
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLaser-driven structural transformations in dextran-graft-PNIPAM copolymer/Au nanoparticles hybrid nanosystem: the role of plasmon heating and attractive optical forces
Oleg A. Yeshchenko *a,
Antonina P. Naumenkoa,
Nataliya V. Kutsevolb and
Iulia I. Harahutsb
*a,
Antonina P. Naumenkoa,
Nataliya V. Kutsevolb and
Iulia I. Harahutsb
aPhysics Department, Taras Shevchenko National University of Kyiv, 60 Volodymyrs'ka str., 01601 Kyiv, Ukraine. E-mail: yes@univ.kiev.ua; oleg.yeshchenko@gmail.com
bChemistry Department, Taras Shevchenko National University of Kyiv, 60 Volodymyrs'ka str., 01601 Kyiv, Ukraine
First published on 14th November 2018
Abstract
Laser induced structural transformations in a dextran grafted-poly(N-isopropylacrylamide) copolymer/Au nanoparticles (D-g-PNIPAM/AuNPs) hybrid nanosystem in water have been observed. The laser induced local plasmonic heating of Au NPs leads to Lower Critical Solution Temperature (LCST) phase transition in D-g-PNIPAM/AuNPs macromolecules accompanied by their shrinking and aggregation. The hysteresis non-reversible character of the structural transformation in D-g-PNIPAM/AuNPs system has been observed at the decrease of laser intensity, i.e. the aggregates remains in solution after the turn-off the laser illumination. This is an essential difference comparing to the case of usual heating–cooling cycles when there is no formation of aggregates and structural transformations are reversible. Such a fundamental difference has been rationalized as the result of action of attractive optical forces arising due to the excitation of surface plasmons in Au NPs. The attractive plasmonic forces facilitate the formation of the aggregates and counteract their destruction. The laser induced structural transformations have been found to be very sensitive to matching conditions of the resonance of the laser light with surface plasmon resonance proving the plasmonic nature of observed phenomena.
1. Introduction
Light interacts with the free electrons in metal nanoparticles (NPs), resonantly exciting their coherent collective oscillations. The phenomenon is known as surface plasmon resonance (SPR). It causes the strong absorption and scattering of incident light by metal NPs.1–3 The plasmonic effects are most pronounced in nanosystems containing NPs of the noble metals, the gold in particular. The surface plasmons in metal NPs decay both radiatively and nonradiatively. The former is responsible for the plasmonic enhancement of the electric field in the vicinity of NPs, whereas the latter decay contributes to particle heating and subsequent heat transfer from the NP to the surrounding medium. Such heating of the metal NPs originating from the conversion of the energy of electron motion to thermal energy is called plasmonic heating.4–8 Since in the NPs the surface/volume ratio is hugely larger than in the bulk, heating of the medium is extremely localized. This is the most remarkable feature of plasmonic heating. Thus, the metal NPs serve as highly efficient localized heat sources at the nanometer-length scale. This unique property promises numerous applications in energy,9–12 chemical catalysis,13,14 protein imaging,15,16 and biomedicine.17–20 The heating effect should have a resonant character, namely the heating should become especially strong under the SPR conditions when the frequency of incident photons is close to the SPR frequency of a metal NP.21,22 Since metal NPs have a very low light emission quantum yield, almost all the absorbed light energy is conversed to the heat energy.Besides the thermal action, the excitation of SPR in metal NPs at laser illumination leads to the mechanical action on them. Light can induce an appearance of significant optical forces acting between the metal NPs.23–28 When two metallic NPs are in close proximity, and the pair is illuminated by laser radiation, there is a laser-induced attractive force between them which depends linearly on the laser power. When arrays of such NPs are suspended in a liquid, this force can be used to promote aggregation. Theoretical studies of this laser-induced attractive force were performed in ref. 25–28, where the strength of plasmon induced force was compared with the competing van der Waals attraction. It has been shown that the plasmonic attractive force has prominent resonant character, i.e. its strength increases strongly when the frequency of laser light is in resonance with SPR in NPs. When the resonance condition is fulfilled the strength of plasmonic force can be considerably higher than the strength of the van der Waals one. The action of attractive optical forces on NPs permits researchers to hold and manipulate them by means of optical tweezers in a variety of techniques.29,30 Such forces are very important at the nanoscale because they may affect strongly the signal of surface enhanced spectroscopy31,32 or promote controllable aggregation of NPs.33–35
The plasmonic heating has been shown to cause the phase transition in surrounding media, in particular in hybrid nanosystems containing the metal NPs and thermosensitive polymers.35–38 The growing progress in nanotechnology and life sciences demonstrates an urgent need for novel advanced hybrid materials composed of biocompatible polymers.39–44 The last decade has seen the publication of hundreds of papers involving poly(N-isopropylacrylamide), (PNIPAM). This interest is driven by the famous lower critical solution temperature (LCST) behavior, where heating an aqueous solution of PNIPAM above 32 °C induces phase separation. This phase transition is associated with the LCST (Lower Critical Solution Temperature) of the thermosensitive polymers. Below this temperature, the solution is homogeneous, the polymer chains are swollen and the polymer exists in water soluble form. At this stage, water and hydrophilic moieties of the polymer are bound to each other. This prevents interactions of the polymer chains and intrapolymer association. Above this temperature, a phase transition takes place. At this stage, the hydrogen bonds between the water molecules and the hydrophilic moieties are disrupted, water is expelled from the polymer chains which lead to their contraction and subsequently they shrink. Hydrophobic interactions among the polymer chains persist and lead to the aggregation of the polymer. However, the complete aggregation only occurs above a critical concentration of free PNIPAM which depends on the molecular weight of the free PNIPAM in solution.45 Below the critical concentration (Guinier regime) the PNIPAM exists in solution in the form of separate macromolecules.45,46
PNIPAM-based polymers are a promising material in medical applications. The unique properties of PNIPAM-based polymers and copolymers indicate them as an innovative drug carrier for drug delivery systems, diagnostic substance carriers, and also as biosensors. The main difficulty in the use of PNIPAM homopolymer in the preparation of delivery systems is its LCST which is around 31–32 °C. Hence LCST for efficient application of the nanosystems based on PNIPAM should be shifted to around 37 °C. Currently, extensive research is going on with regard to the modification of the LCST of the thermosensitive polymers in drug delivery systems for specific targeting of the drugs. On the other hand, recent works point to the high potential of the effect of light-induced plasmonic heating of metal NPs (Au NPs in particular) in hyperthermia treatment at the cellular level.47–50 However, these materials also need appropriate delivery vehicle in order for their controlled release and targeted delivery. In this respect, PNIPAM has been proposed as the stimuli responsive carrier of the NPs.51,52 These applications of PNIPAM/Au NPs hybrids need the knowledge of the peculiarities of the laser-induced phase transition in the PNIPAM/Au NPs system at the nanoscale. The SPR in the Au NPs with its famous sensor properties gives such possibility. The changes in the surrounding lead to change in the spectral characteristics of SPR that can be monitored, e.g. by light extinction spectroscopy. In this regard, it is necessary to mention the recent works36–38 where the laser-induced phase transformations in core–shell Au@PNIPAM NPs at cw laser irradiation36,37 and pulsed laser irradiation38 were studied. The considerable red shift of SPR in Au@PNIPAM NPs was observed in ref. 36 and 37 testifying the formation of aggregates. The formation of aggregates was found to have a reversible character when the SPR scattering spectra relaxed to initial state after turn-off of the laser irradiation.
In present work we study the hybrid nanosystem containing the Au NPs with star-like branched PNIPAM with dextran core ad grafted PNIPAM arms (D-g-PNIPAM). The aim of this work was to study the peculiarities of structural transformations in D-g-PNIPAM/AuNPs nanosystem induced by laser illumination with wavelengths resonant and non-resonant with SPR in Au NPs. To detect such transformations, the behavior of the extinction peak of SPR in Au NPs with variation of laser intensity has been monitored. The LCST phase transition has been detected only at resonant SPR excitation, while non-resonant laser illumination has not lead to any transformation in D-g-PNIPAM/AuNPs system. The resonant plasmon heating of Au NPs leads to LCST transition in D-g-PNIPAM/AuNPs. At the temperature higher than LCST point, an additional low-energy plasmon peak appears in the extinction spectrum indicating the fact of the aggregation of D-g-PNIPAM/AuNPs macromolecules, Fig. 1. At the backward phase transition with decreasing of laser intensity the low-energy plasmon peak of aggregates remains in the extinction spectrum indicating the fact of the existence of aggregates at the temperatures lower than LCST point. The formation of D-g-PNIPAM/AuNPs aggregates at the laser irradiation matches the results reported for Au@PNIPAM core–shell NPs in ref. 36 and 37. However, the aggregation reported in ref. 36 and 37 has reversible character that is dissimilar to the results of our studies. Probably, the causes of this difference may be in following: (i) in our work we use D-g-PNIPAM while in ref. 36 and 37 the simple PNIPAM has been used, (ii) in our work the Au NPs were synthesized in situ in the water solution of D-g-PNIPAM while in ref. 36 and 37 the initially synthesized bare Au NPs have been immersed in solutions of PNIPAM, (iii) in our work we study the small 8 nm Au NPs bound to PNIPAM grafts while in ref. 36 and 37 the large Au@PNIPAM core–shell NPs with gold 100 nm core have been used. Also, the observed behavior of D-g-PNIPAM/AuNPs system at the laser heating is completely opposite to our recent results46 on the LCST phase transition in the same system occurring at their usual heating–cooling cycle when no formation of aggregates has been found and structural transformations have been found to be reversible. The observed difference has been rationalized to be result of the action of laser induced attractive optical forces between the Au NPs that promote the formation of aggregates and counteract their destruction.
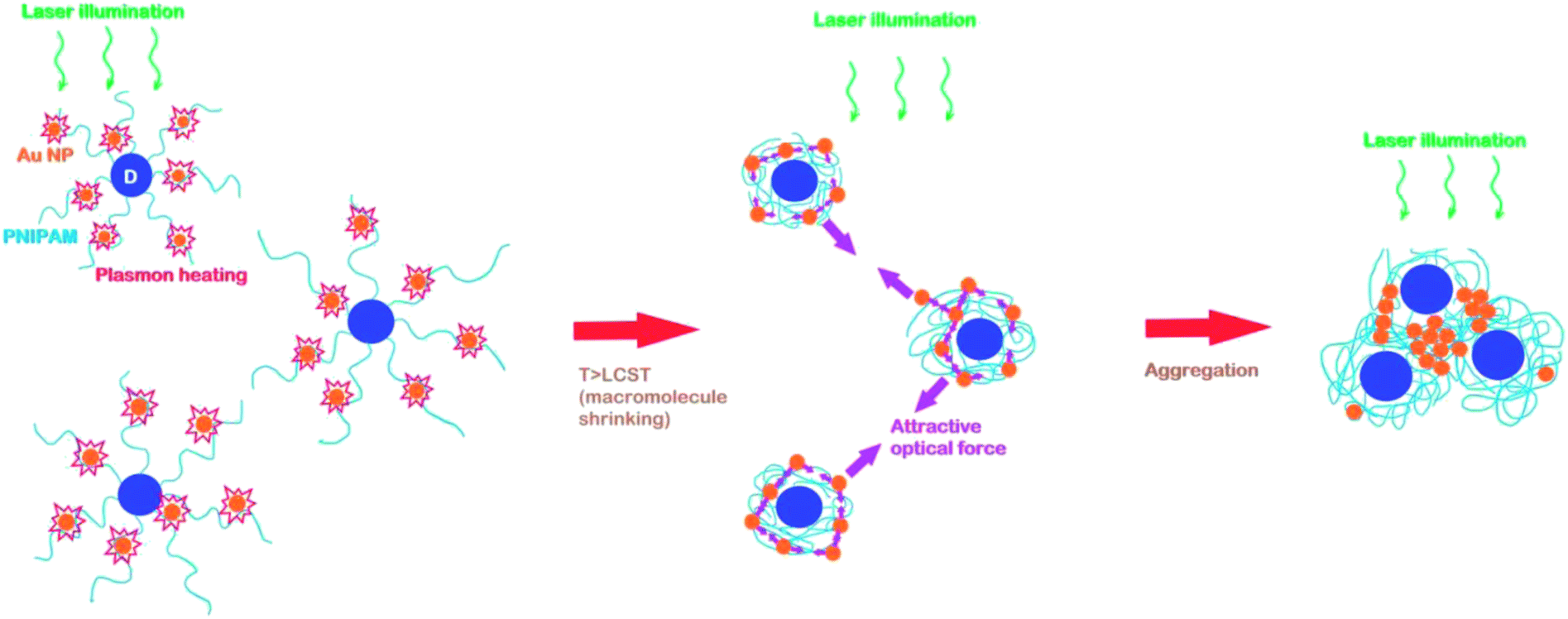 | ||
| Fig. 1 Schematics of laser induced structural transformations in D-g-PNIPAM/AuNPs nanosystem. | ||
2. Experimental
2.1. Synthesis of D-g-PNIPAM/AuNPs nanosystem
Details of the synthesis of studied D-g-PNIPAM/AuNPs hybrid nanosystem have been reported in our recent work.46 Dextran was purchased from Fluka with characteristics given by the manufacturer: Mw = 7 × 104. Cerium(IV) ammonium nitrate (CAN) from Aldrich was used as initiator. Dextran sample and the ceric salt were used without further purification. N-Isopropylacrylamide (NIPAM) from Aldrich was twice re-crystallized from hexane and dried under vacuum at room temperature for 24 h. The number of grafting sites per dextran backbone was pre-determined by molar ratio of acrylamide to cerium ions and it was equal to 15. The reaction path is following. The calculated amount of dextran was dissolved in 100 ml of distilled water. This solution was stirred while removal of the dissolved oxygen was achieved by bubbling a gentle flux of argon for about 20 min. Then Ce(IV)/HNO3 initiator (0.125 N HNO3) was injected to obtain desirable grafts number. NIPAM monomer was added and the polymerization proceeded at room temperature under argon atmosphere for 24 h. The synthesized copolymers were precipitated into a mixture water–methanol, re-dissolved in water and finally freeze-dried.The molecular structure of D-g-PNIPAM polymers and the peculiarities of conformational transition for series of copolymers consisting of dextran core of various size and 15 PNIPAM grafts were reported in ref. 36. The synthesized D-g-PNIPAM copolymer is star-like with low polydispersity. The star-like structure of the synthesized D-g-PNIPAM provides greater stability of colloids comparing to colloids with linear PNIPAM.53 Also, such structure allows to raise the LCST point from 32 °C for linear PNIPAM to 33.8 °C that is closer to the temperature of human body.54
The formation of Au NPs was performed by the reduction of Au ions in aqueous solutions of D-g-PNIPAM copolymer, Fig. 2(a). 0.1 M HAuCl4 aqueous solution was added to 1 ml of polymer solution (C = 1 g l−1) and stirred during 20 min at t = 25 °C. Then, 0.1 M of NaBH4 solution was added drop by drop at stirring. The obtained Au sols were stored in cold dark. The concentration of Au NPs was about 0.086 mg ml−1. The D-g-PNIPAM/AuNPs nanosystem was prepared in polymer solution below the concentration of crossover (Guinier regime) for D-g-PNIPAM. In the theory of diluted polymer solutions, the intrinsic viscosity is used as a criterion for the estimation of a solution concentration regime. Since the size of a macromolecule in the solution determines whether the solution is diluted or moderately concentrated, whereas the intrinsic viscosity is proportional to the macromolecular volume in the solution. The solution is diluted if its volume occupied by macromolecules is much smaller than the total solution volume. With the growth of the polymer concentration, the solution structure changes, by forming a network of intermolecular links, when achieving a critical concentration of overlapping macromolecular coils, C*, the beginning of the so-called crossover region. In the case of flexible-chain polymers, the critical crossover concentration C* can be determined experimentally by the viscometric method:55,56 C* = 1/η, where η is the intrinsic viscosity. As it was reported in our previous paper54 on the synthesis and characterization of D-g-PNIPAM copolymers, the intrinsic viscosity for D-g-PNIPAM sample was equal to 1.28 dl g−1. Thus, C* = 1.25 g dl−1 for sample studied in present work. We have used the concentration of 0.1 g dl−1 that is more than 10 times lower the concentration of crossover C*.
 | ||
| Fig. 2 (a) In situ synthesis of Au NPs in D-g-PNIPAM polymer matrix. (b) The change of color of D-g-PNIPAM/AuNPs aqueous solution from pink to grey after the 532 nm laser treatment of sample. Such change of color proves the fact of aggregation of Au NPs. | ||
2.2. Transmission electron microscopy (TEM)
For the sample preparation 400 mesh Cu grids with plain carbon film were rendered hydrophilic by a glow discharge treatment (Elmo, Cordouan Technologies Bordeaux France). A 5 μl drop was deposited and let adsorbed for 1 min then the excess of solution was removed with a piece of filter paper. TEM measurements were carried on two TEMs, Tecnai G2 or CM12 (FEI, Eindhoven Netherlands) and the images were acquired with a ssCCD Eagle camera on the Tecnai and a Megaview SIS Camera on the CM12. The typical TEM image of D-g-PNIPAM/AuNPs nanosystem obtained at temperature of 25 °C is presented in Fig. 3. It is seen that Au NPs have size of d = (8 ± 3) nm and are spherical in shape. Due to the considerably lower contrast of D-g-PNIPAM macromolecules comparing to Au NPs, the polymer macromolecules are not visible on the TEM image. The size of polymer macromolecules can be estimated as the size of clusters of Au NPs separated from each other. Such estimation gives the size of D-g-PNIPAM macromolecule of d = (40 ± 10) nm. Au NPs were synthesized in dilute aqueous solution of D-g-PNIPAM (polymer concentration was below the concentration of crossover). Thus, the distance between the neighboring PNIPAM macromolecules is larger than the macromolecule size by several times.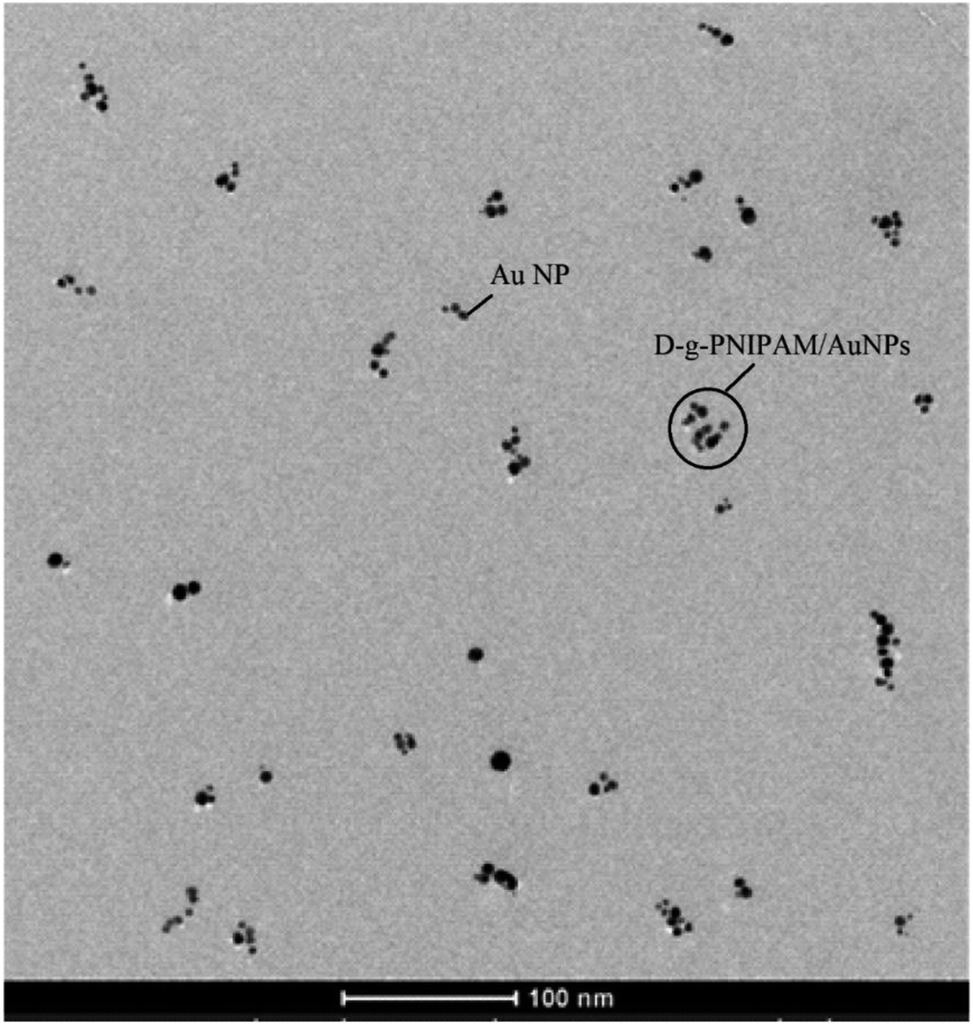 | ||
| Fig. 3 TEM image of D-g-PNIPAM/AuNPs hybrid nanosystem. | ||
2.3. Light extinction spectroscopy
The light extinction spectra were measured using Cary 60 UV-VIS spectrophotometer (Agilent Technologies, Inc). The D-g-PNIPAM/AuNPs and D-g-PNIPAM aqueous solutions were placed in 1 cm × 1 cm × 5 cm polished cell. The spectra were measured both with illumination of the solution by laser beam and without it. The cw diode lasers with wavelengths of 405 nm, 532 nm and 655 nm were used. The laser light with wavelength of 532 nm matches perfectly the resonance with SPR in Au NPs which is at 538 nm. Meanwhile, the laser light with wavelengths of 405 nm and 655 nm supports the non-resonant illumination. During extinction spectra measurement the exciting laser beam and probing beam of Xe-lamp were spatially aligned. Respectively, spectra from the laser illuminated area of the solution were measured. The angle between the laser and Xe-lamp beams was 90°. The extinction spectra were measured at gradual variation of laser intensity in the range of 0–425 W cm−2 in straight and backward directions. The time interval between the measurements of spectra at different laser intensities was 5 min, thus the laser heating and cooling of the sample were carried out quite slowly to equilibrate the temperature of the sample.The spectra of optical density D = log(I0/I) were measured, where I0 is the intensity of probing Xe-lamp light passed through the aqueous solution of D-g-PNIPAM polymer without Au NPs and I is the intensity passed through D-g-PNIPAM/AuNPs solution. Let us note that both spectra of the D-g-PNIPAM without Au NPs and the D-g-PNIPAM/AuNPs hybrid nanosystem were measured at the same laser intensities. Therefore, the spectra presented below are ones of gold NPs affected by the laser induced changes in their D-g-PNIPAM polymer surrounding.
3. Results and discussion
3.1. Extinction spectra of D-g-PNIPAM/AuNPs hybrid nanosystem under laser irradiation: experiment
The extinction spectra of Au NPs embedded in D-g-PNIPAM/AuNPs dilute aqueous solution were measured both with laser illumination and without it, Fig. 4. It is seen that all spectra contain the SPR peak of separate (non-aggregated) Au NPs with maximum at about 538 nm marked as S-peak. One can see that at the absence of laser illumination and at non-resonant with SPR laser illumination at wavelengths of 405 nm and 655 nm the measured spectra coincide with high accuracy. Meanwhile, the resonant with SPR laser illumination at wavelength of 532 nm leads to considerable transformation of the extinction spectrum. The resonant laser illumination leads to considerable decrease of S-peak area and to appearance of additional low-energy peak (marked as A-peak). Note that A-peak appears in the spectrum only at resonant illumination. Such illumination leads to plasmon heating of NPs which causes the LCST phase transition in D-g-PNIPAM macromolecules when the macromolecules shrink. Taking into account this fact as well as the facts that A-peak is at low-energy side from S-peak of separate Au NPs57 and that A-peak area, wavelength and width depend strongly on the laser intensity (in other words on the temperature), as it is discussed below, it is reasonable to assume that A-peak originates from the SPR mode of aggregates of Au NPs. The assumption of the aggregation of Au NPs is proved by a substantial change in color of AuNPs/PNIPAM water solution from pink to grey occurring after the cycle of the 532 nm laser treatment of the sample, Fig. 2(b). Thus, taking into account the considerable difference of the extinction spectra measured at resonant illumination and at non-resonant illumination or without it we can conclude that observed transformations in polymer surrounding of Au NPs have plasmonic nature.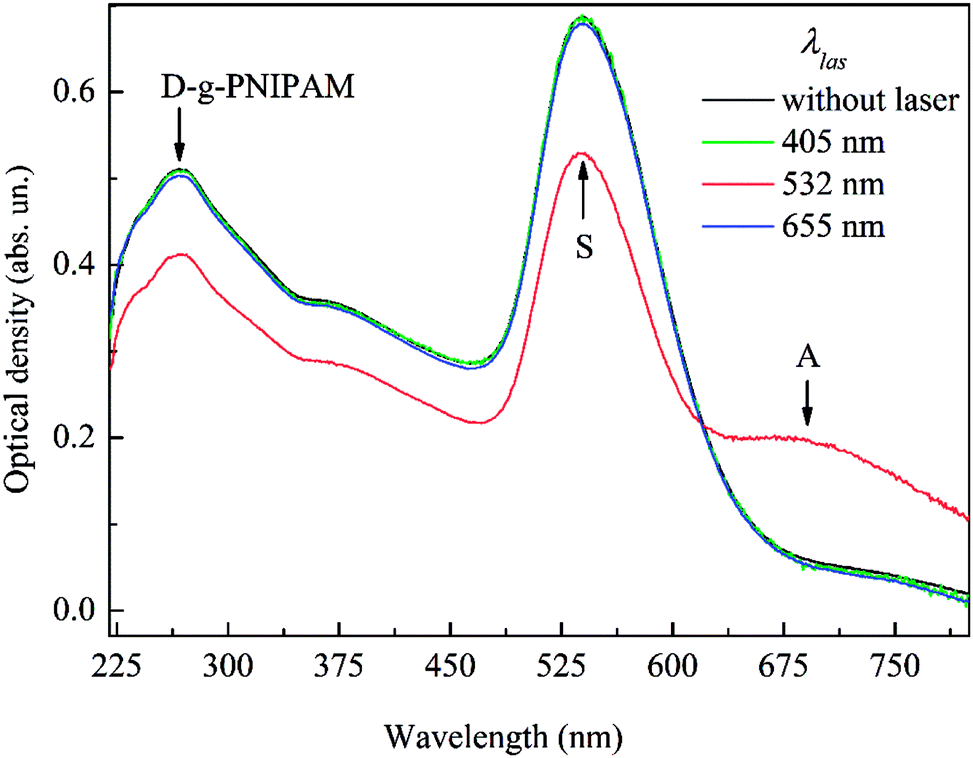 | ||
| Fig. 4 Extinction spectra of Au NPs in D-g-PNIPAM/AuNPs nanosystem after laser illumination with various wavelengths (laser intensity 425 W cm−2) and without illumination. | ||
Besides the plasmonic S and A-peaks of Au NPs, the absorption spectra of D-g-PNIPAM/AuNPs nanohybrid contain the absorption peak of D-g-PNIPAM copolymer with maximum at 268 nm, Fig. 4. One can see that the spectral shape of this peak remains the same for samples treated by laser with various wavelengths and for non-treated ones. This fact indicates that D-g-PNIPAM macromolecules have not been damaged under the laser irradiation retaining their integrity.
Further, we measured the extinction spectra of Au NPs in D-g-PNIPAM/AuNPs nanosystem at resonant laser illumination increasing gradually the intensity of laser beam from 0 to 425 W cm−2 and then gradually decreasing it back to 0. The behavior of the spectrum at the increase of laser intensity is shown in Fig. 5(a), and the behavior occurring at backward decrease of laser intensity is shown in Fig. 5(b). It is seen that the transformation of the spectrum at the increase of laser intensity is much more pronounced than one at decrease of laser intensity. To quantitatively analyze the effects of variation of laser intensity on Au NPs SPR spectral characteristics (area, wavelength and width) in studied nanosystem, the spectra were fitted by the basic Lorentzian peaks. The respective dependencies for two plasmonic peaks are shown in Fig. 6(a)–(c). The main features of the dependences of SPR spectral characteristics on laser intensity are the following.
 | ||
| Fig. 5 Behavior of the extinction spectrum of Au NPs in D-g-PNIPAM/AuNPs nanosystem at the increase of laser intensity from 0 to 425 W cm−2 (a) and at its backward decrease (b). The laser wavelength is 532 nm that matches the laser resonance with SPR in Au NPs. The arrows show the transformation of plasmon S-peak of separate Au NPs and A-peak of aggregated ones. | ||
 | ||
| Fig. 6 Behavior of normalized area (a), spectral shift (b) and normalized width (c) of SPR S-peak of separate Au NPs and A-peak of aggregated ones in D-g-PNIPAM/AuNPs nanosystem at the increase of laser intensity from 0 to 425 W cm−2 and at its backward decrease. The laser wavelength is 532 nm that matches the laser resonance with SPR in Au NPs. The values of SPR peak area, shift and width were taken in relation to the respective values in the absence of laser illumination. The arrows show the direction of process. | ||
At the increase of laser intensity from 0 to 22 W cm−2 the strong (on 20%) decrease of area of high-energy S-peak of separate (non-aggregated) Au NPs and the simultaneous strong (on 27%) increase of area of low-energy A-peak is observed. At further increase of laser intensity more than on an order of magnitude to 425 W cm−2 the area of S-peak decreases and the area of A-peak increases monotonically, but their variation becomes substantially slower. The above results on the behavior of plasmon peaks correlate with the behavior of such SPR spectral characteristics as SPR peaks wavelength and width, Fig. 6(b) and (c). Indeed, the increase of laser intensity from 0 to 22 W cm−2 leads to strong (44 nm) red shift and considerable broadening (on 36%) of low-energy A-peak. Similarly, to behavior of peak area at laser intensity higher than 22 W cm−2 the dependences of A-peak wavelength and width on laser intensity become much weaker remaining monotonic. Meanwhile, the dependences of maximum wavelength and width of S-peak on laser intensity are significantly weaker, being, nevertheless, qualitatively similar to the corresponding dependencies for A-peak. Namely, the sharp blue shift and narrowing of S-peak occur at the increase of laser intensity from 0 to 22 W cm−2 becoming much weaker and remaining monotonic at higher laser intensities.
An important point which has to be considered is the estimation of the temperatures reached in our experiments. High temperatures can lead to the thermal destroying of polymer macromolecules. The local temperature near the Au NPs can be estimated proceeding from the dependence of SPR shift on the laser intensity. Considering the dependences of SPR spectral characteristics on 532 nm laser intensity (Fig. 6), we can conclude that at laser intensity of 22 W cm−2 the LCST transition in D-g-PNIPAM/AuNPs macromolecule occurs. The LCST temperature in studied D-g-PNIPAM/AuNPs nanosystem was determined in our recent work46 to be 33.5 °C. The rate of the refractive index decrease at the increase of the temperature dn/dT for PNIPAM water solution at the temperatures higher than LCST point was taken from ref. 58 to be of −10−4 °C−1. Then taking the SPR shift Δλ of S-peak for non-aggregated Au NPs (Fig. 6(b)) for laser intensities higher than 22 W cm−2 and the calculated rate of SPR shift in Au NPs with the variation of refractive index of surrounding medium Δλ/Δn from our recent work46 we estimated the temperature induced decrease of the refractive index at laser intensity increase from 22 to 425 W cm−2 to be of Δn = −0.0035. Thus, taking the change of Δn = −0.0035 and the value of dn/dT from ref. 46, we estimated the highest temperature reached in our experiments at laser intensity of 425 W cm−2 to be about 70 °C. Let us note that the laser induced rise of temperature reached in our experiments is quite moderate. This fact explains why there is no destruction of D-g-PNIPAM macromolecules under the laser irradiation of the sample that is mentioned above.
3.2. Discussion
Let us discuss the observed behavior of plasmonic S and A-peaks. The increase of laser intensities through the plasmonic heating causes the increase of Au NPs temperature. The heat from NPs is transferred to the surrounding D-g-PNIPAM polymer causing its LCST phase transition in which D-g-PNIPAM macromolecule shrinks. The strong changes of the spectral characteristics of both SPR peaks are apparently caused by the phase transition. Really, the blue shift and narrowing of plasmonic peak of separate Au NPs (S-peak) are similar to those observed for the same D-g-PNIPAM/AuNPs nanosystem at its usual heating.46 The respective behavior was rationalized in ref. 46 as the result of jump-like decrease of the refractive index of PNIPAM polymer during the phase transition. However, let us note that in our previous study46 on the influence of usual heating on D-g-PNIPAM/AuNPs the single plasmonic peak of separate Au NPs is observed in the spectra. Meanwhile, in the case of resonant laser heating an additional plasmonic A-peak appears in the spectra. This peak was above assumed to originate from the SPR in the aggregates of Au NPs. Indeed, the shrinking of the polymer macromolecules due to LCST phase transition leads to sharp decrease of the distance between the Au NPs. However, since the concentration of studied solution is lower than critical concentration needed for macromolecules aggregation, the usual heating does not lead to the formation of their aggregates and, respectively, to the aggregation of the Au NPs.46 Meanwhile, the laser illumination leads not only to the heating of D-g-PNIPAM/AuNPs system but the laser electric field induces the plasmonic dipoles in the Au NPs.The induced dipoles cause an appearance of the attractive forces acting between the NPs as it is discussed in the Introduction. Let us note that there are Au NPs of two types in the solution. First ones are the free Au NPs which are unbound with polymer macromolecules. Second ones are the Au NPs which are bound with polymer. The optical forces attract the Au NPs of both types. Such attraction should lead to the aggregation of Au NPs. Correspondingly, the aggregates of two types should be formed, namely the aggregates unbound with D-g-PNIPAM macromolecules and the aggregates bound with them. However, taking into account that strong changes of A-peak spectral characteristics with variation of laser intensity occur synchronously with LCST phase transition in D-g-PNIPAM/AuNPs macromolecules one can conclude that sufficient aggregation occurs only for Au NPs bound with polymer and the unbound Au NPs aggregate quite slightly. Most probably, this is due to the following. Since the distance between the unbound NPs are quite large, the attractive forces acting between them are too weak to cause their noticeable aggregation. Meanwhile, the optical forces acting between the Au NPs bound with polymer macromolecules strengthen strongly when the macromolecules shrink. Such strong forces attract both the Au NPs within the macromolecule and the Au NPs belonging to different ones. As a result, the attraction should cause both the formation of the aggregates of Au NPs in separate D-g-PNIPAM/AuNPs macromolecules and the aggregation of D-g-PNIPAM/AuNPs macromolecules with the aggregates of Au NPs inside. The formation of Au NPs aggregates causes an appearance of the SPR modes of aggregates which manifest themselves as an additional plasmonic A-peak in the spectra. During the LCST phase transition the separate Au NPs aggregate that leads to sharp increase of the area of A-peak of aggregates and to respective sharp decrease of the area of S-peak of separate NPs. Also, the action of attractive optical forces leads to the fact that during phase transition the aggregates become larger and denser. This leads to stronger plasmonic coupling of the Au NPs in aggregates that causes the increase of frequency (red shift) and increase of width (broadening) of the collective SPR mode of Au NPs aggregates. Considering the sharp dependences of S and A-peaks spectral characteristics on the laser intensity in the range of 0–22 W cm−2, one can conclude that at the intensity of 22 W cm−2 the phase transition in D-g-PNIPAM/AuNPs system is over. Note, however, that at laser intensities higher than 22 W cm−2 the change of spectral characteristics of S and A-peaks are not as strong as during a phase transition, but they remain qualitatively the same. One can assume that this is due to the fact that the increase of laser intensity causes the strengthening of plasmonic attractive forces which make the aggregates larger and denser. Let us note that dependences of the spectral characteristics of both SPR peaks on laser intensity is linear that is accordance with theoretical predictions of the linear dependence of plasmonic optical forces on the laser intensity.26–28
Let us analyze the behavior of the extinction spectrum of Au NPs at the backward decrease of laser intensity from 425 W cm−2 to 0, Fig. 5(b). It is seen that the transformation of the spectrum is considerably slighter than one observed at the increase of laser intensity. The respective behavior of the SPR spectral characteristics occurring at the decrease of laser intensity is presents in Fig. 6. One can see that prominent hysteresis is observed. Indeed, the decrease of laser intensity from 425 W cm−2 to about 22 W cm−2 leads to vanishingly slight variation of plasmon peaks area, spectral position and width. As it is noted above, at 22 W cm−2 the temperature of D-g-PNIPAM/AuNPs system is about 42 °C that is the high limit of LCST phase transition occurring in the temperature interval of 32–42 °C. Thus, at the decrease of temperature down to the LCST point the D-g-PNIPAM/AuNPs aggregates exist almost unchanged. The further decrease of the laser intensity lower than 22 W cm−2 that is equivalent to the decrease of temperature lower than LCST point leads to start of the reverse changes in the area, wavelength and width of plasmonic S and A-peaks, i.e. the increase of area, red shift and broadening of S-peak and simultaneous decrease of area, blue shift and narrowing of A-peak. Such changes in the plasmonic peaks spectral characteristics evidence the fact of destruction of aggregates. Let us note, however, that the changes occurring at the decrease of laser intensity are considerably slighter than ones occurring at its increase. It is seen in Figs. 5(b) and 6(a)–(c) that the decrease of laser intensity back to 0 (absence of laser illumination) leads to non-reversible changes of both SPR peaks, i.e. the hysteresis exists. The cycle of increase-decrease of laser intensity causes the residual changes in area, spectral position and width of plasmonic peaks. Thus, one can conclude that after the finish of laser illumination the quite large amount of D-g-PNIPAM/AuNPs macromolecules remains in the aggregated state. Let us note that an essential difference exists between the behavior of D-g-PNIPAM/AuNPs nanosystem occurring at its usual and laser induced heating–cooling cycles. Namely, the usual heating–cooling cycle does not cause the aggregates formation and leads to reversible structural transformations in this nanosystem.46 Meanwhile, the cycle of laser induced heating–cooling causes the formation of aggregates that remain in the solution after turn-off the laser illumination. The only reason that can cause such a fundamentally different behavior is, in our opinion, the action of optical forces arising due to the excitation of surface plasmons in Au NPs.
4. Conclusions
In conclusion, the extinction spectra of Au nanoparticles in dilute aqueous solution of dextran-grafted-PNIPAM/AuNPs copolymer hybrid macromolecules have been measured at laser illumination intensity variation in the range of 0–425 W cm−2 using the cw diode lasers with wavelengths of 405 nm, 532 nm and 655 nm. The 405 nm and 655 nm laser light is non-resonant with SPR in Au NPs, and 532 nm provides the resonant excitation of SPR. The SPR extinction peaks have been used as an optical sensor of the structural transformations in D-g-PNIPAM/AuNPs nanosystem. It has been shown that illumination with non-resonant laser light does not lead to any observable change in this nanosystem. Meanwhile, the resonant laser illumination leads to strong change of the extinction peak of Au NPs reflecting the fact of structural transformations in D-g-PNIPAM/AuNPs nanosystem caused by laser induced local plasmon heating of Au NPs. The thermally induced LCST structural phase transition in D-g-PNIPAM/AuNPs macromolecule which is accompanied by its shrinking has been detected proceeding from the analysis of SPR peak area, spectral position and width changes occurring at the variation of laser intensity. The LCST phase transition causes the appearance of an additional peak in the extinction spectrum originating from the excitation of collective SPR mode in the aggregates of Au NPs. Such aggregation occurs due to aggregation of D-g-PNIPAM/AuNPs macromolecules. The increase of laser intensity leads to the formation of larger and denser D-g-PNIPAM/AuNPs aggregates. The behavior of SPR peaks at the backward decrease of laser intensity shows the hysteresis non-reversible character of the structural transformations in the studied nanosystem, i.e. the aggregates remains in the solution after the turn-off the laser illumination. An essential difference has been revealed between the behavior of D-g-PNIPAM/AuNPs nanosystem occurring at its usual and laser incused heating–cooling cycles. Unlike the laser induced heating, the usual heating–cooling cycle does not cause the formation of aggregates and leads to reversible structural transformations in this nanosystem. Such fundamental difference has been rationalized as the result of action of attractive optical forces arising due to the excitation of surface plasmons in Au NPs. The attractive plasmonic forces facilitate the formation of the aggregates and counteract their destruction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NATO Science for Peace and Security (SPS) Programme (grant NUKR.SFPP 984617).References
- E. Ozbay, Plasmonics: merging photonics and electronics at nanoscale dimensions, Science, 2006, 311, 189–193 CrossRef CAS PubMed.
- W. L. Barnes, A. Dereux and T. W. Ebbesen, Surface plasmon subwavelength optics, Nature, 2003, 424, 824–830 CrossRef CAS PubMed.
- M. I. Stockman, Nanoplasmonics: past, present, and glimpse into future, Opt. Express, 2011, 19, 22029–22106 CrossRef PubMed.
- A. O. Govorov, W. Zhang, T. Skeini, H. Richardson, J. Lee and N. A. Kotov, Gold nanoparticle ensembles as heaters and actuators: melting and collective plasmon resonances, Nanoscale Res. Lett., 2006, 1, 84–90 CrossRef.
- A. O. Govorov and H. H. Richardson, Generating heat with metal nanoparticles, Nano Today, 2007, 2, 30–38 CrossRef.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Plasmon-induced hot carrier science and technology, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS PubMed.
- G. Baffou and R. Quidant, Thermo-plasmonics: using metallic nanostructures as nano-sources of heat, Laser Photonics Rev., 2013, 7, 171–187 CrossRef CAS.
- Z. Fang, Y. R. Zhen, O. Neumann, A. Polman, F. J. García de Abajo, P. Nordlander and N. J. Halas, Evolution of light-induced vapor generation at a liquid-immersed metallic nanoparticle, Nano Lett., 2013, 13, 1736–1742 CrossRef CAS PubMed.
- B. Choudhuri, A. Mondal, J. C. Dhar, N. K. Singh, T. Goswami and K. K. Chattopadhyay, Enhanced photocurrent from generated photothermal heat in indium nanoparticles embedded TiO2 film, Appl. Phys. Lett., 2013, 102, 233108 CrossRef.
- H. A. Atwater and A. Polman, Plasmonics for improved photovoltaic devices, Nat. Mater., 2010, 9, 205–213 CrossRef CAS PubMed.
- D. Erickson, D. Sinton and D. Psaltis, Optofluidics for energy applications, Nat. Photonics, 2011, 5, 583–590 CrossRef CAS.
- J. A. Schuller, T. Taubner and M. L. Brongersma, Optical antenna thermal emitters, Nat. Photonics, 2009, 3, 658–661 CrossRef CAS.
- J. R. Adleman, D. A. Boyd, D. G. Goodwin and D. Psaltis, Heterogenous catalysis mediated by plasmon heating, Nano Lett., 2009, 9, 4417–4423 CrossRef CAS PubMed.
- P. Christopher, H. L. Xin and S. Linic, Visible-light-enhanced catalytic oxidation reactions on plasmonic silver nanostructures, Nat. Chem., 2011, 3, 467–472 CrossRef CAS PubMed.
- S. Ibrahimkutty, J. Kim, M. Cammarata, F. Ewald, J. Choi, H. Ihee and A. Plech, Ultrafast structural dynamics of the photocleavage of protein hybrid nanoparticles, ACS Nano, 2011, 5, 3788–3794 CrossRef CAS PubMed.
- J. Lee, A. O. Govorov and N. A. Kotov, Nanoparticle assemblies with molecular springs: a nanoscale thermometer, Angew. Chem., Int. Ed., 2005, 44, 7439–7442 CrossRef CAS PubMed.
- Z. Z. J. Lim, J. E. J. Li, C. T. Ng, L. Y. L. Yung and B. H. Bay, Gold nanoparticles in cancer therapy, Acta Pharmacol. Sin., 2011, 32, 983–990 CrossRef CAS PubMed.
- G. von Maltzahn, J.-H. Park, K. Y. Lin, N. Singh, C. Schwöppe, R. Mesters, W. E. Berdel, E. Ruoslahti, M. J. Sailor and S. N. Bhatia, Nanoparticles that communicate in vivo to amplify tumour targeting, Nat. Mater., 2011, 10, 545–552 CrossRef CAS PubMed.
- E. C. Dreaden, M. A. Mackey, X. Huang, B. Kang and M. A. El-Sayed, Beating cancer in multiple ways using nanogold, Chem. Soc. Rev., 2011, 40, 3391–3404 RSC.
- N. S. Abadeer and C. J. Murphy, Recent progress in cancer thermal therapy using gold nanoparticles, J. Phys. Chem. C, 2016, 120, 4691–4716 CrossRef CAS.
- O. A. Yeshchenko and V. V. Kozachenko, Light-induced heating of dense 2D ensemble of gold nanoparticles: dependence on detuning from surface plasmon resonance, J. Nanopart. Res., 2015, 17, 296 CrossRef.
- O. A. Yeshchenko, N. V. Kutsevol and A. P. Naumenko, Light-induced heating of gold nanoparticles in colloidal solution: dependence on detuning from surface plasmon resonance, Plasmonics, 2016, 11, 345–350 CrossRef CAS.
- K. Svoboda and S. M. Block, Optical trapping of metallic Rayleigh particles, Opt. Lett., 1994, 19, 930–932 CrossRef CAS PubMed.
- S. Sato, Y. Harada and Y. Waseda, Optical trapping of microscopic metal particles, Opt. Lett., 1994, 19, 1807–1809 CrossRef CAS PubMed.
- J. R. Arias-González and M. Nieto-Vesperinas, Optical forces on small particles: attractive and repulsive nature and plasmon-resonance conditions, J. Opt. Soc. Am. A, 2003, 20, 1201–1209 CrossRef.
- P. Chu and D. L. Mills, Laser-induced forces in metallic nanosystems: the role of plasmon resonances, Phys. Rev. Lett., 2007, 99, 127401 CrossRef PubMed.
- J. Ng, R. Tang and C. T. Chan, Electrodynamics study of plasmonic bonding and antibonding forces in a biosphere, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 195407 CrossRef.
- A. J. Hallock, P. L. Redmond and L. E. Brus, Optical forces between metallic particles, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1280–1284 CrossRef CAS PubMed.
- A. S. Urban, S. Carretero-Palacios, A. A. Lutich, T. Lohmüller, J. Feldmann and F. Jäckel, Optical trapping and manipulation of plasmonic nanoparticles: fundamentals, applications, and perspectives, Nanoscale, 2014, 6, 4458–4474 RSC.
- Y. Arita, G. Tkachenko, N. McReynolds, N. Marro, W. Edwards, E. R. Kay and K. Dholakia, Invited article: optical trapping of ultrasmooth goldnanoparticles in liquid and air, APL Photonics, 2018, 3, 070801 CrossRef.
- H. Xu and M. Kall, Surface-plasmon-enhanced optical forces in silver nanoaggregates, Phys. Rev. Lett., 2002, 89, 246802 CrossRef PubMed.
- F. Svedberg, Z. Li, H. Xu and M. Kall, Creating hot nanoparticle pairs for surface-enhanced Raman spectroscopy through optical manipulation, Nano Lett., 2006, 6, 2639–2641 CrossRef CAS PubMed.
- D. Fava, M. A. Winnik and E. Kumacheva, Photothermally triggered self-assembly of gold nanorods, Chem. Commun., 2009, 18, 2571–2573 RSC.
- K. Murakoshi and Y. Nakato, Formation of linearly arrayed gold nanoparticles on gold single-crystal surfaces, Adv. Mater., 2000, 12, 791–795 CrossRef CAS.
- T. Ding, V. K. Valev, A. R. Salmon, C. J. Forman, S. K. Smoukov, O. A. Scherman, D. Frenkel and J. J. Baumberg, Light-induced actuating nanotransducers, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5503–5507 CrossRef CAS PubMed.
- I. Aibara, S. Mukai and S. Hashimoto, Plasmonic-heating-induced nanoscale phase separation of free poly(N-isopropylacrylamide) molecules, J. Phys. Chem. C, 2016, 120, 17745–17752 CrossRef CAS.
- I. Aibara, J. Chikazawa, T. Uwada and S. Hashimoto, Localized phase separation of thermoresponsive polymers induced by plasmonic heating, J. Phys. Chem. C, 2016, 121, 22496–22507 CrossRef.
- S. Murphy, S. Jaber, C. Ritchie, M. Karg and P. Mulvaney, Laser flash photolysis of Au-PNIPAM core–shell nanoparticles: dynamics of the shell response, Langmuir, 2016, 32, 12497–12503 CrossRef CAS PubMed.
- M. A. Ward and T. K. Georgiu, Thermoresponsive polymers for biomedical applications, Polymer, 2011, 3, 1215–1242 CAS.
- M. Joglecar and B. G. Trewyn, Polymer-based stimuli responsible nanosystems for biomedical applications, Biotechnol. J., 2013, 8, 931–945 CrossRef PubMed.
- E. Cabane, X. Zhang, K. Langowska, C. G. Palivan and W. Meier, Stimuli responsible polymers and their application in nanomedicine, Biointerphases, 2012, 7, 1–27 CrossRef PubMed.
- C. Gong, T. Qi, X. Wei, Y. Qu, Q. Wu, F. Luo and Z. Qian, Thermosensitive polymeric hydrogels as drug delivery systems, Curr. Med. Chem., 2016, 20, 79–94 CrossRef.
- V. Dal Lago, L. França de Oliveira, K. de Almeida Gonçalves, J. Kobarg and M. B. Cardoso, Size-selective silver nanoparticles: future of biomedical devices with enhanced bactericidal properties, J. Mater. Chem., 2011, 21, 2267–12273 RSC.
- J. F. de Oliveira and M. B. Cardoso, Partial aggregation of silver nanoparticles induced by capping and reducing agents competition, Langmuir, 2014, 30, 4879–4886 CrossRef PubMed.
- S. T. Jones, Z. Walsh-Korb, S. J. Barrow, S. L. Henderson, J. del Barrio and O. A. Scherman, The importance of excess poly(N-isopropylacrylamide) for the aggregation of poly(N-isopropylacrylamide)-coated gold nanoparticles, ACS Nano, 2016, 10, 3158–3165 CrossRef CAS PubMed.
- O. A. Yeshchenko, A. P. Naumenko, N. V. Kutsevol, D. O. Maskova, I. I. Harahuts, V. A. Chumachenko and A. I. Marinin, Anomalous inverse hysteresis of phase transition in thermosensitive dextran-graft-PNIPAM copolymer/Au nanoparticles hybrid nanosystem, J. Phys. Chem. C, 2018, 122, 8003–8010 CrossRef CAS.
- E. C. Dreaden, L. A. Austin, M. A. Mackey and M. A. El-Sayed, Size matters: gold nanoparticles in targeted cancer drug delivery, Ther. Delivery, 2012, 3, 457–478 CrossRef CAS.
- R. Mendes, P. Pedrosa, J. C. Lima, A. R. Fernandes and P. V. Baptista, Photothermal enhancement of chemotherapy in breast cancer by visible irradiation of gold nanoparticles, Sci. Rep., 2017, 7, 10872 CrossRef PubMed.
- R. S. Riley and E. S. Day, Gold nanoparticle-mediated photothermal therapy: applications and opportunities for multimodal cancer treatment, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol., 2017, 9, e1449 CrossRef PubMed.
- I. M. Ibrahim and H. R. Ali, Treatment of natural mammary gland tumors in canines and felines using gold nanorods-assisted plasmonic photothermal therapy to induce tumor apoptosis, Int. J. Nanomed., 2016, 11, 4849–4863 CrossRef PubMed.
- S. A. Abouelmagd, H. Hyun and Y. Yeo, Extracellularly activatable nanocarriers for drug delivery to tumors, Expert Opin. Drug Delivery, 2014, 11(10), 1601–1618 CrossRef CAS PubMed.
- H. Hatakeyama, Recent advances in endogenous and exogenous stimuli-responsive nanocarriers for drug delivery and therapeutics, Chem. Pharm. Bull., 2017, 65, 612–617 CrossRef CAS PubMed.
- N. V. Kutsevol, T. N. Bezuglaya and N. Yu. Bezuglyi, Features of the intramolecular structure of branched polymer systems in solution, J. Struct. Chem., 2014, 55, 575–587 CrossRef.
- V. Chumachenko, N. Kutsevol, Yu. Harahuts, M. Rawiso, A. Marinin and L. Bulavin, Star-like dextran-graft-PNIPAM copolymers. Effect of internal molecular structure on the phase transition, J. Mol. Liq., 2017, 235, 77–82 CrossRef CAS.
- C. Meng Kok and A. Rudin, A semi-empirical method for prediction of critical concentrations for polymer overlap in solution, Eur. Polym. J., 1982, 18, 363–366 CrossRef.
- O. V. Khorolskyi and O. P. Rudenko, Viscometric research of concentration regimes for polyvinyl alcohol solutions, Ukr. J. Phys., 2015, 60, 880–884 CrossRef.
- I. Blakey, Z. Merican and K. J. Thurecht, A method for controlling the aggregation of gold nanoparticles: tuning of optical and spectroscopic properties, Langmuir, 2013, 29, 8266–8274 CrossRef CAS PubMed.
- M. Philipp, R. Aleksandrova, U. Müller, M. Ostermeyer, R. Sanctuary, P. Müller-Buschbaum and J. K. Krüger, Molecular versus macroscopic perspective on the demixing transition of aqueous PNIPAM solutions by studying the dual character of the refractive index, Soft Matter, 2014, 10, 7297–7305 RSC.
| This journal is © The Royal Society of Chemistry 2018 |