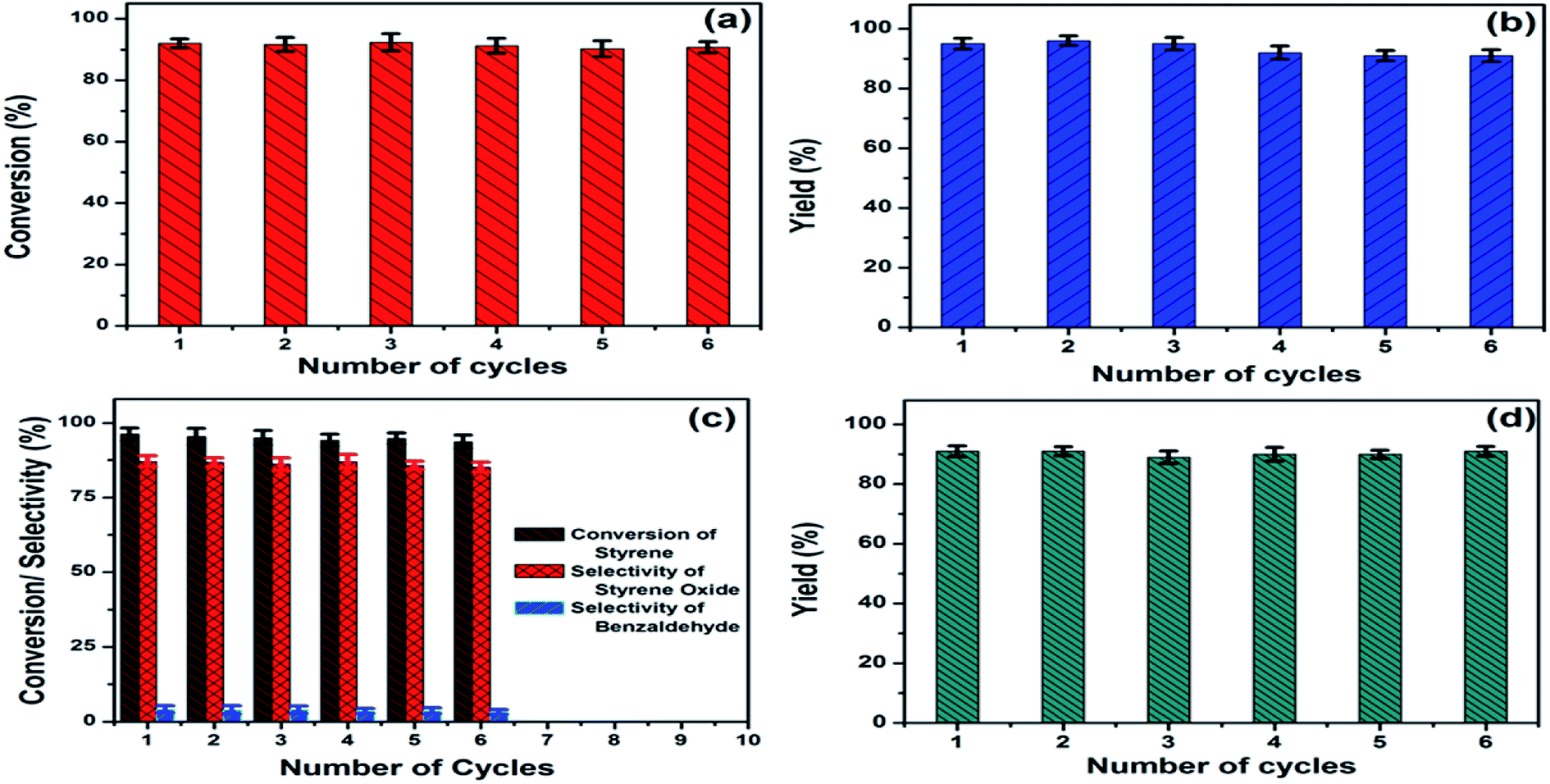 | ||
| Fig. 6 Reusability of (a) (Ag0.05Ni0.95)60RGO40 catalyst for the reduction of 4-NP, and (Ag0.27Ni0.73)37RGO63 catalyst for (b) the synthesis of N,N-diethyl-3-phenylprop-2-yn-1-amine via A3 coupling reaction, (c) the epoxidation reaction of styrene to styrene oxide, and (d) the synthesis of 2-phenyl-2-(4-phenyl-1H-1,2,3-triazole-1-yl)ethanol via Click reaction. | ||
4. Conclusion
Here, we have reported a simple ‘in situ’ co-precipitation reduction technique for the preparation of nanocatalyst containing Ag, Ni, and RGO (AgxNi(1−x))yRGO(100−y). In this catalyst Ag and Ni particles were immobilized on the surface of the nanometer thin RGO sheets. Here, RGO provided a highly conductive support to the catalytically active Ni and Ag nanoparticles. DFT studies were performed to obtain the electronic structure and electronic properties of Ag–Ni-graphene. This study helps us to understand the interfacial interactions and orbital level hybridizations which exist in the interfaces between Ag, Ni, and RGO. These interfacial interactions create electron deficient centres near/on the Ni atoms and also enhance the conductivity of the catalyst. The synergistic effect, which arises from the coexistence of Ag, Ni, and RGO in the catalyst, positively influences the catalytic activity of the synthesized catalysts.Here, it has been demonstrated that combination of Ag, Ni, and RGO in the composition of the catalyst caused significant enhancement of the catalytic activity compared to pure Ag and Ni nanoparticles towards four important reactions (i) reduction of 4-nitrophenol (4-NP) in the presence of NaBH4, (ii) A3 coupling reaction for the synthesis of propargylamines, (iii) epoxidation of styrene, and (iv) ‘Click reaction’ for the synthesis of 1,2,3-triazole derivatives in aqueous medium. (Ag0.05Ni0.95)60RGO40 exhibited highest catalytic activity towards the reduction of 4-nitrophenol with kapp 19.6 1910−3 s−1. In case of the A3 coupling reaction, (Ag0.27Ni0.73)37RGO63 catalyzed reaction produced N,N-diethyl-3-phenylprop-2-yn-1-amine with ∼95% yield. This same catalyst showed ∼96% styrene conversion with ∼89% styrene oxide formation selectivity for epoxidation of styrene reaction, and ∼91% yield for the synthesis of 2-phenyl-2-(4-phenyl-1H-1,2,3-triazol-1-yl)ethanol via Click reaction. The catalyst also demonstrated its easy magnetic recovery from the reaction mixture after completion of the reactions. The recovered catalysts also exhibited a very good reusability.
To the best of our knowledge, this is the first time a catalyst having composition (AgxNi(1−x))yRGO(100−y) which showed the versatile catalytic activity towards the aforesaid reactions with high efficiency and also the investigations on the electronic structures of this catalyst by DFT calculations have been reported. The easy synthetic methodology high catalytic performance with very good reusability and easy magnetic recovery make the synthesize catalyst an attractive catalyst.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr N. N. Ghosh gratefully acknowledges DRDO Jodhpur for VSM analysis. Dr Ghosh is thankful to the Central Sophisticated Instrumentation Facility (CSIF) of BITS Pilani K. K. Birla Goa campus for providing the FESEM and LC-MS facility.Notes and references
- X. Gao, J. Jang and S. Nagase, J. Phys. Chem. C, 2009, 114, 832 CrossRef.
- M. Hu, Z. Yao and X. Wang, Ind. Eng. Chem. Res., 2017, 56, 3477 CrossRef CAS.
- D. Moitra, B. Ghosh, M. Chandel, R. Jani, M. Patra, S. Vadera and N. Ghosh, RSC Adv., 2016, 6, 14090 RSC.
- D. Moitra, M. Chandel, B. K. Ghosh, R. K. Jani, M. K. Patra, S. R. Vadera and N. N. Ghosh, RSC Adv., 2016, 6, 76759 RSC.
- D. Moitra, C. Anand, B. K. Ghosh, M. Chandel and N. N. Ghosh, ACS Appl. Energy Mater., 2018, 1, 464 CrossRef CAS.
- X. Fan, G. Zhang and F. Zhang, Chem. Soc. Rev., 2015, 44, 3023 RSC.
- O. C. Compton and S. T. Nguyen, Small, 2010, 6, 711 CrossRef CAS PubMed.
- K. Hareesh, R. Joshi, D. Sunitha, V. Bhoraskar and S. Dhole, Appl. Surf. Sci., 2016, 389, 1050 CrossRef.
- L. Rout, A. Kumar, R. S. Dhaka, G. N. Reddy, S. Giri and P. Dash, Appl. Catal., A, 2017, 538, 107 CrossRef CAS.
- D. Moitra, B. K. Ghosh, M. Chandel and N. N. Ghosh, RSC Adv., 2016, 6, 97941 RSC.
- T. Aditya, A. Pal and T. Pal, Chem. Commun., 2015, 51, 9410 RSC.
- M. Chandel, D. Moitra, P. Makkar, H. Sinha, H. S. Hora and N. N. Ghosh, RSC Adv., 2018, 8, 27725 RSC.
- H. Hu, J. H. Xin, H. Hu, X. Wang, D. Miao and Y. Liu, J. Mater. Chem. A, 2015, 3, 11157 RSC.
- B. Naik, S. Hazra, V. S. Prasad and N. N. Ghosh, Catal. Commun., 2011, 12, 1104 CrossRef CAS.
- B. K. Ghosh, D. Moitra, M. Chandel, H. Lulla and N. N. Ghosh, Mater. Res. Bull., 2017, 94, 361 CrossRef CAS.
- Y.-g. Wu, M. Wen, Q.-s. Wu and H. Fang, J. Phys. Chem. C, 2014, 118, 6307 CrossRef CAS.
- T. K. Saha and R. Das, ChemistrySelect, 2018, 3, 147 CrossRef CAS.
- N. Hussain and M. R. Das, New J. Chem., 2017, 41, 12756 RSC.
- S. Frindy, A. El Kadib, M. Lahcini, A. Primo and H. García, Catal. Sci. Technol., 2016, 6, 4306 RSC.
- A. M. Munshi, M. Shi, S. P. Thomas, M. Saunders, M. A. Spackman, K. S. Iyer and N. M. Smith, Dalton Trans., 2017, 46, 5133 RSC.
- X. Zhang and A. Corma, Angew. Chem., 2008, 120, 4430 CrossRef.
- K. Namitharan and K. Pitchumani, Eur. J. Org. Chem., 2010, 3, 411 CrossRef.
- N. Salam, A. Sinha, A. S. Roy, P. Mondal, N. R. Jana and S. M. Islam, RSC Adv., 2014, 4, 10001 RSC.
- D. Nepak and D. Srinivas, Appl. Catal., A, 2016, 523, 61 CrossRef CAS.
- Q. Wang, C. Li, J. Bai, W. Sun and J. Wang, J. Inorg. Organomet. Polym. Mater., 2016, 26, 488 CrossRef CAS.
- V. R. Choudhary, R. Jha and P. Jana, Catal. Commun., 2008, 10, 205 CrossRef CAS.
- D.-H. Zhang, H.-B. Li, G.-D. Li and J.-S. Chen, Dalton Trans., 2009, 47, 10527 RSC.
- A. Wang and H. Jing, Dalton Trans., 2014, 43, 1011 RSC.
- B. Sakthivel, D. S. R. Josephine, K. Sethuraman and A. Dhakshinamoorthy, Catal. Commun., 2018, 108, 41 CrossRef CAS.
- A. S. Sharma and H. Kaur, Appl. Catal., A, 2017, 546, 136 CrossRef CAS.
- A. N. Prasad, B. Thirupathi, G. Raju, R. Srinivas and B. M. Reddy, Catal. Sci. Technol., 2012, 2, 1264 RSC.
- H. Naeimi and Z. Ansarian, Appl. Organomet. Chem., 2017, 31, e3796 CrossRef.
- B. K. Ghosh, D. Moitra, M. Chandel, M. K. Patra, S. R. Vadera and N. N. Ghosh, Catal. Lett., 2017, 147, 1061 CrossRef CAS.
- B. K. Ghosh, S. Hazra and N. N. Ghosh, Catal. Commun., 2016, 80, 44 CrossRef CAS.
- C. Wang, D. Ikhlef, S. Kahlal, J.-Y. Saillard and D. Astruc, Coord. Chem. Rev., 2016, 316, 1 CrossRef CAS.
- B. A. Kumar, K. H. V. Reddy, B. Madhav, K. Ramesh and Y. Nageswar, Tetrahedron Lett., 2012, 53, 4595 CrossRef.
- X. Huo, J. Liu, B. Wang, H. Zhang, Z. Yang, X. She and P. Xi, J. Mater. Chem. A, 2013, 1, 651 RSC.
- R. N. Baig and R. S. Varma, Chem. Commun., 2013, 49, 752 RSC.
- D.-H. Zhang, G.-D. Li, J.-X. Li and J.-S. Chen, Chem. Commun., 2008, 29, 3414 RSC.
- X. Wang, Z. Liang, F. Zhang, L. Yang and S. Xu, J. Mater. Sci., 2013, 48, 5899 CrossRef CAS.
- M. Gopiraman, D. Deng, S. Saravanamoorthy, I.-M. Chung and I. S. Kim, RSC Adv., 2018, 8, 3014 RSC.
- W. Hummers and R. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- I. Solovyev, P. Dederichs and V. Anisimov, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 16861 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
- Quantum ESPRESSO, Files: C.pbe-n-rrkjus_psl.1.0.0.UPF, Ag.pbe-n-rrkjus_psl.1.0.0.UPF, and Ni.pbe-n-rrkjus_psl.1.0.0.UPF, https://www.quantum-espresso.org/pseudopotentials, (accessed February 2018).
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188 CrossRef.
- M. Methfessel and A. Paxton, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 3616 CrossRef CAS.
- N. Marzari, D. Vanderbilt, A. De Vita and M. Payne, Phys. Rev. Lett., 1999, 82, 3296 CrossRef CAS.
- M. Zong, Y. Huang, Y. Zhao, X. Sun, C. Qu, D. Luo and J. Zheng, RSC Adv., 2013, 3, 23638 RSC.
- Z. G. Wu, M. Munoz and O. Montero, Adv. Powder Technol., 2010, 21, 165 CrossRef CAS.
- D. Knetsch and W. Groeneveld, Inorg. Chim. Acta, 1973, 7, 81 CrossRef CAS.
- S.-H. Wu and D.-H. Chen, J. Colloid Interface Sci., 2003, 259, 282 CrossRef CAS PubMed.
- T. Zhao, R. Sun, S. Yu, Z. Zhang, L. Zhou, H. Huang and R. Du, Colloids Surf., A, 2010, 366, 197 CrossRef CAS.
- U. Nickel, A. zu Castell, K. Pöppl and S. Schneider, Langmuir, 2000, 16, 9087 CrossRef CAS.
- J. H. Byeon and Y.-W. Kim, Ultrason. Sonochem., 2012, 19, 209 CrossRef CAS PubMed.
- R. Dhanda and M. Kidwai, J. Mater. Chem. A, 2015, 3, 19563 RSC.
- L. Liu, J. Xue, X. Shan, G. He, X. Wang and H. Chen, Catal. Commun., 2016, 75, 13 CrossRef CAS.
- E. Schiavo, A. B. Muñoz-García, V. Barone, A. Vittadini, M. Casarin, D. Forrer and M. Pavone, Chem. Phys. Lett., 2018, 693, 28 CrossRef CAS.
- R. Wyckoff, Crystal structures, 1963, vol. 1, p. 85 Search PubMed.
- F. Cinquini, F. Delbecq and P. Sautet, Phys. Chem. Chem. Phys., 2009, 11, 11546 RSC.
- P. Khomyakov, G. Giovannetti, P. Rusu, G. v. Brocks, J. Van den Brink and P. J. Kelly, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 195425 CrossRef.
- C. Gong, G. Lee, B. Shan, E. M. Vogel, R. M. Wallace and K. Cho, J. Appl. Phys., 2010, 108, 123711 CrossRef.
- Y.-A. Zhu, D. Chen, X.-G. Zhou, P.-O. Åstrand and W.-K. Yuan, Surf. Sci., 2010, 604, 186 CrossRef CAS.
- C. Wei, Z. Li and C.-J. Li, Synlett, 2004, 09, 1472 Search PubMed.
- G.-P. Yong, D. Tian, H.-W. Tong and S.-M. Liu, J. Mol. Catal. A: Chem., 2010, 323, 40 CrossRef CAS.
- W. Yan, R. Wang, Z. Xu, J. Xu, L. Lin, Z. Shen and Y. Zhou, J. Mol. Catal. A: Chem., 2006, 255, 81 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08235a |
| This journal is © The Royal Society of Chemistry 2018 |