
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of novel 1,2,4-thiadiazinane 1,1-dioxides via three component SuFEx type reaction†
Mzilikazi F. Khumaloa,
Ekemini D. Akpan a,
Praveen K. Chinthakindia,
Edikarlos M. Brasila,
Kamal K. Rajbongshia,
Maya M. Makatinib,
Thavendran Govendera,
Hendrik G. Krugera,
Tricia Naicker*a and
Per I. Arvidsson*ac
a,
Praveen K. Chinthakindia,
Edikarlos M. Brasila,
Kamal K. Rajbongshia,
Maya M. Makatinib,
Thavendran Govendera,
Hendrik G. Krugera,
Tricia Naicker*a and
Per I. Arvidsson*ac
aCatalysis and Peptide Research Unit, University of KwaZulu Natal, Durban, South Africa 4001. E-mail: Naickert1@ukzn.ac.za
bMolecular Sciences Institute, School of Chemistry, University of the Witwatersrand, PO Witz 2050, Johannesburg, South Africa
cScience for Life Laboratory, Drug Discovery & Development Platform & Division of Translational Medicine and Chemical Biology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm, Sweden. E-mail: per.arvidsson@scilifelab.se
First published on 7th November 2018
Abstract
Herein, we report the preparation of 1,2,4-thiadiazinane 1,1-dioxides from reaction of β-aminoethane sulfonamides with dichloromethane, dibromomethane and formaldehyde as methylene donors. The β-aminoethane sulfonamides were obtained through sequential Michael addition of amines to α,β-unsaturated ethenesulfonyl fluorides followed by further DBU mediated sulfur(VI) fluoride exchange (SuFEx) reaction with amines at the S–F bond.
The 1,2,4-thiadiazinane 1,1-dioxide motif can be found in many biologically active compounds for vastly different medical conditions. For example, verubecestat (1) has been in phase III clinical trials as a β-amyloid precursor protein cleaving enzyme (BACE 1) inhibitor to treat moderate and prodromal Alzheimer's disease.1 Ribizzi et al. have shown that taurolidine (2) displays cytotoxic activity against certain human tumour cells,2 but primarily it is used as an antibacterial agent.3 In addition, benzothiadiazines (3) are patented as ATP-sensitive potassium channel modulators for the treatment of respiratory, central nervous, and endocrine system disorders.4 1,2,4-Thiadiazinane 1,1-dioxides of this type may be formed by various methods;5–13 most closely related to the present work is the [2 + 2 + 2] sulfa Staudinger cycloaddition of sulfonylchlorides and imines, in which case β-sultams may also be formed through the corresponding [2 + 2] cycloaddition.14,15 α,β-Unsaturated sulfonyl fluorides 4 are so far rarely encountered as starting materials for organic synthesis.16–18 The literature on this reagent describe it as a connector molecule,19 and a warhead in chemical biology.20–22 There are only four publications that, so far, have reported the use of α,β-unsaturated sulfonyl fluoride based compounds as starting materials in organic synthesis.23–26 Based on our earlier experience with the reactivity of aryl α,β-unsaturated sulfonyl fluoride towards various amine nucleophiles17 (Scheme 1), we hypothesized that an α,β-unsaturated sulfonyl fluoride of type 4 can possibly be explored for the synthesis of thiadiazinanes. This hypothesis was based on observation of low amounts of the six-membered product was formed along with the major β-sultam product 5 when p-nitrophenylethenesulfonyl fluoride was subjected to excess methyl amine in methylene chloride as a solvent and triethylamine as additional base at room temperature (Scheme 1).
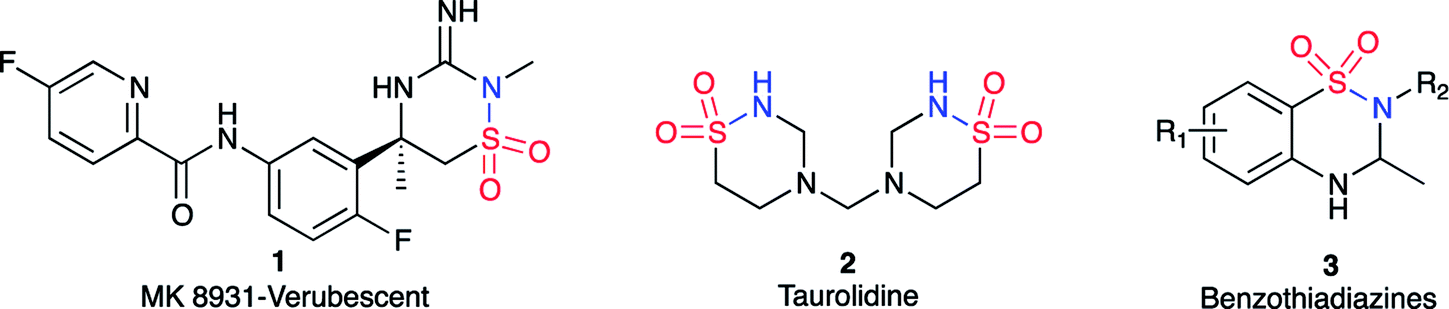
 | ||
| Scheme 1 Formation of 1,2,4-thiadiazinane 1,1-dioxides, along with β-sultams, when aryl ethenesulfonyl fluorides are subjected to large excess of primary amines in DCM as solvent and DBU as catalyst. | ||
The reactivity of dichloromethane (DCM) as a methylene donor was unfamiliar to us at the time, but a literature survey quickly revealed that organic solvents (DMF,27 DMSO,28–30 CHCl3 (ref. 31 and 32) and CH2Cl2 (ref. 33 and 34)) have proved to be more than solvents. DCM has indeed been reported to act as a bis-electrophilic methylene donor in the presence of strong bases and nucleophiles33 (e.g. carboxylic acids,35 thiols,36 amines, etc.). DCM may also form hydrochloride salts,37 aminals,38 and quaternary salts39 when reacted with tertiary and secondary amines. These reactions were reviewed by Mills et al.40 and the kinetics of the reaction of DCM with pyridine was documented by Rudine et al.41 Liu and co-workers reported formation of methylene-bridged 3,3′-bis-(oxazolidin-2-one) through reaction of oxazolidin-2-ones with DCM and sodium hydride.42 Cui et al. reported the synthesis of bispidine with the utilisation of DCM as a C1 unit.43 Dipyrrolidylmethane CH2(pyr)2 and dipiperidylmethane, CH2(pip)2 were synthesized via the condensation of the secondary amine precursors and DCM at room temperature in the absence of light.44 Another reaction of amines with methylene chloride yielded aminals rapidly.45 Matsumoto et al. reported the reaction of DCM with ketones or esters in the presence of secondary amines at high pressure whereby DCM was used as methylene bridge in forming both C–C and C–N bonds.46 Zhang and co-workers also published the formation of simultaneous carbon–carbon bond and carbon-nitrogen bonds whereby DCM acted as a synthon in the presence of 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU) and a copper catalyst.47
Results and discussion
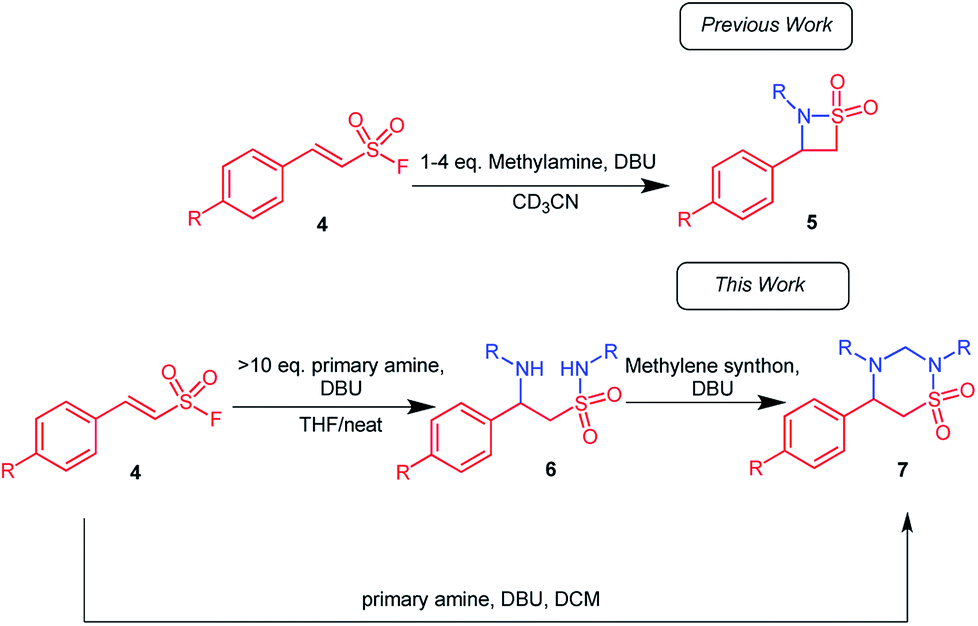
Intrigued by the observed 1,2,4-thiadiazinane 1,1-dioxide formation under certain reaction conditions we decided to investigate the scope of this novel transformation. Herein, we report the optimized synthesis of 1,2,4-thiadiazinane 1,1-dioxides (7) via (para)-aryl substituted ethene sulfonyl fluorides (ESF) with different amines using DCM, formaldehyde, or DBM as the methylene bridge sources and DBU as the catalyst.For the optimization, it was important to understand the reaction sequence involved. Consequently, we first performed a series of experiments to establish that the reaction proceeds via the expected β-aminoethane sulfonamide intermediate 6 (Scheme 1): It was observed that when 10 equiv. of the amine as well as 15 mol% DBU were used, a more polar product formed in small quantity. This product was identified by liquid chromatography mass spectrometry (LC/MS) and confirmed by NMR analysis to be the β-aminoethane sulfonamide intermediate 6. Therefore, in an effort to isolate the β-aminoethane sulfonamide intermediate, methylene chloride was replaced with tetrahydrofuran (THF) as solvent with 30 mol% DBU. The reaction successfully gave the β-aminoethane sulfonamide intermediate in good yields (61–83%) along with traces of the β-sultam at room temperature (Scheme 2).
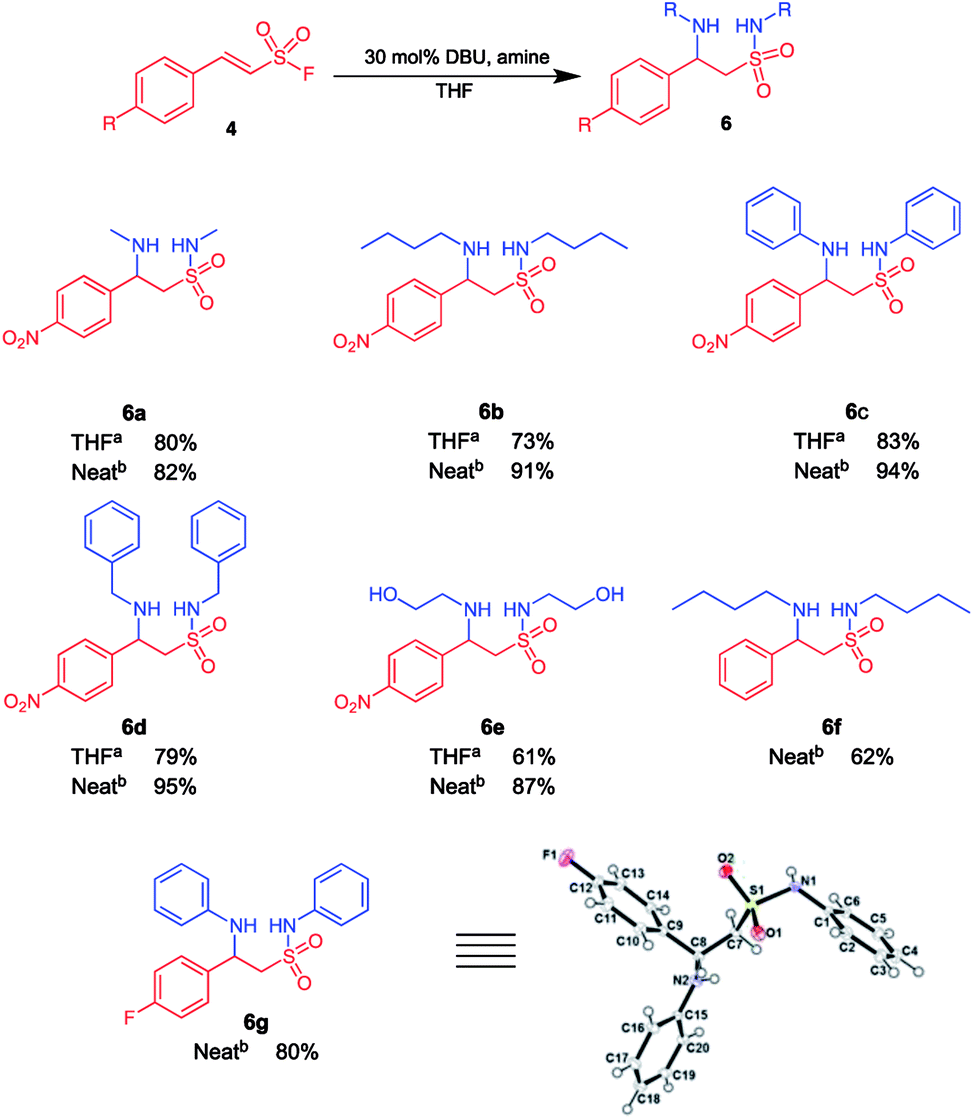 | ||
| Scheme 2 β-Aminoethane sulfonamides may be isolated when the reaction if carried out with THF or neat amine as solvent. a10 Equiv. amine, 30 mol% DBU, 5 ml THF, overnight, room temperature; bneat reaction conditions, 10 equiv. amine, 30 mol% DBU, 3–5 h, room temperature. | ||
A neat reaction that involved para-nitrophenylethenesulfonyl fluoride 4 and an amine in the presence of 30 mol% DBU also successfully yielded the β-aminoethane sulfonamide intermediate in excellent yields (62–95%) that is also depicted in Scheme 2.
We also attempted to ascertain the effect of substituents on the phenyl ring of 4 on the formation of β-aminoethane sulfonamide 6. Compound 6f was isolated in 62% yield from the reaction of (E)-2-phenylethenesulfonyl fluoride with butyl amine (10 equiv.) in the presence of 30 mol% DBU (Scheme 2). This drop in yield could be attributed to the absence of the electron withdrawing nitro substituent, resulting in a less efficient Michael addition and sulfonamide formation. Substrates with electron donating substituents on the aryl ring gave little to no product formation under these conditions; this is in accordance with studies by Qin et al., who concluded that the Michael reaction of secondary amines on β-arylethenesulfonyl fluorides required EWG on the aryl group when performed in THF or DCM.26 Further, the electron withdrawing fluoro substituent on the phenyl ring of 4 was evaluated and compound 6g was isolated in 80% yield using aniline as the amine of choice. All structures of compounds appearing in Scheme 2 was unequivocally deduced from full spectroscopic characterization. In addition, we also obtained crystals of product 6g suitable for single crystal X-ray diffraction.48 Interestingly, the fluorophenyl and phenylamino groups appeared to be attached almost perpendicular to each other with the torsion angle (C1–N1–S1–C7) of −70.12 (11)°.
Having understood the reactions steps involved, we next varied the base in the one pot reaction to obtain the 1,2,4-thiadiazinane 1,1-dioxides 7 according to Scheme 3.
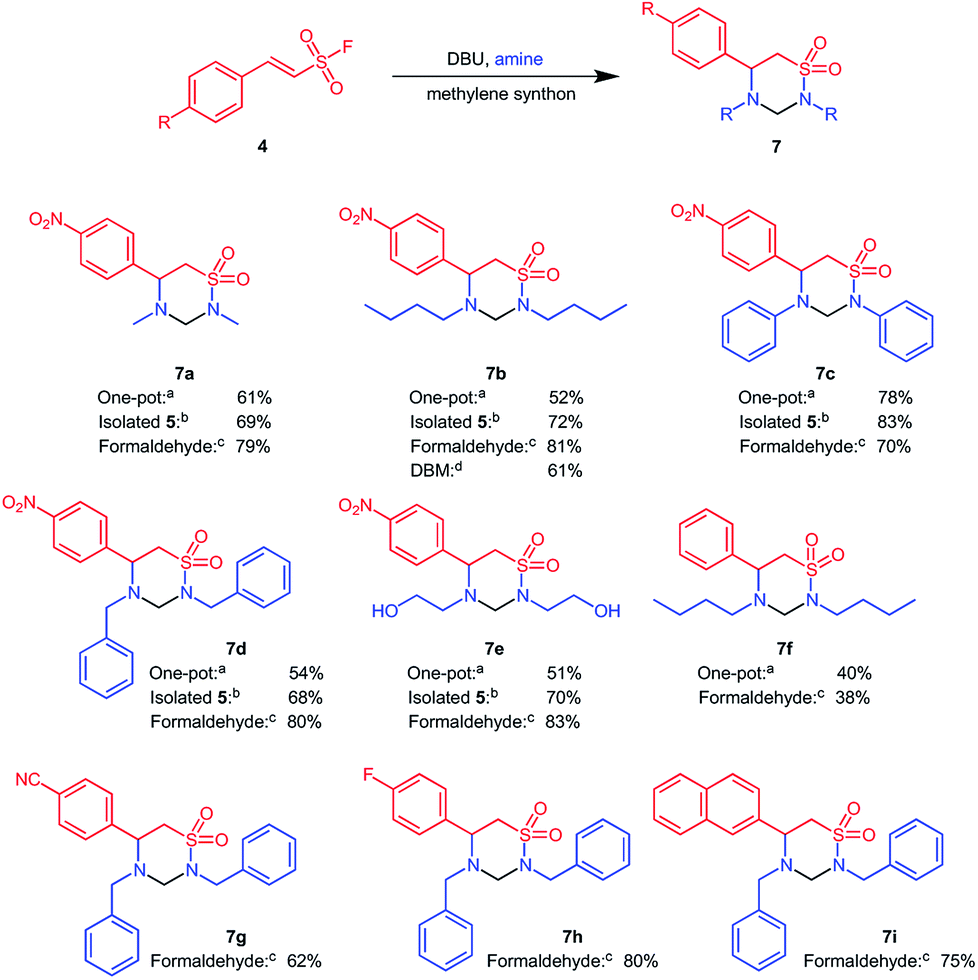 | ||
| Scheme 3 Formation of 1,2,4-thiadiazinane 1,1-dioxides under one-pot conditions in DCM or using a two-step procedure with other methylene donors for cyclization.a(One pot) 10 equiv. amine, 50 mol% DBU, 5.0 ml DCM, reflux; bβ-aminoethane sulfonamide, 20 mol% DBU, 5 ml DCM, reflux; cβ-aminoethane sulfonamide, cat. acetic acid, 1.1 equiv. CH2O, 3 ml EtOH or MeOH microwave 90 °C, 200 W, 5 min; dβ-aminoethane sulfonamide, 20 mol% DBU, 5 ml DBM, reflux. Note for b–d, the yield refers to the final step. | ||
The best conversion was obtained with 50 mol% DBU, whereas 15 and 100 mol% DBU gave comparable conversion values. DBU appeared to be less effective at 100 mol% towards the formation of 1,2,4-thiadiazinane 1,1-dioxide 7. Chen et al. recently showed, with the aid of deuterium labelling experiments, that an excess amount of DBU favours the loss of the sulfonyl group via a benzylic shift.25 This might be associated with the low conversion to the six membered cyclic product observed with stoichiometric amounts of DBU, however the mechanism of the reaction reported herein is not yet fully established.
In the case of 15 mol% DBU, it seemed that the amount of DBU was inadequate for a complete cyclization of the β-amino ethanesulfonamide 6 intermediate, as evidenced from the presence of 6 observed from TLC and via mass spectral analysis of the reaction mixture even after 24 and 48 hours. A range of bases were then screened at 50 mol% but none gave convincing conversions. For example, pyridine gave a conversion of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70 four membered:six membered products after five days of reaction time; hence DBU remained our choice of base. The temperature was varied while keeping 10 equiv. butyl amine and 50 mol% DBU. An 80% conversion towards the thiadiazinane 7 was obtained at reflux in DCM. The best conditions obtained after optimization was 50 mol% DBU; 10 equiv. butyl amine; 24 hours; under reflux in DCM. Increasing the amount of amines above 10 equiv. gave a lower yield; this is probably due to formation of a too harsh reaction mixture that cause loss of sulfonyl group as reported by Chan et al.25
:70 four membered:six membered products after five days of reaction time; hence DBU remained our choice of base. The temperature was varied while keeping 10 equiv. butyl amine and 50 mol% DBU. An 80% conversion towards the thiadiazinane 7 was obtained at reflux in DCM. The best conditions obtained after optimization was 50 mol% DBU; 10 equiv. butyl amine; 24 hours; under reflux in DCM. Increasing the amount of amines above 10 equiv. gave a lower yield; this is probably due to formation of a too harsh reaction mixture that cause loss of sulfonyl group as reported by Chan et al.25
Under the one pot conditions, aniline as a nucleophile achieved a 100% conversion towards the six membered 1,2,4-thiadiazinane 1,1-dioxides 7c, and the product could be isolated in 78% yield. However, methylamine (7a), butyl amine (7c), benzyl amine (7d), and 2-aminoethanol (7e) nucleophiles all gave a mixture of the four membered β-sultam ring and the desired six membered 1,2,4-thiadiazinane 1,1-dioxides (Scheme 3-conditions a). Under this one-pot procedure, (E)-2-phenylethenesulfonyl fluoride and butyl amine resulting in a modest 40% yield of 7f, reflecting a combination of a worse Michael reaction with this substrate (cf. Scheme 2) and the β-sultam formation with n-butylamine. Despite optimization, the one-pot transformation of ESF derivatives to 1,2,4-thiadiazinane 1,1-dioxides continued to yield a mixture of the four membered β-sultam 5 and the desired 6-membered product 7 for all aliphatic amines investigated. We therefore decided to revert to a two-step procedure, in which the intermediate β-aminoethane sulfonamide 6 was isolated before cyclization with DCM as a methylene donor. As mentioned earlier, when DCM was replaced with THF, or ran neat at room temperature, 6 was obtained in optimum quantity. Preceding, 6 was cyclized with DCM whereby the first attempt was conducted at room temperature but yielded only <10% of 7. Thereafter, the cyclization of 6 was carried out using 20 mol% catalytic DBU in DCM under reflux, and offered 1,2,4-thiadiazinane 1,1-dioxides 7 in 68–83% yield within 8–12 h (Scheme 3-conditions b). The two-step procedure proved only marginally more efficient overall than the one-pot procedure, i.e. product 7a was obtained in 56% overall yield vs. 61% in one-pot, product 7b 65% (overall) vs. 52% (one-pot), 7c 78% (overall) vs. 78% (one-pot), 7d 65% (overall) vs. 54% (one-pot), 7e 61% (overall) vs. 51% (one-pot), and 7f 24% (overall) vs. 40% (one-pot). Despite not offering much improved overall yield, the two step procedure simplified purification as a minute amount of the 4-membered β-sultam was formed under these conditions; however, the operational simplicity of the one-pot could make it the preferential protocol.
Thereafter, other methylene donors were evaluated for cyclization of the β-aminoethane sulfonamide 6 to 1,2,4-thiadiazinane 1,1-dioxides. Formaldehyde, in the presence of acetic acid under microwave conditions, successfully cyclized β-aminoethane sulfonamide derivatives 6 to the corresponding 1,2,4-thiadiazinane 1,1-dioxides in 38–83% yield (Scheme 3-conditions c). The ability of dibromomethane (DBM) and chloroform to cyclize 6 to 7 were also compared in relation to DCM. 1,2,4-Thiadiazinane 1,1-dioxide 6b reacted with dibromomethane in the presence of 50 mol% DBU at reflux to give 7b 61% yield (Scheme 3-conditions d). In contrast, reacting 6b under similar conditions in chloroform gave only trace amounts of the corresponding 1,2,4-thiadiazinane 1,1-dioxide after two days as identified by LC/MS. In an attempt to form the 2-oxy derivative of 7, the β-aminoethane sulfonamide 6 was reacted with 1,1-carbonyldiimidazole (CDI) for cyclization; the main product in this case was the activated amine which did not undergo intramolecular cyclization with the sulfonamide functionality.
Overall, we conclude that the best conditions for formation of the desired 1,2,4-thiadiazinane 1,1-dioxides was through isolation of the intermediate 6 followed by ring-closure to 7 with formaldehyde as a methylene donor. We utilized this optimized protocol for the conversion of the two electron withdrawing ESF derivatives 6g and 6h, as well as the naphthyl analogue 6i, to yield the corresponding 1,2,4-thiadiazinane 1,1-dioxides 7g–7i (Scheme 3).
Conclusions
We have explored rarely utilized α,β-unsaturated ethenesulfonyl fluorides to synthesize novel β-aminoethane sulfonamides and developed a convenient procedure for the synthesis of 1,2,4-thiadiazinane 1,1-dioxides in moderate to good yields. Although we initially identified the 6-membered product as a rare example of DCM reactivity as methylene donor, the optimized protocol make use of formaldehyde as one carbon source. We anticipate that the reported methods will aid the synthesis of thiadiazinane derivatives that will find applications as biologically active compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the South African National Research Foundation (Grants 87706, 105213, 105853), Aspen Pharmacare and the Medical Research Council for their support. PKC acknowledges UKZN and The Claude Leon Foundation of South Africa for postdoctoral fellowships.Notes and references
- K. Harati, S. Emmelmann, B. Behr, O. L. E. Goertz, T. Hirsch, N. Kapalschinski, J. Kolbenschlag, I. Stricker, A. Tannapfel, M. Lehnhardt and A. Daigeler, Oncol. Lett., 2016, 11, 1955–1961 CrossRef CAS PubMed.
- I. Ribizzi, W. Darnowski J, A. Goulette F, S. Akhtar M, D. Chatterjee and P. Calabresi, Bone Marrow Transplant., 2002, 29, 313–319 CrossRef CAS PubMed.
- Y. Liu, A.-Q. Zhang, L. Cao, H.-T. Xia and J.-J. Ma, PLoS One, 2013, 8, e79417 CrossRef PubMed.
- S. Boverie, M.-H. Antoine, F. Somers, B. Becker, S. Sebille, R. Ouedraogo, S. Counerotte, B. Pirotte, P. Lebrun and P. de Tullio, J. Med. Chem., 2005, 48, 3492–3503 CrossRef CAS PubMed.
- A. Cherepakha, V. O. Kovtunenko, A. Tolmachev, O. Lukin and K. G. Nazarenko, Synth. Commun., 2011, 41, 1977–1989 CrossRef CAS.
- A. Cherepakha, V. O. Kovtunenko, A. Tolmachev and O. Lukin, Tetrahedron, 2011, 67, 6233–6239 CrossRef CAS.
- G. Dintilhac, D. Arslan, S. Dilly, L. Danober, I. Botez, P. Lestage, B. Pirotte and P. De Tullio, MedChemComm, 2011, 2, 509–523 RSC.
- P. Kundu, A. Mondal and C. Chowdhury, J. Org. Chem., 2016, 81, 6596–6608 CrossRef CAS PubMed.
- A. Cherepakha, V. O. Kovtunenko, A. Tolmachev and O. Lukin, J. Heterocycl. Chem., 2013, 50, 1071–1077 CAS.
- X. Chen, C. Zhu, F. Guo, X. Qiu, Y. Yang, S. Zhang, M. He, S. Parveen, C. Jing, Y. Li and B. Ma, J. Med. Chem., 2010, 53, 8330–8344 CrossRef CAS PubMed.
- W. L. Matier and W. T. Comer, J. Org. Chem., 1974, 39, 3080–3084 CrossRef CAS.
- B. Pirotte, T. Podona, O. Diouf, P. de Tullio, P. Lebrun, L. Dupont, F. Somers, J. Delarge, P. Morain, P. Lestage, J. Lepagnol and M. Spedding, J. Med. Chem., 1998, 41, 2946–2959 CrossRef CAS PubMed.
- P. Francotte, P. de Tullio, E. Goffin, G. Dintilhac, E. Graindorge, P. Fraikin, P. Lestage, L. Danober, J.-Y. Thomas, D.-H. Caignard and B. Pirotte, J. Med. Chem., 2007, 50, 3153–3157 CrossRef CAS PubMed.
- Z. Yang, N. Chen and J. Xu, J. Org. Chem., 2015, 80, 3611–3620 CrossRef CAS PubMed.
- Q. Wu, Z. Yang and J. Xu, Org. Biomol. Chem., 2016, 14, 7258–7267 RSC.
- J. J. Krutak, R. D. Burpitt, W. H. Moore and J. A. Hyatt, J. Org. Chem., 1979, 44, 3847–3858 CrossRef CAS.
- P. K. Chinthakindi, K. B. Govender, A. S. Kumar, H. G. Kruger, T. Govender, T. Naicker and P. I. Arvidsson, Org. Lett., 2017, 19, 480–483 CrossRef CAS PubMed.
- P. K. Chinthakindi and P. I. Arvidsson, Eur. J. Org. Chem., 2018, 2018, 3648–3666 CrossRef CAS.
- A. D. Cat, R. V. Poucke and M. Verbrugghe, J. Org. Chem., 1965, 30, 1498–1502 CrossRef.
- D. E. Fahrney and A. M. Gold, J. Am. Chem. Soc., 1963, 85, 997–1000 CrossRef CAS.
- A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650–2659 RSC.
- H. Mukherjee, J. Debreczeni, J. Breed, S. Tentarelli, B. Aquila, J. Dowling, A. Whitty and N. P. Grimster, Org. Biomol. Chem., 2017, 15(45), 9685–9695 RSC.
- W. E. Truce and F. D. Hoerger, J. Am. Chem. Soc., 1954, 76, 3230–3232 CrossRef CAS.
- D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed.
- X. Chen, G.-F. Zha, G. A. L. Bare, J. Leng, S.-M. Wang and H.-L. Qin, Adv. Synth. Catal., 2017, 359, 3254–3260 CrossRef CAS.
- H. L. Qin, Q. Zheng, G. A. Bare, P. Wu and K. B. Sharpless, Angew. Chem., 2016, 128, 14361–14364 CrossRef.
- S. Ding and N. Jiao, Angew. Chem., Int. Ed., 2012, 51, 9226–9237 CrossRef CAS PubMed.
- X.-F. Wu and K. Natte, Adv. Synth. Catal., 2016, 358, 336–352 CrossRef CAS.
- Z.-K. Wen, X.-H. Liu, Y.-F. Liu and J.-B. Chao, Org. Lett., 2017, 19(21), 5798–5801 CrossRef CAS PubMed.
- J. Magolan, E. Jones-Mensah and M. Karki, Synthesis, 2016, 48, 1421–1436 CrossRef.
- S. N. Gockel and K. L. Hull, Org. Lett., 2015, 17, 3236–3239 CrossRef CAS PubMed.
- J. Ho, J. Zheng, R. Meana-Pañeda, D. G. Truhlar, E. J. Ko, G. P. Savage, C. M. Williams, M. L. Coote and J. Tsanaktsidis, J. Org. Chem., 2013, 78, 6677–6687 CrossRef CAS PubMed.
- Y. Liu, Y. Du and L. Wei, Curr. Org. Chem., 2016, 20, 1656–1658 CrossRef CAS.
- The Application of Dichloromethane and Chloroform as Reagents in Organic Synthesis, ed. A. Tlili and J. Schranck, 2018 Search PubMed.
- F. Lin, Q. Feng, X. Cui and Q. Song, RSC Adv., 2013, 3, 20246–20253 RSC.
- K. Tanaka and K. Ajiki, Org. Lett., 2005, 7, 1537–1539 CrossRef CAS PubMed.
- Y. Wang, P. Li, X. Liang, T. Y. Zhang and J. Ye, Chem. Commun., 2008, 1232–1234 RSC.
- S. H. Hansen and L. Nordholm, J. Chromatogr. A, 1981, 204, 97–101 CrossRef CAS.
- M. H. Abraham, P. L. Grellier, D. V. Prior, J. J. Morris and P. J. Taylor, J. Chem. Soc., Perkin Trans. 2, 1990, 521–529 RSC.
- J. E. Mills, C. A. Maryanoff, R. M. Cosgrove, L. Scott and D. F. McComsey, Org. Prep. Proced. Int., 1984, 16, 97–114 CrossRef CAS.
- A. B. Rudine, M. G. Walter and C. C. Wamser, J. Org. Chem., 2010, 75, 4292–4295 CrossRef CAS PubMed.
- Q. Liu, Y. Zhang, Z. Zhang, T. Liu, L. Shi and G. Zhang, RSC Adv., 2014, 4, 25933–25939 RSC.
- H. Cui, R. Goddard and K. R. Pörschke, J. Phys. Org. Chem., 2012, 25, 814–827 CrossRef CAS.
- S. J. Kyran, S. G. Sanchez, C. J. Arp and D. J. Darensbourg, Organometallics, 2015, 34, 3598–3602 CrossRef CAS.
- J. E. Mills, C. A. Maryanoff, D. F. McComsey, R. C. Stanzione and L. Scott, J. Org. Chem., 1987, 52, 1857–1859 CrossRef CAS.
- K. Matsumoto, Angew. Chem., Int. Ed., 1982, 21, 922 CrossRef.
- D. Yu and Y. Zhang, Adv. Synth. Catal., 2011, 353, 163–169 CrossRef CAS.
- E. Brasil, M. Samipillai, T. Govender, S. Baijnath and T. Naicker, Z. Kristallogr.–New Cryst. Struct., 2018 DOI:10.1515/ncrs-2018-0253 , CCDC 1862841†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1862841. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra07627h |
| This journal is © The Royal Society of Chemistry 2018 |