
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective Michael addition of malonates to α,β-unsaturated ketones catalyzed by 1,2-diphenylethanediamine†
Wei Wang a,
Ling Yeb,
Zhichuan Shia,
Zhigang Zhaoa and
Xuefeng Li*a
a,
Ling Yeb,
Zhichuan Shia,
Zhigang Zhaoa and
Xuefeng Li*a
aCollege of Chemistry and Environment Protection Engineering, Southwest Minzu University, Chengdu 610041, China. E-mail: lixuefeng@swun.edu.cn
bFaculty of Geosciences and Environmental Engineering, Southwest Jiaotong University, Chengdu 610031, China
First published on 12th December 2018
Abstract
A general and highly enantioselective Michael addition of malonates to cinnamones and chalcones has been developed. The commercially available 1,2-diphenylethanediamine could be directly utilized as the organocatalyst to furnish the desired adducts in satisfactory yield (61–99%) and moderate to excellent enantiopurity (65 to >99% ee). β-Ketoester was also a competent donor and was employed to construct densely functionalized cyclohexenones via a tandem Michael-aldol condensation process.
Introduction
The direct Michael addition of stabilized carbon-centered nucleophiles to electron-poor olefins is widely recognized as a highly atom-economic carbon–carbon bond-forming reaction in organic synthesis. Therefore, the development of an enantioselective catalytic protocol for this conversion has constituted an actively pursued field in the past decades.1 Although significant progress has been achieved with metal complexes,1a–c recently the well-designed organocatalyst has played an impressive role in this field as well.1d,1e Particularly, the organocatalytic Michael addition of malonates to α,β-unsaturated ketones will produce versatile adducts, which can be readily converted to the corresponding δ-ketoesters as useful synthetic building blocks after decarboxylation.2 Pioneering work, the first highly enantioselective truly organocatalytic reaction of this type was developed by Jørgensen using an imidazoline catalyst in 2003.2a Subsequently, other organocatalysts such as proline-derived tetrazole,2b,3 metal salts of carboxylic acids,4 phase-transfer catalysts,5 various chiral thioureas,6 proline-derived guanidines,7 primary amines8 and their derivates9 have been introduced to catalyze this reaction. Despite excellent enantiopurities having been achieved in a few cases, nevertheless some of the established approaches suffer from several drawbacks to a certain extent, such as narrow substrate scope and restriction to a special combination of nucleophile and electrophile type. Moreover, among all these well-demonstrated organocatalytic Michael reactions, those untransformed and simple molecules are always not the preferred catalysts as a consequence of inferior enantioselectivity and poorer reactivity, high degrees of optical purity and reactivity need to be achieved in the presence of modified organocatalysts in most cases. As we all know, these employed optimal organocatalysts should usually been prepared from commercially available precursors or naturally occurring compounds after several-step, even multi-step transformation.2,3,5a,5b,6–9 The costly preparative procedure hence impairs the synthetic efficiency and practicality to a certain extent. Therefore, the development of highly general asymmetric Michael addition promoted by simple and commercially available molecules is clearly in high demand.10In this context, chiral vicinal 1,2-diamines, mainly cyclohexane-1,2-diamines (CHDA)11 and 1,2-diphenylethanediamine (DPEN)12 emerged as a class of efficient and commercially available primary amine catalysts.13 These diamines enabled the stereoselective functionalization of a variety of steric-constraint carbonyl compounds, including aliphatic and aromatic ketones,11a–c,12h,12i α-branched substituted aldehydes,11d,11e and α,β-unsaturated carbonyl compounds.11f,12a–g A range of versatile building blocks were smoothly constructed in a highly enantioenriched fashion via enamine,11a,11b,11d,12h iminium,11f,12a–f enamine–iminium11c,11e,12g and dienamine12i activation modes. As part of our continuous efforts in developing asymmetric Michael addition of unactivated α,β-unsaturated ketones,14 we disclosed herein a highly enantioselective Michael addition of malonates to cinnamones2–4,6a–c,8–9 and chalcones5,6d–f,8b,15 catalyzed by a structurally simple DPEN.
Results and discussion
Instead of the often-used CHDA, the moisture- and air-stable, commercially inexpensive DPEN was initially utilized to screen the optimal conditions due to its operational simplicity. Gratifyingly, the Michael reaction between β-naphthyl-substituted cinnamone 1a and diethyl malonate 2a proceeded smoothly to afford the desired adduct 3aa with promising enantiopurity (92% ee) in the presence of acetic acid (Table 1, entry 1). In order to further improve the reactivity, we then turned our attention to examine the effect of other acidic additive. It was revealed that a significant decrease of catalytic capability was observed in the case of stronger acid (entries 2 and 3). Subsequently the model reaction was performed with a range of aromatic carboxylic acids. Although most of aromatic acid furnished 3aa with diminished yield and optical purity (entries 4–7 vs. entry 1), the enhancement of reactivity was fortunately observed with o-phthalic acid and salicylic acid (SA) (entries 8 and 9). In particular, the dicarboxylic acid, o-phthalic acid, gave rise to complete conversion after 144 hours, together with 95% ee.11e,16 The effect of different solvents was successively investigated with SA (entries 10–13). The protic solvent, EtOH, gave the best enantioselectivity and ether led to a considerable improvement of reaction rate. Meanwhile, the model reaction went to completion after 96 hours with maintained enantiomeric excess when exposed to o-phthalic acid in EtOH (entry 14), however, sluggish transformation was detected in ether because of poor solubility of this catalyst system. Moreover, reducing the amount of malonate resulted in substantial decrease of reactivity (entry 15). The model reaction didn't occur in the absence of acidic additive (entry 16). Meanwhile, higher reactivity was observed under neat condition (entry 17).
|
|||||
|---|---|---|---|---|---|
| Entry | Additive | Solvent | Time (h) | Yieldb (%) | eec (%) |
| a Unless otherwise noted, the reaction was performed with 0.2 mmol of 1a, 4 mmol of malonate 2a, 20 mol% (R,R)-DPEN and 40 mol% acid in 1 mL of solvent at rt. TFA = trifluoroacetic acid, TsOH = p-toluenesulfonic acid, BA = benzoic acid, PNBA = p-nitrobenzoic acid, ONBA = o-nitrobenzoic acid, OFBA = o-fluorobenzoic acid, SA = salicylic acid. NR = no reaction.b Isolated yield.c Determined by chiral HPLC.d Conducted with 2 mmol of malonate 2a.e Performed in the absence of acid.f 0.6 mL (4 mmol) malonate 2a was used as the solvent. | |||||
| 1 | HOAc | Toluene | 168 | 86 | 92 |
| 2 | TFA | Toluene | 168 | 79 | 96 |
| 3 | TsOH | Toluene | 168 | 30 | 97 |
| 4 | BA | Toluene | 168 | 82 | 81 |
| 5 | PNBA | Toluene | 168 | 65 | 71 |
| 6 | ONBA | Toluene | 168 | 64 | 72 |
| 7 | OFBA | Toluene | 168 | 77 | 61 |
| 8 | o-Phthalic acid | Toluene | 144 | 95 | 95 |
| 9 | SA | Toluene | 168 | 91 | 88 |
| 10 | SA | CHCl3 | 168 | 91 | 90 |
| 11 | SA | Et2O | 72 | 97 | 90 |
| 12 | SA | THF | 168 | 91 | 88 |
| 13 | SA | EtOH | 168 | 75 | 96 |
| 14 | o-Phthalic acid | EtOH | 96 | 95 | 94 |
| 15d | o-Phthalic acid | EtOH | 168 | 99 | 94 |
| 16e | EtOH | 168 | NR | ||
| 17f | o-Phthalic acid | 30 | 99 | 90 | |
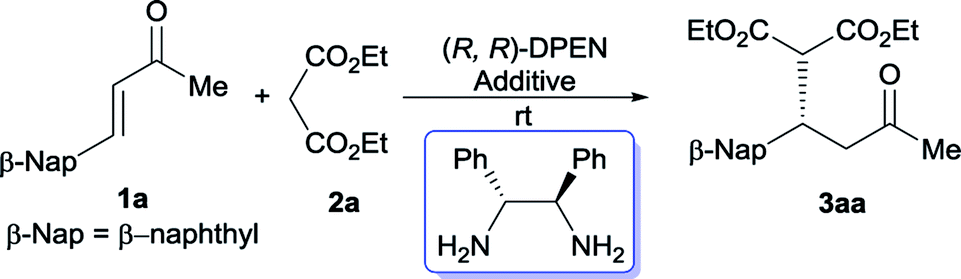
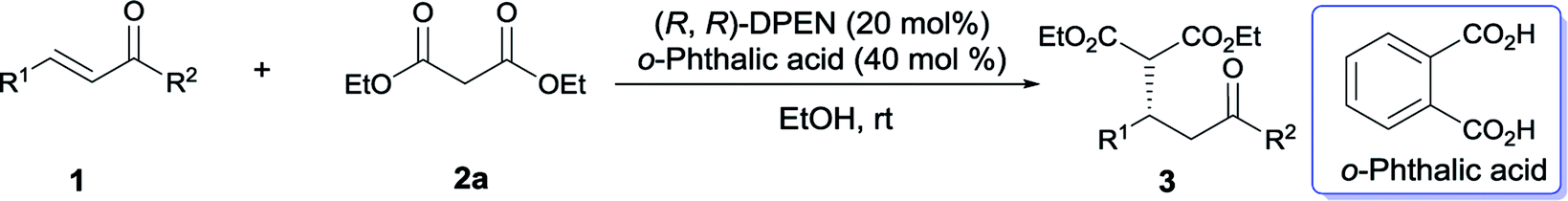
Once the optimal reaction conditions have been established, the substrate scope of this Michael addition was extended to a variety of cinnamones and malonates. As summarized in Table 2, this catalytic approach was not sensitive to the electronic property of cinnamones. The electron-neutral benzylideneacetone 1b reacted properly with diethyl malonate 2a to generate 3ab in synthetically useful yield and good enantioselectivity (Table 2, entry 1). The electron-deficient α,β-unsaturated ketones 1c–1g were well tolerated by this catalytic system and enabled access to the expected adducts 3ac–3ag in a highly enantioselective manner (entries 2–6). Meanwhile, the electron-rich cinnamones 1h and 1i are also suitable acceptors for this conversion (entries 7 and 8). On the other hand, the position of substituent on the phenyl ring exerted negligible affect on this titled Michael reaction. Almost identical isolated yields were obtained in the case of the sterically congested ortho-substituted enone 1d in comparison with the meta-substituted 1e and para-substituted 1f (entry 3 vs. entries 4 and 5). In contrast with bulky α-naphthyl-containing 1j, better catalytic performance in terms of reactivity and enantiocontrol was achieved when β-naphthyl-embedded acceptor 1a was utilized (entry 9 vs. entry 10). The heteroaromatic substrates 1k and 1l served as appropriate acceptors as well, however, a modified condition was required for 1k to achieve synthetically useful conversion (entries 11 and 12). In addition to aromatic substrates, the aliphatic enones 1m and 1n were also compatible with this catalytic strategy, but with slightly poorer reactivity (entries 13 and 14). Notably, variation of R2 ketone substituent indicated that enone 1o possessing a sterically more demanding ethyl group also participated in this conjugate addition (entry 15). Cyclic enones2b,3,4b,6a,6g,7,17 were suitable acceptors as well, generating the corresponding adducts 3ap and 3aq with good enantioselectivities (entries 16 and 17).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | 2 | 3 | Time (h) | Yieldb (%) | eec (%) |
| a Unless otherwise noted, the reaction was performed with 0.2 mmol of 1, 4 mmol of malonate 2a, 20 mol% (R,R)-DPEN and 40 mol% o-phthalic acid in 1 mL of EtOH at rt.b Isolated yield.c Determined by chiral HPLC.d Performed with 40 mol% SA in ether.e 2 mmol of malonate 2a was used. | |||||||
| 1 | Ph | Me (1b) | 2a | 3ab | 168 | 75 | 91 |
| 2 | p-FC6H4 | Me (1c) | 2a | 3ac | 168 | 99 | 95 |
| 3 | o-ClC6H4 | Me (1d) | 2a | 3ad | 168 | 99 | 96 |
| 4 | m-ClC6H4 | Me (1e) | 2a | 3ae | 168 | 99 | 94 |
| 5 | p-ClC6H4 | Me (1f) | 2a | 3af | 168 | 99 | 95 |
| 6 | p-BrC6H4 | Me (1g) | 2a | 3ag | 168 | 70 | 93 |
| 7 | p-MeC6H4 | Me (1h) | 2a | 3ah | 168 | 85 | 94 |
| 8 | p-MeOC6H4 | Me (1i) | 2a | 3ai | 168 | 92 | 96 |
| 9 | 1-Naphthyl | Me (1j) | 2a | 3aj | 168 | 97 | 96 |
| 10 | 2-Naphthyl | Me (1a) | 2a | 3aa | 96 | 95 | 94 |
| 11d | 2-Furanyl | Me (1k) | 2a | 3ak | 168 | 84 | 86 |
| 12 | 2-Thiophenyl | Me (1l) | 2a | 3al | 168 | 97 | 92 |
| 13 | Me | Me (1m) | 2a | 3am | 168 | 70 | 86 |
| 14d | n-Bu | Me (1n) | 2a | 3an | 168 | 65 | 95 |
| 15 | Ph | Et (1o) | 2a | 3ao | 168 | 61 | 91 |
| 16e | –(CH2)3– (1p) | 2a | 3ap | 168 | 71 | 82 | |
| 17 | –(CH2)4– (1q) | 2a | 3aq | 96 | 97 | 87 | |
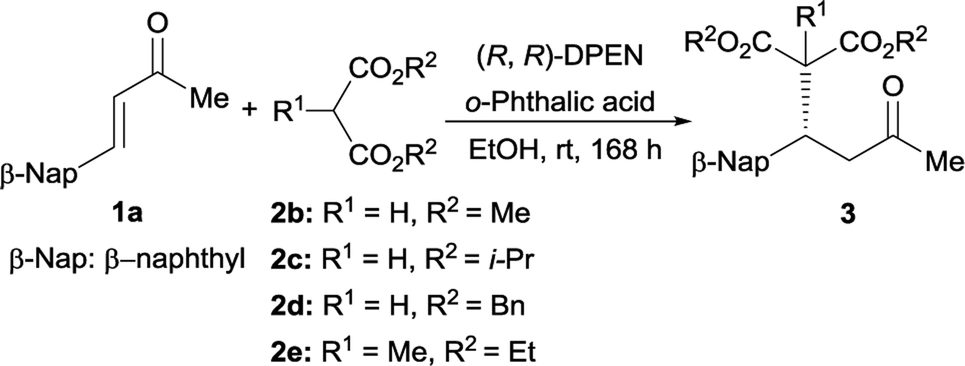
With respect to the donor, good enantiomeric excess was obtained for dimethyl ester 2b, and lower reactivity was detected for diisopropyl ester 2c but without compromising the optical purity (Table 3, entries 1 and 2). In contrast, dibenzyl malonate 2d afforded desired adduct 3da with relatively poorer optical purity (entry 3). Meanwhile, the reaction was totally inert in the case of di-tert-butyl malonate. Moreover, methyl-substituted malonate 2e was also compatible with this catalytic protocol, but relatively lower reactivity was observed (entry 4).
|
||||
|---|---|---|---|---|
| Entry | 2 | 3 | Yieldb (%) | eec (%) |
| a Unless otherwise noted, the reaction was performed with 0.2 mmol of 1a, 4 mmol of malonate 2, 20 mol% (R,R)-DPEN and 40 mol% o-phthalic acid in 1 mL of EtOH at rt for 168 h.b Isolated yield.c Determined by chiral HPLC. | ||||
| 1 | 2b | 3ba | 81 | 90 |
| 2 | 2c | 3ca | 65 | 93 |
| 3 | 2d | 3da | 92 | 74 |
| 4 | 2e | 3ea | 72 | 95 |
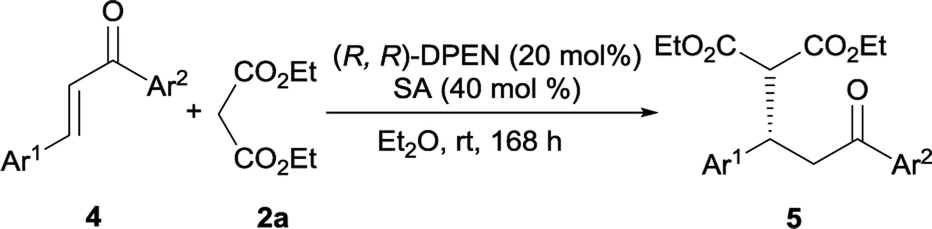
With the exception of cinnamones, our catalytic protocol was also applicable to chalcones, a class of challenging substrates for iminium ion activation.18 Only moderate isolated yield was obtained when performed with o-phthalic acid in EtOH, whereas the reactivity could be effectively improved when conducted with salicylic acid in ether (Table 4, entry 1). Again, this Michael reaction was independent of the electronic nature of substituents on each aromatic ring of chalcones. Both the electron-rich chalcones 4b and 4f, and the electron-poor chalcones 4c, 4g and 4i worked smoothly with diethyl malonate 2a, forming the expected adducts with complete conversion in highly enantioenriched fashion (entries 2, 3, 6, 7 and 9). Only slightly reduced yield was detected for enone 4d bearing a bulky naphthyl group at the β-site, along with 94% ee (entry 4). The heteroaromatic chalcones 4e and 4h underwent clean reactions and gave rise to the desired adducts 5e and 5h in acceptable yields and moderate to excellent enantioselectivities (entries 5 and 8). The absolute configuration of 3 and 5 was confirmed to be S via comparison of HPLC traces and optical rotation value with that of literatures reported.2b,6e
|
|||||
|---|---|---|---|---|---|
| Entry | Ar1 | Ar2 | 5 | Yieldb (%) | eec (%) |
| a Unless otherwise noted, the reaction was performed with 0.2 mmol of 4, 4 mmol of malonate 2a, 20 mol% (R,R)-DPEN, 40 mol% salicylic acid in 1 mL of ether at rt for 168 h.b Isolated yield.c Determined by chiral HPLC.d Carried out with o-phthalic acid in 1 mL of EtOH. | |||||
| 1 | Ph | Ph (4a) | 5a | 75 (55)d | 92 (98)d |
| 2 | p-MeC6H4 | Ph (4b) | 5b | 98 | 98 |
| 3 | p-ClC6H4 | Ph (4c) | 5c | 99 | 94 |
| 4 | 2-Naphthyl | Ph (4d) | 5d | 88 | 94 |
| 5 | 2-Thiophenyl | Ph (4e) | 5e | 83 | 65 |
| 6 | Ph | p-MeC6H4 (4f) | 5f | 99 | 99 |
| 7 | Ph | p-ClC6H4 (4g) | 5g | 99 | >99 |
| 8 | Ph | 2-Thiophenyl (4h) | 5h | 65 | 96 |
| 9 | p-ClC6H4 | p-ClC6H4 (4i) | 5i | 99 | 93 |
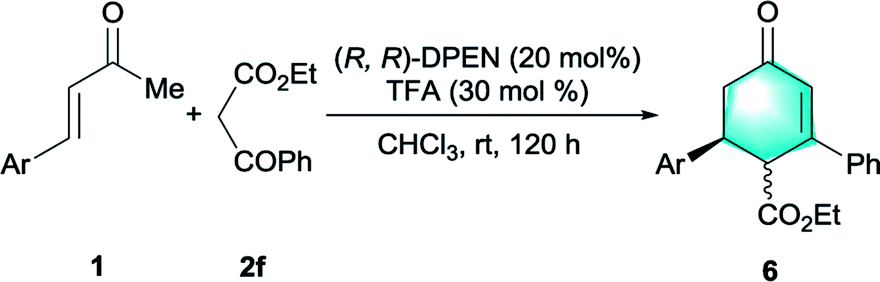
In addition to malonates, we were pleased to find that β-ketoester was also competent donor for this catalytic protocol.19 After further optimization of reaction conditions, we found that the cascade Michael-aldol condensation process between cinnamones 1 and ethyl benzoylacetate 2f readily occurred with 30 mol% of TFA in chloroform, delivering highly functionalized cyclohexenones 6 as an inseparable mixture of diastereomers. (See Table S1 in the ESI†). Both the electron-deficient cinnamones and the electron-rich cinnamones were well tolerated (Table 5, entries 2–4). The bulky naphthyl group-containing enone 1a and the heteroaromatic substrate 1l were compatible with this catalytic protocol as well, leading to the formation of annulated product 6e and 6f with high levels of enantiopurities (entries 5 and 6). The absolute stereochemistry of cyclohexenone 6 was determined to be S via conversion of 6a to known compound after a simple decarboxylation (see eqn S(1) in the ESI†).19b Notably, cyclohexenones and their derivates constituted crucial skeletal components common in enormous natural products and pharmaceutical molecules.20
|
|||||
|---|---|---|---|---|---|
| Entry | Ar | 6 | Yieldb (%) | drc | eed (%) |
| a Unless otherwise noted, the reaction was performed with 0.2 mmol of 1, 0.4 mmol of 2f, 20 mol% (R,R)-DPEN and 30 mol% TFA in 1 mL of CHCl3 at rt for 120 h.b Isolated yield of the diastereomeric mixture.c Diastereomeric ratio (dr) was determined by 1H NMR analysis of the crude mixture; major isomer: trans.d Determined by chiral stationary-phase HPLC. | |||||
| 1 | Ph (1b) | 6a | 97 | 77![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :23 :23 |
96/97 |
| 2 | p-ClC6H4 (1f) | 6b | 97 | 79:21 |
87/87 |
| 3 | p-BrC6H4 (1g) | 6c | 92 | 80:20 |
95/97 |
| 4 | p-MeOC6H4 (1i) | 6d | 99 | 66:34 |
92/90 |
| 5 | 2-Naphthyl (1a) | 6e | 99 | 53:47 |
89/87 |
| 6 | 2-Thiophenyl (1l) | 6f | 94 | 60:40 |
92/90 |
To demonstrate the synthetic potential of this organocatalytic asymmetric process, base-controlled chemoselective conversion of Michael adduct 5a were conducted in the presence of iodine.21 α-Hydroxylation of malonate moiety occurred smoothly to provide α-hydroxyl malonate 7 almost without compromise of enantiopurity, when treated with a catalytic amount of NaOAc (Scheme 1). Moreover, the adduct 5a could be converted to phenyl ester 8 by brief exposure to meta-chloroperoxybenzoic acid (m-CPBA) without deterioration of optical purity. This Baeyer–Villiger oxidation proceeded with exclusive regioselectivity. Lastly, transesterification of crude 8 worked properly with NaBH4 in MeOH to afford methyl ester in 86% yield, albeit a slight deterioration of optical purity was detected.
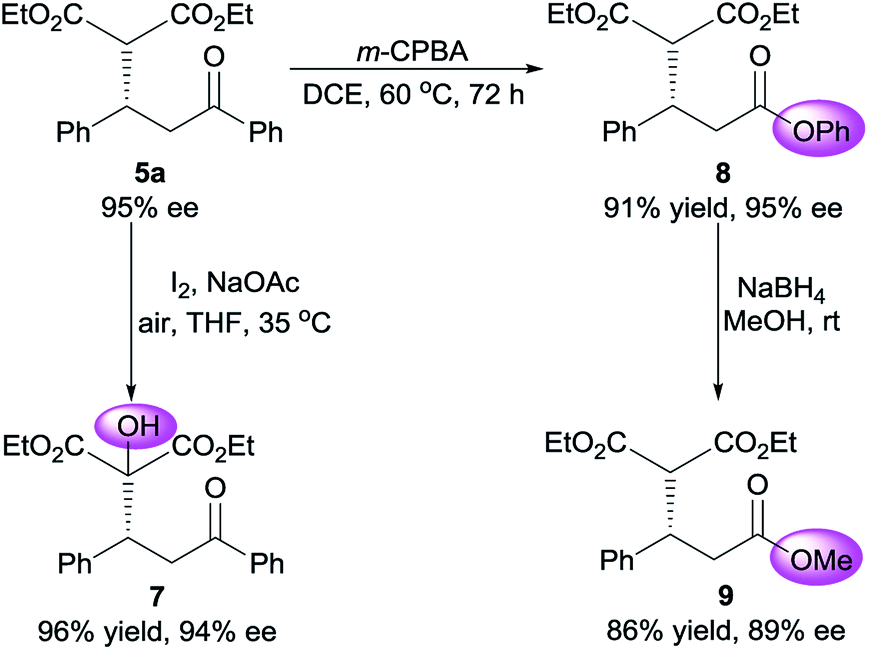 | ||
| Scheme 1 Synthetic transformation of adduct 5a. | ||
To account for the observed stereochemical outcome of this Michael addition, a bifunctional catalytic model was proposed in Scheme 2.12g Initially, benzylideneacetone 1b was activated via formation of iminium ion with one amino group of vicinal diamine catalyst. Another amino group of DPEN could be engaged in hydrogen-bonding interaction with the carbonyl moiety of ethyl malonate. As a result, the donor was restricted to attack Re face of enone, thereby leading to the generation of S-configured adduct 3ab. In the case of ethyl benzoylacetate, the formation of enamine intermediate allowed the following intramolecular aldol reaction to construct cyclohexanone.19b After final dehydration, the cyclohexenone 6a was therefore obtained.
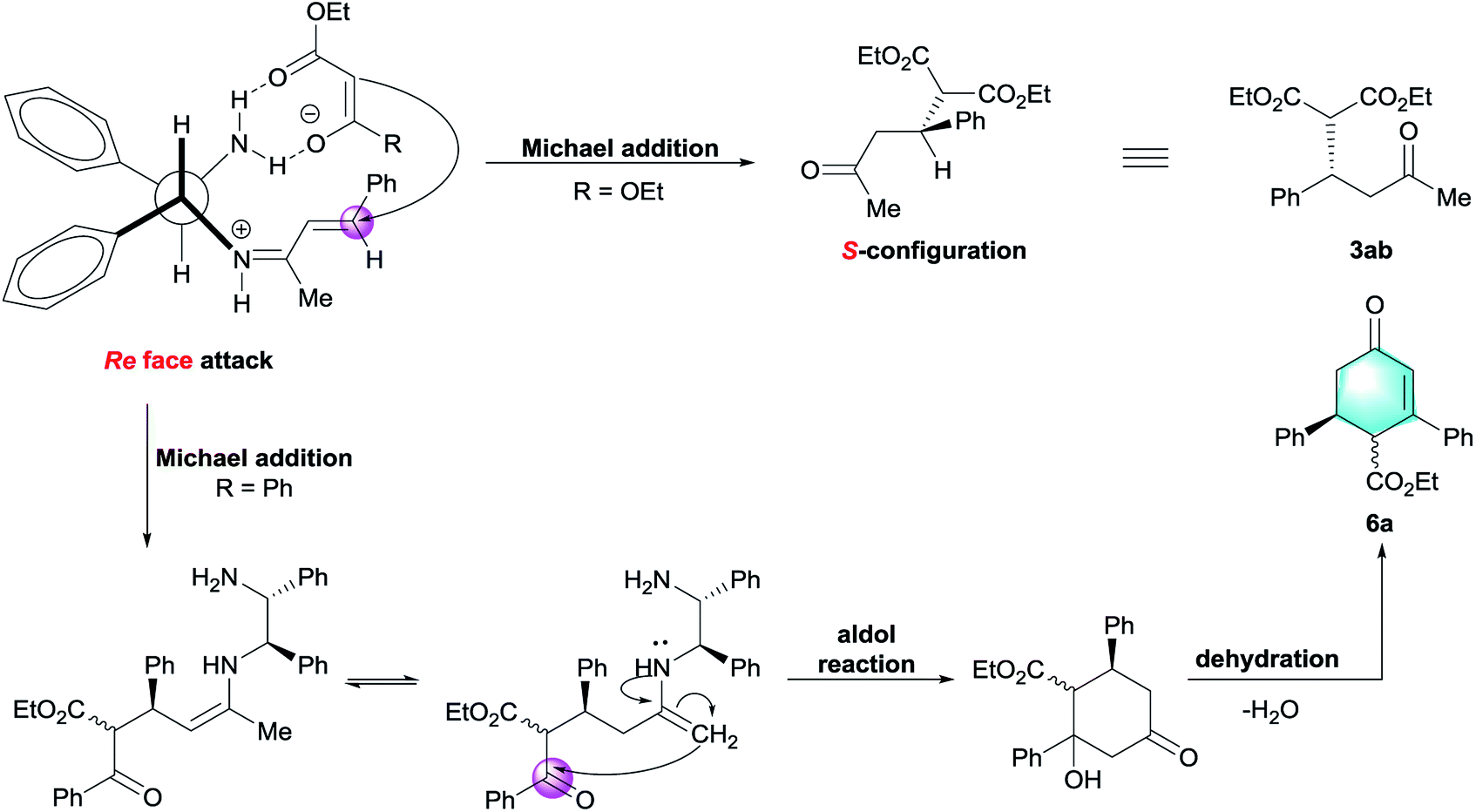 | ||
| Scheme 2 Proposed reaction pathway. | ||
Conclusions
In conclusion, we have developed a general and enantioselective Michael addition of malonate to cinnamones and chalcones. The commercially available DPEN could be utilized as the organocatalyst to furnish the desired adducts in satisfactory yield (61–99%) and moderate to excellent enantiopurity (65 to >99% ee). This catalytic protocol was also applicable to β-ketoester and constructed a densely functionalized cyclohexenone via a tandem Michael-aldol condensation process. Furthermore, profound synthetic manipulation could be performed on the resulting adduct to construct various optically active building blocks.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (No. 21402163) and the Fundamental Research Funds for the Central Universities of Southwest Minzu University (No. 2016NGJPY02). Wang Wei gratefully acknowledges the Graduate Innovation Project of Southwest Minzu University (No. CX2017SZ018).Notes and references
- (a) M. P. Sibi and S. Manyem, Tetrahedron, 2000, 56, 8033 CrossRef CAS; (b) N. Krause and A. Hoffmann-Röder, Synthesis, 2001, 2001, 0171 CrossRef; (c) J. Christoffers, G. Koripelly, A. Rosiak and M. Rössle, Synthesis, 2007, 2007, 1279 CrossRef; (d) S. B. Tsogoeva, Eur. J. Org. Chem., 2007, 2007, 1701 CrossRef; (e) D. Almaşi, D. A. Alonso and C. Nájera, Tetrahedron: Asymmetry, 2007, 18, 299 CrossRef.
- (a) N. Halland, P. S. Aburel and K. A. Jørgensen, Angew. Chem., Int. Ed., 2003, 42, 661 CrossRef CAS PubMed; (b) V. Wascholowski, K. R. Knudsen, C. E. T. Mitchell and S. V. Ley, Chem.–Eur. J., 2008, 14, 6155 CrossRef CAS PubMed.
- K. R. Knudsen, C. E. T. Mitchell and S. V. Ley, Chem. Commun., 2006, 66 RSC.
- (a) M. Yamaguchi, T. Shiraishi and M. Hirama, Angew. Chem., Int. Ed., 1993, 32, 1176 CrossRef; (b) M. Yoshida, M. Narita and S. Hara, J. Org. Chem., 2011, 76, 8513 CrossRef CAS PubMed.
- (a) T. Ooi, D. Ohara, K. Fukumoto and K. Maruoka, Org. Lett., 2005, 7, 3195 CrossRef CAS PubMed; (b) P. Kottala Vijaya, S. Murugesan and A. Siva, Tetrahedron Lett., 2015, 56, 5209 CrossRef CAS; (c) D. Y. Kim, S. C. Huh and S. M. Kim, Tetrahedron Lett., 2001, 42, 6299 CrossRef CAS; (d) R. T. Dere, R. R. Pal, P. S. Patil and M. M. Salunkhe, Tetrahedron Lett., 2003, 44, 5351 CrossRef CAS.
- (a) K. Dudziński, A. M. Pakulska and P. Kwiatkowski, Org. Lett., 2012, 14, 4222 CrossRef PubMed; (b) P. Li, S. Wen, F. Yu, Q. Liu, W. Li, Y. Wang, X. Liang and J. Ye, Org. Lett., 2009, 11, 753 CrossRef CAS PubMed; (c) S.-i. Hirashima, T. Sakai, K. Nakashima, N. Watanabe, Y. Koseki, K. Mukai, Y. Kanada, N. Tada, A. Itoh and T. Miura, Tetrahedron Lett., 2014, 55, 4334 CrossRef CAS; (d) J. Wang, H. Li, L. Zu, W. Jiang, H. Xie, W. Duan and W. Wang, J. Am. Chem. Soc., 2006, 128, 12652 CrossRef CAS PubMed; (e) Y. Liu, X. Wang, X. Wang and W. He, Org. Biomol. Chem., 2014, 12, 3163 RSC; (f) D. Cao, G. Fang, J. Zhang, H. Wang, C. Zheng and G. Zhao, J. Org. Chem., 2016, 81, 9973 CrossRef CAS PubMed; (g) M. Moritaka, N. Miyamae, K. Nakano, Y. Ichikawa and H. Kotsuki, Synlett, 2012, 23, 2554 CrossRef CAS.
- (a) E. Riguet, Tetrahedron Lett., 2009, 50, 4283 CrossRef CAS; (b) S. V. Pansare and R. Lingampally, Org. Biomol. Chem., 2009, 7, 319 RSC.
- (a) Y.-Q. Yang and G. Zhao, Chem.–Eur. J., 2008, 14, 10888 CrossRef CAS PubMed; (b) Z. Mao, Y. Jia, W. Li and R. Wang, J. Org. Chem., 2010, 75, 7428 CrossRef CAS PubMed.
- (a) C. Luo, Y. Jin and D.-M. Du, Org. Biomol. Chem., 2012, 10, 4116 RSC; (b) Y. Kamito, A. Masuda, H. Yuasa, N. Tada, A. Itoh, Y. Koseki and T. Miura, Chem. Lett., 2013, 42, 1151 CrossRef CAS; (c) Y. Kamito, A. Masuda, H. Yuasa, N. Tada, A. Itoh, K. Nakashima, S.-i. Hirashima, Y. Koseki and T. Miura, Tetrahedron: Asymmetry, 2014, 25, 974 CrossRef CAS; (d) H. Huang, Z. Jin, K. Zhu, X. Liang and J. Ye, Angew. Chem., Int. Ed., 2011, 50, 3232 CrossRef CAS PubMed.
- (a) G. Bartoli, M. Bosco, A. Carlone, A. Cavalli, M. Locatelli, A. Mazzanti, P. Ricci, L. Sambri and P. Melchiorre, Angew. Chem., Int. Ed., 2006, 45, 4966 CrossRef CAS PubMed; (b) A. Russo, A. Perfetto and A. Lattanzi, Adv. Synth. Catal., 2009, 351, 3067 CrossRef CAS.
- (a) Y. Liu, J. Wang, Q. Sun and R. Li, Tetrahedron Lett., 2011, 52, 3584 CrossRef CAS; (b) Y. Liu, P. Gao, J. Wang, Q. Sun, Z. Ge and R. Li, Synlett, 2012, 23, 1031 CrossRef CAS; (c) J. Wang, X. Wang, Z. Ge, T. Cheng and R. Li, Chem. Commun., 2010, 46, 1751 RSC; (d) A. Avila, R. Chinchilla, E. Gómez-Bengoa and C. Nájera, Tetrahedron: Asymmetry, 2013, 24, 1531 CrossRef CAS; (e) Y. Inokoishi, N. Sasakura, K. Nakano, Y. Ichikawa and H. Kotsuki, Org. Lett., 2010, 12, 1616 CrossRef CAS PubMed; (f) J. Wang, C. Qi, Z. Ge, T. Cheng and R. Li, Chem. Commun., 2010, 46, 2124 RSC.
- (a) H. Kim, C. Yen, P. Preston and J. Chin, Org. Lett., 2006, 8, 5239 CrossRef CAS PubMed; (b) X. Wang, C. M. Reisinger and B. List, J. Am. Chem. Soc., 2008, 130, 6070 CrossRef CAS; (c) H.-M. Yang, L. Li, K.-Z. Jiang, J.-X. Jiang, G.-Q. Lai and L.-W. Xu, Tetrahedron, 2010, 66, 9708 CrossRef CAS; (d) M. Rogozińska, A. Adamkiewicz and J. Mlynarski, Green Chem., 2011, 13, 1155 RSC; (e) W. Wu, X. Li, H. Huang, X. Yuan, J. Lu, K. Zhu and J. Ye, Angew. Chem., Int. Ed., 2013, 52, 1743 CrossRef CAS PubMed; (f) Y. Wei, S. Wen, Z. Liu, X. Wu, B. Zeng and J. Ye, Org. Lett., 2015, 17, 2732 CrossRef CAS; (g) Y. Liu, X. Liu, M. Wang, P. He, L. Lin and X. Feng, J. Org. Chem., 2012, 77, 4136 CrossRef CAS PubMed; (h) W. Wang, J. Wang, S. Zhou, Q. Sun, Z. Ge, X. Wang and R. Li, Chem. Commun., 2013, 49, 1333 RSC; (i) G. Zhan, Q. He, X. Yuan and Y.-C. Chen, Org. Lett., 2014, 16, 6000 CrossRef CAS.
- (a) Y.-C. Chen, Synlett, 2008, 2008, 1919 CrossRef; (b) L.-W. Xu, J. Luo and Y. Lu, Chem. Commun., 2009, 1807 RSC; (c) P. Melchiorre, Angew. Chem., Int. Ed., 2012, 51, 9748 CrossRef CAS PubMed.
- (a) S. Liu, Q. Wang, L. Ye, Z. Shi, Z. Zhao, X. Yang, K. Ding and X. Li, Tetrahedron, 2016, 72, 5115 CrossRef CAS; (b) Q. Wang, W. Wang, L. Ye, X. Yang, X. Li, Z. Zhao and X. Li, Molecules, 2017, 22, 1096 CrossRef.
- (a) C. Chen, S.-F. Zhu, X.-Y. Wu and Q.-L. Zhou, Tetrahedron: Asymmetry, 2006, 17, 2761 CrossRef CAS; (b) M. Agostinho and S. Kobayashi, J. Am. Chem. Soc., 2008, 130, 2430 CrossRef CAS PubMed; (c) H. Naka, N. Kanase, M. Ueno and Y. Kondo, Chem.–Eur. J., 2008, 14, 5267 CrossRef CAS; (d) D. Chen, Z. Chen, X. Xiao, Z. Yang, L. Lin, X. Liu and X. Feng, Chem.–Eur. J., 2009, 15, 6807 CrossRef CAS.
- Y. Zhu, L. Zhang and S. Luo, J. Am. Chem. Soc., 2016, 138, 3978 CrossRef CAS.
- (a) H. Sasai, T. Arai and M. Shibasaki, J. Am. Chem. Soc., 1994, 116, 1571 CrossRef CAS; (b) H. Sasai, T. Arai, Y. Satow, K. N. Houk and M. Shibasaki, J. Am. Chem. Soc., 1995, 117, 6194 CrossRef CAS; (c) M. Watanabe, K. Murata and T. Ikariya, J. Am. Chem. Soc., 2003, 125, 7508 CrossRef CAS PubMed; (d) N. Mase, M. Fukasawa, N. Kitagawa, F. Shibagaki, N. Noshiro and K. Takabe, Synlett, 2010, 2010, 2340 CrossRef.
- (a) G. Bartoli, M. Bosco, A. Carlone, F. Pesciaioli, L. Sambri and P. Melchiorre, Org. Lett., 2007, 9, 1403 CrossRef CAS PubMed; (b) X. Li, L. Cun, C. Lian, L. Zhong, Y. Chen, J. Liao, J. Zhu and J. Deng, Org. Biomol. Chem., 2008, 6, 349 RSC.
- (a) N. Halland, P. S. Aburel and K. A. Jørgensen, Angew. Chem., Int. Ed., 2004, 43, 1272 CAS; (b) Y.-Q. Yang, Z. Chai, H.-F. Wang, X.-K. Chen, H.-F. Cui, C.-W. Zheng, H. Xiao, P. Li and G. Zhao, Chem.–Eur. J., 2009, 15, 13295 CrossRef CAS PubMed; (c) C. Arróniz, C. Escolano, F. J. Luque, J. Bosch and M. Amat, Org. Biomol. Chem., 2011, 9, 5079 RSC.
- X. Yang, J. Wang and P. Li, Org. Biomol. Chem., 2014, 12, 2499 RSC.
- C.-B. Miao, M. Zhang, Z.-Y. Tian, H.-T. Xi, X.-Q. Sun and H.-T. Yang, J. Org. Chem., 2011, 76, 9809 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and HPLC spectra for all new compounds. See DOI: 10.1039/c8ra07809b |
| This journal is © The Royal Society of Chemistry 2018 |