
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExtraction and isolation of polyhydroxy triterpenoids from Rosa laevigata Michx. fruit with anti-acetylcholinesterase and neuroprotection properties†
Pinyi Gao ab,
Ting Hana,
Mei Jina,
Danqi Lib,
Fuyu Jianga,
Lixin Zhang*b and
Xuegui Liu*ab
ab,
Ting Hana,
Mei Jina,
Danqi Lib,
Fuyu Jianga,
Lixin Zhang*b and
Xuegui Liu*ab
aCollege of Pharmaceutical and Biotechnology Engineering, Shenyang University of Chemical Technology, Shenyang 110142, People's Republic of China. E-mail: gpy1981@163.com; liuxuegui72@163.com
bInstitute of Functional Molecules, Shenyang University of Chemical Technology, Shenyang 110142, China
First published on 14th November 2018
Abstract
Rosa laevigata fruit, at present, is becoming increasingly popular as a functional foodstuff with several nutritional and medicinal properties. To explore the acetylcholinesterase (AChE) inhibitory activity of extracts from the Rosa laevigata Michx. fruit (RLMF), a simple and efficient enrichment purification technology based on microwave-assisted extraction (MAE) and multi vacuum extraction columns (VEC) was applied to screen and identify triterpenoids (TTs) in the RLMF extracts. The MAE conditions were optimized using the Box–Behnken design (BBD) with a quadratic regression model and the response surface method (RSM). The optimum conditions were as follows: ethanol concentration, 69%; extraction time, 12 min; ratio of liquid to raw material, 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mL g−1; and microwave power, 528 W. Under these conditions, the maximum content of triterpenoids reached 62.48 ± 0.25 mg g−1, which was close to the predicted value of 62.69 mg g−1. In addition, two pure polyhydroxy triterpenoids: 2α,3β,19α,23-tetrahydroxyurs-12-en-28-oic acid (1) and 2α,3β,19α,23-tetrahydroxyurs-12-en-28-oic acid-28-O-β-D-glucopyrannoside (2) were isolated and enriched to more than 500 mg by a multi VEC method. Furthermore, the quantities of compounds 1 and 2 from RLMF were 5.36 and 10.37 mg g−1, respectively, as determined using HPLC. These compounds were further assessed for acetylcholinesterase inhibitory and neuroprotection properties. The results showed that 1 and 2 showed potent AChE inhibitory activities with IC50 values of 29.22 and 45.47 μg mL−1, respectively. At high concentration, compounds 1 and 2 produced a 92% and 89% inhibition on the target enzyme, which was consistent with docking results between AChE and each isolate. Moreover, both 1 and 2 exhibited potential neuroprotective activities against H2O2-induced SHSY5Y cell death.
:1 mL g−1; and microwave power, 528 W. Under these conditions, the maximum content of triterpenoids reached 62.48 ± 0.25 mg g−1, which was close to the predicted value of 62.69 mg g−1. In addition, two pure polyhydroxy triterpenoids: 2α,3β,19α,23-tetrahydroxyurs-12-en-28-oic acid (1) and 2α,3β,19α,23-tetrahydroxyurs-12-en-28-oic acid-28-O-β-D-glucopyrannoside (2) were isolated and enriched to more than 500 mg by a multi VEC method. Furthermore, the quantities of compounds 1 and 2 from RLMF were 5.36 and 10.37 mg g−1, respectively, as determined using HPLC. These compounds were further assessed for acetylcholinesterase inhibitory and neuroprotection properties. The results showed that 1 and 2 showed potent AChE inhibitory activities with IC50 values of 29.22 and 45.47 μg mL−1, respectively. At high concentration, compounds 1 and 2 produced a 92% and 89% inhibition on the target enzyme, which was consistent with docking results between AChE and each isolate. Moreover, both 1 and 2 exhibited potential neuroprotective activities against H2O2-induced SHSY5Y cell death.
1. Introduction
During the recent three decades, the population of China has been steadily increasing, with over 200 million Chinese adults aged 60 years and above, accounting for more than 15% of the total population.1 Accordingly, the incidence of Alzheimer's disease (AD) in elderly people is on the rise. Consequently, there has been increasing interest in exploring alternative treatments for AD. Some drugs, especially AChE inhibitors, have been suggested and even applied in the treatment of AD symptoms.2 Physostigmine and galanthamine, along with other natural alkaloids and herbal drugs, have been demonstrated to possess significant therapeutic effects for AD.3Rosa laevigata Michx., belongs to the Rosaceae family of the evergreen climbing plant and is a traditional folk herb widely used in China.4 In the Chinese Pharmacopoeia, its fruits are widely recommended to treat urinary incontinence, urinary frequency, paregoric menstrual irregularities, uterine prolapse, leucorrhea and astringency.5,6 Moreover, Rosa laevigata Michx. fruits can also be used as a health food and food additive, such as a nourishing oral liquid, fruit wine, vinegar, fruit juice and jam. In addition, fruits from this medicinal plant were shown to contain triterpenoids, polysaccharides, flavonoids, organic acid, tannins and other compounds.7 The triterpenoids are the main active components of this plant and are particularly valuable as they possess anti-HIV and anti-tumor effects.8 The increasing interest in the chemical components of RLMF has arisen from need to identify new potential therapeutic agents, such as sapogenins. The conventional methods used to extract triterpenoids, such as boiling, refluxing and heating, are time-consuming, and requires large volumes of solvent and high extraction temperatures and the yields are low. In recent years, a new extraction technique named microwave-assisted extraction (MAE) has been developed and is now widely applied in the extraction of bioactive substances due to its many advantages.9,10 Compared with traditional extraction methods, MAE requires less solvent, shorter extraction duration and offers a higher extraction yield.11 Hence, to obtain a significant amount of TTSs from RLMF, MAE is now considered the method of choice. At the same time, to achieve the maximum extraction yield, the interactive investigation of various extraction factors is performed using response surface methodology (RSM).12 Vacuum extraction columns (VEC) are used to prepare the trace pure sample before HPLC. In our previous work, due to the rapidity and robustness of the method, the VEC was also performed during the isolation and enrichment of the principal constituents of the natural plant in our lab. Besides the silica gel VEC and reversed-phase C18 silica gel VEC, polyamide VEC and D101 macroporous resin VEC were also employed for the separation and purification. For the first time, application of the multi-VECs method coupled with MAE process, provided a rapid and efficient enrichment purification technology to obtain and enrich pure polyhydroxy triterpenoids from the extracts of RLMF.
This aim of this study was to optimize MAE process of extracts TTSs from RLMF by RSM and to investigate the anti-acetylcholinesterase and neuroprotection properties of compounds 1 and 2. Firstly, single-factor experiments were carried out to determine the ranges of different conditions of MAE. Secondly, the MAE process was optimized by RSM. Thirdly, two major polyhydroxy triterpenoids identified as 2α,3β,19α,23-tetrahydroxyurs-12-en-28-oic acid (1) and 2α,3β,19α,23-tetrahydroxyurs-12-en-28-oic acid-28-O-β-D-glucopyrannoside (2) were separated and purified with multi-VECs, and their purity and content were examined by HPLC analysis. RLMF has been previously found to inhibit acetylcholinesterase.13 Here, in vitro AChE inhibitory assay and AChE molecular docking studies of compounds 1 and 2 were performed simultaneously. Finally, the H2O2-induced SH-SY5Y cell death model was used to test the activities of compounds 1 and 2, which indicated that the natural product exhibited protective effects against H2O2-induced injury in human umbilical vein endothelial cells.
2. Materials and methods
2.1. General methods
Extraction is carried out using microwave instrument (Xinyi, China). LC3000 HPLC with UV spectrometer was obtained from TengHong, Beijing, China. Microplate system was acquired from iMark, Japan. NMR spectra of each sample were obtained on spectrometer (AVANCE-600, Bruker Inc., Germany). Macroporous resin (D101) was obtained from Ruida henghui, Beijing, China. Silica gel and polyamide were purchased from Xiya Chemical Industry, Shandong, China. Octadecylsilanized silica gel (ODS) were from Merck, Germany. 5,5′-Dithiobis-(2-nitrobenzoic acid) (DTNB), galanthamine hydrobromide, acetylthiocholine (ATCI) and AChE were gained from MAYA-R, China.2.2. Plant materials
Rosa laevigata Michx. fruits (RLMF) were obtained from Nanning, Guangxi province of China, in October 2016. The specimen (RLSY1609) was stored in the Chemical Technology University, Shenyang, China.2.3. Extraction of TTSs from RLMF
MAE was performed in a microwave bath which allowed variations in the duration of treatment and the irradiation power. For the MAE experiments, one gram of pretreated sample was used for each single extraction and a series of different ethanol concentrations (40–80%), extraction times (4–12 min), ratio of liquid to raw material (10–30 mL g−1) and microwave power (200–600 W). When the extraction was completed, the extracts were centrifuged at 4000 rpm for 20 min to collect the supernatants. The filtrates were diluted with water to 100 mL and 0.1 mL samples were dried in 80 °C. Accurate measurement of the samples was carried out as described above and a standard curve of saponin was constructed. Using the standard curve constructed from the saponin concentration of the samples and the absorbance of the standard curve regression equation, the content of TTSs (mg g−1) was calculated as follows:| TTSs content (mg g−1) = (C × V × N × 1000)/W | (1) |
2.4. Experimental design and statistical analysis
The optimization of extraction conditions were carried out via RSM. The effects of the extraction conditions, such as the ethanol concentration (X1), extraction time (X2), ratio of liquid to raw material (X3) and microwave power (X4), for the extraction content of TTSs, were investigated by the single factor experiments. According to single-factor testing results, preliminary ranges of each factor were used for the subsequent experiment design. Moreover, a four-variable, three-level Box–Behnken design (BBD) was employed in the optimization, as shown in Table S2.† Four independent variables (X1–X4) were studied under three-level.The coded values of the extraction factors were calculated via following equation:
| Xi = (xi − x0)/Δx | (2) |
As shown in Table 1, a matrix consisting of 29 experimental runs was constructed in random order. According to the experiment results, the multiple regression quadratic model used in the second-order polynomial formula is given as:
| Y = β0 + ∑βixi + ∑βixi2 + ∑βijxixj | (3) |
| Run | Coded variable levels | Experimental values | |||
|---|---|---|---|---|---|
| X1/ethanol concentration (%) | X2/extraction time (min) | X3/ratio of liquid to raw material (mL g−1) | X4/microwave power (W) | Content of TTSs (mg g−1) | |
| 1 | 0 | 0 | −1 | 1 | 56.10 |
| 2 | 1 | −1 | 0 | 0 | 57.60 |
| 3 | 0 | 0 | −1 | −1 | 52.80 |
| 4 | 0 | −1 | −1 | 0 | 54.61 |
| 5 | 0 | −1 | 0 | −1 | 53.64 |
| 6 | 0 | 1 | −1 | 0 | 56.40 |
| 7 | 0 | 1 | 0 | −1 | 57.00 |
| 8 | −1 | 0 | 0 | −1 | 52.02 |
| 9 | 0 | 1 | 0 | 1 | 61.26 |
| 10 | −1 | 1 | 0 | 0 | 55.80 |
| 11 | −1 | 0 | 0 | 1 | 55.86 |
| 12 | 0 | 0 | 0 | 0 | 57.84 |
| 13 | 0 | 0 | 0 | 0 | 58.14 |
| 14 | 0 | 1 | 1 | 0 | 59.88 |
| 15 | 0 | 0 | 0 | 0 | 57.72 |
| 16 | −1 | 0 | −1 | 0 | 51.42 |
| 17 | 1 | 0 | −1 | 0 | 58.08 |
| 18 | 0 | 0 | 1 | 1 | 49.40 |
| 19 | 0 | −1 | 0 | 1 | 55.62 |
| 20 | 0 | 0 | 1 | −1 | 55.20 |
| 21 | 1 | 1 | 0 | 0 | 62.40 |
| 22 | −1 | 0 | 1 | 0 | 52.20 |
| 23 | 0 | 0 | 0 | 0 | 57.72 |
| 24 | 0 | 0 | 0 | 0 | 57.84 |
| 25 | −1 | −1 | 0 | 0 | 53.04 |
| 26 | 0 | −1 | 1 | 0 | 55.38 |
| 27 | 1 | 0 | 1 | 0 | 59.58 |
| 28 | 1 | 0 | 0 | −1 | 57.72 |
| 29 | 1 | 0 | 0 | 1 | 62.52 |
The Design-Expert 8.0.6 software was used to design the experimental conditions and statistical analysis of the predicted data. In the regression equations, the statistical significance of the terms was examined by ANOVA for every response. The evaluated adjusted-R2 and R2 were used to verify the suitability of the model. Subsequently, to prove the reliability of statistical experimental design, three additional confirmation experimental runs were carried out under the optimized conditions which gave a maximum analytical response.
2.5. Enrichment and separation of triterpenoids with multi vacuum extraction column chromatography (VEC)
Dried R. laevigata fruit powder (2 kg) was extracted at the following optimal conditions by the RSM: X1: 69%, X2: 12 min, X3: 26:1 mL g−1 and X4: 528 W. After they were concentrated in vacuo, a reddish-brown extract (252 g, 12.6%) was obtained which was dissolved in 5 L of water and then fractionated by dichloromethane (CH2Cl2) (5 L × 3), to obtain CH2Cl2 fractions (25 g, 1.3%). After the removal of the reddish-brown pigment by polyamide (500 g) VEC, the CH2Cl2-soluble extract (15 g) was subjected to purification on the silica gel (120 g) VEC using CH2Cl2–MeOH (100:1 → 5:1) solvent to form fractions E1-E7, from which Fr. E2 (3000 mg) was purified by ODS (50 g) VEC using MeOH/H2O (50:50 → 90:10). E2-3 was further purified by recrystallization to produce compound 1 (520 mg; 0.26 mg g−1). The remaining aqueous extract (200 g) was separated by macroporous resin (D101 3 kg) VEC with EtOH/H2O (v/v = 0:100, 10:90, 50:50, 95:5). The 50% ethanol fraction (53 g) was further separated by polyamide (1000 g) VEC (EtOH/H2O 20:70, 40:60, 60:40, 95:5) to produce 4 fractions (Frs A3-1 → A3-4). A3-3 was further purified by recrystallization to yield compounds 2 (910 mg; 0.455 mg g−1) (Fig. 2).
2.6. HPLC analysis
Compounds 1 and 2 were determined by HPLC with a UV detector at 210 nm via a promosil C18 column (5 μm, 250 mm × 4.6 mm). The mobile phases were consisted of methanol and water (48:52, v/v). The HPLC conditions were set as flow rate of 0.9 mL min−1 and injection volume of 30 μL. As shown in Fig. S3.1,† chromatograms of triterpenoids extracted from R. laevigata were obtained by HPLC analysis according to above conditions. All samples were determined in triplicate.
2.7. AChE inhibitory assay
Using an Ellman assay,14 20 μL compounds 1 and 2 (2, 10, 30, 60, 100, 150, 200 μg mL−1), 140 μL phosphate buffer (0.1 M, pH 7.4) and 15 μL acetylcholinesterase (0.4 U mL−1) were maintained in the each of 96-microtiter well plates for 20 min at 4 °C. Next, 10 μL ATCI and 20 μL DTNB were added, maintained for 30 min at 37 °C. The absorbance was detected at 405 nm via a microplate apparatus. A positive control was carried out using galanthamine. Then, all tests were carried out in triplicate. SPSS statistical software was carried out to calculate the IC50 values. The inhibition rate (%) was calculated by the following formula:| Inhibitory rate (%) = [(A0 − Ac) − (As − Ab)]/(A0 − Ac) × 100% | (4) |
2.8. Computational modeling for AChE inhibitors
2.9. SH-SY5Y neuroblastoma-based assay
The SH-SY5Y neuroblastoma cells were maintained in DMEM, containing 10% FBS, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin at 37 °C with 5% CO2. Compounds 1 and 2 were dissolved in dimethyl sulfoxide (DMSO) to form different concentrations 0 to 100 μM mL−1 (DMSO concentration ≤ 0.01%). Compounds 1, 2 (25, 50 and 100 μM) and cells were incubated at 37 °C for 12 h, after which H2O2 was added to a final concentration of 100 μM, and further incubated for 1.5 h. Cell viability was detected colorimetrically by MTT test.17 The absorbance was measured at 490 nm via a microplate reader. The results of the experimental groups were normalized to the those of control group. Statistical analysis was carried out by analysis of variance (ANOVA) and SPSS statistical software (version 19 for Windows) (*p < 0.05).3. Results and discussion
3.1. Single-factor experiments and results
:1 mL g−1 and X4 at 400 W. As shown in Fig. S5.1a,† when X1 was increased from 40% to 60%, the saponin content was significantly increased and reached a maximum when X1 was 60%. However, the saponin content decreased remarkably when the concentration of X1 increased from 60% to 80%. Thus, X1 of 50–70% was used to extract TTSs in the optimized experiment.:1 mL g−1, 400 W). As shown in Fig. S5.1b,† when X2 was increased from 4 to 10 min, the saponin content increased and reached the maximum at 10 min. But it obvious decreased significantly when X2 was increased to 12 min. Therefore, X2 at 8–12 min was used in the optimized experiments.:1, 15:1, 20:1, 25:1 and 30:1 mL g−1. Other extraction conditions were set as follows: the X1 was 60%, the X2 was 8 min, and the X4 was 400 W. As shown in Fig. S5.1c,† the saponin content increased obviously when the X3 increased from 10:1 mL g−1 to 25:1 mL g−1. When X3 was 25:1 mL g−1, the saponin content reached its the maximum. But the saponin content decreased remarkably when X3 was increased from 25:1 to 30:1 mL g−1. Hence, the X4 of 20:1–30:1 mL g−1 was selected as optimal for the BBD experiment.:1 mL g−1). As shown in Fig. S5.1d,† the saponin content increased markedly when X4 was increased from 200 to 500 W and it reached maximum at 500 W. However, the results showed that it decreased from 500 to 600 W. Therefore, the X4 of 400–600 W was regarded as optimal for the BBD investigation.3.2. Optimization of the extraction parameters of MAE using BBD
| Y = 57.85 + 3.13X1 + 1.91X2 + 1.02X3 + 1.86X4 + 0.15X1X2 + 0.18X1X3 + 0.24X1X4 + 0.68X2X3 + 0.57X2X4 + 0.22X3X4 + 0.63X12 − 0.079X22 − 1.53X32 − 0.51X42 | (5) |
The results of data analysis using the BBD are presented in Table 2. The variance (ANOVA) analysis of the significance of the quadratic polynomial models4 of the content of TTSs showed that the coefficients of linear (X1, X2, X3 and X4) and quadratic coefficients (X32) were highly significant (P ≤ 0.0001) with regard to the content of TTSs. In addition, statistical analysis of the quadratic coefficients (X12) with a small p-value (P < 0.05) revealed that they were significant. The p-value (P < 0.0001) polynomial model confirmed the suitability of this model to determine the content of TTSs. Moreover, the correlation coefficient of determination (R2) of 0.9742 indicated that the calculated model, the R2-pred of 0.8535 and the R2-adj of 0.9484 showed a close relationship between the experimental data and the theoretical values. At the same time, the lack of fit (0.0051) in the model exhibited significant difference and the small values of the coefficient of variation (C.V.) (1.18%) suggested that the model was effective in analyzing the experimental data on the extraction of TTSs from RLMF by the MAE method.
| Source | Sum of squares | df | Mean square | F value | P-value prob > F | Significance |
|---|---|---|---|---|---|---|
| Model | 236.71 | 14 | 16.91 | 37.78 | <0.0001 | ** |
| X1 | 117.56 | 1 | 117.56 | 262.72 | <0.0001 | ** |
| X2 | 43.55 | 1 | 43.55 | 97.32 | <0.0001 | ** |
| X3 | 12.48 | 1 | 12.48 | 27.90 | 0.0001 | ** |
| X4 | 41.74 | 1 | 41.74 | 93.27 | <0.0001 | ** |
| X1X2 | 1.04 | 1 | 1.04 | 2.32 | 0.1496 | |
| X1X3 | 0.13 | 1 | 0.13 | 0.29 | 0.5989 | |
| X1X4 | 0.23 | 1 | 0.23 | 0.51 | 0.4848 | |
| X2X3 | 1.82 | 1 | 1.82 | 4.07 | 0.0632 | |
| X2X4 | 1.30 | 1 | 1.30 | 2.90 | 0.1104 | |
| X3X4 | 0.20 | 1 | 0.20 | 0.45 | 0.5121 | |
| X12 | 2.54 | 1 | 2.54 | 5.68 | 0.0319 | * |
| X22 | 0.040 | 1 | 0.040 | 0.089 | 0.7694 | |
| X32 | 15.10 | 1 | 15.10 | 33.76 | <0.0001 | ** |
| X42 | 1.71 | 1 | 1.71 | 3.82 | 0.0708 | |
| Residual | 6.26 | 14 | 0.45 | |||
| Lack of fit | 6.15 | 10 | 0.61 | 20.82 | 0.0051 | ** |
| Pure error | 0.12 | 4 | 0.030 | |||
| Cor total | 242.98 | 28 |
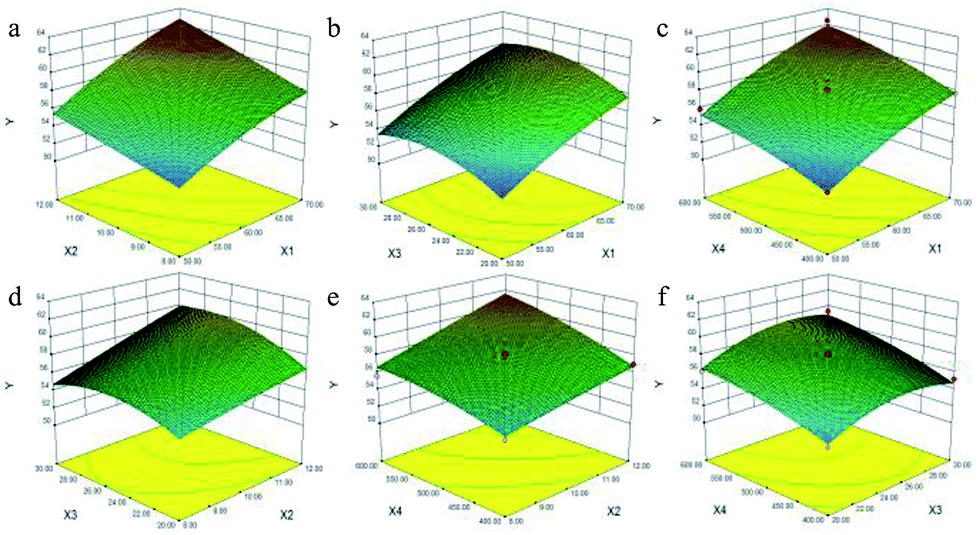 | ||
| Fig. 1 Response surface plots displaying the interaction effects of X1 and X2 (a), X1 and X3 (b), X1 and X4 (c), X2 and X3 (d), X2 and X4 (e), X3 and X4 (f) on the saponin content yield of Rosa laevigata Michx. fruits. X1: ethanol concentration; X2: extraction time; X3: ratio of liquid to raw material; X4: microwave power. | ||
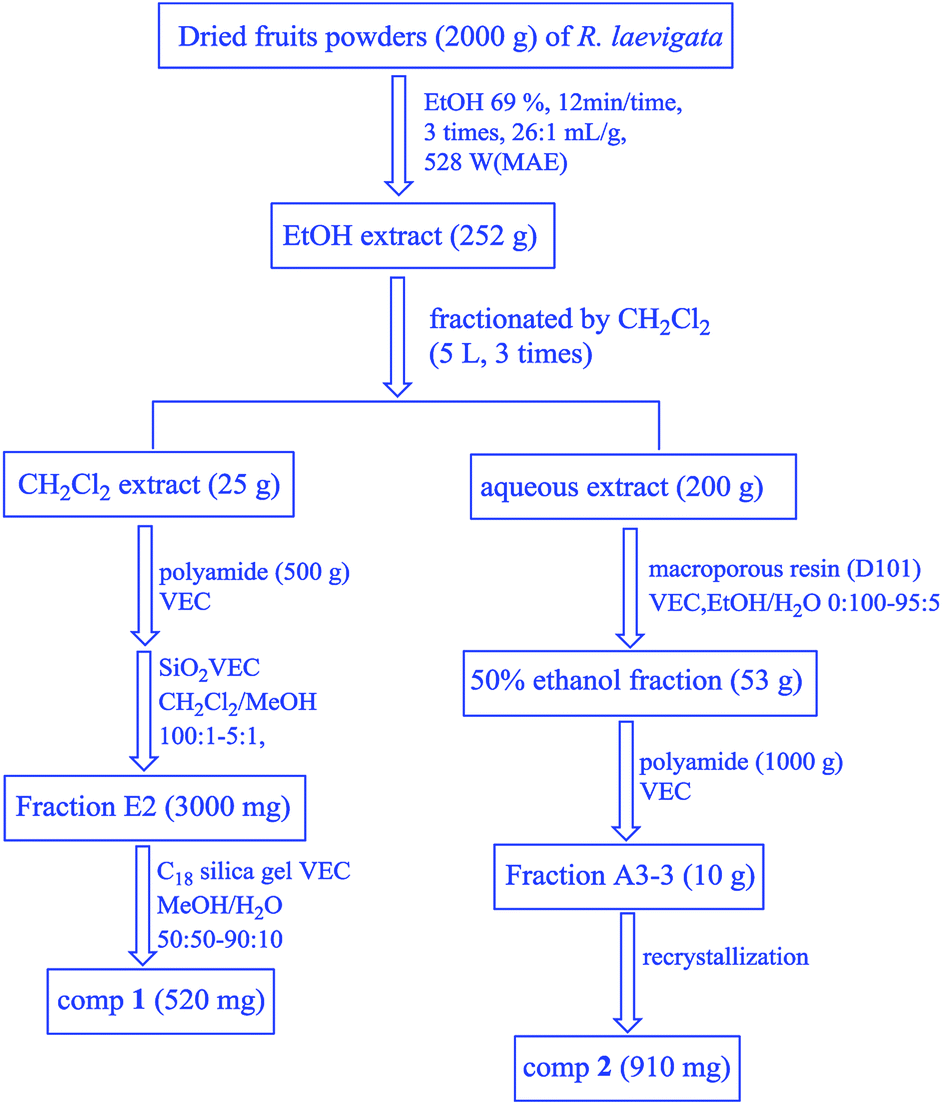 | ||
| Fig. 2 The scheme for the isolation of compounds 1 and 2 from the fruits of R. laevigata. | ||
Fig. 1a and S2.1a† shows the effects of X1 and X2 on the content of TTSs calculated using eqn (5) at a fixed X3 (25:1 mL g−1) and X4 (500 W). From the two figures, the maximum content of TTSs was obtained when X1 and X2 were 70% and 12 min, respectively.
The 3-D plot shown in Fig. 1b and the contour plot shown in Fig. S2.1b,† for two fixed variables X2 of 10 min and X4 of 500 W, showed that the content of TTSs increased as the ratio of liquid to raw material increased from 20:1 to 26:1 mL g−1, but beyond 26:1 mL g−1, the content of TTSs increased parallel to the concentration of ethanol.
As shown in Fig. 1c and S2.1c,† it was evident that the content of TTSs increased rapidly as the X1 increased from 50% to 70%, but the increase was slowly as the value of X4 increased.
The 3-D plot and the contour plot for the independent factors X2 and X3 when both X1 and X4 were zero are shown in Fig. 1d. A significant increase in the content of TTSs was observed when the extraction duration was increased from 8 to 12 min, and the content of TTSs increased with an increase in the X3 from 20:1 to 26:1 mL g−1, but decreased beyond 26:1 mL g−1.
From the 3-D plot in shown in Fig. 1e and the contour plot shown in Fig. S2.1e,† it can be concluded that the content of TTSs increased rapidly with an increase in X2 from 8 to 12 min, and an increase in the X4 ranging from 400 to 600 W.
Fig. 1f and S2.1f† show the 3-D response surface plot and the contour plot for variations in the X3 and X4. The content of TTSs increased linearly with X4 of 400–500 W whereas it decreased with the increasing ratio of liquid to raw material from 26:1 to 30:1 mL g−1. The maximum content of TTSs was achieved at ethanol concentration of 69%, extraction time of 12 min; a ratio of liquid to raw material of 26:1 mL g−1 and microwave power of 528 W.
:1 mL g−1; X4 of 528 W. An experimental value of 62.48 ± 0.25 mg g−1 obtained from the verification experiments was consistent with the predicted value of 62.69 mg g−1 (Table S3†). Therefore, the predictive model and its formula (5) were confirmed to be suitable for optimizing the process of TTSs extraction from Rosa laevigata Michx. fruit (RLMF) using the microwave-assisted extraction (MAE) technique.3.3. Determination of compounds
Compound 1 was characterized by ESIMS, 1H and 13C NMR spectroscopies. The molecular formula C30H48O6 was inferred from the ESI-MS spectrum: m/z 527 [M + Na]+ and m/z 1031 [2M + Na]+, indicating that it had a MW of 504 dalton and seven degrees of double-bond equivalence. The 1H NMR spectrum of 1 showed six tertiary methyl groups at δH ppm: 1.62 (3H, s), 1.48 (3H, s), 1.10 (3H, s), 1.09 (3H, d, J = 6.8 Hz), 1.07 (3H, s), and 1.04 (3H, s), a olefinic proton at δH 5.55 (1H, brs), two oxygen-bearing methylene protons at δH 4.17 (1H, d, J = 10.8 Hz) and 4.15 (1H, d, J = 10.8 Hz), and two oxygen-bearing methine protons at δH 4.23 (1H, dt, J = 9.6, 4.2 Hz) and 3.69 (1H, d, J = 9.6 Hz). The 13C NMR spectrum exhibited 30 carbon resonances (Table S1†), and further information of compound 1 was obtained from the DEPT-135 experiment, including functional group signals of four carbons bearing oxygen δC: 66.5 (23-CH2–O), 68.8 (2-CH–O), 72.6 (19-C–O), and 78.3 (3-CH–O), a double bond group δC: 139.9 (13-C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH) and 127.9 (12-CHC) and a carbonyl group 180.6 (28-CO). Furthermore, there were six methyls at δC 14.3 (CH3-24), 16.7 (CH3-25), 17.2 (CH3-26), 24.6 (CH3-27), 27.0 (CH3-29), and 17.3 (CH3-30), eight methylenes δC 47.8 (CH2-1), 18.6 (CH2-6), 33.1 (CH2-7), 24.1 (CH2-11), 29.2 (CH2-15), 26.3 (CH2-16), 26.9 (CH2-21), and 38.3 (CH2-22), four methines δC: 47.9 (CH-5), 47.8 (CH-9), 54.5 (CH-18), and 42.3 (CH-20), and five quaternary carbons δC: 42.1 (C-4), 40.4 (C-8), 38.3 (C-10), 42.1 (C-14), and 48.3 (C-17), which suggested that compound 1 is 2α,3β,19α,23-tetrahydroxyurs-12-en-28-oic acid.6
CH) and 127.9 (12-CHC) and a carbonyl group 180.6 (28-CO). Furthermore, there were six methyls at δC 14.3 (CH3-24), 16.7 (CH3-25), 17.2 (CH3-26), 24.6 (CH3-27), 27.0 (CH3-29), and 17.3 (CH3-30), eight methylenes δC 47.8 (CH2-1), 18.6 (CH2-6), 33.1 (CH2-7), 24.1 (CH2-11), 29.2 (CH2-15), 26.3 (CH2-16), 26.9 (CH2-21), and 38.3 (CH2-22), four methines δC: 47.9 (CH-5), 47.8 (CH-9), 54.5 (CH-18), and 42.3 (CH-20), and five quaternary carbons δC: 42.1 (C-4), 40.4 (C-8), 38.3 (C-10), 42.1 (C-14), and 48.3 (C-17), which suggested that compound 1 is 2α,3β,19α,23-tetrahydroxyurs-12-en-28-oic acid.6
Compound 2 was also characterised by ESIMS, 1H and 13C NMR spectroscopies. The molecular formula of compound 2 C36H58O11 was inferred from the ESI-MS spectrum: [M + Na]+ peak at m/z 689 (the major fragment ion) and [2M + Na]+ peak at m/z 1355, demonstrating that it had a MW of 666 and eight degrees of double-bond equivalence. A comparison of NMR data (Table S1†) suggested that compounds 1 and 2 had the same triterpenoid skeleton, except for the presence of one additional C6H10O5 sugar unit in compound 2, and this was confirmed from its molecular formula. Furthermore, the glycosidic site was determined from the upfield shift of the carbonyl carbon signal (C-28) in the 13C NMR from 180.5 (CO) to 178.5 (CO), indicating that C-28 was assigned to the linkage site between the triterpenoid skeleton and the sugar.6
To further determine the structure of the sugar, compound 2 was analyzed by acid hydrolysis followed by PMP derivatization prior to HPLC, and comparison with an standard sample by HPLC analysis with UV detector. The HPLC profile (Fig. S3.1†) showed that the PMP derivative of 2 had similar retention properties with the derivative of β-D-glucose. Hence, compound 2 was identified as 2α,3β,19α,23-tetrahydroxyurs-12-en-28-oic acid-28-O-β-D-glucopyrannoside.
3.4. Method validation
To analyze the MAE method, several factors such as linearity, reproducibility, recovery, precision and stability were examined using the optimized factors. Compounds 1 and 2 were examined by comparison with the chromatograms and retention times of the standard samples. Fig. 1 shows that the retention time of 1 and 2 purified from RLMF was consistent with the that of standard 1 and 2 (tR1 7.0 min, tR2 32.0 min).3.5. Sample analysis
Rosa laevigata Michx. fruits (RLMF) were collected from Nanning, Guangxi province, P. R. China, in October 2016. Compounds 1 and 2 were extracted with optimized MAE conditions and characterized using the HPLC method. The quantities of the extracts were calculated by determining the peak area. The result indicated that their quantities in the fruit were 5.36 and 10.37 mg g−1, respectively.3.6 AChE inhibitory effects
Our preliminary in vitro screening study indicated that the 69% EtOH extract from the fruits of R. laevigata showed potent AChE inhibitory activity, with IC50 value of 36.33 μg mL−1, in which galanthamine, a well-known natural AChE inhibitor, was used as a positive control with an IC50 value of 36.30 μg mL−1 which is consistent with that reported in our previous study.13 Furthermore, its CH2Cl2-soluble extract fraction showed stronger AChE inhibitory activity, with an IC50 value of 26.371 μg mL−1. Compound 1, the main constituent of the CH2Cl2-soluble active fraction, showed potent AChE inhibitory activity with an IC50 value of 29.22 μg mL−1, which was stronger compared to compound 2 (IC50 value of 45.47 μg mL−1) from the aqueous active fraction. At a concentration of 2–200 μg mL−1 for both compounds, different active fractions, exhibited varying degrees of inhibition (Fig. 3). Compounds 1 and 2 showed significant AChE inhibitory activity, of 92% and 89%, respectively. Furthermore, the results showed that a dose–response effect was observed in each concentration curve of different samples.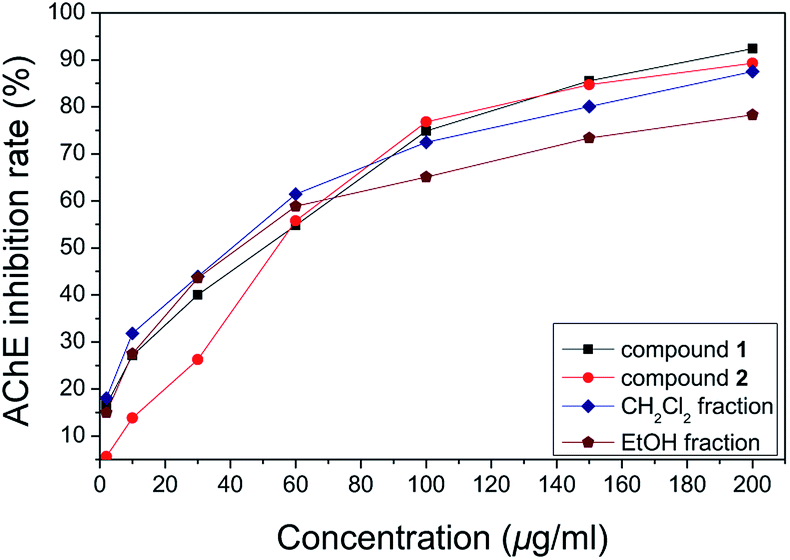 | ||
| Fig. 3 The curve plot showing a dose AChE inhibition rate in each concentration curve of different samples. | ||
3.7 Interactions of bioactive compounds with AChE protein
Interactions between compounds 1 and 2 with AChE protein were carried out via molecular modeling (Fig. 4). The experiments showed that the 23-OH of 1 and the catalytic residues (PHE 288 and ARG 289) were in close proximity, and exhibited hydrogen bonding interactions (distance 2.29 and 2.20 Å). The H-bond interactions were situated at positions of the Try 70, Tyr 334 and 3,23-OH (distance 2.35 and 2.66 Å) as well as those of Try 70, Trp 84, Asn 85, Ser 122 and glycosyl of 2 (distance 1.69, 2.48, 2.05, 1.60 and 1.52 Å). Compounds 1 and 2 were found in the aromatic pocket formed by Tyr334, Phe330 and Trp279. Moreover, PHE290, His440, and ILE287 were found to essential active residues.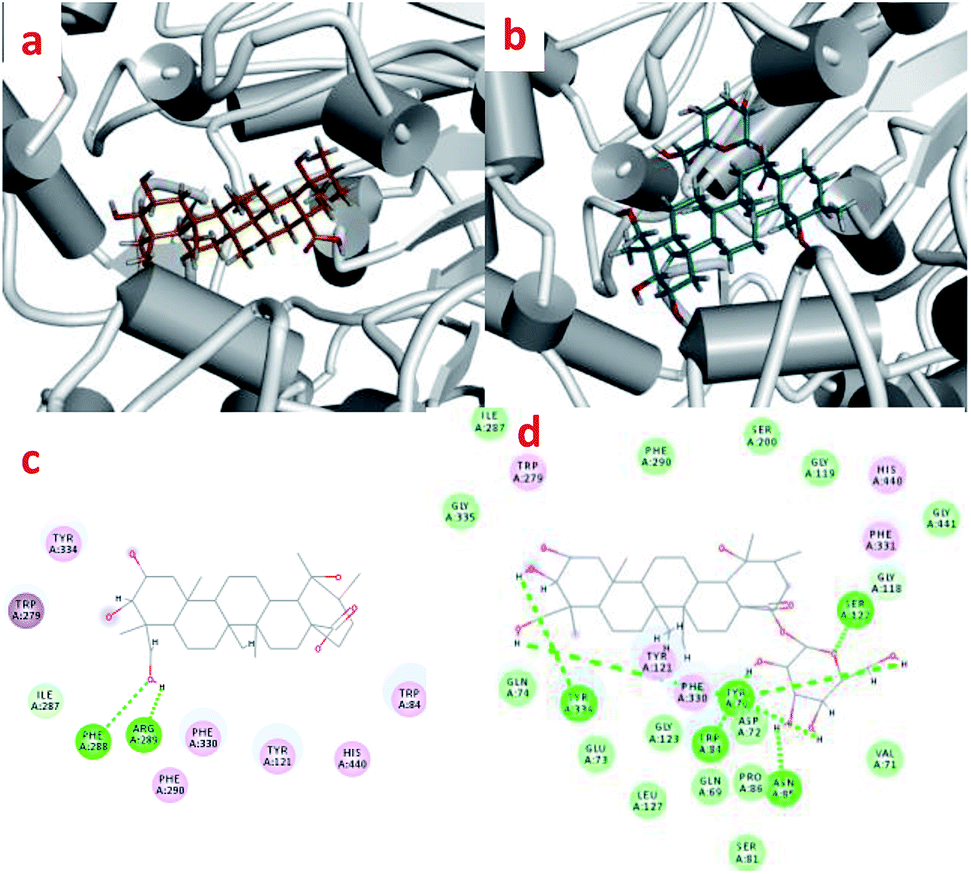 | ||
| Fig. 4 The enzyme–ligand binding interactions of compounds 1 and 2. | ||
3.8 Protective effect of SH-SY5Y neuroblastoma cells
According to previous studies, the SH-SY5Y cell line is an efficient model for investigating the mechanisms of neurodegeneration induced by H2O2.17 The protective effects of compounds 1 and 2 were examined via the MTT test. As shown in Fig. 5, cell viability following exposure to H2O2 (100 μM) was 56.64 ± 37.65%. Cell viabilities at 25 μM, 50 μM and 100 μM of compound 1 or 2 were 60.53 ± 8.34%, 56.89 ± 9.37%, 63.15 ± 5.33% and 58.83 ± 8.95%, 59.93 ± 5.21%, 61.53 ± 1.52%, respectively. The results of the cell growth are shown in Fig. 6. In addition, the phase contrast microscope was used to analyze the morphological features of the cells. We found that treatment of cells with H2O2 (100 M) for 1.5 h led to cells loss, synapse injury, and shrinkage compared to the control group. Interestingly, pretreatment with different concentrations of compound 1 or 2 for 12 h before adding H2O2 significantly ameliorated the cell injury. It can be deduced that cell growth is in accordance with the cell viability results. Thus, compounds 1 and 2 showed protective effects against H2O2-induced SH-SY5Y neuroblastoma cell death.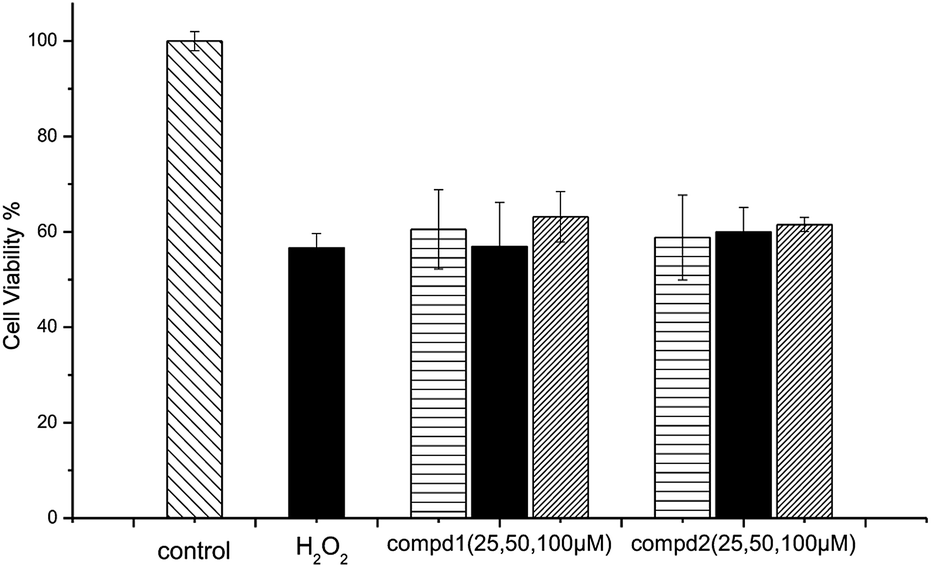 | ||
| Fig. 5 Protective effects of compounds 1 and 2 against H2O2-induced SH-SY5Y cell death. | ||
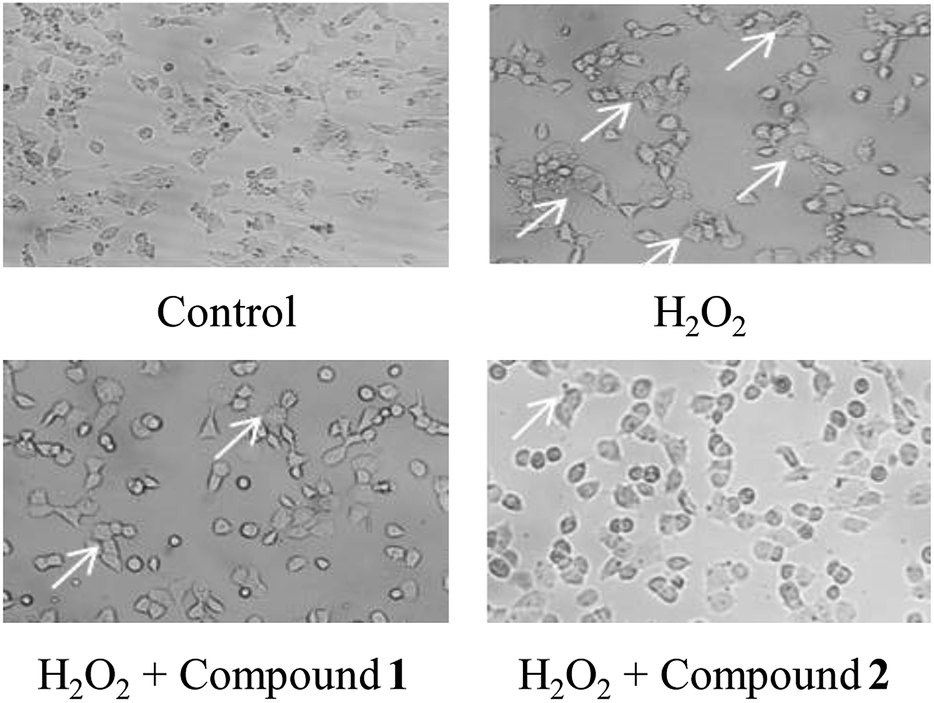 | ||
| Fig. 6 Meliorative effects of compounds 1 and 2 with H2O2 induced morphological changes in SH-SY5Y cells. | ||
4. Conclusions
In this study, two major polyhydroxy triterpenoids (2α,3β,19α,23-tetrahydroxyurs-12-en-28-oic acid and 2α,3β,19α,23-tetrahydroxyurs-12-en-28-oic acid-28-O-β-D-glucopyrannoside) were purified from triterpenoids (TTs) of Rosa laevigata Michx. fruit using microwave-assisted extraction (MAE) technology coupled with multi vacuum extraction columns (VEC) method. Based on the single-parameter experiments, response surface optimization (RSM), which reduces the extraction duration, amount of solvent and raw material needed, and was applied to optimize the four experimental variables (ethanol concentration, 69%; extraction time, 12 min; ratio of liquid to raw material, 26:1 mL g−1; and microwave power, 528 W). Under these optimum conditions, the experimental content of TTSs was in accordance with the predicted value of 62.69 mg g−1. Further analysis of the purified compounds 1 and 2 revealed that their quantities in the fruit were 5.36 and 10.37 mg g−1, respectively. To the best of our knowledge, this is the first report on enrichment of TTSs, the principal constituents of R. laevigata using rapid extraction techniques. In vitro assays demonstrated that the acetylcholinesterase inhibitory activity of compound 1 (with an IC50 value of 29.22 μg mL−1) was stronger than compound 2 (with an IC50 value of 45.47 μg mL−1). At a concentration of 2–200 μg mL−1, a dose–response effect was observed in the concentration curves of each isolate. Docking studies were performed which demonstrated to the presence of interaction between compounds 1 and 2 with AChE protein. Both compounds 1 and 2 (at the concentration of 25–100 μM) exhibited significant protective effects against H2O2-induced cell death of SH-SY5Y neuroblastoma cells. Collectively, the two active polyhydroxy triterpenoids 1 and 2 were characterized and found to possess acetylcholinesterase inhibitory efforts and neuroprotection functions during nontoxic condition. These findings imply that the two compounds are potential bioactive agents that can be used to develop drugs for AD treatment.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully thank National Natural Science Foundation of China (81703377), Natural Science Foundation from Liaoning province of China (20170540723) and the Key R & D and Technology Transfer Program from Shenyang of Liaoning province of China (Y17-0-001).References
- Z. Sui, C. Qi, Y. Huang, S. Ma, X. Wang, G. Le and Z. Sun, Aqueous extracts from asparagus stems prevent memory impairments in scopolamine-treated mice, Food Funct., 2017, 8, 1460–1467 RSC.
- A. Ciaramella, F. Salani, F. Bizzoni, M. D. Orfei, C. Caltagirone, G. Spalletta and P. Bossù, Myeloid dendritic cells are decreased in peripheral blood of Alzheimer's disease patients in association with disease progression and severity of depressive symptoms, J. Neuroinflammation, 2016, 13, 18 CrossRef PubMed.
- S. Pacifico, S. Piccolella, A. Lettieri, P. Nocera, F. Bollino and M. Catauro, A metabolic profiling approach to an Italian sage leaf extract (SoA541) defines its antioxidant and anti-acetylcholinesterase properties, J. Funct. Foods, 2017, 29, 1–9 CrossRef CAS.
- J. Q. Yuan, X. Z. Yang, J. H. Miao, C. P. Tang, C. Q. Ke, J. B. Zhang, X. J. Ma and Y. Ye, New Triterpene Glucosides from the Roots of Rosa laevigata Michx, Molecules, 2008, 13, 2229–2237 CrossRef CAS PubMed.
- Y. T. Liu, B. N. Lu, L. N. Xu, L. H. Yin, X. N. Wang, J. Y. Peng and K. X. Liu, The antioxidant activity and hypolipidemic activity of the total flavonoids from the fruit of Rosa laevigata michx, Natural Science, 2010, 2, 175–183 CrossRef CAS.
- X. G. Liu, W. C. Zhang, M. Jin, Z. Y. Wu, X. K. Mu and P. Y. Gao, Isolation and identification of triterpenes of fruit of Rosa laevigata michx, J. Shenyang Pharm. Univ., 2013, 11, 851–857 Search PubMed.
- H. Yan, B. X. Han, Q. Y. Wu, M. Z. Jiang and Z. Z. Gui, Rapid Detection of Rosa laevigata polysaccharide content by near-Infrared spectroscopy, Spectrochim. Acta, Part A, 2011, 79(1), 179–184 CrossRef CAS PubMed.
- Y. Zhuang, Y. Z. He, B. Wang, Q. S. Chen, X. L. Zhou and J. Q. Yuan, Triterpenes from Rosa Laevigata Michx. and its activity, J. Chin. Med. Mater., 2012, 35, 162–164 CAS.
- T. Wu, J. Yan, R. Liu, M. F. Marcone, H. A. Aisa and R. Tsao, Optimization of microwave assisted extraction of phenolics from potato and its downstream waste using orthogonal array design, Food Chem., 2012, 133, 1292–1298 CrossRef CAS.
- W. Routray and V. Orsat, Microwave-Assisted Extraction of Flavonoids: A Review, Food Bioprocess Technol., 2012, 5, 409–424 CrossRef CAS.
- J. G. Gujara, S. J. Wagha and V. G. Gaikar, Experimental and modeling studies on microwave-assisted extraction of thymol from seeds of Trachyspermum ammi (TA), Sep. Purif. Technol., 2010, 70, 257–264 CrossRef.
- H. Zhang, J. Birch, C. Xie, H. Yang, G. Dias, L. Kong and A. E. Bekhit, Optimization of extraction parameters of antioxidant activity of extracts from New Zealand and Chinese Asparagus officinalis L root cultivars, Ind. Crops Prod., 2018, 119, 191–200 CrossRef CAS.
- P. Y. Gao, M. Wang, X. G. Liu, Y. X. Gao, J. L. Li, Z. X. Zhang, H. W. Lin and S. J. Song, Triterpenes from the fruits of Rosa laevigata with acetylcholinesterase and Aβ-aggregation inhibitory activities, RSC Adv., 2016, 6, 2431–2435 RSC.
- K. Rhee, d. M. M. Van, K. Ingkaninan and R. Verpoorte, Screening for acetylcholinesterase inhibitors from Amaryllidaceae using silica gel thin-layer chromatography in combination with bioactivity staining, J. Chromatogr. A, 2001, 915, 217–223 CrossRef PubMed.
- G. Lotfy, M. M. Said, E. S. El Ashry, E. S. El Tamany, A. Al-Dhfyan, Y. M. Abdel Aziz and A. Barakat, Synthesis of new spirooxindole-pyrrolothiazole derivatives: Anti-cancer activity and molecular docking, Bioorg. Med. Chem., 2017, 25(4), 1514–1523 CrossRef CAS PubMed.
- H. M. Greenblatt, C. Guillou, D. Guénard, A. Argaman, S. Botti, B. Badet, C. Thal, I. Silman and J. L. Sussman, The Complex of a Bivalent Derivative of Galanthamine with Torpedo Acetylcholinesterase Displays Drastic Deformation of the Active-Site Gorge: Implications for Structure-Based Drug Design, J. Am. Chem. Soc., 2004, 126, 15405–15411 CrossRef CAS PubMed.
- X. L. Hu, Y. X. Niu, Q. Zhang, X. Tian, L. Y. Gao, L. P. Guo, W. H. Meng and Q. C. Zhao, Neuroprotective effects of Kukoamine B against hydrogen peroxide-induced apoptosis and potential mechanisms in SH-SY5Y cells, Environ. Toxicol. Pharmacol., 2015, 40, 230–240 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07930g |
| This journal is © The Royal Society of Chemistry 2018 |