
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Long non-coding RNA XIST promotes proliferation, autophagy and inhibits apoptosis by regulating microRNA-30c/ATG5 axis in gastric cancer
Mingjian Liu†
a,
Hongbo Zhao†a,
Min Shengb and
Hui Li *c
*c
aDigestive Department, The Central Hospital of Shanxian County, 274300, Shanxian County, Heze City, China
bDigestive Department, Heze Municipal Hospital, 274000, Heze City, China
cDepartment of Oncology, Heze Municipal Hospital, No. 2888, Cao Zhou Road, Peony District, 274000, Heze City, China. E-mail: zhajzscq1@163.com; Tel: +86-530-561-3306
First published on 7th November 2018
Abstract
Background: Gastric cancer (GC) is a great threat to human health and life. Long non-coding RNA X inactive-specific transcript (XIST) and microRNA-30c (miR-30c) function as crucial players in the tumorigenesis of GC. Bioinformatics analysis suggests that miR-30c has a chance to interact with XIST and autophagy related 5 (ATG5). Moreover, ATG5 has been identified as a target of miR-30c in human intestinal epithelial cells. Hence, whether XIST could regulate cell proliferation, apoptosis and autophagy by miR-30c/ATG5 axis was further investigated in GC. Methods: The levels of XIST, miR-30c and ATG5 mRNA were measured by RT-qPCR assay. ATG5, p62, LC3-I, and LC3-II protein expression was detected by western blot assay. The relationships of XIST, miR-30c and ATG5 were examined by luciferase, RNA immunoprecipitation (RIP) and RNA pull down assays. Cell proliferation was assessed by MTS assay. Cell apoptotic rate was determined using flow cytometry. ATG5 protein expression in tissues was measured by immunohistochemistry (IHC) assay. Results: XIST was highly expressed in GC tissues and cell lines. XIST knockdown suppressed proliferation, autophagy and promoted apoptosis in GC cells. XIST inhibited miR-30c expression by direct interaction in GC cells. Furthermore, miR-30c depletion abrogated XIST deficiency-mediated anti-proliferation, pro-apoptosis and anti-autophagy effects in GC cells. Additionally, ATG5 was a target of miR-30c and XIST promoted ATG5 expression by sequestering miR-30c from ATG5 in GC cells. Conclusion: XIST knockdown suppressed proliferation, autophagy and induced apoptosis through regulating miR-30c/ATG5 axis in GC cells, hinting at the potential value of XIST in the management of GC.
Introduction
Gastric cancer (GC) is the fourth most common cancer and the third leading cause of cancer-related death among men worldwide with an estimated 631![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 new cases and 469000 deaths in 2012 alone.1 Also, the morbidity and mortality of GC ranks fifth among women globally.1 Most GC cases are diagnosed at advanced stages and the prognosis for patients with advanced GC is poor.2,3 The lack of early diagnosis and effective therapy is the major reason for the deaths of GC patients, concomitant with the massive healthcare costs.4–6 Hence, it is imperative to have a deep insight into the etiology of GC so as to discover some effective management strategies.
300 new cases and 469000 deaths in 2012 alone.1 Also, the morbidity and mortality of GC ranks fifth among women globally.1 Most GC cases are diagnosed at advanced stages and the prognosis for patients with advanced GC is poor.2,3 The lack of early diagnosis and effective therapy is the major reason for the deaths of GC patients, concomitant with the massive healthcare costs.4–6 Hence, it is imperative to have a deep insight into the etiology of GC so as to discover some effective management strategies.
Long non-coding RNAs (lncRNAs), a class of long non-coding transcripts of more than 200 nucleotides (nt) in length, have been identified as crucial players in various cellular and physiological processes including proliferation, apoptosis and autophagy.7–9 Also, mounting evidence shows that numerous lncRNAs are dysregulated in GC and closely associated with the pathogenesis of GC.10 LncRNA X inactive-specific transcript (XIST), located on the X inactivation center (XIC), has been reported to be involved in the tumorigenesis and progression of some cancers.11 For instance, lncRNA XIST downregulation resulted in the reduction of cell proliferative, migratory and invasive abilities in vitro and growth inhibition of xenograft tumors in vivo in non-small cell lung cancer (NSCLC).12,13 However, Du et al. showed that lncRNA XIST inhibited cell proliferation and metastasis by regulating microRNA-23a/Raf kinase inhibitory protein (RKIP) pathway in prostate cancer.14 Previous studies also manifested that XIST was abnormally expressed in GC and implicated in the development and progression of GC.15,16
MicroRNAs (miRNAs) are a group of small endogenous non-coding RNAs that can induce post-transcriptional repression or degradation of target mRNAs.17 Over the past decades, plenty of miRNAs have been reported to be vital players in the tumorigenesis and progression of GC.18,19 Moreover, some miRNAs have potential values in the diagnosis, treatment and prognosis for GC.20 MicroRNA-30c (miR-30c), a member of miR-30 family, plays crucial roles in multiple biological processes such as adipogenesis, lipid metabolism, cell proliferation and differentiation.21 Also, prior reports pointed out that miR-30c could regulate the development and progression of some cancers including GC.21,22
In the present study, we demonstrated that XIST promoted cell proliferation and autophagy and hampered cell apoptosis by regulating miR-30c/autophagy related 5 (ATG5) axis in GC.
Materials and methods
Clinical samples and cell culture
A total of 20 primary GC patients (12 males and 8 females, age: 50–70 years old) who underwent radical gastrectomy were recruited from our hospital between March 2017 and October 2017. All enrolled patients did not receive any treatment prior to surgery. GC tissues and corresponding adjacent normal tissues located at least 5 cm away from the primary tumors were stored at −80 °C. For immunohistochemistry (IHC) analysis, the fresh samples were fixed with formalin and embedded with paraffin. The written informed consents were obtained from all patients prior to surgery. Also, our study was approved by the Ethics Committee of the Central Hospital of Shanxian County.GC cell lines BGC-823 and SGC-7901 and human embryonic kidney 293 (HEK293T) were purchased from China Center for Type Culture Collection (Wuhan, China). The immortalized human gastric epithelial cells (GES-1) were obtained from Shanghai Yubo Biological Technology Co., Ltd (Shanghai, China). GES-1, BGC-823, SGC-7901 and HEK293T cells were maintained in Dulbecco's Modified Eagle Medium (DMEM; Thermo Fisher Scientific, Rockford, IL, USA) supplemented with 10% fetal bovine serum (FBS, Thermo Fisher Scientific).
Reagent and cell transfection
MiR-30c mimic (miR-30c), miR-30c inhibitor (anti-miR-30c) and their corresponding negative controls (miR-NC, anti-miR-NC), small interference RNA (siRNA) targeting XIST (si-XIST) and its control (si-NC) were purchased from GenePharma Co., Ltd (Shanghai, China). The pcDNA-XIST (XIST) overexpression plasmid was purchased from Hanbio Biotechnology Co., Ltd (Shanghai, China). These oligonucleotides or plasmids were transfected into GC cells using Lipofectamine 2000 reagent (Thermo Fisher Scientific) referring to manufacturer's instructions.RT-qPCR assay
Total RNA was extracted from GC cells using Trizol reagent (Thermo Fisher Scientific). Then, TaqMan MicroRNA Assays for miR-30c and RNU6B (Thermo Fisher Scientific) were used to measure miR-30c level with RNU6B as the endogenous control. For expression detection of XIST and ATG5 mRNA, RNA was reversely transcribed into cDNA using GoScript Reverse Transcription System (Promega, Madison, WI, USA) and then real time PCR analysis was performed using iQ™ SYBR® Green Supermix (Bio-Rad Laboratories, Philadelphia, PA, USA) with GAPDH as the internal control. The primers to amplify XIST, ATG5, and GAPDH were listed as below: ATG5, 5′-CAACTTGTTTCACGCTATATCAGG-3′ (forward) and 5′-CACTTTGTCAGTTACCAACGTCA-3′ (reverse); XIST, 5′-GCATAACTCGGCTTAGGGCT-3′ (forward) and 5′-TCCTCTGCCTGACCTGCTAT-3′ (reverse); GAPDH, 5′-GACTCCACTCACGGCAAATTCA-3′ (forward) and 5′-TCGCTCCTGGAAGATGGTGAT-3′ (reverse).MTS assay
Cell proliferative ability was assessed by MTS assay using CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega) according to the protocols of manufacturer. Briefly, cells were seeded into 96-well plates and transfected with corresponding miRNAs or siRNAs. At the indicated time points (0, 24, 48, 72 h) after transfection, CellTiter 96® AQueous One Solution Reagent (20 μl) was added into 96-well plates (100 μl medium). After 3 h of incubation at 37 °C, cell absorbance was determined at 490 nm.Cell apoptotic assay
Cell apoptotic rate was detected using FITC Annexin V Apoptosis Detection Kit I (BD Biosciences, San Jose, CA, USA) following the protocols of manufacturer at 48 h post transfection. Briefly, harvested cells were resuspended in 1× binding buffer at the concentration of 106 cells per ml. Then, 5 μl of FITC Annexin V and 5 μl of propidium iodide (PI) solution was added into 100 μl of cell suspension. After 15 min of incubation at room temperature in the dark, 400 μl of 1× binding buffer was added into each sample. Finally, cell apoptotic rate was determined using a FACScan® flow cytometry (BD Biosciences).Western blot assay
GC cells were lysed using RIPA Lysis and Extraction Buffer (Thermo Fisher Scientific) supplemented with protease inhibitors (Thermo Fisher Scientific). Then, protein concentration was determined by Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Next, an equal amount of protein (30 μg per lane) was separated on SDS-PAGE, blotted onto polyvinylidene fluoride (PVDF) membranes (Millipore, Bedford, MA, USA) and blocked with 5% nonfat milk. Subsequently, the membranes were incubated overnight at 4 °C with primary antibodies against p62, LC3B (LC3-II and LC3-I), ATG5, and β-actin. On the next day, the membranes were probed for 1 h at room temperature with horseradish peroxidase (HRP)-conjugated secondary antibody. Finally, protein bands were visualized using Pierce™ ECL Western Blotting Substrate (Thermo Fisher Scientific) with the intensity analysis via Quantity One software (Bio-Rad Laboratories). All antibodies were obtained from Abcam Inc. (Cambridge, UK).Luciferase reporter assay
Partial fragments of lncRNA XIST and ATG5 3′UTR containing miR-30c binding sites were subcloned into psiCHECK-2 luciferase vector to produce XIST-WT and ATG5-WT reporters, respectively. Also, XIST-MUT and ATG5-MUT reporters with mutant miR-30c binding sites were constructed. All luciferase reporters were generated by Hanbio Biotechnology Co., Ltd. Then, these luciferase reporters were transfected into HEK293T cells along with corresponding miRNA mimics or overexpression plasmids, respectively. At 48 h post transfection, luciferase activities were examined through a dual luciferase reporter assay kit (Promega).RNA immunoprecipitation (RIP) assay
RIP assay was carried out using EZ-Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit (Millipore) referring to the protocols of manufacturer at 48 h after transfection. Briefly, cells were lysed using RIP lysis buffer, and then incubated with magnetic beads conjugated with antibody against IgG (Millipore) or argonaute2 (Ago2, Millipore). After purification, immunoprecipitated RNA was subjected to RT-qPCR analysis to detect the levels of XIST and ATG5 mRNA in IgG or Ago2 immunoprecipitation complex.RNA pull down assay
RNA pull down assay was conducted as previously described.23 Biotin-labeled wild type miR-30c (Bio-miR-30c-WT) and mutant type miR-30c (Bio-miR-30c-MUT), biotin-labeled wild or mutant type XIST probe (Bio-probe-XIST-WT or Bio-probe-XIST-MUT) and a control probe Bio-NC were purchased from Sangon Inc. (Shanghai, China). Briefly, cells were seeded in 10 cm culture dish and then transfected with Bio-miR-30c-WT, Bio-miR-30c-MUT, Bio-probe-XIST-WT, Bio-probe-XIST-MUT and Bio-NC. Forty eight hours post transfection, cells were collected and lysed. Then, cell lysates were co-incubated overnight at 4 °C with Streptavidin-Dyna beads, followed by RNA isolation. Finally, enriched miR-30c and XIST levels were detected by RT-qPCR assay.Immunohistochemistry (IHC) assay
Formalin-fixed and paraffin-embedded tissues were subjected to the sequential treatment of deparaffination, rehydration, antigen retrieval, and inactivation of endogenous peroxidase, blockade of non-specific signals. Next, tissues slices were incubated overnight at 4 °C with anti-ATG5 antibody (Abcam) and then treated for HRP-conjugated secondary antibody for 1 h at room temperature, followed by the visualization using diaminobenzidin (DAB) reagent. After counterstaining with haematoxylin, the slices were photophaged using a microscope.Statistical analysis
All experiments were repeated at least three times with the quantitative data expressing as mean ± standard deviation. Difference analysis was performed by Student's t-test (for 2 group data) and one-way analysis of variance (ANOVA) (for more than 3 group data) with P value less than 0.05 as statistically significant.Results
XIST was highly expressed in GC tissues and cell lines
Firstly, the expression pattern of XIST was explored in GC tissues and cell lines. RT-qPCR assay revealed that XIST level was strikingly upregulated in 20 cases of GC tissues compared with adjacent normal tissues (Fig. 1A). Also, a notable increase of XIST expression was observed in two GC cells (BGC-823 and SGC-7901) as compared to that in immortalized human gastric epithelial cells (GES-1) (Fig. 1B).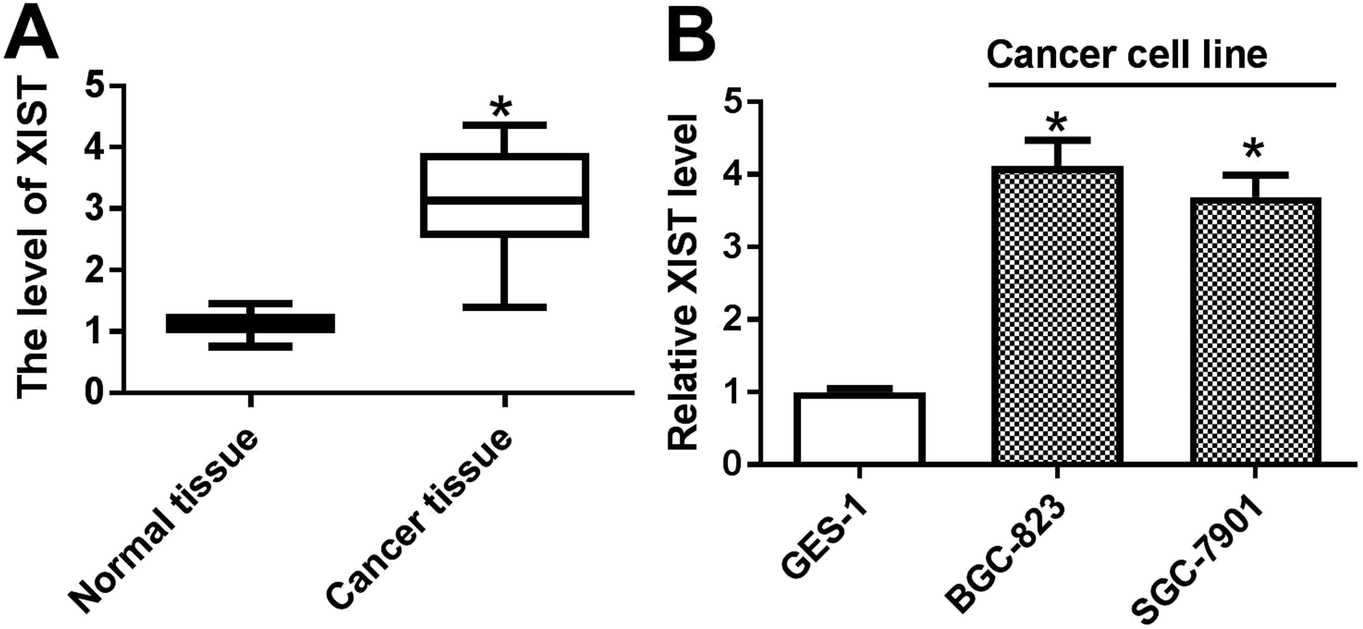 | ||
| Fig. 1 XIST was highly expressed in GC tissues and cell lines. (A and B) RT-qPCR assay was performed to measure the expression of XIST in 20 pairs of GC tissues and adjacent normal tissues (A), cell lines (GES-1, BGC-823 and SGC-7901) (B). *P < 0.05. | ||
XIST knockdown suppressed proliferation, autophagy and promoted apoptosis in GC cells
Next, RT-qPCR assay further validated that the transfection of si-XIST resulted in the significant reduction of XIST level in BGC-823 and SGC-7901 cells, meaning that si-XIST could be used for the subsequent loss-of-function explorations (Fig. 2A). Then, MTS assay showed that si-XIST-mediated XIST silence triggered a marked downregulation of cell proliferative ability in BGC-823 and SGC-7901 cells (Fig. 2B and C). Also, cell apoptotic rate was strikingly increased in BGC-823 and SGC-7901 cells transfected with si-XIST compared with si-NC-transfected cells (Fig. 2D). Moreover, we further demonstrated that XIST knockdown inhibited cell autophagy, as evidenced by reduced LC3-II (the autophagosomal form)/LC3-I (the free cytosolic form) ratio and increased p62 protein expression in si-XIST-transfected BGC-823 and SGC-7901 cells relative to si-NC-transfected control cells (Fig. 2E and F). In a word, these data disclosed that the depletion of XIST inhibited proliferation, autophagy and induced apoptosis in GC cells. | ||
| Fig. 2 XIST knockdown suppressed proliferation, autophagy and promoted apoptosis in GC cells. (A–F) BGC-823 and SGC-7901 cells were transfected with si-NC or si-XIST. (A) XIST level was measured by RT-qPCR assay at 48 h post transfection. (B and C) Cell proliferative capacity was evaluated by MTS assay at 0, 24, 48, 72 h after transfection. (D) Cell apoptotic rate was detected using a flow cytometry at 48 h upon transfection. (E and F) Protein levels of p62, LC3-I and LC3-II were examined by western blot assay at 48 h following transfection. *P < 0.05. | ||
XIST directly interacted with miR-30c
Following bioinformatics analysis by StarBase online website indicates that XIST has a chance to interact with miR-30c (Fig. 3A). To further confirm the prediction, XIST-WT luciferase reporter containing miR-30c binding sites and XIST-MUT reporter with mutant miR-30c binding sites were generated. Luciferase reporter assay showed that the transfection of miR-30c mimic resulted in the conspicuous downregulation of luciferase activity of XIST-WT reporter, but had no influence on luciferase activity of XIST-MUT reporter, hinting the interaction between XIST and miR-30c by predicted binding sites (Fig. 3B). Ago2, a core protein component of RNA-induced silencing complex (RISC), plays a key role in small RNA (including miRNA)-mediated gene silencing.24,25 Consequently, RIP assay was performed to test whether XIST could interact with miR-30c in space. Results showed that the introduction of miR-30c mimic resulted in the substantial enrichment of XIST in Ago2 immunoprecipitation complex versus IgG control group (Fig. 3C), hinting the interaction of XIST and miR-30c. Also, RNA pull down assay further manifested that biotin-labeled wild type XIST probe (Bio-probe-XIST-WT) could pull down more miR-30c than Bio-NC control probe, while mutant type XIST probe (Bio-probe-XIST-MUT) had no impact on miR-30c enrichment (Fig. 3D). Also, XIST was significantly enriched by Bio-miR-30c-WT, but not by Bio-miR-30c-MUT (Fig. 3E). Taken together, these results showed that XIST could directly bind with miR-30c. | ||
| Fig. 3 XIST directly interacted with miR-30c. (A) Bioinformatics prediction analysis showing the putative binding sites between XIST and miR-30c, and the mutant sites in XIST-MUT luciferase reporter. (B) HEK293T cells were co-transfected with XIST-WT or XIST-MUT luciferase reporter and miR-30c mimic or miR-NC, followed by the measurement of luciferase activities at 48 h after transfection. (C) BGC-823 cells were transfected with miR-30c mimic or miR-NC. At 48 h post transfection, RIP and RT-qPCR assays were performed to detect XIST enrichment level in IgG or Ago2 immunoprecipitation complex. (D) BGC-823 cells were transfected with Bio-NC probe, Bio-probe-XIST-WT or Bio-probe-XIST-MUT. At 48 h after transfection, RNA pull down and RT-qPCR assays were conducted to measure miR-30c level pulled down the above probes. (E) BGC-823 cells were transfected with Bio-NC probe, Bio-miR-30c-WT, or Bio-miR-30c-MUT. At 48 h post transfection, RNA pull down and RT-qPCR assays were carried out to determine XIST level pulled down the above biotin-labeled oligonucleotides. *P < 0.05. | ||
XIST inhibited miR-30c expression in GC cells
Next, RT-qPCR assay demonstrated that miR-30c level was notably reduced in GC tissues (n = 20) and cells compared with matching controls (Fig. 4A and B). Also, miR-30c level was negatively associated with XIST level in 20 cases of GC tissues (Fig. 4C). Transfection efficiency analysis revealed that XIST level was remarkably upregulated in BGC-823 transfected with XIST overexpression plasmid, but was dramatically downregulated in si-XIST-transfected SGC-7901 cells (Fig. 4D). Moreover, following RT-qPCR assay showed that XIST overexpression resulted in the notable reduction of miR-30c level in BGC-823 cells, whereas XIST knockdown induced miR-30c expression in SGC-7901 cells (Fig. 4E). That was to day, XIST inhibited miR-30c expression in GC cells. | ||
| Fig. 4 XIST inhibited miR-30c expression in GC cells. (A and B) RT-qPCR assay was conducted to determine XIST level in 20 pairs of GC tissues and adjacent normal tissues (A), cell lines (GES-1, BGC-823 and SGC-7901) (B). (C) Correlation analysis between miR-30c and XIST in 20 cases of GC tissues. (D and E) BGC-823 cells were transfected with pcDNA3.1 empty vector or pcDNA-XIST overexpression plasmid, and SGC-7901 cells were transfected with si-NC or si-XIST. At 48 h after transfection, the levels of XIST and miR-30c were measured by RT-qPCR assay. *P < 0.05. | ||
The depletion of miR-30c abrogated XIST deficiency-mediated anti-proliferation, pro-apoptosis and anti-autophagy effects in GC cells
Next, MTS assay further evinced that the introduction of miR-30c inhibitor resulted in the remarkable increase of cell proliferative ability in XIST-silenced BGC-823 and SGC-7901 cells (Fig. 5A). Also, cell apoptotic rate was noticeably reduced in si-XIST-transfected BGC-823 and SGC-7901 cells following the downregulation of miR-30c level (Fig. 5B). Additionally, the depletion of miR-30c alleviated the inhibitory effect of XIST knockdown on GC cell autophagy, as evidenced by improved LC3-II/LC3-I ratio and reduced p62 protein expression in BGC-823 and SGC-7901 cells co-transfected with si-XIST and anti-miR-30c than that in cells transfected with si-XIST and anti-miR-NC (Fig. 5C). In a word, these data revealed that the downregulation of miR-30c markedly weakened XIST knockdown-mediated anti-proliferation, pro-apoptosis and anti-autophagy effects in GC cells. | ||
| Fig. 5 The depletion of miR-30c abrogated XIST deficiency-mediated anti-proliferation, pro-apoptosis and anti-autophagy effects in GC cells. (A–C) BGC-823 and SGC-7901 cells were transfected with si-NC, si-XIST, si-XIST + anti-miR-NC, si-XIST + anti-miR-30c. (A) Cell proliferative ability was assessed by MTS assay at 0, 24, 48, 72 h post transfection. (B and C) Cell apoptotic rate and protein levels of p62, LC3-I and LC3-II were determined at 48 h after transfection. *P < 0.05. | ||
ATG5 was a target of miR-30c
Subsequent bioinformatics examination by Targetscan online website revealed that there existed several complementary sites between miR-30c and ATG5 3′UTR (Fig. 6A). Luciferase reporter assay further showed that the luciferase activity of ATG5-WT reporter was notably reduced in miR-30c-overexpressed cells, while miR-30c overexpression had no effect on luciferase activity of ATG5-MUT reporter (Fig. 6B), implying the specificity of miR-30c and ATG5 3′UTR binding. RIP assay further showed that miR-30c upregulation resulted in the copious enrichment of ATG5 in Ago2 immunoprecipitation complex (Fig. 6C), suggesting the interaction between miR-30c and ATG5. RT-qPCR assay further revealed that ATG5 expression was strikingly upregulated in 20 cases of GC tissues relative to adjacent normal tissues (Fig. 6D). Also, IHC assay showed that ATG5 was highly expressed in GC tissues (Fig. 6E). Moreover, higher ATG5 mRNA level was observed in BGC-823 and SGC-7901 cells compared with GES-1 cells (Fig. 6F). Correlation analysis revealed that miR-30c level was reversely associated with ATG5 mRNA level in 20 cases of GC tissues (Fig. 6G). Additionally, RT-qPCR assay further verified that the introduction of miR-30c mimic resulted in the obvious upregulation of miR-30c level in BGC-823 cells, and the transfection of miR-30c inhibitor triggered the noticeable downregulation of miR-30c level in SGC-7901 cells (Fig. 6H). Following western blot assay further showed that ATG5 level was remarkably reduced in miR-30c-enforced BGC-823 cells, but was markedly elevated in miR-30c-depleted SGC-7901 cells (Fig. 6I). In conclusion, these data disclosed that ATG5 was a target of miR-30c. | ||
| Fig. 6 ATG5 was a target of miR-30c. (A) Predicted binding sites between miR-30c and ATG5 3′UTR together with the mutant sites in ATG5-MUT luciferase reporter. (B) HEK293T cells were co-transfected with ATG5-WT or ATG5-MUT luciferase reporter and miR-30c mimic or miR-NC, followed by the determination of luciferase activities at 48 h after transfection. (C) BGC-823 cells were transfected with miR-30c mimic or miR-NC. At 48 h post transfection, RIP and RT-qPCR assays were performed to detect ATG5 enrichment level in IgG or Ago2 immunoprecipitation complex. (D) ATG5 mRNA level was measured through RT-qPCR assay in 20 pairs of GC tissues and adjacent normal tissues. (E) IHC analysis of ATG5 level in GC tissues and matching normal tissues. (F) ATG5 mRNA level was measured through RT-qPCR assay in GES-1, BGC-823 and SGC-7901 cells. (G) Correlation analysis between miR-30c and ATG5 mRNA in 20 cases of GC tissues. (H and I) BGC-823 cells were transfected with miR-30c mimic or miR-NC, and SGC-7901 cells were transfected with anti-miR-30c or anti-miR-NC for 48 h. Then, miR-30c level and ATG5 protein level were determined by RT-qPCR and western blot assays, respectively. *P < 0.05. | ||
XIST promoted ATG5 expression by relieving miR-30a-mediated inhibitory effect on ATG5 in GC cells
Next, we further demonstrated that ATG5 mRNA level was positively associated with XIST level in 20 cases of GC tissues (Fig. 7A). Also, luciferase reporter assay showed that XIST overexpression abrogated the inhibitory effect of miR-30c on luciferase activity of ATG5-WT reporter in HEK293T cells (Fig. 7B). Moreover, the ectopic expression of XIST resulted in the upregulation of ATG5 protein level in BGC-823 cells, while this effect of XIST was markedly weakened by miR-30c (Fig. 7C). Conversely, ATG5 protein level was notably reduced in XIST-silenced SGC-7901 cells and the introduction of miR-30c inhibitor abolished XIST knockdown-mediated ATG5 downregulation in SGC-7901 cells (Fig. 7D). That was to say, XIST could function as a competing endogenous RNA (ceRNA) of miR-30c to segregate miR-30c from ATG5, resulting in the downregulation of miR-30c level and the upregulation of ATG5 level in GC cells. | ||
| Fig. 7 XIST promoted ATG5 expression by relieving miR-30a-mediated inhibitory effect on ATG5 in GC cells. (A) Correlation analysis between ATG5 mRNA and XIST in 20 cases of GC tissues. (B) HEK293T cells with ATG5-WT reporter transfection was also transfected with miR-NC, miR-30c mimic, miR-30c mimic + pcDNA 3.1 empty vector, miR-30c mimic + pcDNA-XIST overexpression plasmid, followed by the detection of luciferase activities at 48 h post transfection. (C) BGC-823 cells were transfected with pcDNA 3.1 empty vector, pcDNA-XIST overexpression plasmid, pcDNA-XIST overexpression plasmid + miR-NC, or pcDNA-XIST overexpression plasmid + miR-30c mimic, followed by the measurement of ATG5 protein level at 48 h following transfection. (D) SGC-7901 cells were transfected with si-NC, si-XIST, si-XIST + anti-miR-NC, si-XIST + anti-miR-30c. At 48 h post transfection, ATG5 protein level was determined by western blot assay. *P < 0.05. | ||
Discussion
GC is a major contributor to the global cancer burden with about 50% of GC cases and deaths occur in China.26,27 Recently, accumulating studies showed that non-coding RNAs including lncRNAs and miRNAs could function as potential tumor markers or therapeutic targets in GC.28,29 LncRNA XIST has been reported to be an oncogenic factor in GC.15,16,30 For instance, Ma et al. showed that XIST facilitated cell cycle progression and cell invasion and impeded cell apoptosis by regulating microRNA-497/metastasis-associated in colon cancer1 (MACC1) axis in GC.15 Chen et al. demonstrated that the depletion of XIST suppressed cell proliferation, migration and invasion in vitro and hampered tumor growth and metastasis in vivo through regulating microRNA-101/enhancer of zeste homolog 2 (EZH2) in GC.16 Also, higher XIST expression was positively correlated with advanced disease status and poor prognosis in GC.16 Moreover, Sun et al. reported that XIST silence resulted in the reduction of cell autophagic activity by upregulating microRNA-17 and downregulating ATG7 in NSCLC.31 Additionally, a previous document pointed out that XIST could exert oncogenic effects by upregulating TGF-β1 in GC.30 and TGF-β1 could facilitate GC cell invasion by inducing autophagy,32 further hinting the close link between XIST and cell autophagy in GC. However, the effect of XIST on cell autophagy has not been examined in GC.Hence, the roles and molecular mechanisms of XIST in GC cell proliferation, apoptosis and autophagy were further explored. Consistent with prior reports,15,16,30 XIST was highly expressed in GC tissues and cell lines and XIST knockdown suppressed proliferation and triggered apoptosis in GC cells. Moreover, our study disclosed that XIST depletion inhibited autophagy in GC cells.
Recently, emerging evidence suggests that lncRNAs can act as ceRNAs of specific miRNAs to regulate the expression of miRNAs and downstream target mRNAs in GC.33,34 Consequently, bioinformatics analysis by StarBase online website was conducted to seek for miRNAs that could bind with XIST with the result presenting that miR-30c has several complementary sites with XIST. Also, the interaction of XIST and miR-30c was further authenticated by succedent luciferase reporter assay, RIP assay and RNA pull down assay.
MicroRNA-30 (miR-30) family, composed of miR-30a, miR-30b, miR-30c, miR-30d, miR-30e five members, has been identified as vital players in the development of tissues and organs and the pathogenesis of various diseases including cancers.35 MiR-30c has been widely reported to be a tumor suppressor in multiple malignancies such as hepatocellular cancer,36 glioblastoma37 and prostate cancer.38 However, Dobson et al. pointed out that miR-30c could facilitate the invasion of metastatic breast cancer cells by targeting nephroblastoma overexpressed gene-3 (NOV/CCN3).39
Mu et al. showed that pre-miR-30c A/G polymorphism was related with increased GC risk by altering mature miR-30c expression in a Chinese population, hinting the crucial roles of miR-30c in pathogenesis of GC.40 Moreover, Cao et al. demonstrated that the enforced expression of miR-30c inhibited cell migration and invasion through targeting metastasis-associated protein 1 in GC.22 Our study manifested that miR-30c level was markedly downregulated in GC tissues and cells, which was in line with previous reports.22,41 Also, we further demonstrated that miR-30c level was negatively associated with XIST level in GC tissues and XIST inhibited miR-30c expression in GC cells. Moreover, the downregulation of miR-30c weakened XIST loss-mediated anti-proliferation, pro-apoptosis and anti-autophagy effects in GC cells.
Then, ATG5 was identified as a target of miR-30c by bioinformatics analysis of Targetscan online website, luciferase reporter assay and RIP assay. Similarly, Nguyen et al. showed that the enforced expression of miR-30c blocked adherent-invasive Escherichia coli (AIEC)-induced autophagic responses by targeting ATG5 and ATG16L1 in human intestinal epithelial cells.42
ATG5, a crucial protein in the autophagic pathway, has been identified as a vital player in the development of some cancers such as cervical cancer,43 prostate cancer44 and colorectal cancer.45 Also, earlier studies pointed out that ATG5 expression was remarkably upregulated in GC tissues.46,47 Moreover, ATG5 expression was associated with advanced disease status and poor prognosis of GC.47 Additionally, Ye et al. demonstrated that 3,30-diindolylmethane hampered the proliferation of GC cells in vitro and in vivo by downregulating miR-30e and upregulating ATG5.48
Our study further manifested that ATG5 expression was significantly elevated in GC tissues and cells, which was in accordance with prior studies.46,47 Moreover, ATG5 expression was negatively associated with miR-30c expression and positively associated with XIST expression in GC tissues. And, miR-30c inhibited ATG5 expression in GC cells. Additionally, XIST could function as a ceRNA of miR-30c to reduce miR-30c expression and increase ATG5 expression in GC cells.
Conclusion
Taken together, our data show that XIST promoted proliferation, autophagy and blocked apoptosis through downregulating miR-30c and upregulating ATG5 in GC cells, providing some potential therapeutic targets or diagnostic markers for GC and deepening our understanding on the vital roles of lncRNA–miRNA functional networks in cancers. However, we only explored the roles of XIST/miR-30c/ATG5 axis in the proliferation, apoptosis, and autophagy of GC cells (BGC-823 and SGC-7901). The influence of XIST, miR-30c and ATG5, alone or in combination, on the development (including migration and invasion) of GC needs to further investigated in other GC cells and xenograft models.Funding
None.Consent for publication
Not applicable.Detailed ethical statement
All experiments were performed in accordance with NIH Guidelines. Experiments was approved by the Ethics Committee of the Central Hospital of Shanxian County. Informed consents were obtained from human participants of this study.Conflicts of interest
The authors declare that they have no financial or non-financial conflicts of interest.Acknowledgements
Not applicable.References
- L. A. Torre, F. Bray, R. L. Siegel, J. Ferlay, J. Lortettieulent and A. Jemal, Ca-Cancer J. Clin., 2015, 65, 87–108 CrossRef PubMed.
- J. A. Ajani, T. A. D'Amico, K. Almhanna, D. J. Bentrem, J. Chao, P. Das, C. S. Denlinger, P. Fanta, F. Farjah and C. S. Fuchs, J. Natl. Compr. Cancer Network, 2016, 14, 1286–1312 CrossRef.
- V. Pasechnikov, S. Chukov, E. Fedorov, I. Kikuste and M. Leja, World J. Gastroenterol., 2014, 20, 13842 CrossRef PubMed.
- J. H. Kim, S. S. Kim, J. H. Lee, D. H. Jung, D. Y. Cheung, W. C. Chung and S. H. Park, J. Gastric Cancer, 2018, 18, 82–89 CrossRef PubMed.
- Z. Song, Y. Wu, J. Yang, D. Yang and X. Fang, Tumor Biol., 2017, 39, 1010428317714626 Search PubMed.
- C. E. Van, X. Sagaert, B. Topal, K. Haustermans and H. Prenen, Lancet, 2016, 57, 2654–2664 Search PubMed.
- A. Bhan, M. Soleimani and S. S. Mandal, Long Noncoding RNA (lncRNA): Functions in Health and Disease, Wiley-VCH Verlag GmbH & Co. KGaA, 2017 Search PubMed.
- P. G. Maass, F. C. Luft and S. Bähring, J. Mol. Med., 2014, 92, 337–346 CrossRef CAS PubMed.
- Z. Xu, Y. Yan, L. Qian and Z. Gong, Oncol. Rep., 2017, 37, 1359–1366 CrossRef CAS PubMed.
- X. Y. Fang, H. F. Pan, R. X. Leng and D. Q. Ye, Cancer Lett., 2015, 356, 357–366 CrossRef CAS PubMed.
- S. M. Weakley, H. Wang, Q. Yao and C. Chen, World J. Surg., 2011, 35, 1751–1756 CrossRef PubMed.
- J. Fang, C. C. Sun and C. Gong, Biochem. Biophys. Res. Commun., 2016, 478, 811–817 CrossRef CAS PubMed.
- H. Wang, Q. Shen, X. Zhang, C. Yang, S. Cui, Y. Sun, L. Wang, X. Fan and S. Xu, Cell. Physiol. Biochem., 2017, 41, 2221–2229 CrossRef CAS PubMed.
- Y. Du, X. D. Weng, L. Wang, X. H. Liu, H. C. Zhu, J. Guo, J. Z. Ning and C. C. Xiao, Oncotarget, 2017, 8, 94358–94370 Search PubMed.
- L. Ma, Y. Zhou, X. Luo, H. Gao, X. Deng and Y. Jiang, Oncotarget, 2017, 8, 4125–4135 Search PubMed.
- D. L. Chen, H. Q. Ju, Y. X. Lu, L. Z. Chen, Z. L. Zeng, D. S. Zhang, H. Y. Luo, W. Feng, M. Z. Qiu and D. S. Wang, J. Exp. Clin. Cancer Res., 2016, 35, 142 CrossRef PubMed.
- D. P. Bartel, Cell, 2018, 173, 20–51 CrossRef CAS PubMed.
- V. Y. Shin and K. M. Chu, World J. Gastroenterol., 2014, 20, 10432–10439 CrossRef CAS PubMed.
- F. Tong, C. Peng, Y. Yuan, S. Xia, R. Lai and S. Liu, Dig. Dis. Sci., 2014, 59, 24–30 CrossRef CAS PubMed.
- K. C. D. S. Oliveira, T. M. T. Araújo, C. I. Albuquerque, G. A. Barata, C. O. Gigek, M. F. Leal, F. Wisnieski, A. S. Khayat and P. P. D. AssumpçãO, World J. Gastroenterol., 2016, 22, 7951–7962 CrossRef PubMed.
- S. Irani and M. M. Hussain, Curr. Opin. Lipidol., 2015, 26, 139–146 CrossRef CAS PubMed.
- J. M. Cao, G. Z. Li, M. Han, H. L. Xu and K. M. Huang, Biomed. Pharmacother., 2017, 93, 554–560 CrossRef CAS PubMed.
- P. Phatak and J. Donahue, Bio-Protoc., 2017, 7(9), e2253 Search PubMed.
- T. Kawamata and Y. Tomari, Trends Biochem. Sci., 2010, 35, 368–376 CrossRef CAS PubMed.
- K. T. Gagnon, L. Li, Y. Chu, B. A. Janowski and D. R. Corey, Cell Rep., 2014, 6, 211 CrossRef CAS PubMed.
- S. J. Winawer, J. Dig. Dis., 2010, 6, 107–109 Search PubMed.
- Y. Nie, K. Wu, J. Yu, Q. Liang, X. Cai, Y. Shang, J. Zhou, K. Pan, L. Sun and J. Fang, Expert Rev. Gastroenterol. Hepatol., 2017, 11, 651–661 CrossRef CAS PubMed.
- J. Wang, Y. X. Song and Z. N. Wang, Gene, 2015, 560, 1–8 CrossRef CAS PubMed.
- P. F. Li, S. C. Chen, X. Tian, X. M. Jiang, Y. F. Shao, B. X. Xiao and J. M. Guo, World J. Gastroenterol., 2014, 20, 5411–5419 CrossRef CAS PubMed.
- Q. Zhang, B. Chen, P. Liu and J. Yang, J. Cell. Biochem., 2018, 119, 2787–2796 CrossRef CAS PubMed.
- W. Sun, Y. Zu, X. Fu and Y. Deng, Oncol. Rep., 2017, 38, 3347–3354 Search PubMed.
- Z. Xiang and D. Bing, Eur. Rev. Med. Pharmacol. Sci., 2017, 21, 1013–1019 Search PubMed.
- L. L. Guo, C. H. Song, P. Wang, L. P. Dai, J. Y. Zhang and K. J. Wang, World J. Gastroenterol., 2015, 21, 11680–11687 CrossRef CAS PubMed.
- Y. Hu, H. Tian, J. Xu and J. Y. Fang, Briefings Funct. Genomics, 2016, 15, 266–273 CrossRef CAS PubMed.
- L. Mao, S. Liu, L. Hu, L. Jia, H. Wang, M. Guo, C. Chen, Y. Liu and L. Xu, BioMed Res. Int., 2018, 2018, 9623412 Search PubMed.
- W. Wu, X. Zhang, Y. Liao, W. Zhang, H. Cheng, Z. Deng, J. Shen, Q. Yuan, Y. Zhang and W. Shen, Am. J. Cancer Res., 2015, 5, 1435–1446 CAS.
- S. Liu, X. Li and S. Zhuang, Oncol. Res., 2018 DOI:10.3727/096504018X15193506006164.
- J. Zhang, X. Wang, Y. Wang, R. Peng, Z. Lin, Y. Wang, B. Hu, J. Wang and G. Shi, Oncol. Lett., 2017, 14, 363–368 CrossRef CAS PubMed.
- J. R. Dobson, H. Taipaleenmaki, Y. J. Hu, D. Hong, A. J. van Wijnen, J. L. Stein, G. S. Stein, J. B. Lian and J. Pratap, Cancer Cell Int., 2014, 14, 73 CrossRef PubMed.
- Y. P. Mu and X. L. Su, Med. Oncol., 2012, 29, 1723–1732 CrossRef CAS PubMed.
- T. Ueda, S. Volinia, H. Okumura, M. Shimizu, C. Taccioli, S. Rossi, H. Alder, C. G. Liu, N. Oue and W. Yasui, Lancet Oncol., 2010, 11, 106–107 CrossRef.
- H. T. Nguyen, G. Dalmasso, S. Müller, J. Carrière, F. Seibold and A. Darfeuillemichaud, Gastroenterology, 2014, 146, 508–519 CrossRef CAS PubMed.
- S. Leng, Y. Hao, D. Du, S. Xie, L. Hong, H. Gu, X. Zhu, J. Zhang, D. Fan and H. F. Kung, Int. J. Cancer, 2013, 133, 2781–2790 CAS.
- A. Bommareddy, E. R. Hahm, D. Xiao, A. A. Powolny, A. L. Fisher, Y. Jiang and S. V. Singh, Cancer Res., 2009, 69, 3704–3712 CrossRef CAS PubMed.
- S. J. Won, C. H. Yen, H. S. Liu, S. Y. Wu, S. H. Lan, Y. F. Jiangshieh, C. N. Lin and C. L. Su, J. Cell. Physiol., 2015, 230, 930–946 CrossRef CAS PubMed.
- Q. H. Cao, F. Liu, Z. L. Yang, X. H. Fu, Z. H. Yang, Q. Liu, L. Wang, X. B. Wan and X. J. Fan, Am. J. Transl. Res., 2016, 8, 3831–3847 CAS.
- J. Ge, Z. Chen, J. Huang, J. Chen, W. Yuan, Z. Deng and Z. Chen, PLoS One, 2014, 9, e110293 CrossRef PubMed.
- Y. Ye, Y. Fang, W. Xu, Q. Wang, J. Zhou and R. Lu, Biochem. Pharmacol., 2016, 115, 77–84 CrossRef CAS PubMed.
Footnote |
| † These author contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2018 |