
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual-generation dendritic mesoporous silica nanoparticles for co-delivery and kinetically sequential drug release†
Yanxin Liuab,
Baolin Huangac,
Jiaoyang Zhuab,
Kailin Fenga,
Yuan Yuan *ab and
Changsheng Liu*ab
*ab and
Changsheng Liu*ab
aKey Laboratory for Ultrafine Materials of Ministry of Education, East China University of Science and Technology, Shanghai 200237, P. R. China. E-mail: yyuan@ecust.edu.cn; Fax: +86-21-54283420; Tel: +86-21-64251308
bState Key Laboratory of Bioreactor Engineering, East China University of Science and Technology, Shanghai 200237, P. R. China
cSchool of Life Sciences, Guangzhou University, Guangzhou 510006, P. R. China
First published on 5th December 2018
Abstract
Although multi-drug synergetic therapy is increasingly important in clinical application, sophisticated delivery systems with the ability to deliver multiple drugs and realize sequential release with independently tunable kinetics at different stages are highly desirable. In this study, a dual-generation mesoporous silica nanoparticle (DAMSN) with three-dimensional dendrimer-like structure as an adaptable dual drug delivery system is developed. The DAMSN was synthesized via a heterogeneous interfacial reaction and was of uniformly spherical morphology (150–170 nm) with dendritic structures and hierarchical pores (inner pore, 3.5 nm; outer pore, 8.3 nm). And the inner generation of DAMSN was modified with 3-aminopropyltriethoxysilane (APTMS). The IBU and BSA as model drugs were loaded into the inner generation via covalent conjugation and the outer generation by electrostatic adsorption, respectively. Intriguingly, DAMSN underwent a rapid bio-degradation for about 4 days, partly due to its center-radial dendritic channel structure. The release results showed that IBU was of a typical two-phase release profile with almost zero release in the first 12 h and more sustained release for the following 88 h, while BSA was sustained over a long period of 100 h. Notably, the release behaviors of both drugs can be independently tailored by changing the intrinsic properties of the DAMSN. In addition, DAMSN exhibited good bio-compatibility. These results indicated that the dual-generation, dendrimer-like MSN structure could spatiotemporally present different drugs to realize sequential drug release, and has potential use in the field of tissue engineering and regenerative medicine.
1 Introduction
With the fast development of science and technology, many attempts have been made to improve the well-being of life in the field of biomedical science.1,2 Currently, various advanced delivery systems are popularly developed to enhance the therapeutic effect of drugs/proteins.3–6 But, using only a single drug may often not be able to achieve effective therapeutic efficacy,7,8 especially for recalcitrant diseases that have developed drug resistances or have temporal progression through different phases. Moreover, some these formidable diseases often have a series of complex stages, which usually need different kinds of drugs to work synergistically.9 For these cases, combination therapies with spatiotemporally optimized multi-drug regimens can profoundly improve the biological effect.10 As reported, the sequential treatments of erlotinib prior to doxorubicin,11 siRNA followed by a small molecule,12 have shown remarkable improvements over simultaneous administration. Unfortunately, till now, it is still a great challenge to formulate a spatiotemporal treatment with controlled sequential release, as there is a significant overlap between their drug release kinetics, especially in the initial phase of release.13Currently, the concept of dual-drug co-delivery systems based on nanoparticle structures has attracted great attention.14,15 Diverse nano-scale materials have been designed to build co-delivery systems, such as polymeric micelles, porous scaffolds, and biodegradable nanogels.16–18 Among them, mesoporous silica nanoparticles (MSNs), with tunable particle size and pore structure, high specific surface area, and potential capability of modification, appear to be most attractive.19–21 Specifically, a kind of three-dimensional dendrimer-like MSNs (especially the multi-generation dendritic hierarchical structure), has been developed and gradually has attracted a lot of interest.22,23 The dendritic structure of MSNs could not only efficiently improve the degradation rate by increasing the surface area exposed to the dissolution media, but also be a potential route to co-drug delivery, where drugs of different sorts and sizes could be loaded simultaneously by controlling the pore size of the dendritic hierarchical structure. Inspired by the above, we hypothesize that these two- or multi-generation dendritic MSNs should be a promising nanocarrier for co-loading, co-delivery and combined therapy due to the favorable structural character of hierarchical pores. However, current studies mainly focus on improving the complex synthesis method of MSNs and exploring its applications in some areas like separation and catalysis. Till now, seldom attention has been paid to its application in drug delivery systems, especially dual/multi-delivery discovery.24,25
In this study, we aim to develop a novel kinetically, discrete co-delivery, dual-generation dendritic mesoporous silica nanoparticle (DAMSN) for co-delivery of a small molecule model drug IBU and a big molecule model drug BSA (as shown in Fig. 1A). To achieve well-defined release sequence of IBU and BSA, the pore size of the inner and outer generation were controlled about 3 nm and 8 nm, respectively. In order to avoid the interdiffusion of the drug loaded in the inner generation during the second generation self-assembly, we first modified the surface of the inner generation with –NH2 and hence loaded IBU via chemical graft; and then BSA was loaded into the second generation by physical adsorption. Under physiological environment, the release of the IBU in inner generation is initially suppressed, while the BSA in outer generation is continuously released from the initial stage, thus realize sequential release and dual-drug synergistic therapy (as shown in Fig. 1B). On the basis of the studies above, we found that the particle size of DAMSN could be conveniently increased (or decreased) by adjusting the reaction time and temperature of the second generation. As previously demonstrated, the location of the drug could affect its release order and kinetics; thus when changing the depth of the second generation, the release behavior of IBU and BSA could be furthermore affected. In this way, the sequential drug release of DAMSN could be realized at will more controllable.
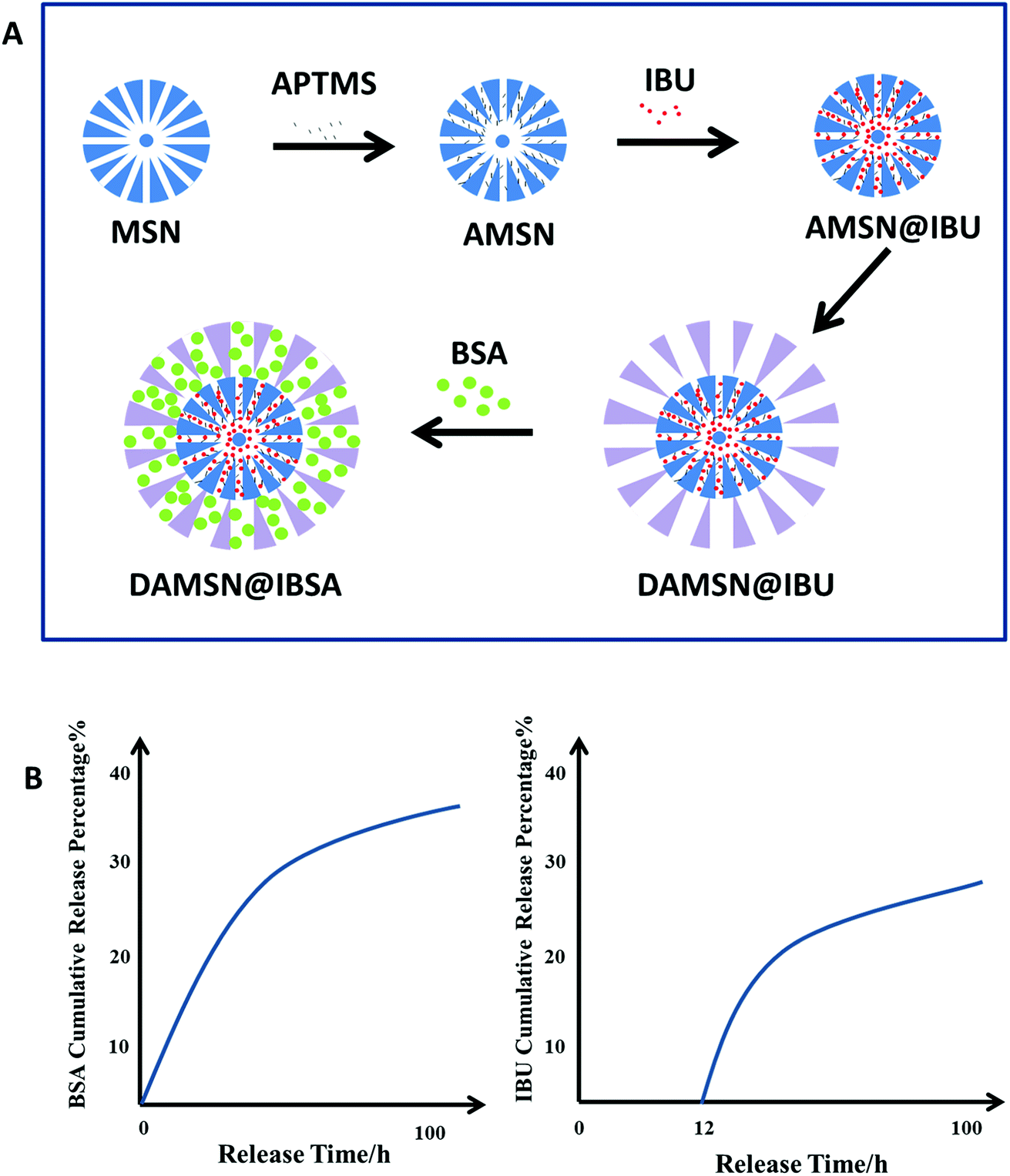 | ||
| Fig. 1 (A) Schematic synthesis of DAMSN@IBSA carrier. (a) Synthesis of single-generation mesoporous silica nanoparticle (MSN) and amino-modified MSN (AMSN). (b) IBU was loaded into AMSN (AMSN@IBU). (c) Synthesis of the dual-generation carrier (DAMSN@IBU). (d) BSA was loaded into DAMSN@IBU (DAMSN@IBSA). (B) The simulative release profiles of BSA and IBU reflected the effect of dual-generation and inner amino-modification. | ||
2 Materials and methods
2.1 Materials
Cetyltrimethylammonium chloride (CTAC), trimethylamine (TEA) and cyclohexane were purchased from Shanghai Lingfeng Chemical Reagent Co. Ltd. China. Tetraethylorthosilicate (TEOS) was obtained from Sinopharm Chemical Reagent Co. Ltd. China. 3-Aminopropyltriethoxysilane (APTMS) was purchased from J&K Scientific Co. Ltd. China. α-Methyl-4-(isobutyl)phenylacetic acid (IBU) was bought from Alfa Aesar Co. Ltd. China. N-Hydroxysuccinimide (NHS) and 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC) were obtained from Sigma Aldrich Chemicals Co. Ltd. China. Ammonium-nitrate (NH4NO3) was received from Xilong Scientific Co. Ltd. China. Decahydronaphthalene was purchased from Aladdin Co. Ltd. China. Dimethyl formamide (DMF) and dimethyl sulphoxide (DMSO) were bought from Shanghai Titan Scientific Co. Ltd. China. Bovine serum albumin (BSA) was obtained from Shanghai Yeasen Biotechnology Co. Ltd. China. 3-(4,5-Dimethyl-2-thiazolyl)-2,5-di-phenyl-2-tetrazolium bromide (MTT) was received from Life Technology. USA. The chemicals used were of analytical grade and without further purification.2.2 Synthesis of single-generation mesoporous silica nanoparticles (MSN)
MSN nanoparticles were synthesized via a heterogeneously interfacial reaction according to a previous report.19 Firstly, 24 mL of CTAC–H2O solution (25 wt%) was prepared in a 100 mL round bottom flask, then 0.18 g of TEA and 36 mL of deionized water were added. The mixture was gently stirred at 60 °C for 1 h to form micelles. After that, 20 mL of TEOS–decahydronaphthalene solution (20 vt%) was slowly dropped into the flask, and the reaction was kept under a continuous stirring of 230 rpm in a 60 °C water bath for 12 h. Subsequently, the aqueous phase was collected, centrifuged, washed, and calcined at 600 °C to remove the template agent. The final powder was obtained and named as MSN. In addition, to compare the pore sizes of different generations, another kind of single-generation nanoparticle named as MSN2 was also synthesized. The synthesis method of MSN2 was the same as that of MSN, but the only difference was that the upper organic phase was changed from TEOS–decahydronaphthalene solution to TEOS–cyclohexane solution.2.3 Amino-modified of MSN (AMSN)
0.7 g of MSN was dispersed in 70 mL of toluene, then 0.7 mL of APTMS was added rapidly. The reaction was kept under condensation reflux and should proceed for more than 9 h at 60 °C to ensure that the amino groups were well linked to MSN particles. After the reaction, the product was collected, centrifuged, washed with ethanol twice to remove the unreacted modifier, and dried at 80 °C. The obtained powder was named as AMSN. This step of modification was the basis of IBU loading, through which IBU (with reactive carboxyl groups) could covalently conjugate to the delivery by an amide bond.262.4 Synthesis of the second generation (DAMSN)
The second generation was designed to load another kind of drug (different from the inner generation), in order to realize co-delivery and dual-drug synergy. The synthesis process was similar to that of MSN. In this part, the AMSN obtained earlier were used as cores for the new nanoparticles (DAMSN) and the upper organic phase was changed from decahydronaphthalene to cyclohexane in order to formulate bigger pores. 20 mL of CTAC–H2O (20 wt%), 0.15 g of TEA and 36 mL of deionized water were fully mixed by stirring at 60 °C for 30 min. Then AMSN powder was added and dispersed, and the mixture was stirred for another 20 min. Subsequently, 50 mL of TEOS–cyclohexane solution (7 vt%) was slowly dropped into the flask, and the reaction was kept in a 60 °C water bath for 10 h. After the reaction was over, the nanoparticles were centrifuged, washed with ethanol, and extracted with ammonium-nitrate (NH4NO3)–ethanol solution (0.75 wt%) twice to remove the template. Finally the product was collected and dried at 37 °C, and the powder obtained was named as DAMSN. In addition, if the cores used were changed from AMSN to MSN, the product obtained was accordingly named as DMSN, and this kind of nanoparticles without inner modification was used as a control in the following experiments.The upper organic phase on the one hand provides a storage medium for TEOS, on the other hand assembles with the surfactant molecules to create emulsion micelles, which could further play a role of mesoscaled template to synthesize MSNs.27 In fact, besides decahydronaphthalene and cyclohexane, there are also some other reagents could be used in synthesizing mesoporous silica nanoparticles. For example, previous studies have reported using 1-octadecene as the upper organic phase to obtain MSNs with pore size of 2.8 nm.19 And dodecane was also be used to synthesize inorganic porous monoliths.28
2.5 Characterization
In this work, DAMSN prepared at 60 °C was mainly set as a representation to study the characteristics of this novel drug delivery. The morphologies and sizes of the samples were observed by field emission scanning electron microscope (FE-SEM, S4800, HITACHI, Japan) and transmission electron microscopy (TEM, JEM2100, HITACHI, Japan). The nanoparticles (MSN, AMSN, DMSN and DAMSN) were dispersed in ethanol (0.5 mg mL−1) and dropped onto a 400 mesh copper grid, air-dried, followed by field emission scanning electron microscope and transmission electron microscopy imaging. The mesoporous structure was determined by small angle X-ray diffraction (SAXRD, Rotating Anode X-ray Powder Diffractometer, Japan) and N2 adsorption/desorption measurement device (Tristar 3000, Micromeritics, USA). The samples no less than 100 mg were lyophilized and sent for test. And the specific surface area and pore sizes were examined by the Barrett–Joyner–Halenda (BET) measurement. The Fourier Transform Infrared (FTIR) spectra were obtained to analyze the chemical structure of surface by ATR (Nicolet 6700, Thermo Fisher, USA). The samples to be tested were mixed with potassium bromide powder, fully ground and then put into the infrared spectrometer to analyze. The surface potential of the nanoparticles were measured by Zeta potential analyzer (Nicomp 380 ZLS, PSS, USA). The weight loss was measured by thermal gravimetric analysis (TGA, STA409PC, NETZSCH, Germany). For the measurement of zeta potential, the material was dispersed into PBS at 1 mg mL−1 and then the pH was adjusted to 7.4. And as for the TGA test, the materials were put into the machine and programing heated at 20 °C min−1 in the nitrogen atmosphere. In vitro degradation of the material was measured by a weight loss test as follows. Firstly, a certain amount of material was weighed and dispersed in a polyethylene bottle at 1 mg/1 mL (material/PBS) at 37 °C. Then during the degradation, the material was taken out at a specific time, dried and weighed. The weight loss rate of the material was calculated according to the following formula:| Weight loss (%) = [(W0 − Wt)/W0] × 100% |
As for the following drug release experiment, the release behavior of IBU and BSA were measured by an enzyme-linked immune detector (SPECTRA max 384, Molecular Devices, USA).
2.6 Drug loading
In this study, the delivery system was designed to load two different kinds of drugs in the two generations respectively. The small molecule drug (IBU) was loaded in the inner generation by chemical graft and the big molecule drug (BSA) was loaded in the outer generation by physical absorption.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were added to activate the carboxyl groups so that the chemical grafting between IBU and AMSN could be much easier to build.30 After stirring at 40 °C for about 30 min, 200 mg of AMSN was added, and then continuously stirred for another 24 h. The product was then centrifuged, washed with ethanol, dried at 40 °C, and named as AMSN@IBU. To illustrate the chemical grafting effect, IBU was also loaded in MSN as a control, using the same method above. And in this part, besides ethanol, two other kinds of solvent (DMF and DMSO) were also chosen to explore a better drug loading effect.
:1) were added to activate the carboxyl groups so that the chemical grafting between IBU and AMSN could be much easier to build.30 After stirring at 40 °C for about 30 min, 200 mg of AMSN was added, and then continuously stirred for another 24 h. The product was then centrifuged, washed with ethanol, dried at 40 °C, and named as AMSN@IBU. To illustrate the chemical grafting effect, IBU was also loaded in MSN as a control, using the same method above. And in this part, besides ethanol, two other kinds of solvent (DMF and DMSO) were also chosen to explore a better drug loading effect.2.7 In vitro drug release
In this experiment, IBU and BSA were loaded into DAMSN and other nanoparticles of the synthesis process to investigate the in vitro release in the PBS buffer solution.2.8 Cytotoxicity assay
The cytotoxicity of synthesized materials (DMSN and DAMSN) was investigated by MTT assays. BMSC cells (bone marrow mesenchymal stem cells) were incubated in 24-well plates containing α-MEM culture media with a destiny of 1 × 104 cells per well at 37 °C under 5% CO2 for 24 h. After that, the cell media was removed and the cells were washed twice by PBS. Then 1 mL of the material samples at two different concentrations (100 μg mL−1 and 200 μg mL−1) was added into each well respectively. At the same time cells without materials were set as blank control groups. The plates containing cells were then cultured at the same condition as above for 1 day and 3 days, respectively. After that, 100 μL of MTT solution was added to each well, and the cells were continuously cultured for another 4 h. The media was carefully discarded and 1 mL of DMSO was added to each well followed by incubated for 10–15 min. Finally 200 μL of the solution in each well was transferred to a 96-well plate and the value of optical density (OD) was measured at 492 nm through a microplate reader. The cytotoxicity of the leachable was also been investigated and the test was almost the same as the above. DAMSN nanoparticles were dispersed in culture media at different concentrations from 10 μg mL−1 to 1000 μg mL−1 and immersed for 48 h. After that, these culture media samples were collected and added into the plates containing cells, followed by cultured for 48 h. After adding MTT solution and DMSO, the value of optical density (OD) was also measured at 492 nm through a microplate reader.3 Results and discussions
3.1 Characterization of the fabricated DAMSN nanoparticles
In this study, we aimed to synthesize DAMSN with dendritic structures and hierarchical pores, which could simultaneously load two model drugs IBU and BSA (as shown in Fig. 1A). For convenience and brevity, the nomenclatures of the nanoparticles were outlined in Table 1. The morphologies and characteristics of MSN, AMSN, DMSN and DAMSN were investigated by SEM and TEM, and shown in Fig. 2. It can be found from Fig. 2A–D that all the synthesized nanoparticles were of regular spherical shape with uniform sizes and diameters. These nanoparticles were all well dispersed and showed no obvious aggregation, which endowed the fabricated nanoparticles with the ability of close contact with guest drugs, and thus effectively improved the loading rate of drugs.| Nanoparticle architecture | Drug loaded | Abbreviation |
|---|---|---|
| Mesoporous silica nanoparticle | — | MSN |
| Amino-modified MSN | — | AMSN |
| Dual-generation MSN | — | DMSN |
| Inner amino-modified DMSN | — | DAMSN |
| MSN | IBU | MSN@IBU |
| AMSN | IBU | AMSN@IBU |
| DAMSN | IBU | DAMSN@IBU |
| DMSN | BSA | DMSN@BSA |
| DAMSN | BSA | DAMSN@BSA |
| DAMSN | IBU&BSA | DAMSN@IBSA |
 | ||
| Fig. 2 (A–D) SEM images of MSN (A), AMSN (B), DMSN (C) and DAMSN (D). Scale bars = 500 nm. (a–d) TEM images of MSN (a), AMSN (b), DMSN (c) and DAMSN (d). Scale bars = 50 nm. | ||
As revealed in the TEM pictures (as shown in Fig. 2a–d), both MSN and DMSN exhibited uniform spherical structures containing center-radial channels and complete pores. However, a little difference still existed that the particle size of MSN (50–60 nm) was slightly smaller than that of DMSN (90–100 nm). The reason for this difference should be that MSN was a single generation nanoparticle while DMSN was of double-generation. Meanwhile, as compared with MSN, AMSN showed no significant difference on the aspect of structures and sizes (as shown in Fig. 2b). But it can be observed that the dispersity of AMSN was somehow decreased. This phenomenon may be related to the strong electrostatic attraction of amino groups on the AMSN. Similarly, DAMSN (as shown in Fig. 2d) also showed a little agglomeration, but was still of regular spherical morphology. In addition, it can be observed that DAMSN exhibited obvious double-generation mesoporous structures with obviously increased particle size (150–170 nm). It is worthwhile to mention that the sizes of MSN, AMSN, DMSN and DAMSN all corresponded well with the data observed by SEM.
To further investigate the influence of the intrinsic properties of DAMSN on drug release behavior, a series of DAMSNs were also synthesized by controlling the reaction time and temperature of the second generation. Besides the kind of DAMSN mentioned above and investigated through the whole study (as shown in Fig. 2D and d), the other three kinds of nanoparticles (DAMSN ∼45 °C 4 h, DAMSN ∼50 °C 6 h and DAMSN ∼65 °C 15 h) were also synthesized here, of which the reaction temperatures were 45 °C, 50 °C and 65 °C respectively and the reaction times were correspondingly increased (4 h, 6 h and 15 h). As revealed in Fig. 3, it could be easily found that the DAMSNs were of uniformly spherical structures which were just the same as that in Fig. 2D(d), and the other morphologies and characteristics were also similar to the kind of DAMSN (60 °C, 10 h). However, with the increase of temperature and reaction time, the particle sizes were obviously increased from 65 nm to 200 nm. As envisaged, when the second generation was synthesized at a higher temperature and sustained for a longer time, the self-assembly reaction could be finished more completely, through which the nanoparticles could further grow and the particle size was accordingly increased. And by adjusting this characteristic (for example increasing the particle size), the location of the loaded drugs could be also adjusted; thus the release speed of the outer loaded drug should be slowed and the release beginning of the inner loaded drug should be postponed too. In this way, we speculated that the co-release behavior of DAMSN could be further controlled at will, which was proved by the following drug release experiments.
 | ||
| Fig. 3 (A–C) SEM images of DAMSNs of different reaction conditions ∼45 °C 4 h (A), ∼50 °C 6 h (B) and 65 °C 15 h (C). Scale bars = 100 nm. (a–c) TEM images of DAMSNs of different reaction conditions. Scale bars = 20 nm. | ||
The structure of MSN, AMSN and DAMSN were preliminarily explored by small angle X-ray diffraction (SAXRD). As revealed in Fig. 4A, MSN and AMSN both exhibited obvious diffraction peak at 1.11°, which suggested the same channel structure with uniform mesoporous. And after the amino-modification, the regularity of mesoporous was a little weakened (which was also proved by N2 sorption and desorption results below), so that the diffraction peak of AMSN was accordingly a bit weaker than that of MSN. Furthermore, it could be found that DAMSN had a diffraction peak at 0.93°, which was not observed in the curves of MSN and AMSN. This result proved that the mesostructure of DAMSN was a little different from the other two, also indicating that the second generation was synthesized successfully.
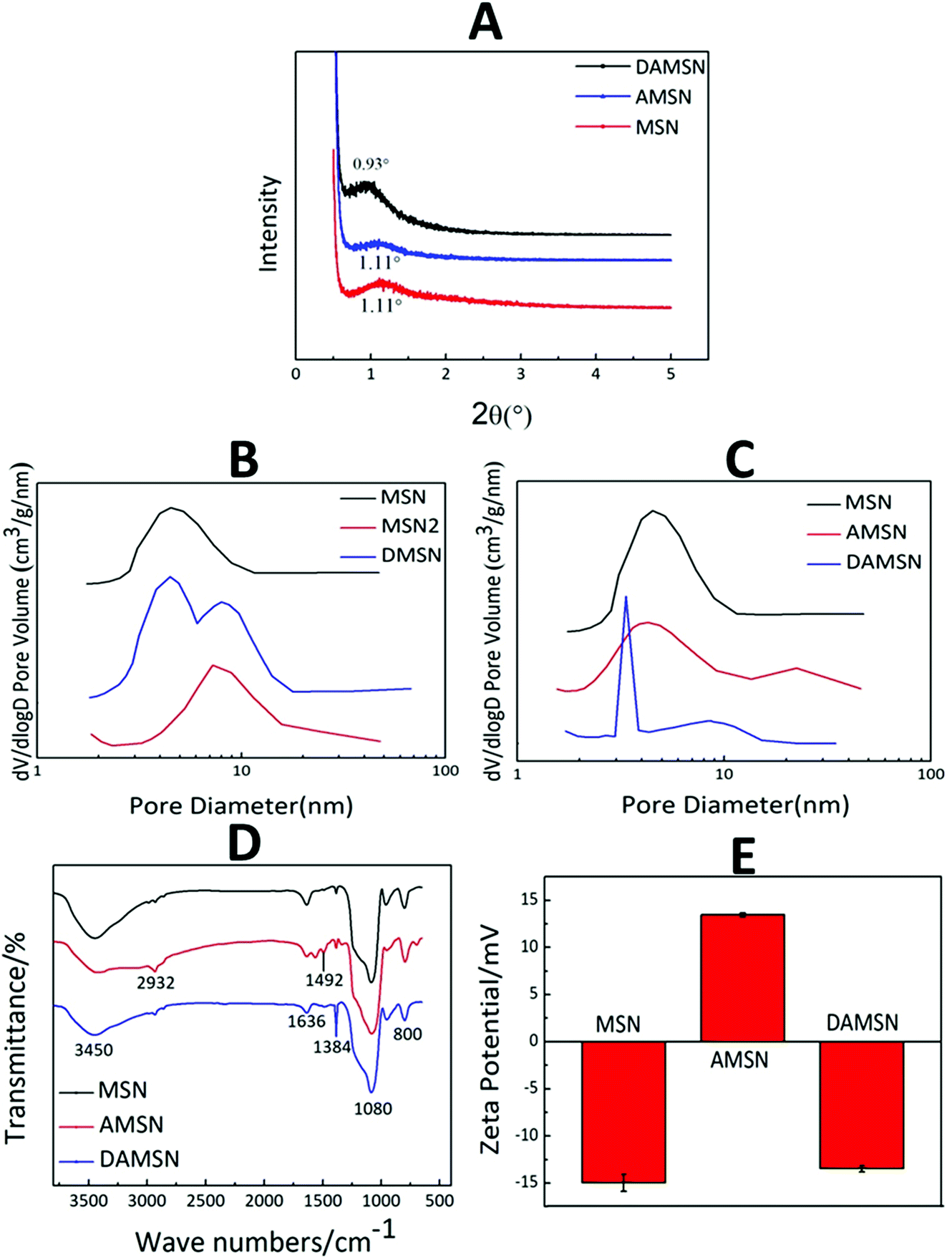 | ||
| Fig. 4 (A) The SAXRD patterns of MSN, AMSN and DAMSN. (B and C) N2 sorption and desorption results of MSN, MSN2, DMSN, AMSN and DAMSN. (D) FTIR spectra and (E) zeta potentials of MSN, AMSN and DAMSN. | ||
The mesoporous structures of all the MSNs were also measured by N2 sorption and desorption (as shown in Fig. 4B, C and Table 2). According to the self-assembly mechanism, the upper organic phase can expand the volume of the micelle and thus adjust the pore size of the synthesized nanoparticles. It could be found that the organic phase for synthesizing MSN2 was the same as that for synthesizing the second generation of DAMSN and DMSN, therefore MSN2 represented the big pore structure and MSN represented the small pore structure. As revealed in Fig. 4B, the obtained MSN exhibited a pore diameter of 4.5 nm, while MSN2 showed a pore diameter of 8 nm. By replacing the upper organic phase, DMSN with two pore sizes were synthesized, and the diameters of the inner and the outer generation were almost the same as that of MSN and MSN2, respectively. This result indicated that the two-generation structure could be successfully synthesized, and the sizes of pores did relate to the varieties of organic phase. It is worthwhile to note that the inner pore size of DAMSN (3.5 nm) was smaller than that of AMSN (4.1 nm) (as shown in Table 2). This phenomenon should be related to the re-hydrolyzation and re-polymerization of the remained APTMS in the pores of AMSN, which could accumulate in the pores and resulted in a reduction of the inner pore size. Furthermore, it can be found that the specific surface area of AMSN (253.1 m2 g−1) was smaller than that of MSN (640 m2 g−1) (as shown in Table 2) and the pore diameter of AMSN was also slightly smaller than that of MSN (as shown in Fig. 4C), demonstrating that the amino-modification did have great impacts on the structure of MSN. Combining the analysis above, it can be inferred that some excess APTMS molecules might exist in the pores of AMSN and thus also decreased the pore diameter and specific surface area. As a result of the four measurements above, it is evident that DAMSN was of regular spherical morphology with dual-generation and did possess dendritic channel structures and hierarchical pores (inner pore, 3.5 nm; outer pore, 8.3 nm), which could simultaneously load various sized therapeutic drugs. Inspired by characteristics above, these dendrimer-like silica nanoparticles should be a potential carrier.
| Material | Pore diameter/nm | Specific surface area/m2 g−1 |
|---|---|---|
| MSN | 4.5 | 640.0 |
| AMSN | 4.1 | 253.1 |
| MSN2 | 8.0 | 727.7 |
| DMSN | 4.5/8.1 | 529.5 |
| DAMSN | 3.5/8.3 | 307.5 |
As mentioned above, in order to construct chemical graft with the guest drug, the inner generation of DAMSN was modified with APTMS to introduce amino groups. The successful amino-modification was confirmed by FTIR spectra (as shown in Fig. 4D). As shown, all samples (MSN, AMSN and DAMSN) exhibited obvious absorption peaks at 1080 cm−1 and 800 cm−1, which were assigned to the Si–O asymmetric stretching vibration and symmetrical stretching vibration, respectively. The absorption peaks at 2932 cm−1 and 1492 cm−1 corresponded to the stretching vibration peak and flexural vibration peak of C–C bond were observed on the amino-modified AMSN and DAMSN. However, there were no C–C bond absorption peaks in the FTIR spectra of MSN. The appearance of C–C bond peak in AMSN proved that MSN was successfully modified with the silane coupling agent (APTMS). And as for DAMSN, the density of C–C peaks was slightly weakened.
The surface potentials of MSN, AMSN and DAMSN were also investigated. As shown in Fig. 4E, the surface of MSN, due to the presence of large amounts of silanol groups (Si–OH), showed negative potential. Interestingly, after the amino-modification (AMSN), a negative–positive charge reversal occurred, proving the presence of amino groups on the surface of AMSN. However, after the self-assembly of the second generation, the surface potential of DAMSN with a large number of silanol groups became negative again. In summary, these results demonstrated that the amino-modification was achieved upon MSN. And in this way, the inner-modified generation could further generate covalent conjugations with the guest drug (IBU), which could on the one hand enhance the spatial distribution; on the other hand postpone the inner drug (IBU) release during the therapy.
It is known to all that an ideal degradation performance of the designed dual-generation carrier is necessary in clinical application. The amounts of material mass loss and Si ion released were both measured (as shown in Fig. 5A and B) to investigate degradation behavior. It could be found that the mass loss of DAMSN in buffer solution was slightly slower than that of DMSN. As revealed in Fig. 5A, DAMSN and DMSN both reached the maximum value of mass loss at about 4 days and after that the weights of these two materials remained unchanged. However, there was still Si ion released after 4 days (as shown in Fig. 5B), indicating that the collapse of the structure mainly concentrated on this stage and the degradation still continued thereafter without mass loss. These results demonstrated that the DAMSN and DMSN with dendritic structure, compared with the traditional MSNs, possessed a fast degradation performance. Studies have also suggested that with the degradation of mesoporous silica materials, the concentration of Si ion would increase and the main existing form of Si ion was Si(OH)4.32 Researches have also suggested that Si ion dissolved from bioactive materials could contribute to cell proliferation and promote osteogenic differentiation through activating osteogenic gene expression.33 Based on our analysis above, an EDS experiment was also taken in this study as a complement. With the degradation of outer generation, Si ion gradually released into PBS, therefore the content ratio of it would be decreased. As revealed in Fig. 5C, after degradation for 3 days, the content ratio of Si element was smaller than that of 0 day and the content of N element was increased. Furthermore, with the degradation going on, the inner generation was also destroyed and the modified amino group would also release into the solution along with the Si element, so the content ratio of elements would decrease again. These results once more confirmed the degradation process of DAMSN.
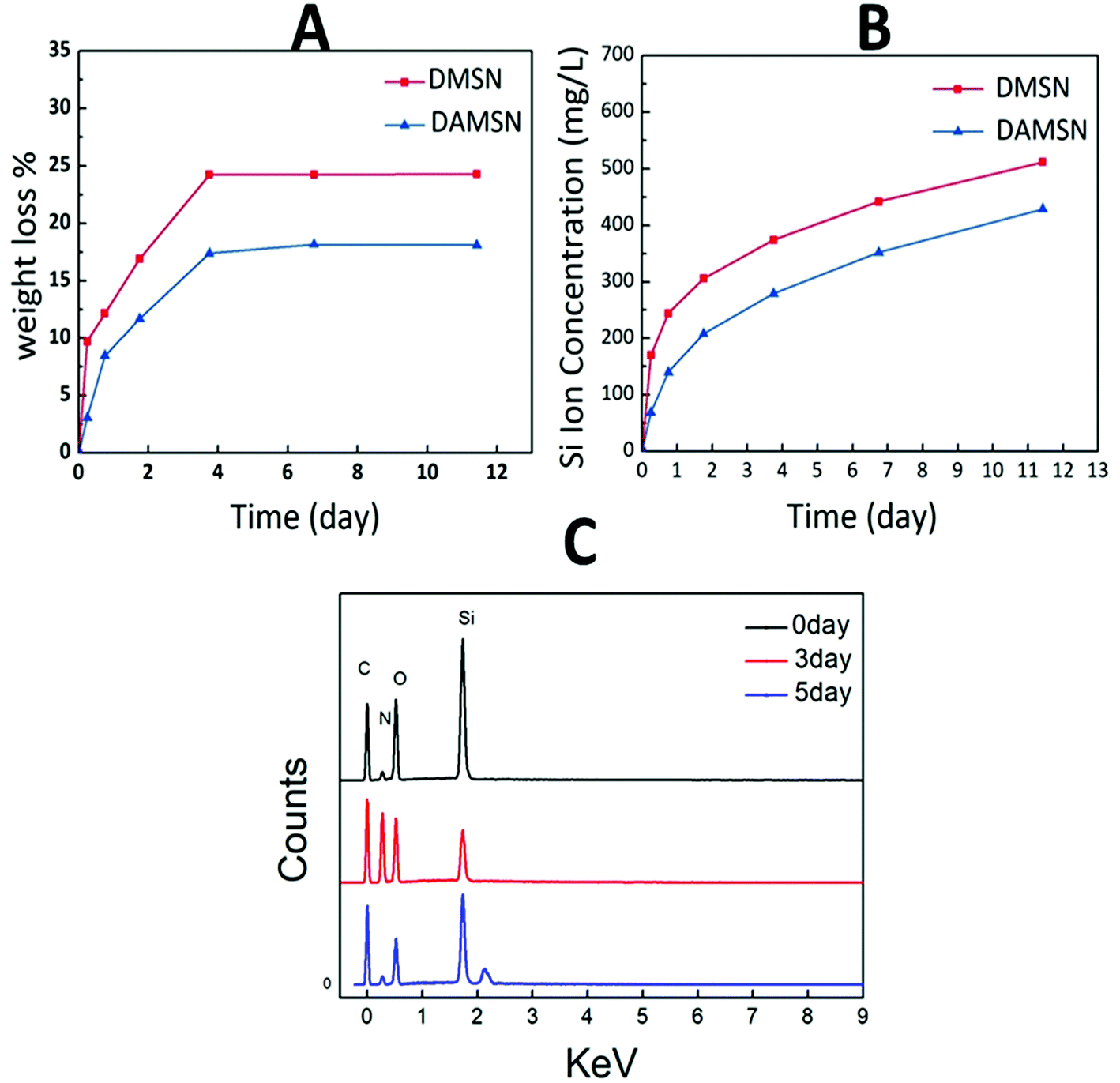 | ||
| Fig. 5 (A) Weight loss and (B) Si ion release profile of DMSN and DAMSN in PBS.·(C) The EDS patterns of DAMSN during degradation. | ||
3.2 Drug loading and release
In this study, we posited that controlling interdiffusion during the second-generation assembly process would allow us to embed different drugs in different regions at will. Furthermore, the location of the drug and loading method would govern its release order and kinetics. Herein the small IBU and big BSA were chosen as models to investigate the co-loading and release ability of this designed carrier. The molecular size of IBU was 0.6–1 nm which was suitable for placement in MSNs, and furthermore it contained carboxyl groups which could react with amino groups. BSA was one of the most frequently used proteins in the studies of macromolecular drug transportation and the molecular diameter of BSA was about 3.8 nm.34 To achieve such purpose above, IBU was introduced into the inner generation via chemical graft; and BSA was loaded in the outer generation by physical adsorption. Firstly we compared the FTIR spectra of AMSN and AMSN@IBU (as shown in Fig. 6A) to prove the successful conjugation of IBU and AMSN. Except for some absorption peaks mentioned in Fig. 4D (Si–O bond and C–C bond), some new absorption peaks also appeared. Specifically, AMSN and AMSN@IBU both exhibited an absorption peak around 1652 cm−1, which was assigned to the carbon–nitrogen bond (C–N). Moreover, the intensity of this peak increased with the loading of IBU. Furthermore, it was found that an absorption peak at 1724 cm−1 was appeared on AMSN@IBU, which was corresponded to the carbonyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of IBU. These results confirmed that IBU could be successfully introduced into the carrier by the reaction between the carboxyl groups of IBU and the amino groups of AMSN. As revealed in Fig. 6C, IBU was introduced into the inner generation of DAMSN by constructing covalent conjugation. The carboxyl groups (–COOH) of IBU would react with the amino groups of DAMSN through amidation reaction. The obtained amide bonds between loaded drugs and delivery would be much stronger than the physical adsorption of usual ways.35 In this way, IBU could be effectively reserved and protected from diffusion, thus keeping its location in the spatiotemporal administration.
O) of IBU. These results confirmed that IBU could be successfully introduced into the carrier by the reaction between the carboxyl groups of IBU and the amino groups of AMSN. As revealed in Fig. 6C, IBU was introduced into the inner generation of DAMSN by constructing covalent conjugation. The carboxyl groups (–COOH) of IBU would react with the amino groups of DAMSN through amidation reaction. The obtained amide bonds between loaded drugs and delivery would be much stronger than the physical adsorption of usual ways.35 In this way, IBU could be effectively reserved and protected from diffusion, thus keeping its location in the spatiotemporal administration.
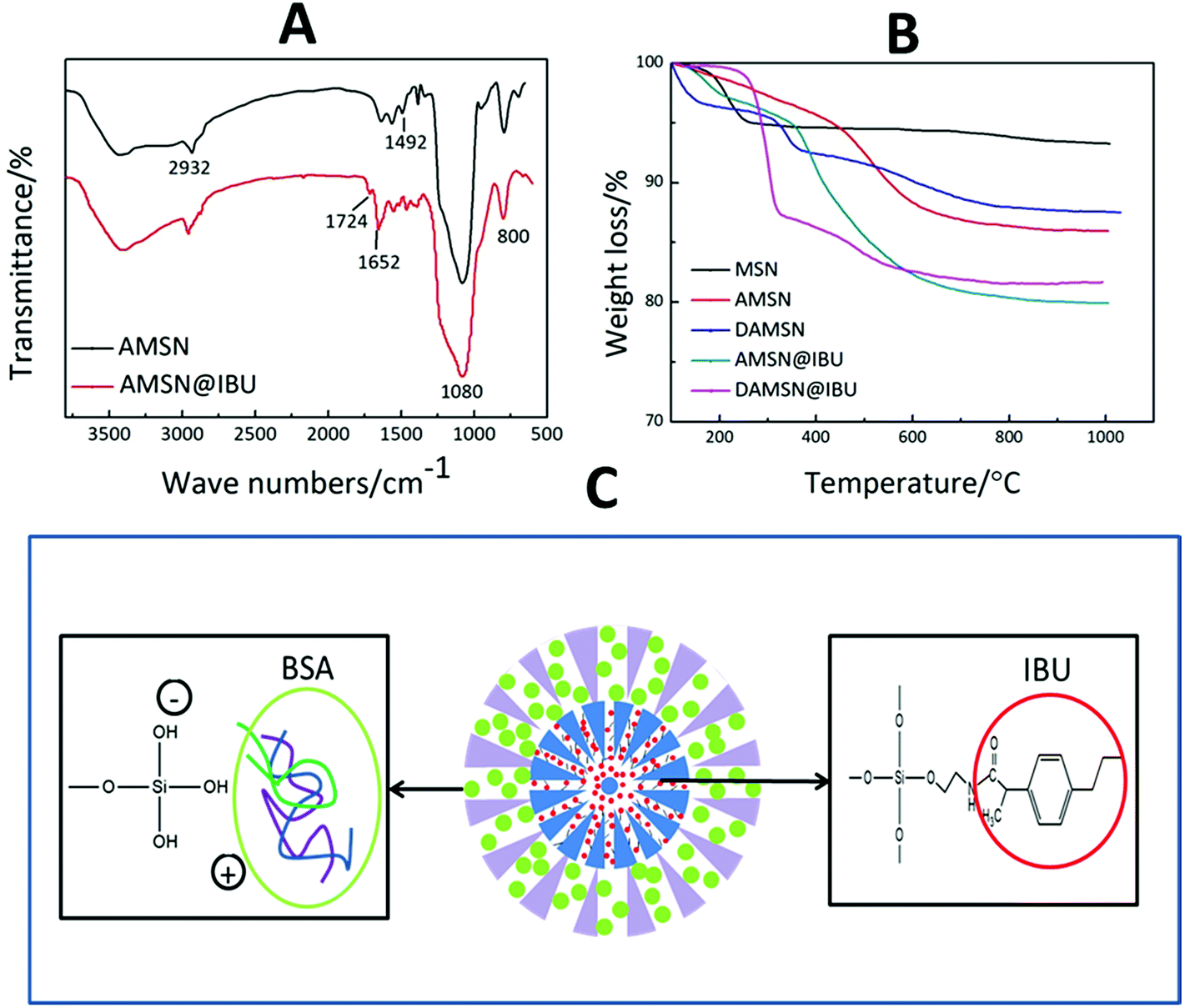 | ||
| Fig. 6 (A) FTIR spectra of AMSN and AMSN@IBU. (B) Thermal gravimetric analysis of MSN, AMSN, DAMSN, AMSN@IBU and DAMSN@IBU. (C) Schematic interaction of loaded drugs and delivery. | ||
Based on our analysis above, TGA was also taken in this study as a complement. Silica-based material demonstrates excellent heat resistant property, so that only organics could be destroyed with the increasing of temperature. As revealed in Fig. 6B, excluding the influence of small amount of water, the weight loss of AMSN was higher than that of MSN without modification, which had once again proved that the amino-modification was successful. Moreover, it could be found that the weight loss of AMSN@IBU and DAMSN@IBU at 1000 °C was 19.94% and 18.34% respectively. By calculating, the IBU loading rates of AMSN and DAMSN could be inferred to be around 23.71% and 22.58%, which were almost the same as the results obtained in the drug loading experiments (as shown in Table 3). Therefore IBU was once more confirmed to be successfully introduced into this kind of drug delivery.
| Material | Solvent | Drug loading rate/wt% |
|---|---|---|
| AMSN | Alcohol | 11.4 |
| AMSN | DMF | 19.4 |
| AMSN | DMSO | 25.1 |
| MSN | DMSO | 14.3 |
| DAMSN | DMSO | 20.1 |
Secondly, we further studied three different organic solvents (ethanol, DMF and DMSO) to choose a better one for IBU loading, and the loading capacities were shown in Table 3. When DMSO was used, the loading rate was obviously increased, which may be related to its better dissolving ability of IBU. In experiment, we found that the nanoparticles and IBU all dispersed more completely in DMSO, thus making sure an easier and completer reaction between drugs and carriers truly happened. As compared to the physical absorption, this kind of chemical graft could lead to stronger connections between drugs and carriers. This recapitulation could also be proved in Table 3, where the IBU loading rate (in DMSO) was increased by about 1.75 times after amino-modification (from MSN to AMSN). As previously demonstrated, with the chemical graft, IBU could avoid diffusion during the synthesis of the second generation. Through our further study, the IBU loading capacity of dual-generation nanoparticle (DAMSN) exhibited a little decrease than that of single-generation (AMSN). Because there was still some physical adsorption existing, IBU would somehow diffuse when the outer generation was synthesized (just like drug release).
Different from IBU, the macromolecular protein BSA was loaded after synthesis of the second generation. To meet the spatiotemporal administration needs, BSA was loaded in the outer generation without amino-modification. As revealed in Fig. 6C, BSA introduced into DAMSN mainly depended on physical adsorption. The positive groups of the amino acid of BSA could generate electrostatic adsorption with the negative hydroxyl groups of DAMSN. Previous studies suggested that the van der Waals force and hydrogen bonds may also exist in this process. Large pore size and big specific surface area of this kind of mesoporous delivery could somehow reduce the transportation resistance of BSA and promote protein adsorption capacity.36 For an understanding of the inner-modification impact on the outer loading, DMSN and DAMSN were used here. The BSA loading rates in DMSN and DAMSN were 29.5 and 25.8 wt% respectively, which suggested that the inner amino-modification had little effect on BSA loading in outer pores. Furthermore, the co-loading of these two therapeutic drugs was successfully achieved and the spatial distribution was also built efficiently.
Next we investigated the single-drug release behavior, including BSA and IBU. As revealed in Fig. 7A, the BSA release profile from DAMSN was similar to that from DMSN, which indicated that the inner-modification had little effect on the release of outer BSA. It could be further found that BSA was quickly released from the very beginning, which behaved as a pioneer in the dual-drug combination therapy. As for another drug (IBU), its release profiles from these three materials (MSN@IBU, AMSN@IBU and DAMSN@IBU) in PBS (pH = 7.4) were shown in Fig. 7B. It can be observed that in MSN, the release amount had reached more than 70% in the initial 10 h. With the amino-modification (AMSN), IBU released only 25% at the first 10 h, and the later release was also very slow. It is desirable to find that the chemical graft could not only restrict the diffusion of loaded-drug during synthesis of the second generation but also have significant improvement on restraining the burst release. According to the original plan that sequential release behavior was designed for, the release of BSA should be started immediately and IBU be delayed. To this end, we combined the dual-generation structure and inner chemical graft together to build the spatiotemporal administration and further achieve controlled release. It could be found that the IBU release from dual-generation nanoparticles (DAMSN) was obviously slower than that from the single-generation one, which should be attributed to the barrier effect of the dual-generation structures. Most importantly, the initial release of IBU did be limited, which started after 12 h.
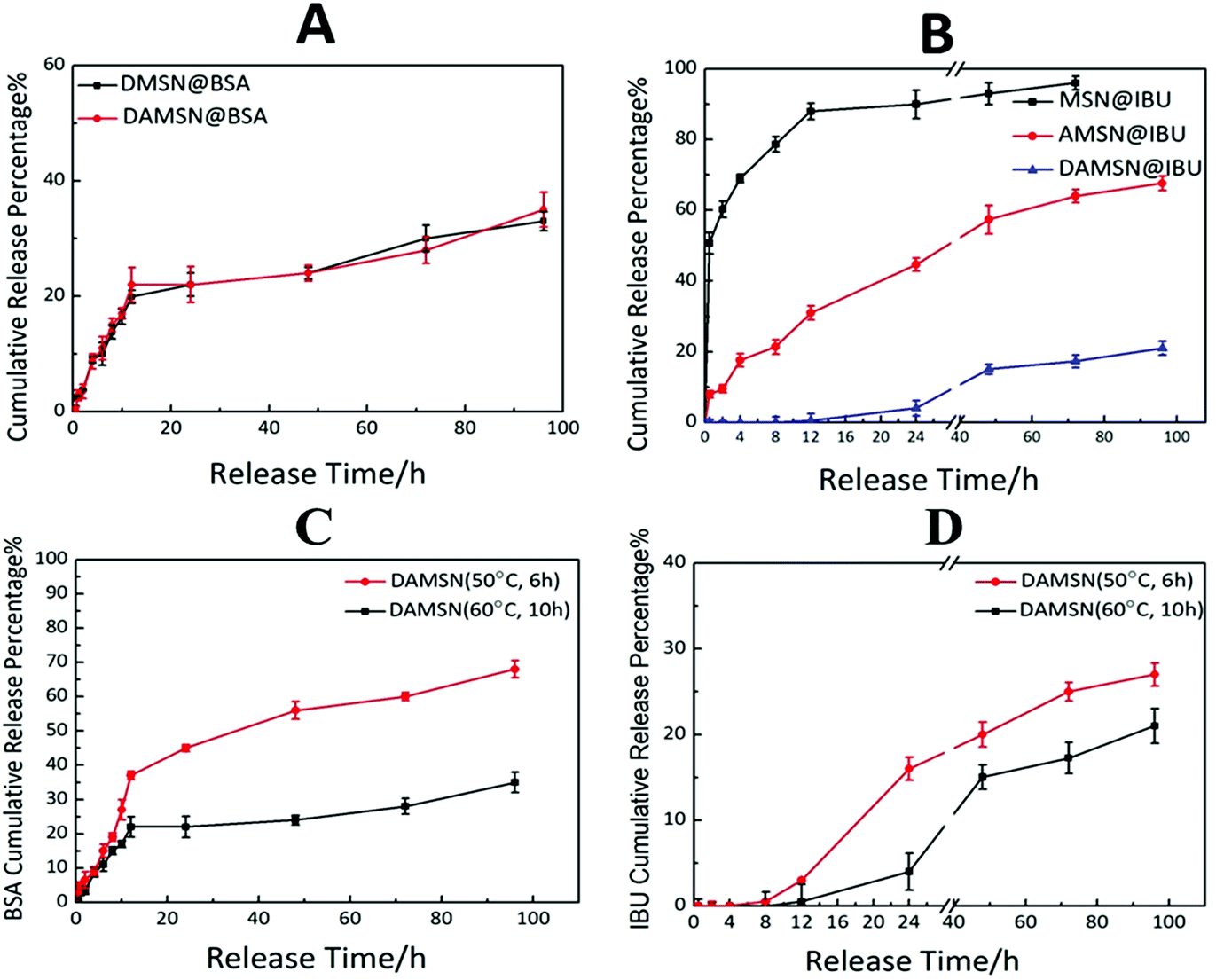 | ||
| Fig. 7 (A) The release profile of BSA from DMSN@BSA and DAMSN@BSA in PBS solution. (B) The release profile of IBU from MSN@IBU, AMSN@IBU and DAMSN@IBU in PBS solution. (C) The release profile of BSA from DAMSN@IBSA (50 °C, 6 h) and DAMSN@IBSA (60 °C, 10 h) in PBS solution. (D) The release profile of IBU from DAMSN@IBSA (50 °C, 6 h) and DAMSN@IBSA (60 °C, 10 h) in PBS solution. | ||
Finally the co-release of BSA and IBU was investigated. While on the basis of the previous confirmation that single-drug release was controlled, we hypothesized that by kinetically freezing the diffusion of drugs in inner-generation during the second-generation assembly via chemical graft, we would be able to govern the sequence of their release. As expected, DAMSN exhibited significant differences between the release of IBU and BSA (as shown in Fig. 8A). The release of BSA was very quick in the initial stage and sustained for 100 h, while IBU exhibited a typical two-phase release with almost zero release in the first 12 h and more sustained release for the following 88 h. Based on our analysis thus far, dual-therapeutic release kinetics without simultaneous release in the initial phase was successfully achieved, depending on the spatiotemporal distribution constructed by the dual-generation structure and chemical graft between IBU and inner pores. Overall it is evident that drugs in outer generation was firstly released while in inner generation was initially suppressed and later released (as shown in Fig. 8B), suggesting this designed DAMSN-based co-delivery system could realize an efficient sequential drug release to promote the long-term therapeutic effect.
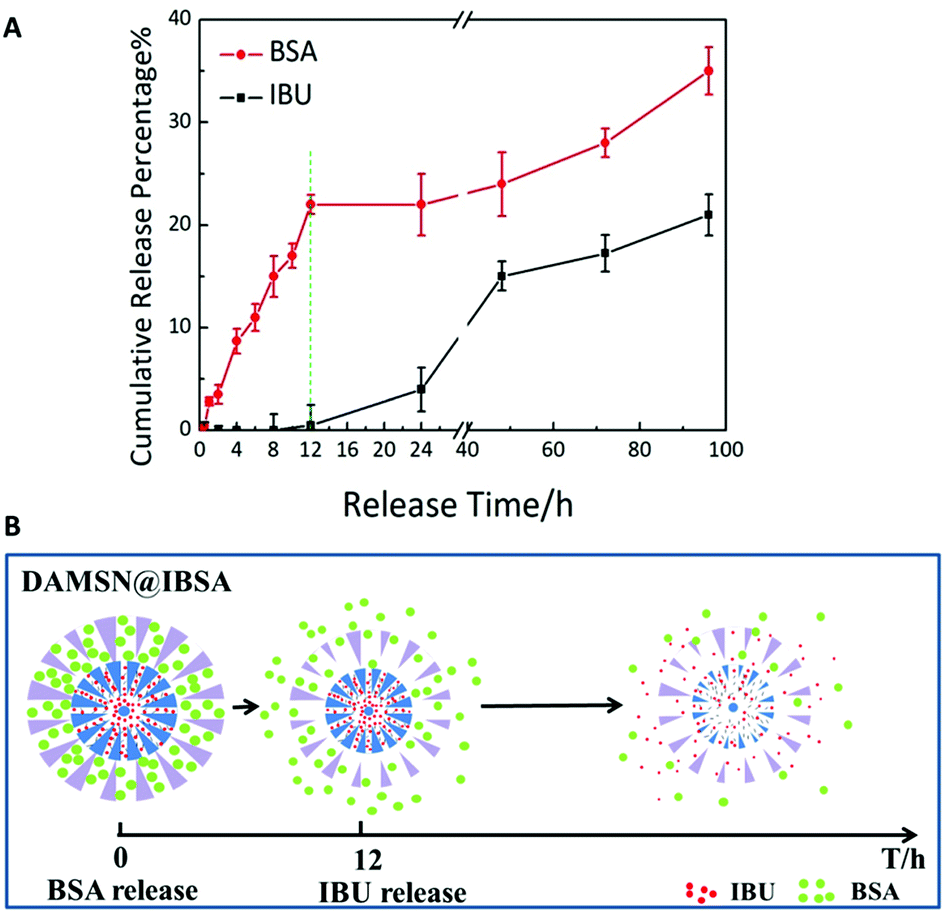 | ||
| Fig. 8 (A) The sequential release profile of IBU and BSA from DAMSN@IBSA in PBS solution. (B) Schematic release of DAMSN@IBSA and the sequential release behavior of IBU and BSA. | ||
On the basis of the studies above, we further investigated the influence of the intrinsic properties (particle size) of DAMSN on drug release behavior. Two kinds of DAMSNs of different reaction conditions were chosen to compare their drug release behavior, the particle sizes of which were around 90 nm and 160 nm (as shown in Fig. 3B and 2D), respectively. It was found in Fig. 7C and D that the small size nanoparticle (50 °C, 6 h) also exhibited good controlled sequential drug release behavior of IBU and BSA. BSA was firstly released in the initial stage and after about 8 h the release of IBU was soon started, of which the co-release tendency was similar to that with bigger particle size (60 °C, 10 h). However, along with the reduce of the depth of the second generation, BSA from DAMSN (50 °C, 6 h) was released much more quickly, which had already reached 50% in the first 40 h and the release of IBU was also started earlier (at the first 8 h). It could be inferred that when the growth of second generation was decreased by changing the reaction condition, the particle size of nanoparticle was accordingly reduced and its ability of drug loading was also lessened, meanwhile the drugs were also more easily to release. With the adjusting of the particle size, the release behavior of BSA and IBU were both tuned. In this way, we could further control the release behavior of DAMSNs by setting different reaction conditions to realize more tunable application.
3.3 In vitro biocompatibility assays
Good bio-compatibility is a significant prerequisite for a potential drug carrier, so the MTT assays were adopted in this study to evaluate the cytotoxicity of DAMSN. As revealed in Fig. 9A, there was no statistical difference between DAMSN and DMSN, which may due to their similar composition and structure. Moreover it also suggested that the amino-modification had little impact on the cytotoxicity of this material. Furthermore, the cell viability at different concentrations of designed nanoparticles was also measured. No matter the concentration was 0.1 mg mL−1 or 0.2 mg mL−1, the data results both showed no obvious cytotoxicity, as compared to control groups. It is evident that DAMSN truly had no cytotoxicity to BMSC cells at the test concentration, thus could be a potential choice for clinical application. Furthermore the cytotoxicity of the leachable was also been investigated, because the leachable could somehow contain the degradation product of DAMSN and reflect information of the metabolic process of this kind of delivery. As can be seen from Fig. 9B, the cell viabilities were entirely over 80% at all designed concentration and in a range of lower concentrations, DAMSN showed much obvious biocompatibility. Moreover, with the increasing of concentration (from 10 μg mL−1 to 200 μg mL−1), the cell proliferation was promoted, which may be contributed to the effect of Si ion. Previous studies have reported that the leachable of some bioactive materials containing Si ion could activate certain genes in osteoblasts that are associated with new bone formation.37,38 Combined with the above results, DAMSN also revealed good biocompatibility and truly promoted cell proliferation.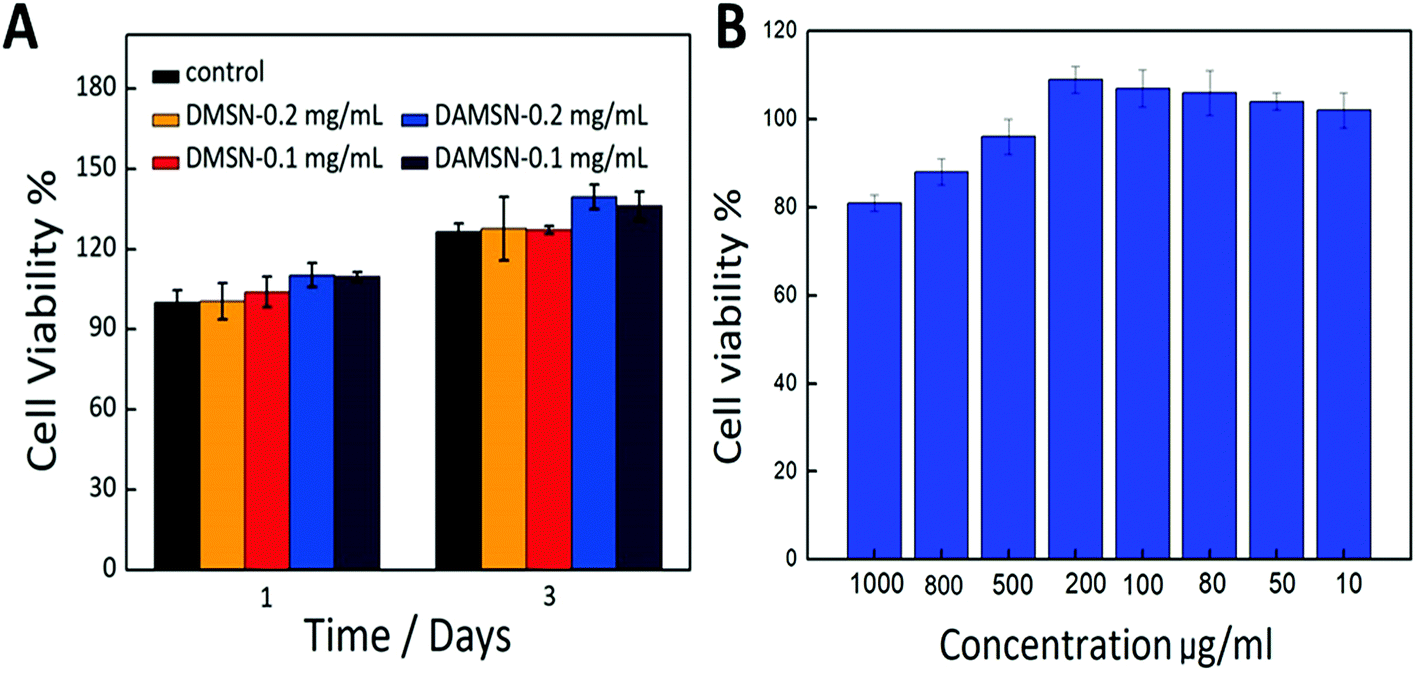 | ||
| Fig. 9 (A) The in vitro cytotoxicity of DAMSN and DMSN at concentrations of 0.1 mg mL−1 and 0.2 mg mL−1 on BMSC cells by the MTT assay. (B) The in vitro cytotoxicity of the leachable of DAMSN at different concentrations on BMSC cells by MTT assay. | ||
To further investigate the potential application of this material (DAMSN) in the field of bone regeneration, an ALP (alkaline phosphatase) activity experiment was also carried out. As revealed in the ESI (as shown in Fig. S-1†), after co-cultured with BMSC cells for 7 days and 14 days respectively, DAMSN and DMSN both showed obvious osteogenic capacities. And it could be observed that DAMSN had a stronger effect on accelerating the ALP expression during the differentiation of the BMSCs. Furthermore, co-culturing with the material for 14 days had more obvious effect on promoting bone formation than 7 days. In addition, it could also be found that the concentrations of the material had little effect on the ALP activity. These results demonstrated that this fabricated drug carrier could also be applied in bone reconstruction field.
4 Conclusion
In this study, we have designed a kind of DAMSN-based co-delivery system to realize kinetically controlled sequential drug release. The DAMSN was synthesized via a heterogeneously interfacial reaction and further modified with APTMS. IBU was loaded in the inner generation by chemical graft and BSA was loaded in the outer generation by physical adsorption. DAMSN exhibited uniformly spherical morphology, center-radial dendritic channels, complete hierarchical pore structures, good biocompatibility, and desirable biodegradation rate. Most importantly, this carrier could realize controlled sequential drug release. The release of BSA was sustained over a long period while IBU was of typical two-phase release with almost zero release in initial stage and more sustained release for the following time. And by adjusting the reaction conditions, the intrinsic properties of DAMSN could be easily tuned to realize a more controlled drug release at will. In summary, the designed DAMSN is a potential material for co-delivery and sequential drug release.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors wish to express their gratitude to the financial supports from National Natural Science Foundation of China (No. 31330028, No. 31470924), the National Natural Science Foundation of China for Innovative Research Groups (No. 51621002), Leading talents in Shanghai in 2017, 111 Project (B14018) and the Fundamental Research Funds for the Central Universities (222201718002).References
- H. Feng, D. Chu, Z. Li, Z. Guo, L. Jin, B. Fan, J. Zhang and J. Li, RSC Adv., 2018, 8, 25949–25954 RSC.
- G. Choi, T. H. Kim, J. M. Oh and J. H. Choy, Coord. Chem. Rev., 2018, 359, 32–51 CrossRef CAS.
- M. Liong, J. Lu, M. Kovochich, T. Xia, S. G. Ruehm, A. E. Nel, F. Tamanoi and J. I. Zink, ACS Nano, 2008, 2, 889–896 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed.
- J. Ji, L. Li, J. Chen and X. Yi, Polym. Bull., 2012, 68, 1721 CrossRef CAS.
- S. Samanta, L. Pradhan and D. Bahadur, New J. Chem., 2018, 42, 2804–2814 RSC.
- J. Tao, Z. Tan, L. Diao, Z. Ji, J. Zhu, W. Chen and Y. Hu, RSC Adv., 2018, 8, 21735–21744 RSC.
- Y. Lin, X. He, D. Zhou, L. Li, J. Sun and X. Jiang, RSC Adv., 2018, 8, 23768–23779 RSC.
- H. Li, H. Yu, C. Zhu, J. Hu, M. Du, F. Zhang and D. Yang, RSC Adv., 2016, 6, 94160–94169 RSC.
- C. Vakifahmetoglu, D. Zeydanli, V. C. Ozalp, B. A. Borsa and G. D. Soraru, Mater. Des., 2017, 142, 138 CrossRef.
- M. J. Lee, A. S. Ye, A. K. Gardino, A. M. Heijink, P. K. Sorger, G. Macbeath and M. B. Yaffe, Cell, 2012, 149, 780–794 CrossRef CAS PubMed.
- J. A. Macdiarmid, N. B. Amaro-Mugridge, J. Madrid-Weiss, I. Sedliarou, S. Wetzel, K. Kochar, V. N. Brahmbhatt, L. Phillips, S. T. Pattison and C. Petti, Nat. Biotechnol., 2009, 27, 643–651 CrossRef CAS PubMed.
- B. B. Hsu, K. S. Jamieson, S. R. Hagerman, E. Holler, J. Y. Ljubimova and P. T. Hammond, Angew. Chem., Int. Ed. Engl., 2014, 53, 8093–8098 CrossRef CAS PubMed.
- A. Wang and B. J. Edwards, Int. J. Heat Mass Transfer, 2016, 103, 997–1007 CrossRef CAS.
- X. Du, B. Shi, J. Liang, J. Bi, S. Dai and S. Z. Qiao, Adv. Mater., 2013, 25, 5981–5985 CrossRef CAS PubMed.
- L. Zhu, F. Perche, T. Wang and V. P. Torchilin, Biomaterials, 2014, 35, 4213–4222 CrossRef CAS PubMed.
- M. Farokhi, F. Mottaghitalab, M. A. Shokrgozar, K. L. Ou, C. Mao and H. Hosseinkhani, J. Controlled Release, 2016, 225, 152–169 CrossRef CAS PubMed.
- A. V. Kabanov and S. V. Vinogradov, Angew. Chem., Int. Ed. Engl., 2009, 48, 5418–5429 CrossRef CAS PubMed.
- D. Shen, J. Yang, X. Li, L. Zhou, R. Zhang, W. Li, L. Chen, R. Wang, F. Zhang and D. Zhao, Nano Lett., 2014, 14, 923–932 CrossRef CAS PubMed.
- Y. Wang, J. Wang, Y. Yang, Y. Sun, Y. Yuan, Y. Li and C. Liu, Colloids Surf., B, 2017, 153, 272–279 CrossRef CAS PubMed.
- L. Cheng, B. G. Trewyn, D. M. Jeftinija, K. Jeftinija, S. Xu, S. Jeftinija and V. S.-Y. Lin, J. Am. Chem. Soc., 2003, 125, 4451–4459 CrossRef PubMed.
- X. Du, B. Shi, J. Liang, J. Bi, S. Dai and S. Z. Qiao, Adv. Mater., 2013, 25, 5981–5985 CrossRef CAS PubMed.
- J. Khandare, M. Calderón, N. M. Dagia and R. Haag, Chem. Soc. Rev., 2012, 41, 2824–2848 RSC.
- D. S. Moon and J. K. Lee, Langmuir, 2012, 28, 12341–12347 CrossRef CAS PubMed.
- N. J. Carroll, S. Pylypenko, P. B. Atanassov and D. N. Petsev, Langmuir, 2009, 25, 13540–13544 CrossRef CAS PubMed.
- T. Li, X. Chen, Y. Liu, L. Fan, L. Lin, Y. Xu, S. Chen and J. Shao, Eur. J. Pharm. Sci., 2017, 96, 456–463 CrossRef CAS PubMed.
- A. Monnier, F. Schüth, Q. Huo, D. Kumar, D. Margolese, R. S. Maxwell, G. D. Stucky, M. Krishnamurty, P. Petroff and A. Firouzi, Science, 1993, 261, 1299–1303 CrossRef CAS PubMed.
- F. Carn, A. Colin, M.-F. Achard, H. Deleuze, E. Sellier, M. Birot and R. n. Backov, J. Mater. Chem., 2004, 14, 1370 RSC.
- J. Lu, J. Wei, Y. Yan, H. Li, J. Jia, S. Wei, H. Guo, T. Xiao and C. Liu, J. Mater. Sci.: Mater. Med., 2011, 22, 607–615 CrossRef CAS PubMed.
- L. S. Jang and H. K. Keng, Biomed. Microdevices, 2008, 10, 203–211 CrossRef CAS PubMed.
- Y. Li, F. Song, Y. Guo, L. Cheng and Q. Chen, J. Nanomater., 2018, 2018, 1–10 Search PubMed.
- S. Stephanie, D. R. Bert, V. Mattias, V. G. Stefaan, J. Sandra, S. J. Won and L. Jean-Pierre, J. Nanomater., 2018, 7390618 Search PubMed.
- I. D. Xynos, A. J. Edgar, L. D. Buttery, L. L. Hench and J. M. Polak, Biochem. Biophys. Res. Commun., 2000, 276, 461–465 CrossRef CAS PubMed.
- D. C. Carter and J. X. Ho, Adv. Protein Chem., 1994, 45, 153–176 CrossRef CAS PubMed.
- P. Zhao, M. Liu, H. Lin, X. Sun, Y. Li and S. Yan, Journal of Medical Biotechnology, 2017, 1, 1–5 Search PubMed.
- D. Kwon, B. G. Cha, Y. Cho, J. Min, E. B. Park, S. J. Kang and J. Kim, Nano Lett., 2017, 17, 2747–2756 CrossRef CAS PubMed.
- R. C. Bielby, I. S. Christodoulou, R. S. Pryce, W. J. Radford, L. L. Hench and J. M. Polak, Tissue Eng. Part A, 2004, 10, 1018–1026 CrossRef CAS PubMed.
- I. D. Xynos, A. J. Edgar, L. D. Buttery, L. L. Hench and J. M. Polak, J. Biomed. Mater. Res., 2015, 55, 151–157 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07849a |
| This journal is © The Royal Society of Chemistry 2018 |