
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and characterization of chionodracine-derived antimicrobial peptides with enhanced activity against drug-resistant human pathogens
Cristina Olivieri†
ad,
Francesca Bugli†b,
Giulia Menchinellib,
Gianluigi Vegliacd,
Francesco Buonocore a,
Giuseppe Scapigliatia,
Valentina Stocchia,
Francesca Ceccaccie,
Massimiliano Papif,
Maurizio Sanguinetti*b and
Fernando Porcelli*a
a,
Giuseppe Scapigliatia,
Valentina Stocchia,
Francesca Ceccaccie,
Massimiliano Papif,
Maurizio Sanguinetti*b and
Fernando Porcelli*a
aDepartment for Innovation in Biological, Agrofood and Forest Systems, University of Tuscia, L.go dell'Universita' snc, 01100 Viterbo, Italy. E-mail: porcelli@unitus.it; Tel: +390761357041
bMicrobiology Institute, Catholic University of Sacred Heart, Rome, Italy. E-mail: maurizio.sanguinetti@unicatt.it
cDepartment of Chemistry, University of Minnesota, Minneapolis, 55455 USA
dDepartment of Biochemistry, Molecular Biology and Biophysics, University of Minnesota, Minneapolis, 55455 USA
eCNR – Institute of Chemical Methodologies, Sezione Meccanismi di Reazione UOS of Rome, Rome, Italy
fPhysics Institute, Catholic University of Sacred Heart, Rome, Italy
First published on 12th December 2018
Abstract
Starting from the sequence of the amphipathic α-helix of chionodracine (Cnd, 22 amino acids), we designed a series of mutants to increase Cnd's antimicrobial activity and selectivity toward prokaryotic cells and drug-resistant bacterial pathogens. We characterized these new Cnd-derived peptides using fluorescence, CD spectroscopy, and transmission electron microscopy, studying their interactions with synthetic lipid vesicles and assaying their biological function against E. faecium, S. aureus, K. pneumoniae, A. baumannii, P. aeruginosa, and Enterobacter sp. Upon interaction with model membranes, these new peptides with higher net charges and hydrophobic moments adopt a helical conformation similar to Cnd. Notably, they display a low cytotoxic activity against human primary cells, a low hemolytic activity, but a significantly high bactericidal activity against drug-resistant bacterial pathogens. The low values of micromolar minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) make these Cnd-derived peptides potential templates to develop antimicrobial agents against drug-resistant human pathogens.
Introduction
Antimicrobial peptides (AMPs) are antibiotic agents found in microorganisms, plants, and animals that are promising alternatives to conventional antibiotics.1,2 AMPs represent the first line of the innate immune system defense against pathogens and usually show a broad spectrum of action against fungi, bacteria, and protozoans. AMPs can interact with and insert into the anionic cell membranes of microorganisms3–5 usually performing their functions via cell membrane disruption.6 An important class of AMPs is composed by short peptides (less than 50 amino acids) that fold into stable amphipathic α-helical structures in the presence of membranes,7–12 although other conformations, such as β-peptides or more complex folds, are also found in nature.13,14 The amphipathic character of AMPs structures plays a pivotal role in facilitating their interactions with anionic groups present on the surface of the bacterial cell membrane and its consequent disruption. The polar, positively charged face of amphipathic AMPs drives their binding to the negative surface of the bacterial membrane via electrostatic forces; while the nonpolar face facilitates the insertion of the peptide into the membrane bilayer, changing membrane's permeability.15,16 These small, linear peptides are particularly interesting since they can be easily chemically synthesized, and their mode of action can be modulated by specific mutations to increase their selective toxicity toward pathogens without damaging eukaryotic cells. More importantly, their potency is directly correlated to the ratio of hydrophobic and hydrophilic (anionic and cationic) residues into the amphipathic structure of the peptide.17–19 Specifically, the positively charged residues are important for AMPs interaction with the negatively charged bacterial membrane, while the hydrophobic residues are responsible for peptide insertion and the disruption of the cell membrane via pore formation or other molecular mechanisms.20Piscidins, or moronecidins, constitute an important family of amphipathic α-helical antimicrobial peptides identified in teleost fish.21–23 The first member of this family, piscidin-1, was originally found in the mast cells, skin, gills and gastrointestinal tract of hybrid striped bass.21 Piscidins are synthetized as immature pro-peptides (∼80 amino acids) and then cleaved into active, cationic peptides (∼22 amino acids) with potent antimicrobial activity, but also elevated cytotoxicity toward eukaryotic cells.24
In a previous study, we isolated and determined the primary structure of chionodracine (Cnd), a 22-residue antimicrobial peptide from the gills of the Antarctic teleost Chionodraco hamatus.25 Using fluorescence studies, we showed that Cnd disrupts the outer membrane of E. coli and Psychrobacter sp. TAD 1. In vitro assays showed that Cnd binds lipid vesicles of different compositions with a preference toward negatively charged phospholipid head-groups.26 Cnd is essentially unstructured in aqueous solution and undergoes a disorder-to-order transition, folding into an amphipathic α-helix in the presence of phospholipids. Cnd has thus emerged as a very promising AMP due to its ability to interact and disrupt natural and synthetic charged membranes, even at low concentrations (∼1 μM), but unfortunately it was not able to kill human related bacterial pathogens.
In this paper, we exploited the short amphipathic α-helix of Cnd as a template to design a series of Cnd derivatives with increased antimicrobial activity and selectivity toward prokaryotic cells, while maintaining Cnd's three-dimensional structure. To study their conformation and membrane association, we used fluorescence and CD spectroscopy in membrane models, mimicking prokaryotic and eukaryotic cell membranes. We tested their antibacterial activity against the ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species),27 a group of several drug-resistant human pathogens, which are the leading cause of nosocomial infections worldwide. These new peptides displayed minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) values ranging between 0.8 and 10.0 μM, with low cytotoxic and hemolytic activity making them optimal candidates to develop antimicrobial agents toward human pathogens.
Materials and methods
1. Reagents
All peptides (<95%) were purchased from United Biosystem Inc. USA. Peptide concentrations were determined before each sample preparation by UV light absorption at 280 nm (ε280 = 6990 M−1 cm−1). All lipids were purchased from Avanti Polar Lipids (Alabaster, AL, USA).2. Preparation of vesicle
LUVs (Large Unilamellar Vesicles) were prepared as previously reported.26 Briefly, the lipids in chloroform were dried under nitrogen and then overnight under high vacuum. The lipid film was then resuspended in 1 mL of buffer (20 mM phosphate buffer at pH 7.4 with 150 mMNaCl and 0.8 mM EDTA), subjected to 5 freeze–thaw cycles and vortexed. LUVs were prepared by extrusion through a polycarbonate membrane with an Avanti Polar mini-extruder (20 times through two-stacked polycarbonate membranes with pore sizes of 100 nm). LUVs were composed of 100% POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) and of a 70/30 (w/w) combination of POPC/POPG (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol).3. Steady-state fluorescence experiments
Steady-state fluorescence measurements were performed using a PerkinElmer LS55 spectrometer. All the experiments were conducted at 298 K in a thermostatic cell holder.where Fobs and Fo are the observed fluorescence at a given concentration of peptide and the fluorescence of ANS in the absence of peptide, respectively.
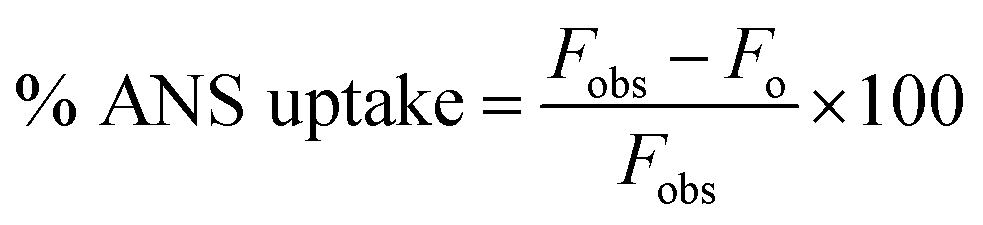
where Kx is the mole fraction partition constant, and [L] and [W] the molar concentrations of lipids and water (55.3 M), respectively.
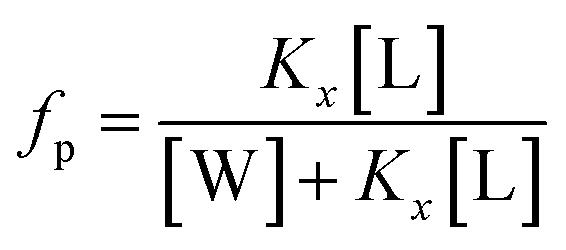
The mole fraction partition coefficient, Kx, was defined according to Wimley and White:31
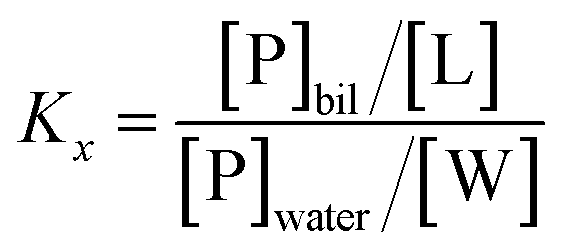
The values of Kx were calculated by plotting fp vs. [L] and fitting the experimental data using the GraphPad Prism 6 software package (GraphPad Software Inc.) to the equation:
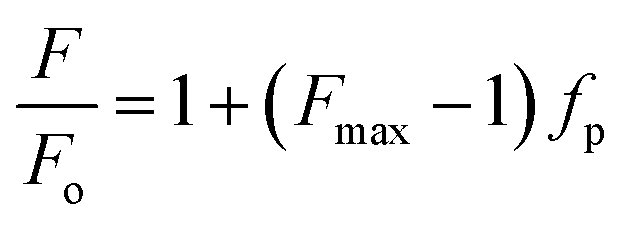
where F and Fo are, respectively, the fluorescence in the presence and absence of the quencher Q, and KSV is the Stern–Volmer constant for the collisional quenching process.24 All the experiments were performed in quadruplicate.
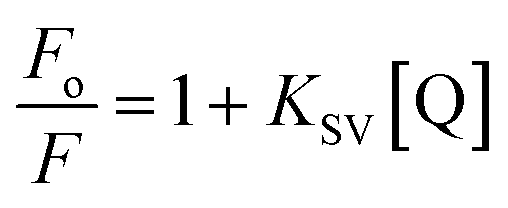
4. Secondary structure determination: CD spectroscopy
Secondary structures of the three peptides were determined in PBS at pH 7.4 in the absence and presence of POPC LUVs (60 μM). Circular Dichroism (CD) spectra were recorded in the 190–260 nm spectral range at 298 K on a J 715 JASCO spectropolarimeter equipped with a Peltier device for temperature control (0.5 cm path length quartz cuvette). The reported CD spectra are the average of 16 scans obtained with an instrument scanning speed of 20 nm min−1, a response time of 8 s, a bandwidth of 1.0 nm and a step size of 0.1 nm. A stock solution of LUVs (10 mM) was used to obtain a lipid/peptide ratio of 20. Contributions from the buffer and LUVs were removed by subtracting their spectra in the absence of peptides. The obtained data in millidegrees (θ) were converted to mean molar ellipticities (deg cm2 dmol−1).5. Hemolytic activity assays
The hemolytic assay was performed as indicated by Belokoneva et al.32 Briefly, a 2.5% (v/v) suspension (in PBS) of human red blood cells from healthy donors was incubated with serial dilutions of the three mutant peptides. Red blood cells were counted by a hemocytometer and adjusted to a concentration of approximately 8.0 × 106 cell per mL. Erythrocytes were then incubated at 310 K for 2 h with different concentrations of the peptides (from 50 to 3.0 μM with five dilutions) or with 10% solution of Triton X-100 (positive control) or PBS (negative control). Each point was acquired in triplicate. The supernatant was separated from the pellet by centrifugation at 1500 × g for 5 min, and the absorbance was measured at 570 nm. The OD relative to that of the positive control defined the percentage of hemolysis, and the data were expressed as the mean ± SD.6. Cytotoxicity assays
Normal primary human fibroblasts (FB789), kindly provided by M. Stefanini (Institute of Molecular Genetics, Pavia, Italy),33,34 were grown in minimal essential medium (MEM) containing 15% fetal calf serum (FCS) and 40 μg mL−1 gentamycin at 37 °C in a humidified 5% CO2 atmosphere.Cultures were passaged every 2–3 days, and the cytotoxicity of the three peptides was determined by measuring the intracellular adenosine triphosphate (ATP) levels by the luciferase-based ATPlite assay (PerkinElmer) according to the manufacturer's instructions. Cells were seeded on 96-well microplates at a density of 3 × 103 cell per well in 100 μL of medium for 12 or 24 h at 310 K in a humidified 5% CO2 atmosphere. Serial 2-fold dilutions of peptide solutions (starting from 100 μM) in water were added; wells containing cells in normal medium with no peptides served as negative controls, while wells with 10% water served controls for the medium in which the peptides were diluted and wells with NaN3 served as positive controls. After 12/24 h, the cells were lysed, and the lysates were transferred into opaque well plates (OptiPlate-96, PerkinElmer). The amount of emitted light, linearly correlated with ATP concentration,35 was measured with a microplate luminometer (Victor II PerkinElmer) for 10 minutes in the dark. Three replicates per treatment were performed from three different experiments. Cell viability values were reported as the percent values (in Relative Luminescence Units (RLUs)) of treated samples with respect to untreated cells and were expressed as the mean ± SD of RLUs measured from each well.
7. Bacterial strains and culture conditions
The antimicrobial activity of the peptides was determined against a panel of 70 clinical isolates with known resistance profiles, including 10 KPC producers, 10 ESBL Escherichia coli, 10 XDR Acinetobacter baumannii, 10 MDR Pseudomonas aeruginosa, 10 MRSA, 10 MRSE, and 10 VRE. All the isolates were retrieved from frozen glycerol stocks, streaked on fresh Trypticase soy agar with 5% sheep blood plate (bioMerieux), incubated at 310 K for 18 h and sub-cultured to provide fresh colonies.8. Determination of MICs and MBCs
Minimum inhibitory concentrations (MIC) were determined for liquid growth inhibition assays by standard procedures (CLSI)36 using serial dilutions of the peptides in a 96-well flat-bottom Microtiter® plate. The peptides were dissolved in buffer and diluted in Mueller–Hinton broth to reach a final concentration of 200 μg mL−1. Logarithmic phase bacterial cultures were suspended in saline solution to achieve a turbidity equivalent to that of a 0.5 McFarland standard and then diluted to a final concentration of 1–2 × 105 CFU mL−1. Each well contained 100 μL of diluted compounds suspended in LB medium and 100 μL of bacterial suspension. The final concentration of the peptides ranged from 0.097 to 100 μg mL−1. The MIC is the lowest concentration of the peptide that completely inhibited growth during 24 h of incubation at 310 K. To determine the minimum bactericidal concentration (MBC), we plated an aliquot (100 μL) from the wells with no visible microbial growth onto Trypticase soy agar plates with 5% sheep blood (bioMerieux) and incubated them at 310 K overnight. All tests were performed in triplicate in two different experimental sessions, and for each series of experiments, both positive (no peptide) and negative (no bacteria) controls were included.379. Time-kill curves
One strain of each species was randomly selected to undergo time-kill assays. Mueller–Hinton broth was inoculated with the bacterial suspension and diluted to reach a final concentration of 1 × 105 CFU mL−1. The three peptides were tested at 0.5× (sub-inhibitory concentration), 1× and 2× MICs. Control experiments without active peptides (bacterial growth curve) were simultaneously conducted with the time-kill studies. The plates were incubated for 18 h at 310 K. Plates with each of the bacteria/peptide concentrations were sampled at 0, 2, 6, 8 and 24 h from the time of peptide addition. Aliquots of 100 μL were serially diluted and then plated onto Trypticase soy agar plates with 5% sheep blood (bioMerieux). Bacterial counts (expressed as log10 CFU mL−1) were quantified after an incubation for 18–24 h at 310 K. The log10 value of the viable colony count was determined at each time interval. The lower limit of quantification in this model was 10 CFU mL−1. Bacterial concentrations < 1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log10 CFU mL−1 were counted as 1.0log10 CFU mL−1. Bactericidal activity was defined as a minimum of a 3 log10 CFU mL−1 reduction in bacterial counts from the starting concentration. Strains that experienced bactericidal activity at any point during time-kill assays but then regrew by ≥2log10 CFU mL−1 were defined as having regrowth. The experiments were performed in triplicate on two separate days for each tested isolate.38,39
log10 CFU mL−1 were counted as 1.0log10 CFU mL−1. Bactericidal activity was defined as a minimum of a 3 log10 CFU mL−1 reduction in bacterial counts from the starting concentration. Strains that experienced bactericidal activity at any point during time-kill assays but then regrew by ≥2log10 CFU mL−1 were defined as having regrowth. The experiments were performed in triplicate on two separate days for each tested isolate.38,39
10. Transmission electron microscopy
Bacteria were fixed in glutaraldehyde (2.5%), deposited on a carbon grid, negatively stained (2% uranyl acetate) and imaged by TEM (Zeiss, Libra 120, Germany) as reported previously.40Results
1. Chionodracine analogs design
The new peptides were designed starting from the sequence of Cnd, (Fig. 1). In dodecylphosphocholine (DPC) micelles, Cnd adopts an α-helical structure typical of cationic AMPs, with the hydrophobic face pointing toward the lipid environment and the hydrophilic face toward the solvent.26 Most AMPs carry a net positive charge (ranging from +2 and +11) since the positive charge is crucial for interactions with the negatively charged membranes of bacteria.41 For these new analogs, we modified the Cnd peptide sequence by introducing charged residues (i.e., lysines) to increase their amphipathicity. Three different Cnd analogs, KS-Cnd, KH-Cnd and KSH-Cnd, were designed directly from the Cnd primary sequence by fitting the residues to an α-helical template and evaluating the average charge and the number of hydrophobic residues to obtain amphipathic helices. The peptide sequences, along with their hydrophobic moment (μH) and the helical wheel representation of the amphipathic structures obtained using HeliQuest42 are reported in Fig. 1. The mutations were strategically placed to increase the positive charge and hydrophobic moment to enhance the peptides' affinity for anionic membranes,15,43–45 without modifying the original structure as the amphipathic helix conformation is central for the antimicrobial activity.46 For the KS-Cnd mutant, Ser11 and Ser22 were replaced by two lysines; for KH-Cnd, histidines 4, 15, and 17 were all replaced by lysines, and for KSH-Cnd, histidines and serines were all replaced by lysines. The overall charge for wild-type Cnd is +2; while for KS-Cnd, KH-Cnd and KSH-Cnd analogs is +4, +5, and +7, respectively. The mean charge per residue was increased from ∼0.09 for Cnd to ∼0.32 for KSH-Cnd. Since an increase in charge beyond +7 does not increase peptides' antimicrobial activity,17 we did not further modify these peptides. The average number of hydrophobic residues (V, I, L, W, and F) per residue was ∼0.41, corresponding to 9 hydrophobic residues. The helical wheel projection (Fig. 1) demonstrates the amphipathic nature of the peptides with well-defined hydrophilic (blue/gray) and hydrophobic (yellow) faces.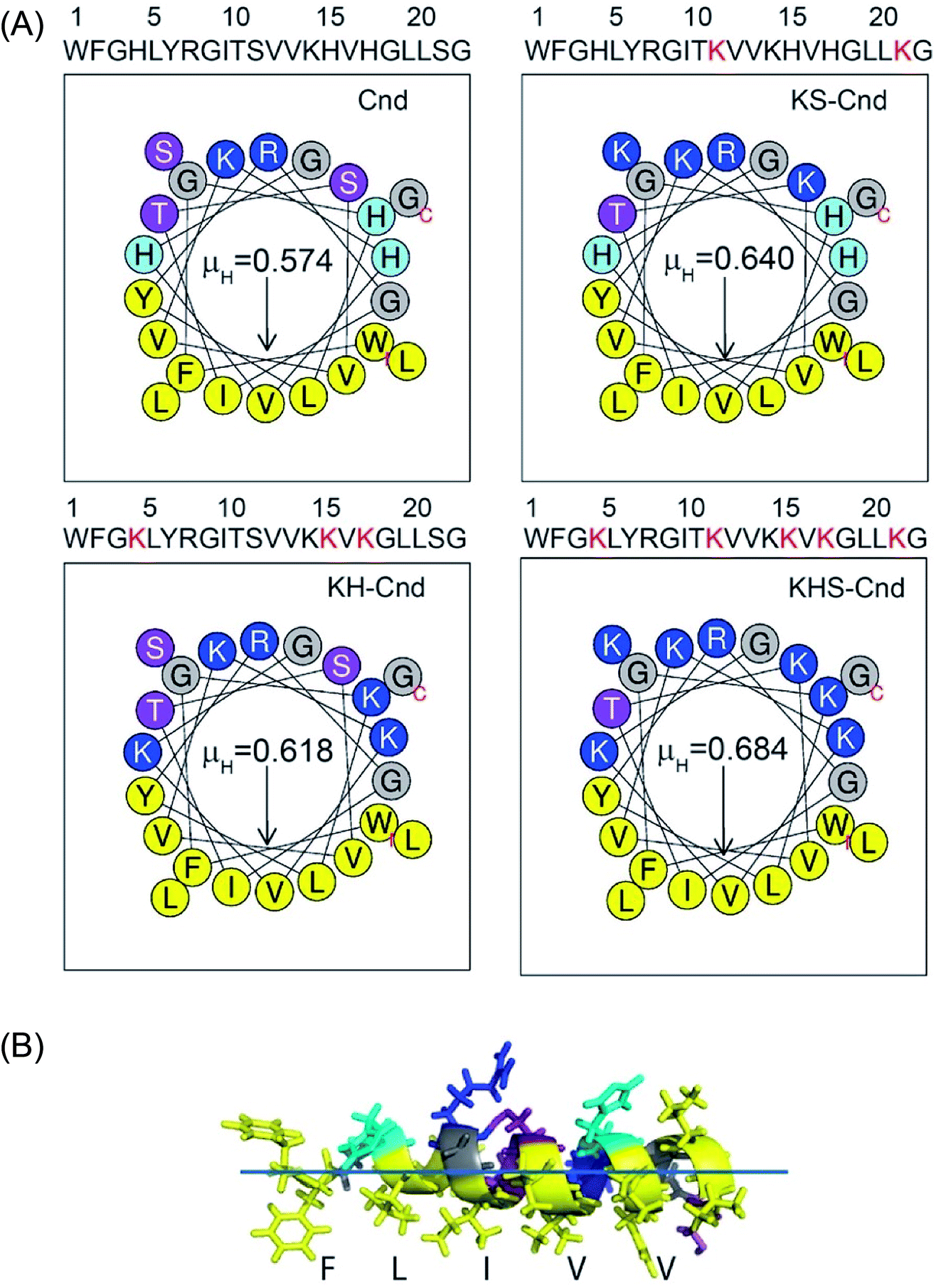 | ||
| Fig. 1 (A) Sequence and helical wheel representation of amphipathic structures of Cnd and its mutants using HeliQuest (http://heliquest.ipmc.cnrs.fr). The arrows indicate the magnitude and direction of the hydrophobic moment (μH). Nonpolar and aromatic residues are yellow, charged residues are blue, polar uncharged residues are indigo, and glycines are gray. (B) Representative NMR structure of Cnd obtained in DPC micelles showing the asymmetric distribution of the amino acids to form an amphipathic helix. | ||
To evaluate the propensity of Cnd and its derivatives to proteolytic digestion, we carried out in silico tests using the Peptide Cutter tool available on the ExPASY Server (https://web.expasy.org/peptide_cutter/) that predicts potential cleavage sites cleaved by proteases.47 This analysis did not reveal any substantial differences between the wild-type Cnd and its three mutants. According to the prediction, mutants KH-Cnd and KHS-Cnd are more resistant to the chymotrypsin-low specificity, but less resistant to trypsin cleavage with respect to the wild-type.
2. Outer membrane permeability assay
To assess the membranolytic activity of all peptides, we performed an ANS fluorescence assay using the E. coli BL21 strain (DE3). Due to its hydrophobic nature, ANS is not able to pass through the intact bacterial outer membrane, resulting in very weak fluorescence signal. Upon disruption of the outer membrane, ANS penetrates the cell and the hydrophobic environment of the periplasmic space causes a drastic increase in the fluorescence band with a blue-shift from 510 to 450 nm (Fig. 2A–D). A rapid increase in ANS fluorescence indicates that all the Cnd analogs disrupt the outer membrane of E. coli within a few minutes. Fig. 2E reports the percentage of ANS uptake as a function of peptide concentration. From these data, it is apparent that in presence of KHS-Cnd and KH-Cnd. At a concentration of 1.0 μM the percentage of ANS uptake is ∼60% and ∼50% higher than that of Cnd (∼40%) and KS-Cnd ∼25%. These effects increase in a dose-dependent manner, suggesting that the increased positive charge and hydrophobic moment for KHS-Cnd augment its ability to disrupt membranes with respect to KH-Cnd, Cnd, and KS-Cnd peptides.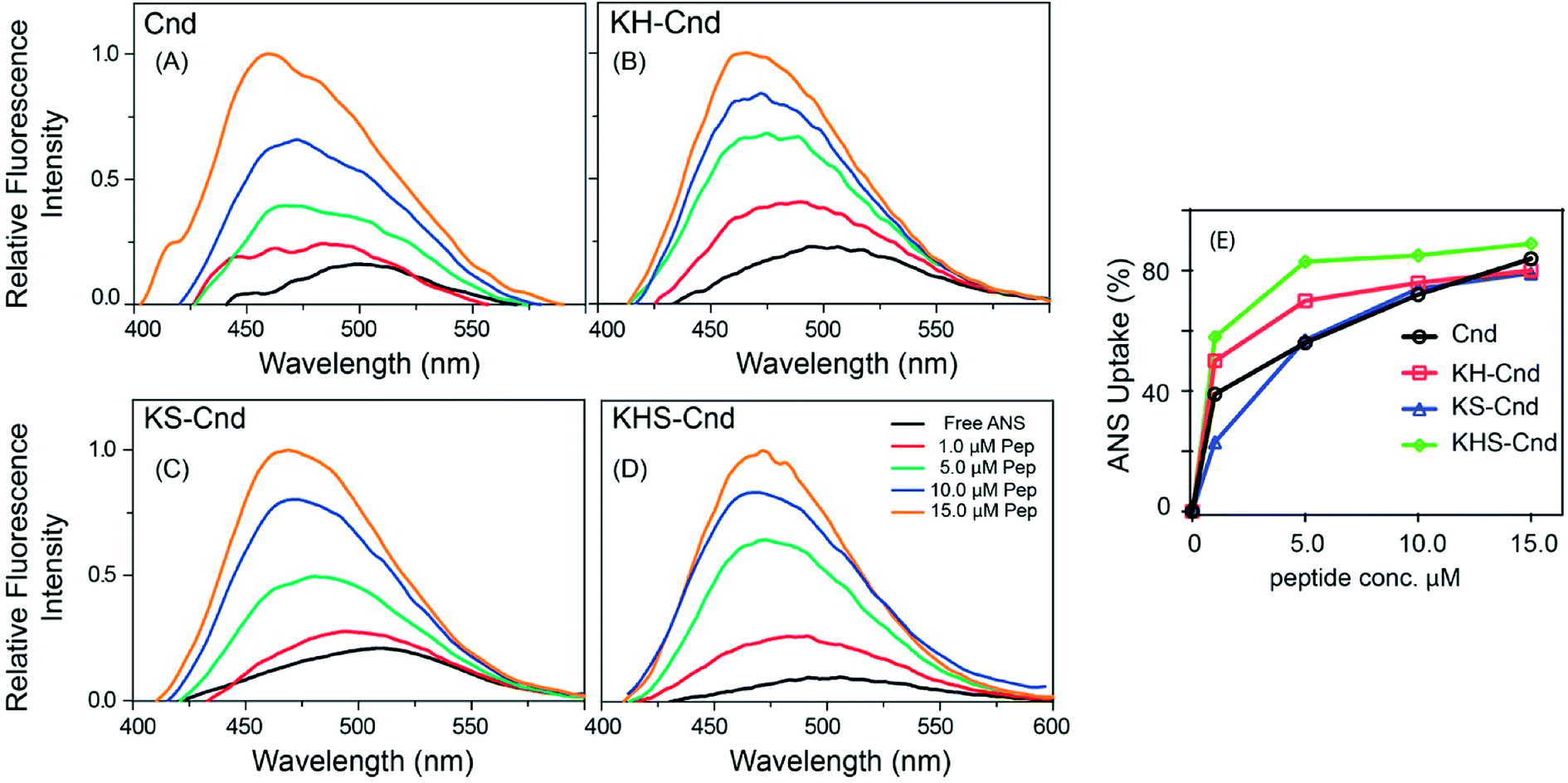 | ||
| Fig. 2 (A–D) Permeabilization of E. coli outer membrane by Cnd and Cnd analogs. (E) Percentage of ANS uptake as a function of the peptide concentration. The experiments were performed in triplicate and representative results are shown. | ||
3. Membrane partitioning studies
Many antimicrobial peptides elicit their activity by interacting with membranes, and the difference in the membrane compositions of prokaryotic and eukaryotic cells is central to their selectivity. A pre-requisite for AMP to be used in therapy is the selective toxicity only toward the pathogen and not toward the host cells. To establish the relative selectivity of the Cnd analogs for different membrane compositions, we conducted membrane partitioning assays. Specifically, we assessed and quantified the interactions of wild-type and mutant Cnd peptides with model membranes that mimic the phospholipid compositions of both bacterial and mammalian natural membranes. Large unilamellar vesicles (LUVs) exclusively composed of POPC, a zwitterionic lipid with no net charge, were chosen to mimic mammalian membranes, while POPC/POPG (70/30) LUVs were selected to mimic the bacterial membrane, as POPG is negatively charged.48 The POPC/POPG and POPC mimics do not completely describe the compositional complexity of bacterial and plasma membranes but are used here to evaluate the preference of Cnd peptides for prokaryotic or eukaryotic membranes. Upon vesicles addition, a blue-shift and an increase in fluorescence emission were observed indicating that Trp-1 was in a less polar environment, directly involved in the interaction with lipid vesicles with a decreased flexibility. This fluorescence behavior is due to the 1La state of tryptophan that is solvent-sensitive and involves in the transition the polar nitrogen atom.49 As an example, in Fig. 3 are reported the spectra obtained for the titration of KH-Cnd with POPC and POPC/POPG vesicles. We analyzed the binding isotherms derived from the titration of these peptides with increasing amounts of LUVs (Fig. 3) to estimate the mole fraction partitioning coefficients (Kx).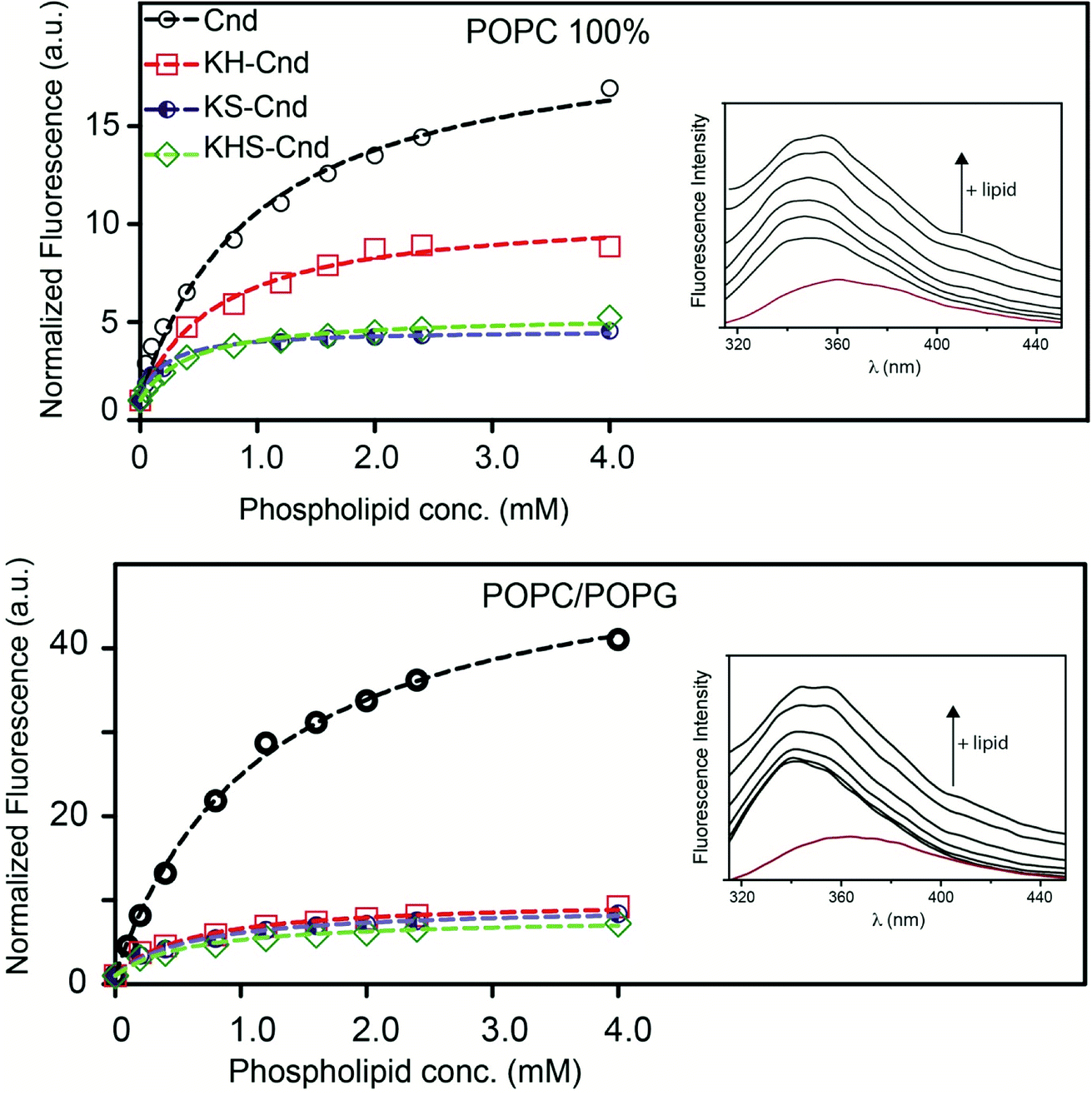 | ||
| Fig. 3 Representative steady state emission spectra for KH-Cnd in presence of lipid vesicles of POPC and POPC/POPG (the red spectrum is of KH-Cnd in buffer) and binding isotherms (298 K) for Cnd analogs interacting with LUVs displaying different lipid compositions. The fluorescence of tryptophan was measured as lipid vesicles were added to samples containing 1.0 μM peptide. Samples were equilibrated for 10 minutes after each addition. The experiments were performed in quadruplicate and representative results are shown. All data were fitted to the partition model reported in Section 3.2. | ||
The Kx values are reported in Table 1 together with ΔG values, calculated using the expression ΔG = −RTlnKx, and the selectivity ratio, calculated as the ratio between the Kx measured for peptides in POPC/POPG and POPC. Higher values of selectivity ratio mean a preference of peptide for POPC/POPG vesicles that mimic the bacterial cell. These results have been confirmed by the antimicrobial tests (Section 8 of results).
| Peptide | Lipid mixture | Kx (×104) | ΔG (kJ mol−1) | Selectivity ratio |
|---|---|---|---|---|
| Cnd | POPC (100%) | 3.43 ± 0.26 | −25.9 | 1.43 |
| POPC/POPG (70/30 w/w) | 4.91 ± 0.29 | −26.7 | ||
| KS-Cnd | POPC (100%) | 8.59 ± 0.77 | −28.1 | 1.79 |
| POPC/POPG (70/30 w/w) | 15.36 ± 0.66 | −29.6 | ||
| KH-Cnd | POPC (100%) | 14.09 ± 0.71 | −29.4 | 1.21 |
| POPC/POPG (70/30 w/w) | 17.07 ± 0.87 | −29.8 | ||
| KHS-Cnd | POPC (100%) | 9.33 ± 0.92 | −28.3 | 2.39 |
| POPC/POPG (70/30 w/w) | 22.3 ± 1.5 | −30.5 |
For all peptides, we found that Kx increases for the POPC/POPG LUVs, indicating a higher propensity to interact with negatively charged lipid bilayers that mimic microbial membranes. Moreover, the maximum of the Trp-1 emission spectrum shifted from 356 to 348 nm in the presence of this lipid vesicles composition, indicating the insertion of Trp-1 and the hydrophobic portion of the peptides at the bilayer interface.26 Furthermore, the partitioning data suggest that the three Cnd analogs (with higher Kx) interact with POPC/POPG LUVs with higher affinity than the wild-type Cnd. This feature should increase their antimicrobial activity and selectivity toward bacterial cell membranes. These biophysical data are in good agreement with cellular assays carried out with the Cnd analogs (see below). Collectively, these results stress the importance of the interplay between the compositions of both bilayers and peptides for peptide–lipid interactions.
4. Fluorescence quenching experiments
We carried out fluorescence quenching experiments using iodide as quencher for Trp-1 to further elucidate the nature of the peptide–membrane interactions and investigate the exposure of Trp-1 to the aqueous environment. Fig. 4 shows the Stern–Volmer plots for the quenching of the Trp residue (Trp-1) in the three mutants upon titration with potassium iodide (KI), both in the absence (filled circles) and presence of lipid vesicles of different compositions. To avoid the presence of free peptide in solution, the peptide/lipid molar ratio was 1:100. All data were corrected for dilution and fluorophore absorption.
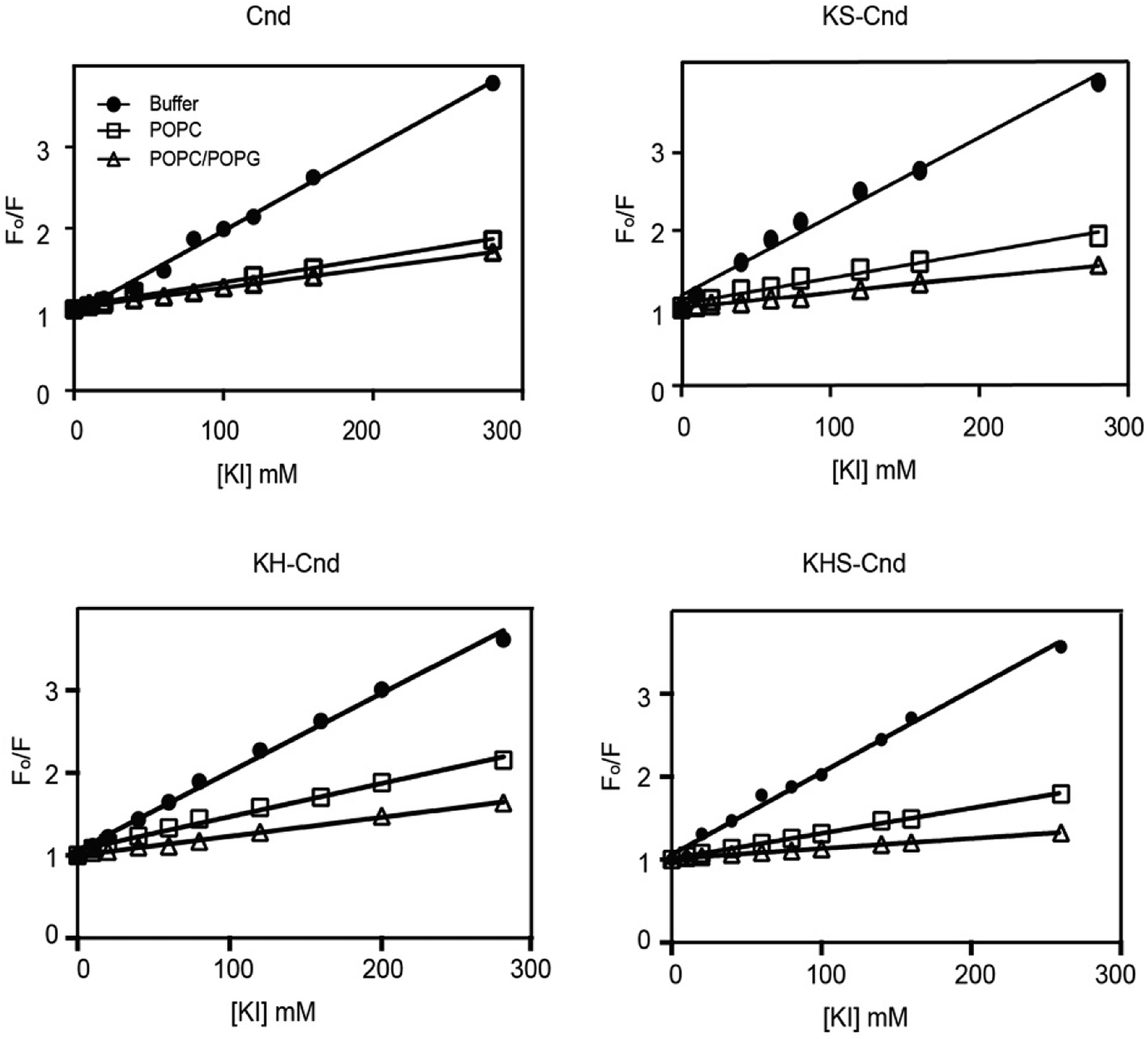 | ||
| Fig. 4 Stern–Volmer plot obtained from the fluorescence quenching of Trp-1 in Cnd and Cnd-analogs by KI in aqueous buffer (filled circle) and in the presence of lipid vesicles of POPC (open square) and POPC/POPG (open triangle). The peptide to lipid molar ratio was 1:100. The experiments were performed in quadruplicate and representative results are shown. | ||
The linear trends indicate that the quenching mechanism is collisional (dynamic). The slopes obtained in the absence of LUVs were higher than those in their presence, supporting the formation of strong interactions between peptides and LUVs. From these plots, we estimated the values of Stern–Volmer constants, KSV, and the percentage of quenching (Table 2).
| POPC (100%) | POPC/POPG (70/30 w/w) | Buffer | ||||
|---|---|---|---|---|---|---|
| KSV (M−1) | % quenching | KSV (M−1) | % quenching | KSV (M−1) | % quenching | |
| Cnd | 2.9 ± 0.1 | 28.4 | 2.5 ± 0.1 | 23.6 | 10.4 ± 0.3 | 100 |
| KS-Cnd | 3.3 ± 0.2 | 32.6 | 2.0 ± 0.1 | 22.1 | 9.0 ± 0.3 | 100 |
| KH-Cnd | 4.0 ± 0.2 | 42.4 | 2.3 ± 0.1 | 24.1 | 9.5 ± 0.2 | 100 |
| KHS-Cnd | 3.0 ± 0.1 | 14.8 | 1.2 ± 0.1 | 17.9 | 9.9 ± 0.1 | 100 |
Smaller KSV values suggest that the Trp residue has a greater solvent protection and a putative strong interaction with the lipid bilayer. By comparing the KSV values for Cnd and the Cnd analogs, we observed a 3-fold decrease going from buffer (i.e., absence of membrane mimetic system) to POPC and POPC/POPG, which is indicative of strong peptide–lipid interactions and suggest that Trp-1 is not solvent-exposed. However, a little quenching is still detected in presence of vesicles suggesting that Trp-1 interacts with the membrane layer but lies in the interface region.
5. Secondary structure in POPC LUVs
To determine the structural properties of these peptides, we performed CD experiments in aqueous solution in the absence and presence of POPC LUVs at a lipid/peptide ratio of ∼20. In aqueous solution, the CD spectra for the three peptides display a strong negative band at approximately 200 nm, which is indicative of a random coil conformation (Fig. 5) in agreement with previous results for the Cnd.26 Upon addition of POPC LUVs, a positive band at ∼192 nm and negative minima at ∼208 and ∼222 nm appear in the CD spectra, suggesting that the peptides adopt an ordered α-helical conformation. These findings are consistent with previous results showing that almost all linear antimicrobial peptides are unstructured in water solution and adopt the correct folding in presence of biological membranes.50 The estimation of the α-helical content has been carried out on DichroWeb51 using CDSSTR and SELCON3.52 Both analysis confirmed the correct folding of peptides upon interaction with LUVs. SELCON3 revealed the same percentage of helical structure for Cnd and mutants while CDSSTR reported a decrease of 4% for mutants with respect to Cnd.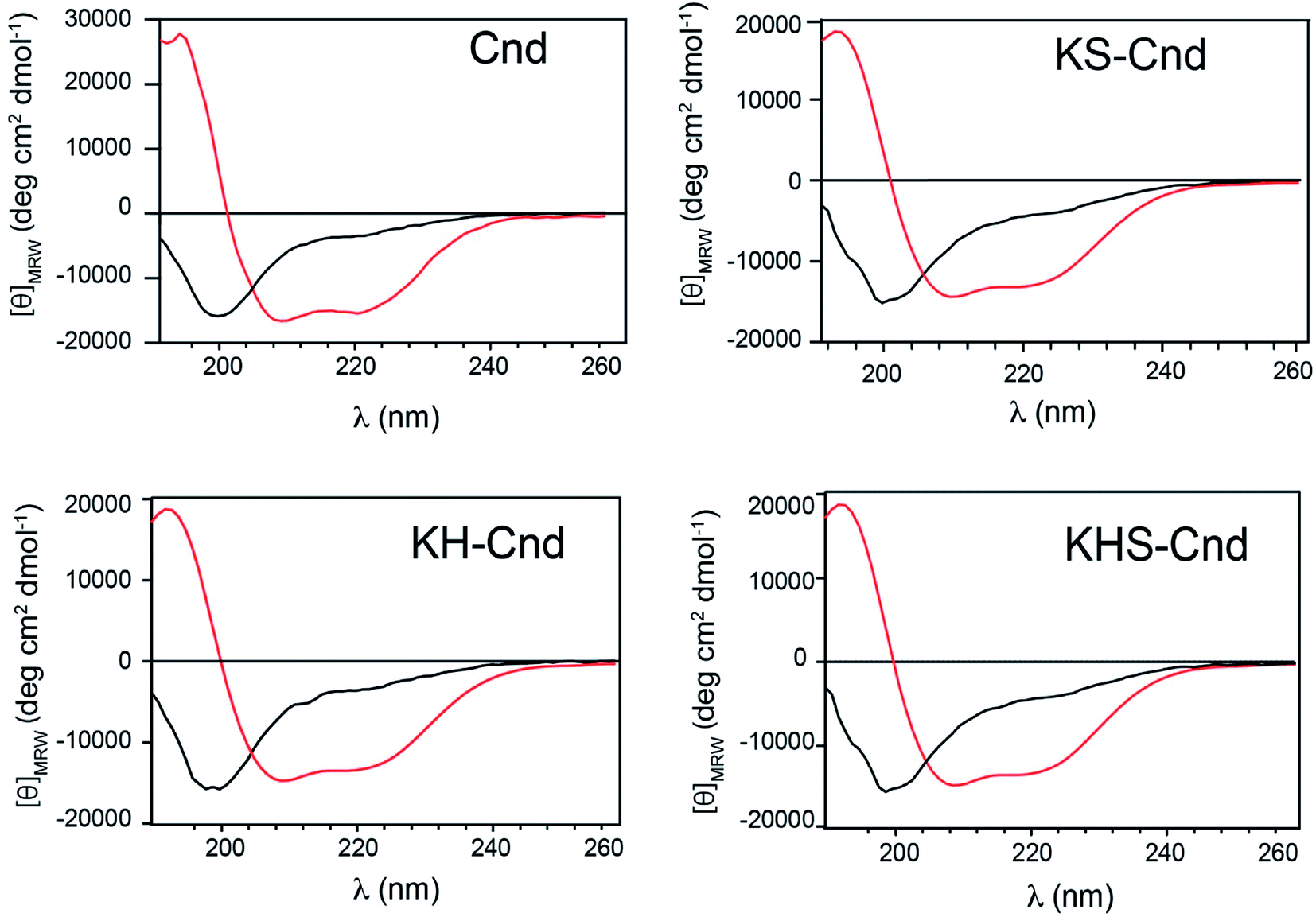 | ||
| Fig. 5 CD spectra of Cnd and Cnd-analogs in PBS buffer, at pH 7.4 and T = 298 K. The free peptides are represented by the black dichroic profiles and the lipid-bound peptides (POPC LUVs) by the red profiles. | ||
6. Hemolytic activity
The hemolytic effect was tested to determine the toxicity against human erythrocytes. The 100% of hemolysis was the value of absorbance when the cells have been incubated with 10% Triton X-100 solution, while the negative control (0% hemolysis) was considered the value obtained with the cells incubated in PBS. Five different peptide concentrations were used starting from 50 μM peptide stock solution with successive dilutions (Fig. 6). For each point, we estimated the ratio between the obtained absorbance value and the 100% of hemolysis.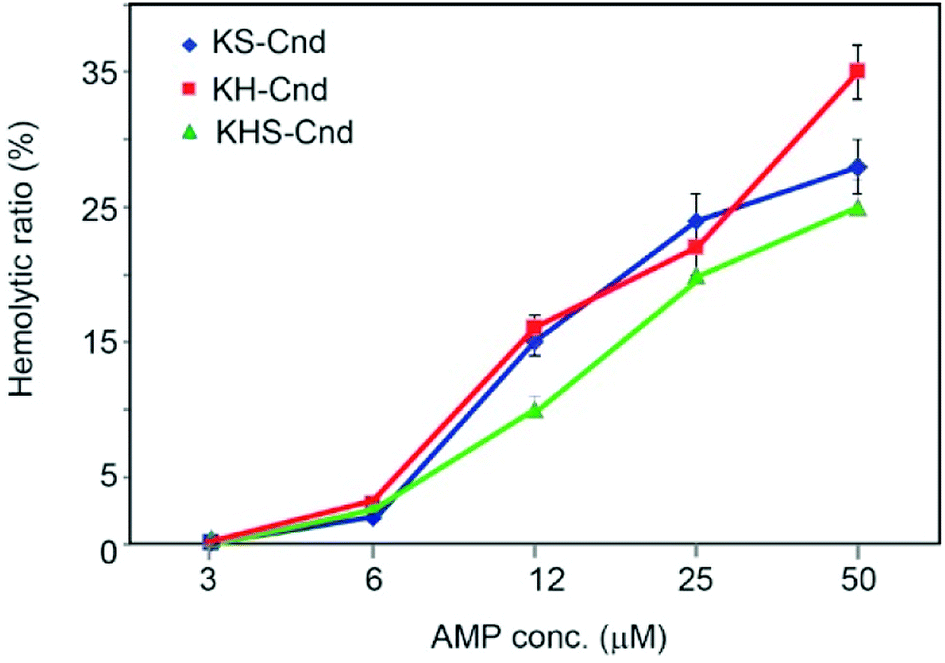 | ||
| Fig. 6 Hemolytic activity of the three peptides against human erythrocytes. Five different successive dilutions starting from 50 μM were tested. The values represent the mean ± SD (n = 3). A sample using 10% Triton X-100 was employed as a positive control and was considered to represent 100% of hemolysis. | ||
The percentage of hemolysis is very low (about 2%) for all three peptides at 6 μM concentration. At a concentration of 12 μM, we observed an increase of hemolysis to ∼15%, which reaches a value of 30% at 50 μM. These values are higher than those obtained for Cnd at the same peptide concentrations,25 but the hemolytic effect at the MIC and MBC concentrations needed to kill human bacterial pathogens (see below) is substantially low. From Fig. 6 we were able to evaluate the minimal hemolytic concentration (MHC), defined as the concentration of peptide that caused 10% hemolysis of human red blood cells, for the three peptides.53 The values are 8 μM, 9 μM and 13 μM for KS-Cnd, KH-Cnd and KHS_Cnd, respectively.
7. Cytotoxicity assays against human primary cell line
We studied the effects of Cnd mutants peptides on primary human fibroblasts (FB789 cells) (Fig. 7). Both at 12 and 24 hours, the survival rates were very low for all peptides at concentrations above 6 μM. However, at 3 μM we were able to retrieve approximately 100% cell viability for both KH-Cnd and KS-Cnd and ∼70% for KHS-Cnd after 24 hours. This concentration is sufficiently high to kill most of the tested human pathogen bacteria (see next section), without being cytotoxic to human healthy cells. Note that the cell viability in this assay can be higher than the initial value (up to 150%) as previously reported in some cases for this standard assay.54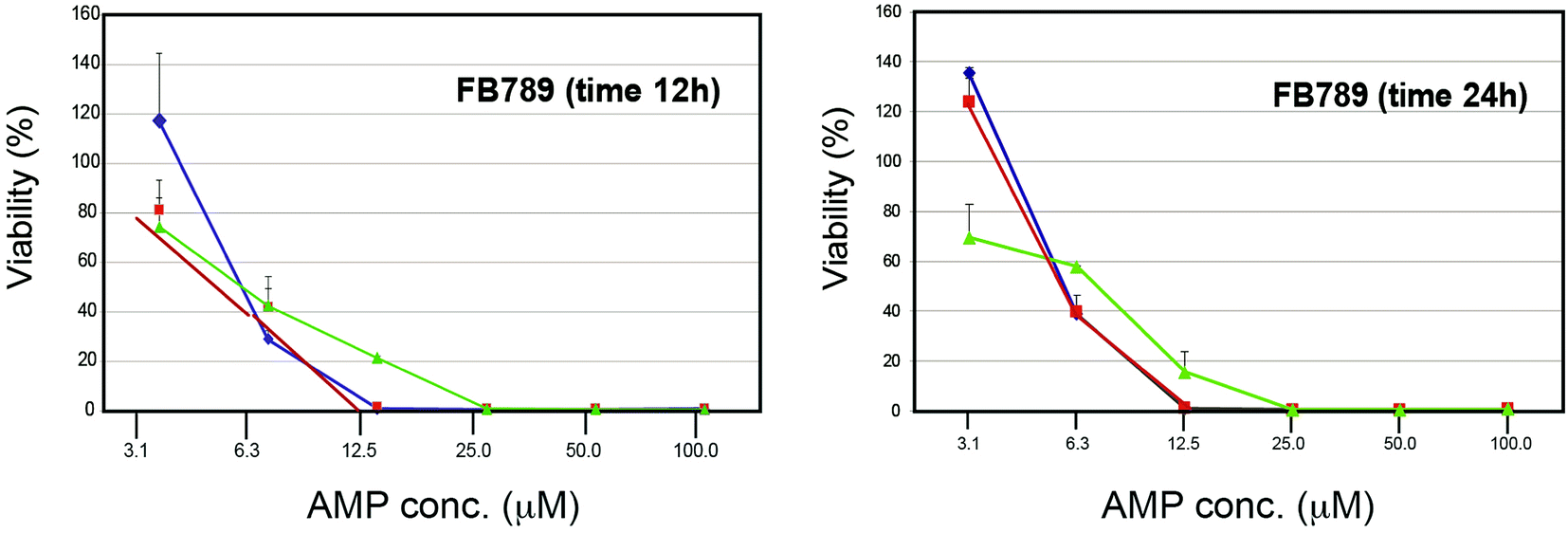 | ||
| Fig. 7 Dose–response cytotoxicity of the three peptides (KH-Cnd, red, KS-Cnd, blue, and KHS-Cnd, green) towards FB789 human cell line after 12 and 24 hours. | ||
8. In vitro antimicrobial activities against multidrug-resistant (MDR) bacteria
The antimicrobial activities of all peptides against different Gram-negative and Gram-positive bacterial strains are summarized in Table 3.| Strain and agent | MIC (μM) | MBC (μM) | ||
|---|---|---|---|---|
| Median | Range | Median | Range | |
| KPC producer | ||||
| Cnd | 21.0 | 10.0–21.0 | 41.0 | 21.0–41.0 |
| KS-Cnd | 1.2 | 1.2–2.5 | 7.4 | 2.5–9.8 |
| KH-Cnd | 1.3 | 0.6–2.6 | 2.6 | 2.6–10.3 |
| KSH-Cnd | 0.6 | 0.3–1.2 | 2.5 | 2.4–2.5 |
|
||||
| ESBL E. coli | ||||
| Cnd | 5.2 | 5.2–10.3 | 20.6 | 10.3–20.6 |
| KS-Cnd | 2.5 | 1.3–5.9 | 3.7 | 2.5–4.9 |
| KH-Cnd | 1.3 | 1.3–2.6 | 3.9 | 2.6–10.3 |
| KSH-Cnd | 1.2 | 0.6–2.5 | 2.5 | 1.2–2.5 |
|
||||
| XDR A. baumannii | ||||
| Cnd | 5.2 | 5.2–10.3 | 10.3 | 10.3–20.6 |
| KS-Cnd | 1.2 | 0.6–1.2 | 2.5 | 2.5–2.5 |
| KH-Cnd | 1.3 | 0.6–2.6 | 3.9 | 1.3–5.1 |
| KSH-Cnd | 0.6 | 0.6–1.2 | 2.5 | 1.2–2.5 |
|
||||
| MDR P. aeruginosa | ||||
| Cnd | 10.3 | 10.3–20.6 | 20.6 | 10.3–20.6 |
| KS-Cnd | 4.9 | 2.5–9.8 | 14.7 | 4.9–19.6 |
| KH-Cnd | 2.6 | 1.3–5.1 | 5.1 | 5.1–5.1 |
| KSH-Cnd | 1.3 | 1.2–5.0 | 5.0 | 2.5–5.0 |
|
||||
| MRSA | ||||
| Cnd | 20.6 | 20.6–41.2 | 41.2 | 20.6–41.2 |
| KS-Cnd | 19.6 | 9.8–39.3 | 39.3 | 19.6–39.3 |
| KH-Cnd | 10.3 | 5.1–20.5 | 20.5 | 20.5–20.5 |
| KSH-Cnd | 9.9 | 5.0–19.9 | 19.9 | 9.9–19.9 |
|
||||
| MRSE | ||||
| Cnd | 10.3 | 5.2–10.3 | 20.6 | 20.6–41.2 |
| KS-Cnd | 2.5 | 1.2–4.9 | 4.9 | 4.9–4.9 |
| KH-Cnd | 2.6 | 1.3–5.1 | 5.1 | 2.6–103 |
| KSH-Cnd | 1.2 | 0.6–2.5 | 3.7 | 2.5–5.0 |
|
||||
| VRE spp. | ||||
| Cnd | 10.3 | 5.2–10.3 | 20.6 | 10.3–20.6 |
| KS-Cnd | 1.2 | 0.6–2.5 | 4.9 | 2.5–4.9 |
| KH-Cnd | 2.6 | 1.3–5.1 | 5.1 | 2.6–5.1 |
| KSH-Cnd | 1.2 | 0.6–2.5 | 2.5 | 2.5–4.9 |
The MIC values of all modified peptides were significantly lower than those of Cnd, indicating an overall improvement of the antimicrobial activity for these mutants. Moreover, we found that all peptides had a stronger activity against Gram-negative compared to Gram-positive strains. The MIC values indicated that KSH-Cnd has the highest antibacterial activity against both Gram-negative and Gram-positive bacteria. These results are in agreement with the ability of KSH-Cnd to damage the integrity of the bacterial membrane more than Cnd, KH-Cnd and KS-Cnd. This peptide has a higher positive charge and hydrophobic moment compared to the other two mutants, which contributes to its improved membrane bacterial degradation. Notably, the three Cnd-derivative peptides exhibited high antibacterial activity against all tested Gram-negative bacterial strains. KSH-Cnd was particularly active against Klebsiella pneumoniae carbapenemase (KPC) producers and A. baumannii, with MIC = 0.6 μM or 1.5 μg mL−1, whereas it displayed a MIC value of 1.2 μM (3.12 μg mL−1) for an extended spectrum of β-lactamase E. coli producers (ESBL) and MDR P. aeruginosa bacteria. In contrast, the MIC values of Cnd toward these Gram-negative bacteria were all significantly higher than those obtained for the Cnd's mutants, ranging from 5.0 to 21.0 μM (12.5 to 50 μg mL−1). Interestingly, the three designed peptides displayed higher MIC values against Gram-positive compared to the Gram-negative bacteria, with the highest values (25–50 μg mL−1 or 10.0–20.0 μM) against methicillin-resistant Staphylococcus aureus (MRSA). Moreover, KSH-Cnd showed the best antibacterial properties against Gram-positive bacteria with a MIC of 1.2 μM (3.12 μg mL−1) for methicillin-resistant Staphylococcus epidermidis (MRSE) and E. faecalis (a vancomycin-resistant Enterococcus, VRE). The Cnd peptide displayed MIC values between 10.0–20.0 μM (25 and 50 μg mL−1) for all Gram-positive tested bacterial isolates. Additionally, we measured the MBC values to assess whether the mutant peptides are bactericidal or bacteriostatic. The MBC values obtained are reported in Table 3 and further support that these newly designed peptides possess a bactericidal effect against all the Gram-negative and Gram-positive bacteria tested. Note that these peptides are considered bactericidal when their MBC is equal to or four-fold greater than their MIC.55
9. Time-killing curves
To monitor cell viability versus time, we challenged a representative clinical isolate for each species used in this work with MIC/2, MIC and 2× MIC (Table 4) of KS-Cnd, KH-Cnd, and KSH-Cnd, respectively.| μM | E. coli | K. pneumoniae | A. baumannii | P. aeruginosa | S. aureus | S. epidermidis | E. faecalis |
|---|---|---|---|---|---|---|---|
| KS-Cnd | |||||||
| 2× MIC | 5.0 | 2.4 | 2.4 | 9.8 | 39.2 | 4.9 | 2.4 |
| MIC | 2.5 | 1.2 | 1.2 | 4.9 | 19.6 | 2.5 | 1.2 |
| MIC/2 | 1.25 | 0.6 | 0.6 | 2.45 | 9.8 | 1.25 | 0.6 |
|
|||||||
| KH-Cnd | |||||||
| 2× MIC | 2.6 | 2.6 | 2.6 | 5.2 | 20.6 | 5.2 | 5.2 |
| MIC | 1.3 | 1.3 | 1.3 | 2.6 | 10.3 | 2.6 | 2.6 |
| MIC/2 | 0.65 | 0.65 | 0.65 | 1.3 | 5.15 | 1.3 | 1.3 |
|
|||||||
| KSH-Cnd | |||||||
| 2× MIC | 2.4 | 1.2 | 1.2 | 2.6 | 19.8 | 2.4 | 2.4 |
| MIC | 1.2 | 0.6 | 0.6 | 1.3 | 9.9 | 1.2 | 1.2 |
| MIC/2 | 0.6 | 0.3 | 0.3 | 0.65 | 4.95 | 0.6 | 0.6 |
Time-kill curves are shown in Fig. 8. All the peptides displayed concentration-dependent and time-dependent bactericidal activity against all bacterial strains, with faster killing kinetics at higher concentrations. The bactericidal effects of all peptides had faster kinetics and higher potency for Gram-negative than Gram-positive bacteria. Bacterial regrowth occurred when strains were incubated with peptides at 0.5× MIC. Among Gram-negative isolates, the colonies of an extensively drug-resistant (XDR) A. baumannii strain were almost completely killed at 1× MIC after 2 h of KH-Cnd challenge. A. baumannii is an opportunistic nosocomial pathogen that is commonly resistant to multiple antibiotics. This clinical isolate appears to be the most rapidly killed by KSH-Cnd peptide. The MIC of KSH-Cnd against A. baumannii is in fact the lowest, along with that against K. pneumoniae. As expected therefore, the lower the MIC, the better the bactericidal activity in term of CFU log decreases. Several advantages derive from the rapid bactericidal activity of AMPs, among them, the ability to limit the spread of infection, as well as the containment of the onset of drug resistance phenotype. Moreover, the rapidity with which the peptide inhibits A. baumannii viability could potentially reduce the duration of treatment. On the other hand, the treatment with both KS-Cnd and KH-Cnd produced a significant reduction in the number of viable P. aeruginosa, K. pneumonia, and E. coli bacteria at 1× MIC after 6 h of exposure, exhibiting slower bactericidal kinetics. Changes in the density (log CFU mL−1) over time from the starting inoculum density for Gram-positive strains were evident after 8 h of challenge for all peptides at 1× MIC, with practically complete killing of E. faecalis and S. aureus achieved at 24 hours and nearly complete killing for S. epidermidis.
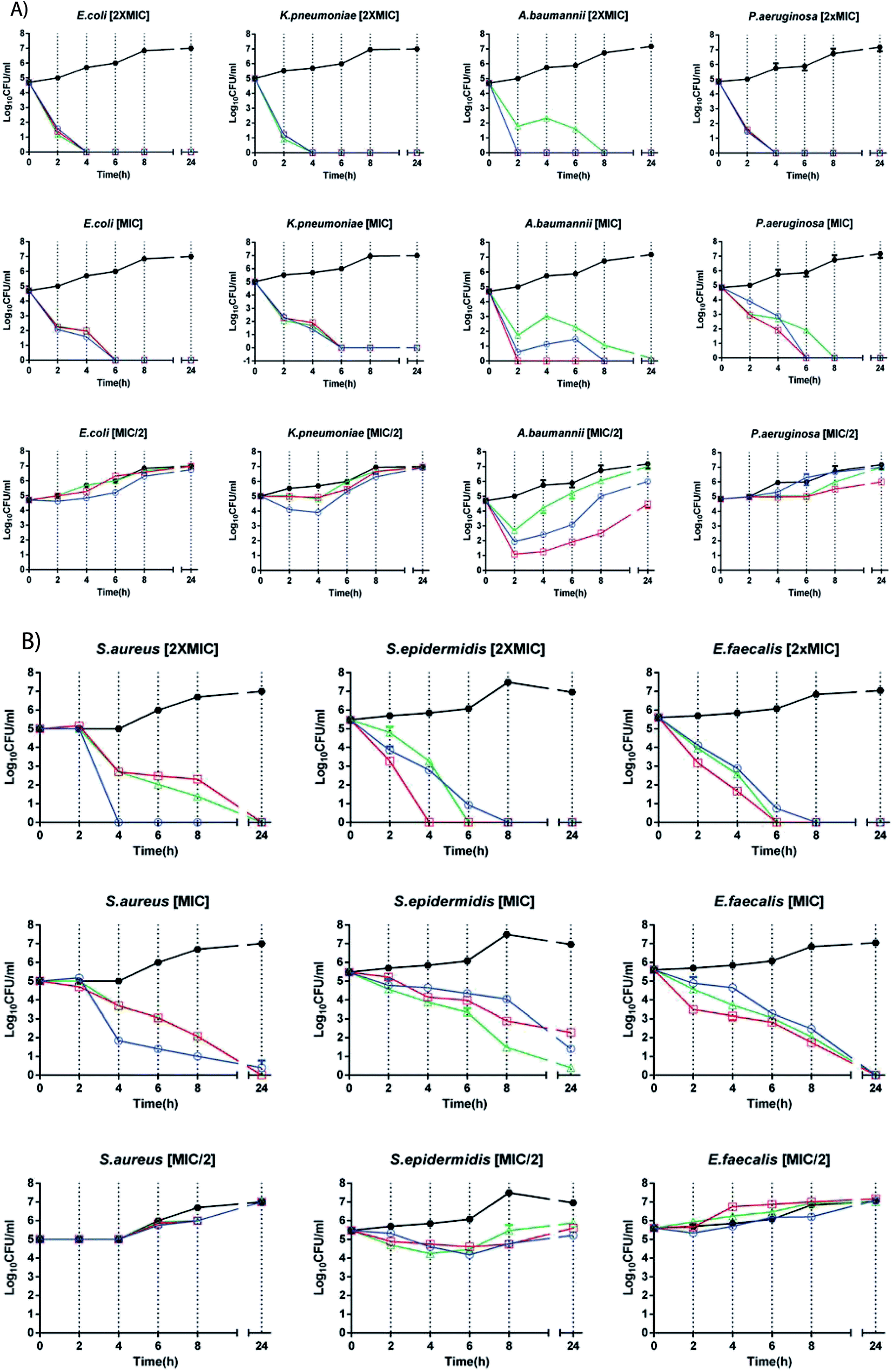 | ||
| Fig. 8 Time-kill studies with MIC/2, MIC and 2× MIC concentrations of KS-Cnd, KH-Cnd and KSH-Cnd peptides applied to seven strains, each representing one of the following species: (A) E. coli, K. pneumoniae, A. baumannii, P. aeruginosa, (B) S. aureus, S. epidermidis, and E. faecalis (open circle-blue: KS-Cnd, square-red: KH-Cnd, triangle-green: KSH-Cnd, and filled circle-black: control). | ||
10. Therapeutic index
The therapeutic index (TI) is defined as the ratio between the MHC of peptides over MIC and is a widely used parameter to evaluate the cell specificity and selectivity of pharmacological agents. Larger values for TI indicate greater specificity of peptide towards bacterial cell. In Table 5 are reported the TI calculated for the three new derived antimicrobial peptides.| TI | E. coli | K. pneumoniae | A. baumannii | P. aeruginosa | S. aureus | S. epidermidis | E. faecalis |
|---|---|---|---|---|---|---|---|
| KS-Cnd | 3.3 | 6.6 | 6.5 | 1.6 | 0.4 | 3.3 | 6.5 |
| KH-Cnd | 7.0 | 7.0 | 7.0 | 3.5 | 0.9 | 3.5 | 3.5 |
| KSH-Cnd | 10.5 | 21.0 | 21.0 | 10.0 | 1.3 | 10.5 | 10.6 |
KSH-Cnd showed the highest values of TI with an increase of 2–6 time with respect KS-Cnd and KH-Cnd. The least efficient TI is reported for S. aureus with poor values ranging from 0.4 to 1.3 but showing a slight increase going from KS-Cnd to KSH-Cnd. For K. pneumoniae and A. baumannii we obtained the highest specificity with a TI of 21 for KSH-Cnd. TI values seem to be influenced from the net charge of peptides: increasing the net charge increases the value of TI and the efficacy of peptide. To further evaluate the global activity of peptides we calculated the geometric mean (GM) of MIC for Gram-positive and Gram-negative bacteria and the mean therapeutic index TIM as reported in Table 6.
| Peptides | GM (μM) Gram-negative | GM (μM) Gram-positive | TIM Gram-negative | TIM Gram-positive |
|---|---|---|---|---|
| KS-Cnd | 2.5 | 7.8 | 3.2 | 1.0 |
| KH-Cnd | 1.6 | 5.2 | 5.6 | 1.7 |
| KSH-Cnd | 0.9 | 4.1 | 14.4 | 3.2 |
Peptides displayed higher activity against Gram-negative bacteria species than Gram-positive with about a 5-fold increase in the value of therapeutic index TIM for KSH-Cnd.
11. Transmission electron microscopy (TEM)
To visualize the morphology of bacterial cells treated with KSH-Cnd at 1× MIC, we utilized a TEM analysis. Since KSH-Cnd was the most effective in the in vitro assays, we choose this peptide and one representative strain of each Gram-negative and Gram-positive bacteria to study the changes in the integrity of the bacterial membranes after peptide treatment. Untreated S. epidermidis and E. coli (Fig. 9a and b, respectively) control cells showed an intact surface and dense internal structure, with their cytoplasmic content equally distributed and filling completely the internal space surrounded by the bacterial cell wall. However, after 60 min of treatment with KSH-Cnd, we observed a significant disruption of the E. coli and S. epidermidis cell membranes. Bacterial envelope damage was more evident for E. coli than S. epidermidis bacteria. The cell membrane of E. coli was clearly disrupted, and pores with large diameters were observed (red arrows in Fig. 9d). Cell surface integrity in S. epidermidis was also affected by peptide treatment (red arrows in Fig. 9c).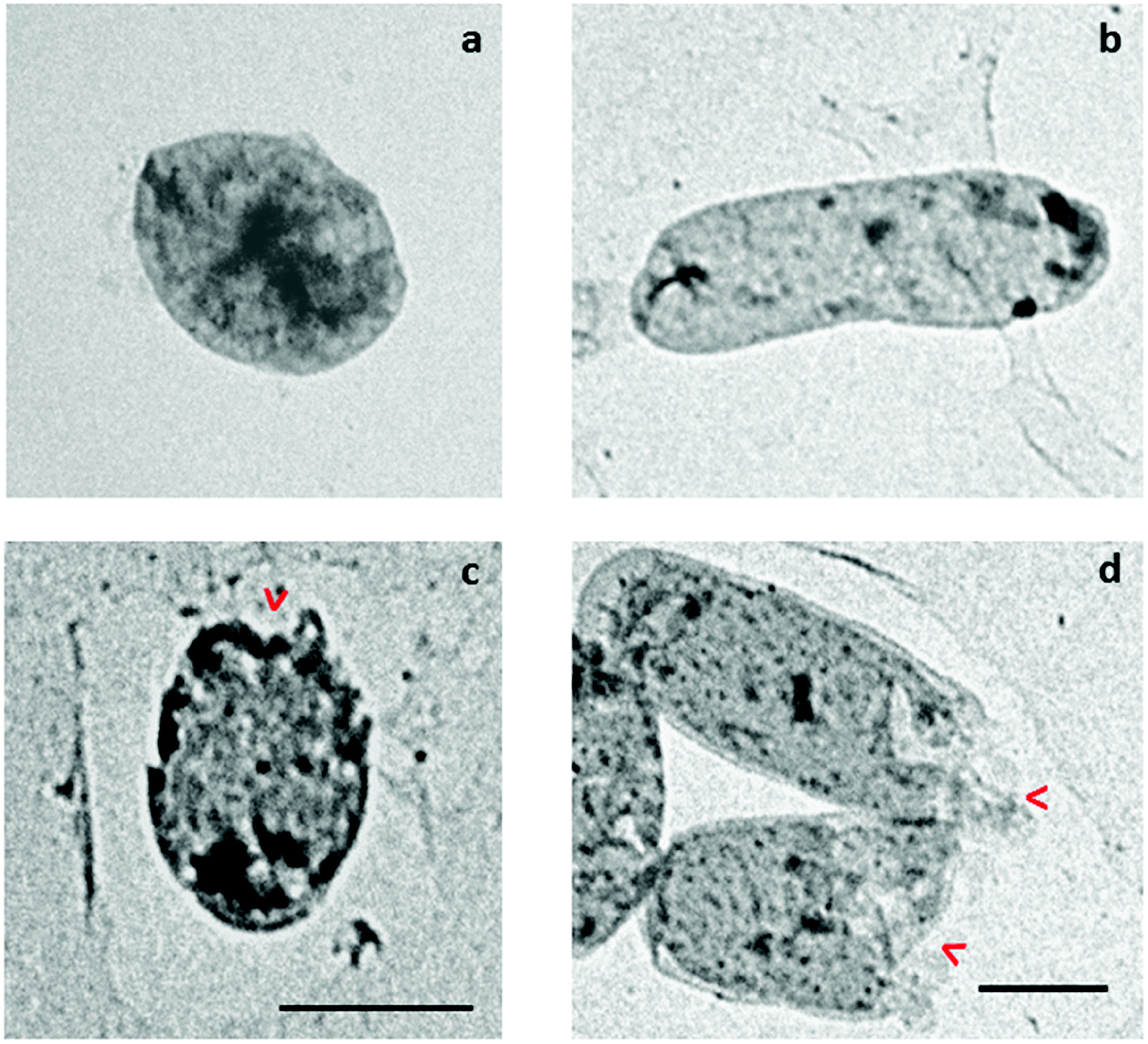 | ||
| Fig. 9 Transmission electron microscopy (TEM) micrographs of S. epidermidis and E. coli cells treated with KSH-Cnd peptide at 1× MIC for approximately 1 h. (a) Untreated S. epidermidis, (b) untreated E. coli, (c) KSH-Cnd peptide-treated S. epidermidis, and (d) KSH-Cnd peptide-treated E. coli. Both scale bars represent 1 μm. | ||
Discussion
In the last few decades, the number of infections caused by antibiotic-resistant bacteria has increased dramatically, calling for new and more powerful antimicrobial agents (Center for Disease Control, https://www.cdc.gov/). Therefore, AMPs and their synthetic analogs have been receiving considerable interest as alternatives to conventional antibiotic treatments.56–61In this work, we explored the possibility to improve the potency and selectivity of Cnd designing new analogs. We changed charge and hydrophobic moments of Cnd wild-type to steer their binding preference toward the negatively charged bacterial membranes. While engineering positive charges increases the interactions with bacterial membranes, fine-tuning the hydrophobic moment influences both the antibacterial activity and the cytotoxicity.43,48,62
Since the Cnd forms an amphipathic α-helix upon interacting with phospholipid membranes, we first substituted the Ser residues with lysines (KS-Cnd) on the hydrophilic face of the helix. In this case, we obtained a peptide with a slight increase in both potency and selectivity (approximately 20%) in vitro with the exception of MRSA. Importantly, our biological assays revealed a significant increase of the peptide potency, corresponding to a marked decrease of both MIC and MBC. When we substituted the His with Lys residues (KH-Cnd), the peptide potency increased four times with respect to Cnd; however, this peptide displayed lower selectivity. The MIC and MBC values were similar to the first analog. Of note, the activity toward MRSA increased by 50%.
The most promising analog was obtained by changing both Ser and His residues into Lys (KSH-Cnd). In this case, the affinity for the membrane increased and the selectivity increased by 40%, which corresponded to a decrease in MIC ranging from 2 times for the MRSA to 30 times for KPC producer. The MIC values indicated that Cnd-analogs shows antibacterial activity against both Gram-negative and Gram-positive bacteria, but higher MIC values were determined against Gram-positive bacteria compared to Gram-negative. These results are also supported from the TIM values that are higher for Gram-negative strains. This behavior can be interpreted in relation to the affinity of diverse peptides for the molecular composition of Gram-negative or Gram-positive bacteria surfaces. The Gram-positive bacteria envelope consists of a single phospholipid membrane followed by a thick peptidoglycan layer enriched in negatively charged teichoic acids. Gram-negative bacteria possess an inner cytoplasmic membrane surrounded by a thin peptidoglycan layer and an outer membrane containing negative lipopolysaccharides (LPS). The external membrane, which is the peptide target, makes Gram-negative bacteria more susceptible to the antimicrobial effect exerted by these peptides. This is particularly relevant considering that the increase of antibiotic-resistant health care-associated infections is particularly evident and difficult to manage for MDR Gram-negative bacteria. Despite all three Cnd-derived peptides have increased their antibacterial properties, both the MIC values and the kinetics of the time kill curves show that the KSH-Cnd peptide, with the higher positive charge and hydrophobic moment, is the most effective as also shown by the in vivo assays. Taken together, the current data demonstrate that optimization of the amphipathic character of a template peptide like Cnd is able to significantly enhance in vitro antimicrobial spectrum against the most common clinical isolates of ESKAPE pathogens. Peptides were also analyzed considering their TI and TIM. The goal in developing new antimicrobial agents is to find molecules showing high antimicrobial activity and low cytotoxicity, i.e. high values of TI. In this study the values of TI calculated for different strains and the TIM calculated for Gram-negative and Gram-positive strains revealed that increasing the charge and modulating the hydrophobic moment improve the therapeutic potential of different Cnd-analogs.
Conclusions
In conclusion, we designed three new Cnd analogs (KS-Cnd, KH-Cnd and KSH-Cnd) that exhibit increased and broad activity against highly drug-resistant strains of diverse Gram-negative and Gram-positive bacteria. Binding energies and spectroscopic studies using membrane-mimicking systems proved to be good modelling to predict the antimicrobial activity of peptides. Moreover, our biological assays revealed that these Cnd-derived peptides exhibit strong activity against all tested bacteria, a very low toxicity against human fibroblast cell lines and low hemolytic effects at concentrations needed to kill human bacterial pathogens.The antimicrobial activity was consistent with the hypothesis that increasing the net positive charge and the hydrophobic moment improves the affinity of these peptides for bacterial membranes. Time-killing tests indicated that the peptides had dose- and time-dependent killing potency and that at MIC concentrations they could kill most tested strains within 2–6 h, whereas at higher concentrations (2× MIC), the killing rates increased markedly. Furthermore, hemolytic effects were virtually undetectable at 3 μM and very low at 6 μM for all three peptides, and no cytotoxic effects were observed against human primary cell lines at this concentration. The prevalence of MDR/XDR bacteria requires the development of anti-infective countermeasures. It is increasingly evident that the membrane-targeting antimicrobial mechanisms of AMPs make them likely candidates for overcoming bacterial drug resistance properties. Based on our data, Cnd-derived peptides constitute excellent templates for the development of possible alternative antimicrobial agents.
Ethical statement
All methods used in this work are in accordance with guidelines and regulations approved by the Ethics Committee at the Università Cattolica del Sacro Cuore, Fondazione Policlinico Universitario Agostino Gemelli IRCCS (Protocol No. 0040288/16). Informed consent was waived by the Institutional Review Board of the Università Cattolica.Author contributions
F. P., C. O., G. V., F. B. designed and guided all aspects of this work. F. B., G. S. and V. S. performed the biological in vitro test. C. O., F. P. designed and performed all the fluorescence experiments. F. B., G. M. M. S. performed and designed the antimicrobial experiments, F. C. performed CD spectroscopy, M. P. performed TEM. All authors contribute to manuscript preparation. G. V. and F. P. wrote the final version.Conflicts of interest
The authors declare no conflicts of interest.Abbreviations
| AMP | Antimicrobial peptide |
| ANS | 1-Aminonaphthalene-8-sulfonic acid |
| CD | Circular dichroism |
| Cnd | Chionodracine |
| DPC | Dodecylphosphocholine |
| EDTA | Ethylenediaminetetraacetic acid |
| ESBL | Extended spectrum beta-lactamase |
| FBS | Fetal bovine serum |
| KPC | Klebsiella pneumoniae carbapenemase |
| LB | Luria–Bertani |
| LUV | Large unilamellar vesicles |
| MBC | Minimum bactericidal concentration |
| MDR | Multidrug resistant |
| MHC | Minimal hemolytic concentration |
| MIC | Minimum inhibitory concentration |
| MRSE | Methicillin resistant Staphylococcus epidermidis |
| NMR | Nuclear magnetic resonance |
| OD | Optical density |
| PBS | Phosphate buffered saline |
| PG | Phosphatidylglycerol |
| Polem | Emission polarizer |
| Polex | Excitation polarizer |
| POPC | 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine |
| POPG | 1-Palmitoyl-2-oleoyl-sn-glycero-3-(1′-rac-glycerol) sodium salt |
| RLU | Relative luminescence unit |
| SD | Standard deviation |
| TEM | Transmission electron microscopy |
| VRE | Vancomycin resistant Enterococcus |
| XDR | Extensively drug-resistant |
Acknowledgements
This research was supported by the “Departments of Excellence-2018” Program (Dipartimenti di Eccellenza) of the Italian Ministry of Education, University and Research, DIBAF-Department of University of Tuscia, Project “Landscape 4.0 – food, wellbeing and environment”.References
- T. Takahashi and R. L. Gallo, Dermatol. Clin., 2017, 35, 39–50 CrossRef CAS PubMed.
- H. G. Boman, J. Intern. Med., 2003, 254, 197–215 CrossRef CAS.
- Z. Zheng, N. Tharmalingam, Q. Liu, E. Jayamani, W. Kim, B. B. Fuchs, R. Zhang, A. Vilcinskas and E. Mylonakis, Antimicrob. Agents Chemother., 2017, 61, e00686-17 CrossRef PubMed.
- J. Xie, Q. Zhao, S. Li, Z. Yan, J. Li, Y. Li, L. Mou, B. Zhang, W. Yang, X. Miao, X. Jiang and R. Wang, Chem. Biol. Drug Des., 2017, 90(5), 690–702 CrossRef CAS PubMed.
- V. S. de Paula and A. P. Valente, Molecules, 2018, 23(8), 2040 CrossRef PubMed.
- J. D. Hale and R. E. Hancock, Expert Rev. Anti-Infect. Ther., 2007, 5, 951–959 CrossRef CAS PubMed.
- R. F. Epand, L. Maloy, A. Ramamoorthy and R. M. Epand, Biophys. J., 2010, 98, 2564–2573 CrossRef CAS PubMed.
- F. Porcelli, R. Verardi, L. Shi, K. A. Henzler-Wildman, A. Ramamoorthy and G. Veglia, Biochemistry, 2008, 47, 5565–5572 CrossRef CAS.
- F. Porcelli, B. A. Buck-Koehntop, S. Thennarasu, A. Ramamoorthy and G. Veglia, Biochemistry, 2006, 45, 5793–5799 CrossRef CAS PubMed.
- F. Porcelli, A. Ramamoorthy, G. Barany and G. Veglia, Methods Mol. Biol., 2013, 1063, 159–180 CrossRef CAS PubMed.
- B. Deslouches, J. D. Steckbeck, J. K. Craigo, Y. Doi, J. L. Burns and R. C. Montelaro, Antimicrob. Agents Chemother., 2015, 59, 1329–1333 CrossRef CAS PubMed.
- I. Marcotte, K. L. Wegener, Y. H. Lam, B. C. Chia, M. R. de Planque, J. H. Bowie, M. Auger and F. Separovic, Chem. Phys. Lipids, 2003, 122, 107–120 CrossRef CAS PubMed.
- R. F. Epand, T. L. Raguse, S. H. Gellman and R. M. Epand, Biochemistry, 2004, 43, 9527–9535 CrossRef CAS PubMed.
- R. Omidvar, Y. Xia, F. Porcelli, H. Bohlmann and G. Veglia, Biochim. Biophys. Acta, 2016, 1864, 1739–1747 CrossRef CAS PubMed.
- S. H. White and W. C. Wimley, Biochim. Biophys. Acta, 1998, 1376, 339–352 CrossRef CAS.
- J. Koehler, N. Woetzel, R. Staritzbichler, C. R. Sanders and J. Meiler, Proteins, 2009, 76, 13–29 CrossRef CAS PubMed.
- I. Zelezetsky and A. Tossi, Biochim. Biophys. Acta, 2006, 1758, 1436–1449 CrossRef CAS.
- W. C. Wimley, ACS Chem. Biol., 2010, 5, 905–917 CrossRef CAS.
- M. A. Sani and F. Separovic, Acc. Chem. Res., 2016, 49, 1130–1138 CrossRef CAS PubMed.
- K. A. Brogden, Nat. Rev. Microbiol., 2005, 3, 238–250 CrossRef CAS.
- E. J. Noga and U. Silphaduang, Drug News Perspect., 2003, 16, 87–92 CrossRef CAS.
- E. Y. Chekmenev, B. S. Vollmar, K. T. Forseth, M. N. Manion, S. M. Jones, T. J. Wagner, R. M. Endicott, B. P. Kyriss, L. M. Homem, M. Pate, J. He, J. Raines, P. L. Gor'kov, W. W. Brey, D. J. Mitchell, A. J. Auman, M. J. Ellard-Ivey, J. Blazyk and M. Cotten, Biochim. Biophys. Acta, 2006, 1758, 1359–1372 CrossRef CAS.
- S.-A. Lee, Y. K. Kim, S. S. Lim, W. L. Zhu, H. Ko, S. Y. Shin, K.-S. Hahm and Y. Kim, Biochemistry, 2007, 46, 3653–3663 CrossRef CAS.
- N. G. Park, U. Silphaduang, H. S. Moon, J. K. Seo, J. Corrales and E. J. Noga, Biochemistry, 2011, 50, 3288–3299 CrossRef CAS PubMed.
- F. Buonocore, E. Randelli, D. Casani, S. Picchietti, M. C. Belardinelli, D. de Pascale, C. De Santi and G. Scapigliati, Fish Shellfish Immunol., 2012, 33, 1183–1191 CrossRef CAS PubMed.
- C. Olivieri, F. Buonocore, S. Picchietti, A. R. Taddei, C. Bernini, G. Scapigliati, A. A. Dicke, V. V. Vostrikov, G. Veglia and F. Porcelli, Biochim. Biophys. Acta, 2015, 1848, 1285–1293 CrossRef CAS.
- L. B. Rice, Infect. Control Hosp. Epidemiol., 2010, 31(suppl. 1), S7–S10 CrossRef PubMed.
- P. N. Domadia, A. Bhunia, A. Ramamoorthy and S. Bhattacharjya, J. Am. Chem. Soc., 2010, 132, 18417–18428 CrossRef CAS PubMed.
- A. S. Ladokhin, S. Jayasinghe and S. H. White, Anal. Biochem., 2000, 285, 235–245 CrossRef CAS PubMed.
- A. S. Ladokhin, Methods Enzymol., 2009, 466, 19–42 CAS.
- W. C. Wimley and S. H. White, Biochemistry, 1993, 32, 6307–6312 CrossRef CAS.
- O. S. Belokoneva, E. Villegas, G. Corzo, L. Dai and T. Nakajima, Biochim. Biophys. Acta, 2003, 1617, 22–30 CrossRef CAS.
- S. Filippi, P. Latini, M. Frontini, F. Palitti, J. M. Egly and L. Proietti-De-Santis, EMBO J., 2008, 27, 2545–2556 CrossRef CAS PubMed.
- P. Latini, M. Frontini, M. Caputo, J. Gregan, L. Cipak, S. Filippi, V. Kumar, R. Velez-Cruz, M. Stefanini and L. Proietti-De-Santis, Cell Cycle, 2011, 10, 3719–3730 CrossRef CAS PubMed.
- I. A. Cree and P. E. Andreotti, Toxicol. In Vitro, 1997, 11, 553–556 CrossRef CAS PubMed.
- CLSI document M07-A9 Vol. 32 No. 2 Replaces M07-A8 Vol. 29 No. 2 Wayne, PA: Clinical and Laboratory Standards Institute; 2012./CLSI. Performance Standards for Antimicrobial Susceptibility Testing. Vol. Twenty-fifth Informational Supplement. Wayne, PA: Clinical Laboratory Standards Institute, (2015):M100-S25.
- V. Straniero, M. Pallavicini, G. Chiodini, C. Zanotto, L. Volonte, A. Radaelli, C. Bolchi, L. Fumagalli, M. Sanguinetti, G. Menchinelli, G. Delogu, B. Battah, C. De Giuli Morghen and E. Valoti, Eur. J. Med. Chem., 2016, 120, 227–243 CrossRef CAS PubMed.
- P. Verma, in Antimicrobial Susceptibility Testing Protocols, ed. L. Steele-Moore, A. C. Goodwin and R. Schwalbe, CRC Press, 2007, pp. 275–298, DOI:10.1201/9781420014495.ch12.
- P. Lagerback, W. W. Khine, C. G. Giske and T. Tangden, J. Antimicrob. Chemother., 2016, 71, 2321–2325 CrossRef CAS PubMed.
- V. Palmieri, D. Lucchetti, I. Gatto, A. Maiorana, M. Marcantoni, G. Maulucci, M. Papi, R. Pola, M. De Spirito and A. Sgambato, J. Nanopart. Res., 2014, 16, 2583 CrossRef.
- N. Y. Yount and M. R. Yeaman, Biochim. Biophys. Acta, 2006, 1758, 1373–1386 CrossRef CAS PubMed.
- R. Gautier, D. Douguet, B. Antonny and G. Drin, Bioinformatics, 2008, 24, 2101–2102 CrossRef CAS PubMed.
- M. Dathe, T. Wieprecht, H. Nikolenko, L. Handel, W. L. Maloy, D. L. MacDonald, M. Beyermann and M. Bienert, FEBS Lett., 1997, 403, 208–212 CrossRef CAS PubMed.
- T. Wieprecht, M. Dathe, E. Krause, M. Beyermann, W. L. Maloy, D. L. MacDonald and M. Bienert, FEBS Lett., 1997, 417, 135–140 CrossRef CAS.
- T. M. Postma and R. M. J. Liskamp, RSC Adv., 2016, 6, 94840–94844 RSC.
- Y. Chen, C. T. Mant, S. W. Farmer, R. E. Hancock, M. L. Vasil and R. S. Hodges, J. Biol. Chem., 2005, 280, 12316–12329 CrossRef CAS.
- E. Gasteiger, A. Gattiker, C. Hoogland, I. Ivanyi, R. D. Appel and A. Bairoch, Nucleic Acids Res., 2003, 31, 3784–3788 CrossRef CAS.
- M. Dathe, H. Nikolenko, J. Meyer, M. Beyermann and M. Bienert, FEBS Lett., 2001, 501, 146–150 CrossRef CAS.
- O. K. Abou-Zied, A. Boarbour, N. A. Al-Sharji and K. Philip, RSC Adv., 2015, 5, 14214–14220 RSC.
- N. Harmouche, C. Aisenbrey, F. Porcelli, Y. Xia, S. E. D. Nelson, X. Chen, J. Raya, L. Vermeer, C. Aparicio, G. Veglia, S. U. Gorr and B. Bechinger, Biochemistry, 2017, 56, 4269–4278 CrossRef CAS.
- L. Whitmore and B. A. Wallace, Biopolymers, 2008, 89, 392–400 CrossRef CAS.
- N. Sreerama and R. W. Woody, Anal. Biochem., 2000, 287, 252–260 CrossRef CAS PubMed.
- S. K. Zhang, J. W. Song, F. Gong, S. B. Li, H. Y. Chang, H. M. Xie, H. W. Gao, Y. X. Tan and S. P. Ji, Sci. Rep., 2016, 6, 27394 CrossRef CAS PubMed.
- X. Wu, Z. Wang, X. Li, Y. Fan, G. He, Y. Wan, C. Yu, J. Tang, M. Li, X. Zhang, H. Zhang, R. Xiang, Y. Pan, Y. Liu, L. Lu and L. Yang, Antimicrob. Agents Chemother., 2014, 58, 5342–5349 CrossRef PubMed.
- M. E. Levison and J. H. Levison, Infect. Dis. Clin. North Am., 2009, 23, 791–815 CrossRef.
- J. A. Masso-Silva and G. Diamond, Pharmaceuticals, 2014, 7, 265–310 CrossRef.
- G. Roscia, C. Falciani, L. Bracci and A. Pini, Curr. Protein Pept. Sci., 2013, 14, 641–649 CrossRef CAS.
- H. Moravej, Z. Moravej, M. Yazdanparast, M. Heiat, A. Mirhosseini, M. Moosazadeh Moghaddam and R. Mirnejad, Microb. Drug Resist., 2018, 24, 747–767 CrossRef CAS PubMed.
- R. Hirsch, J. Wiesner, A. Marker, Y. Pfeifer, A. Bauer, P. E. Hammann and A. Vilcinskas, J. Antimicrob. Chemother., 2018 DOI:10.1093/jac/dky386.
- A. Hollmann, M. Martinez, P. Maturana, L. C. Semorile and P. C. Maffia, Front. Chem., 2018, 6, 204 CrossRef PubMed.
- J. O'Neill, Tackling drug-resistant infections globally: Final report and recommendations Wellcome Trust, 2016 Search PubMed.
- T. Wieprecht, M. Dathe, M. Beyermann, E. Krause, W. L. Maloy, D. L. MacDonald and M. Bienert, Biochemistry, 1997, 36, 6124–6132 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work |
| This journal is © The Royal Society of Chemistry 2018 |