
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctionalization of boron-doped diamond with a push–pull chromophore via Sonogashira and CuAAC chemistry†
Jorne Raymakers a,
Hana Krysovab,
Anna Artemenkoc,
Jan Čermákc,
Shannon S. Nicleya,
Pieter Verstappena,
Sam Gielena,
Alexander Kromkac,
Ken Haenena,
Ladislav Kavanb,
Wouter Maes*a and
Bohuslav Rezek*cd
a,
Hana Krysovab,
Anna Artemenkoc,
Jan Čermákc,
Shannon S. Nicleya,
Pieter Verstappena,
Sam Gielena,
Alexander Kromkac,
Ken Haenena,
Ladislav Kavanb,
Wouter Maes*a and
Bohuslav Rezek*cd
aUHasselt – Hasselt University, Institute for Materials Research (IMO), IMEC vzw, IMOMEC, Agoralaan 1, 3590 Diepenbeek, Belgium. E-mail: wouter.maes@uhasselt.be
bJ. Heyrovsky Institute of Physical Chemistry, Czech Academy of Sciences, Dolejskova 3, 18223 Prague 8, Czech Republic
cInstitute of Physics, Czech Academy of Sciences, Cukrovarnická 10, 162 00 Prague 6, Czech Republic. E-mail: rezek@fzu.cz
dFaculty of Electrical Engineering, Czech Technical University in Prague, Technická 2, 166 25 Prague 6, Czech Republic
First published on 26th September 2018
Abstract
Improving the performance of p-type photoelectrodes represents a key challenge toward significant advancement in the field of tandem dye-sensitized solar cells. Herein, we demonstrate the application of boron-doped nanocrystalline diamond (B:NCD) thin films, covalently functionalized with a dithienopyrrole–benzothiadiazole push–pull chromophore, as alternative photocathodes. First, a primary functional handle is introduced on H-terminated diamond via electrochemical diazonium grafting. Afterwards, Sonogashira cross-coupling and Cu(I) catalyzed azide–alkyne cycloaddition (CuAAC) reactions are employed to attach the chromophore, enabling the comparison of the degree of surface functionalization and the importance of the employed linker at the diamond-dye interface. X-ray photoelectron spectroscopy shows that surface functionalization via CuAAC results in a slightly higher chromophore coverage compared to the Sonogashira cross-coupling. However, photocurrents and photovoltages, obtained by photoelectrochemical and Kelvin probe measurements, are approximately three times larger on photocathodes functionalized via Sonogashira cross-coupling. Surface functionalization via Sonogashira cross-coupling is thus considered the preferential method for the development of diamond-based hybrid photovoltaics.
1. Introduction
Dye-sensitized solar cells (DSSCs) are considered as one of the prominent technologies in the field of emerging photovoltaics, next to perovskite, quantum dots and organic solar cells. After three decades of optimization, record efficiencies up to 14.3% under 1 sun illumination have been achieved.1,2 The most efficient and most intensively studied DSSCs employ n-type electrodes such as TiO2 as the photoanode with platinum or graphene as counter electrode. These photoelectrochemically ‘silent’ counter electrodes do not contribute to the generation of photocurrent, but merely serve to transfer the electrons to the redox electrolyte.3,4 Further progress can be made by replacing these cathode materials with photoactive ones toward the development of tandem or p,n-DSSCs. Via careful design of the (organic) chromophores anchored on both electrodes, more light can be absorbed. Theoretically, this could increase the maximum efficiency to 43% (in comparison with 33% for single junction devices).5 Unfortunately, reports on these tandem devices are very rare and the efficiencies obtained are hardly comparable with those of classical n-type DSSCs,6–8 which can mainly be ascribed to the lower performance of the photocathode materials. Until now, the best performing photocathodes are based on p-type NiO sensitized with perylene–thiophene–triphenylamine (PMI-6T-TPA) dyes and tris(acetylacetonato)-iron(III/II) ([Fe(acac)3]0/1–) as redox couple.9 Nevertheless, this only resulted in efficiencies of 2.5% and a current density of 7.65 mA cm−2 at 1 sun illumination. In order to grasp the full potential of tandem DSSCs, further improvements on these p-type photoelectrodes are needed.Diamond is a promising alternative photocathode material, especially due to the development of techniques such as microwave plasma enhanced chemical vapor deposition (MWPECVD).10 CVD allows the growth of diamond thin films on a broad range of substrates over large areas (even m2) and at a relatively low cost from a mixture of hydrogen and methane.11 By introducing trimethylborane to the gas mixture, diamond can be converted from an insulating material to one with either semiconducting or even metallic conductivity, depending on the concentration of boron.12 Boron-doped nanocrystalline diamond (B:NCD) thin films exhibit a good optical transparency for light with wavelengths from 300 to 1000 nm.13 This means that by using diamond as a transparent electrode, the attached (organic) dyes are protected from harmful UV light, as it is mostly absorbed by the diamond itself. When compared with NiO, there are several other advantages in utilizing B:NCD as an electrode material: (i) the chemical inertness of diamond is beneficial when corrosive electrolyte solutions such as I3−/I− are being employed.14 (ii) B:NCD has a hole diffusion coefficient (2–30 cm2 s−1) that is eight orders of magnitude higher than that for NiO (4 × 10−8 cm2 s−1).15 (iii) Diamond surfaces, and H-terminated ones in particular, can be readily functionalized through diazonium chemistry,16–18 photochemical grafting,19,20 or plasma treatment.21,22 In a subsequent step, light-harvesting molecules can be covalently attached to the surface, leading to the formation of carbon–carbon bonds between the dye and the electrode.23–26 These covalent bonds are stronger than the coordination bonds formed with NiO and should lead to improved stability of the diamond–dye interface.
Pioneering research on the application of p-type diamond in photovoltaics was performed in 1999 via the sensitization of boron-doped diamond in an electrolyte solution containing tris(bipyridine)ruthenium(II) (Ru(bpy)32+).27 In 2008, Zhong et al. reported the functionalization of diamond with bithiophene-C60 (2TC60) via a combination of diazonium grafting and Suzuki cross-coupling.28 This resulted in a current density of 150 nA cm−2, which was later optimized to 4–6 μA cm−2 under 1 sun (100 mW cm−2) illumination.29 In 2014, cis-bis(isothiocyanato)bis(2,2′-bipyridyl-4,4′-dicarboxylato)ruthenium(II) (N3 dye) was covalently attached to B:NCD via a combination of diazonium, Suzuki, and EDC-NHS chemistry.30 Despite suitable energy level alignment, photocurrents were limited to 25 nA cm−2 under 1 sun illumination. Later that year, the Suzuki cross-coupling efficiency was improved by employing electron-rich biarylmonodentate phosphine ligands. A surface coverage of 0.56 monolayers was achieved for an organic molecular wire (Br-CPDT-Fur) and photocurrents of 150 nA cm−2 under 0.15 sun illumination were generated.31 Translation of this approach from thin film to a diamond foam electrode resulted in a 3-fold increase in photocurrent. After illumination for 1 day at 1 sun, the photocurrent further increased to 15–22 μA cm−2.32 However, the ‘aging-activation’ leading to this champion photoelectrochemical performance is not fully understood. More recently, Krysova et al. compared the non-covalent and covalent surface modification of B:NCD with 4-(bis(4-[5-(2,2-dicyanovinyl)thiophene-2-yl]phenyl)amino)benzoic acid (P1 dye).33,34 They concluded that covalent functionalization is preferred as the incident photon-to-current conversion efficiency (IPCE) values were approximately 4 times higher than those achieved via the non-covalent approach.
Herein, we further contribute to the development of p-type diamond as a photocathode material through covalent surface functionalization with a specific light-harvesting molecule using a combination of diazonium and Sonogashira chemistry. The Sonogashira cross-coupling is a widely employed reaction for the synthesis of conjugated molecules and polymers due to its broad scope, simplicity and facile monomer synthesis.35 We previously optimized this reaction on B:NCD surfaces with special emphasis on obtaining a high coverage in combination with minimal surface impurities, both crucial parameters to obtain efficient photovoltaics.36 In the presented work, a novel organic donor–acceptor chromophore, bearing an alkyne functional group, was synthesized. The alkyne moiety was introduced on the donor part of the chromophore to enable fast hole injection into the valence band of diamond after exciton generation. On the other hand, the acceptor unit will be in close contact to the electrolyte solution, ensuring a good electron transfer to the redox couple and thereby minimizing charge recombination. The working principle of p-type DSSCs is illustrated in Fig. 1.
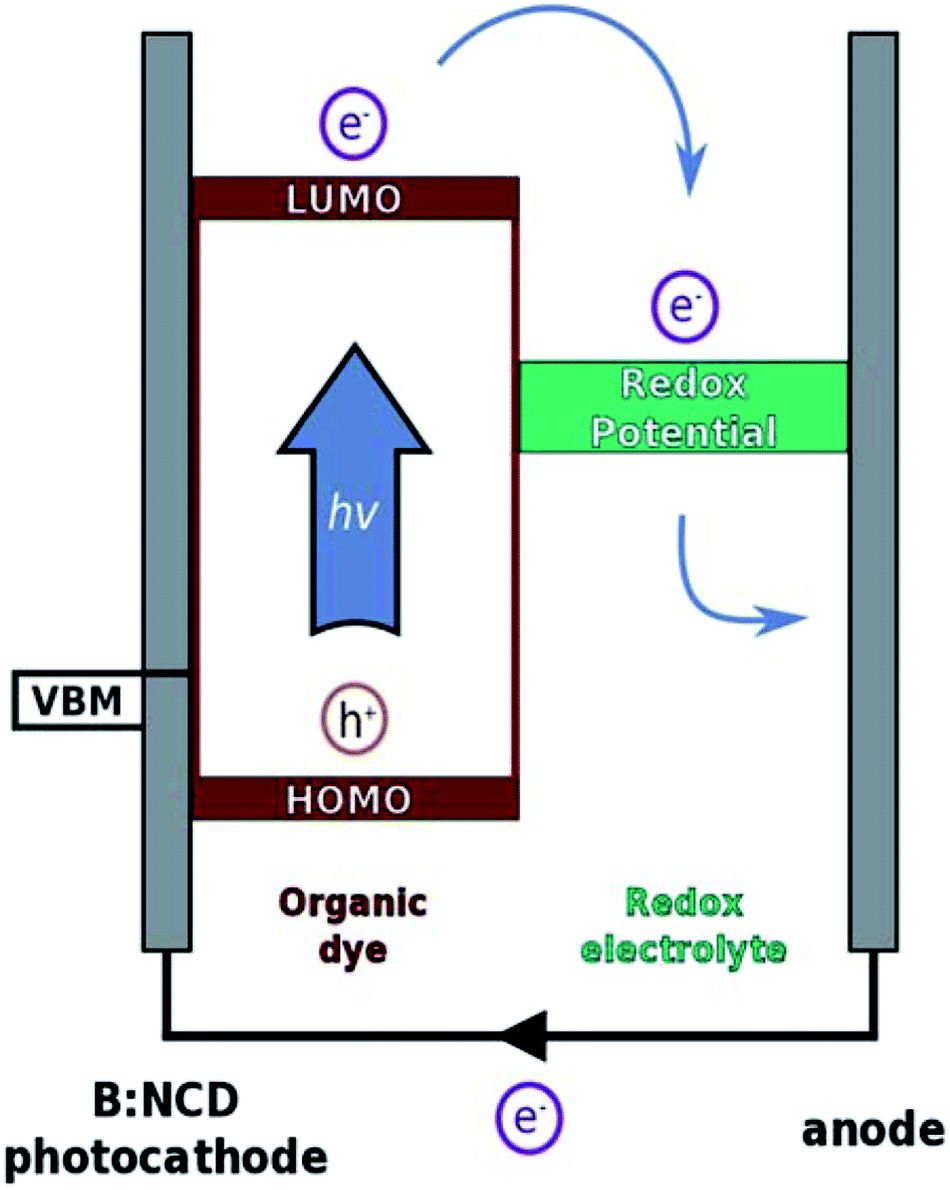 | ||
| Fig. 1 Working principle of a p-type dye DSSC. Under solar illumination, an electron is excited from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) of the dye. This excited electron will result in a reduction of the redox electrolyte, which will diffuse to the counter electrode, where it is re-oxidized. Finally, the electron is transferred back to the dye via an external circuit, which produces a current and leads to regeneration of the dye. (VBM = valence band maximum) | ||
In addition to Sonogashira cross-coupling, alkynylated chromophores can also be attached onto azidophenyl functionalized diamond surfaces via the Cu(I) catalyzed azide–alkyne cycloaddition (CuAAC).37,38 Comparing these two approaches provides additional insights into the influence of employing different linkers on the structural and electronic properties and the potentially detrimental influence of metal residues on the electrode surface.
2. Experimental section
2.1 Materials and instrumentation
All chemicals used were of reagent grade and no further purification was performed prior to use. Sodium nitrate, 4-iodoaniline, 4-azidoaniline hydrochloride, tris(3-hydroxypropyltriazolylmethyl)amine (THPTA), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA), sodium diethyldithiocarbamate, copper(I) bromide and copper(I) iodide were purchased from Sigma-Aldrich. Chloroform (CHCl3), dry tetrahydrofuran (THF), dry N,N-dimethylformamide (DMF), dry toluene, triethylamine (TEA), diisopropylamine (DIPA), n-butyl lithium (2.5 M solution in n-hexane; n-BuLi), trimethyltin chloride, hexylamine, anhydrous copper chloride, sodium tert-butoxide, tetrabutylammonium fluoride (1.0 M solution in THF; TBAF) and glacial acetic acid were purchased from Acros Organics. 2,1,3-Benzothiadiazole, 3-bromothiophene, N-iodosuccinimide, (trimethylsilyl)acetylene and (±)-2,2′-bis(diphenylphosphino)-1,1′-dinaphthalene (BINAP) were purchased from Fluorochem. Tris(dibenzylideneacetone)dipalladium(0) Pd2(dba)3, tri(ortho-tolyl)phosphine and trans-dichlorobis(triphenylphosphine)palladium(II) (Pd(PPh3)2Cl2) were purchased from Strem Chemicals. All solvents employed for rinsing were of HPLC quality.Preparative (recycling) size exclusion chromatography (prep-SEC) was performed on a JAI LC-9110 NEXT system equipped with JAIGEL 1H, 2H, and 3H columns (eluent CHCl3, flow rate 3.5 mL min−1). Nuclear magnetic resonance (NMR) chemical shifts (δ, in ppm) were determined relative to the residual CHCl3 (7.26 ppm) absorption or the 13C resonance shift of CDCl3 (77.16 ppm). High resolution electrospray ionization mass spectrometry (ESI-MS) was performed using an LTQ Orbitrap Velos Pro mass spectrometer equipped with an atmospheric pressure ionization source operating in the nebulizer assisted electrospray mode. The instrument was calibrated in the m/z range 220–2000 using a standard solution containing caffeine, MRFA and Ultramark 1621. UV-Vis absorption spectroscopy measurements were performed on a VARIAN Cary 5000 UV-Vis spectrophotometer at a scan rate of 600 nm min−1. The film for the UV-Vis absorption measurement was prepared by drop casting a solution of the chromophore in chloroform on a quartz substrate. The solid-state UV-Vis absorption spectrum was used to estimate the optical gap (from the wavelength at the intersection of the tangent line drawn at the low energy side of the absorption spectrum with the baseline: Eg (eV) = 1240/(wavelength in nm)). Electrochemical measurements (cyclic voltammetry) were performed with an Eco Chemie Autolab PGSTAT 30 potentiostat/galvanostat using a three-electrode microcell with a platinum working electrode, a platinum counter electrode, and a Ag/AgNO3 reference electrode (silver wire dipped in a solution of 0.01 M AgNO3 and 0.1 M NBu4PF6 in anhydrous acetonitrile). The reference electrode was calibrated against ferrocene/ferrocenium as an external standard. Samples were prepared by dip coating the platinum working electrode in the chromophore solution. The CV measurements were done on the resulting films with 0.1 M NBu4PF6 in anhydrous acetonitrile as electrolyte solution. To prevent air from entering the system, the experiments were carried out under a curtain of argon. Cyclic voltammograms were recorded at a scan rate of 100 mV s−1. For the conversion of V to eV, the onset potentials of the first oxidation/reduction peaks were used and referenced to ferrocene/ferrocenium, which has an ionization potential of −4.98 eV vs. vacuum.
X-ray photoelectron spectroscopy (XPS) was used to determine the chemical composition of the diamond surfaces using an XPS spectrometer (Kratos, AXIS Supra) equipped with a hemispherical analyzer and a monochromatic Al Kα X-ray source (1486.6 eV). The XPS spectra were acquired from an area of 0.7 × 0.3 mm2 with a takeoff angle of 90°. The survey XPS spectra were recorded with a pass energy of 80 eV, whereas the high-resolution spectrum scans were recorded with a pass energy of 20 eV. The obtained XPS spectra were calibrated on 283.8 eV binding energy (sp3 carbon phase).39 The CasaXPS software with implemented linear baseline and Gaussian line shapes was used for spectral processing. XPS peak positions were determined with an accuracy of ±0.2 eV.
Atomic force microscopy (AFM) and Kelvin probe force microscopy (KPFM) measurements were performed on an NTEGRA Prima system (NT-MDT) with a Pt/Cr coated probe (BudgetSensors Multi75E-G) using a value of dz = 0 nm and an AC voltage of 2 V. The obtained data were line-corrected and root-mean-square (RMS) values were evaluated from the images using the NT-MDT software.
Macroscopic Kelvin probe measurements were performed on a scanning Kelvin probe system (KP Technologies) using a gold coated probe (tip of 2 mm in diameter) at a distance defined by a gradient of 300. Illumination was provided by a cold light source KL2500 (Schott) with a halogen lamp set to full power (250 W). The light was introduced by a light guide to the sample (within 5 cm distance). The photovoltage was calculated as the difference between the potential values (averaged for 18 s) with and without illumination. The error bars correspond to the standard deviation of the averaged potential values.
Photoelectrochemical measurements were performed in a Ar-saturated 0.1 M Na2SO4 solution containing 5 mM methyl viologen (MV2+) as an electron carrier in a three-electrode glass cell equipped with a quartz optical window for illumination of the B:NCD working electrode. The counter electrode was platinum and the reference electrode was Ag/AgCl (sat. KCl). The glass cell was placed in a dark room and controlled by a potentiostat (Autolab, Ecochemie, B.V. with NOVA software). The white light source was an Oriel xenon lamp, model 6269. The incident light intensity was focused and calibrated with a standard Si photodiode (PV Measurements, Inc. USA). The incident white light intensity was 90 mW cm2 (which is roughly 0.9 sun).
2.2 Dye synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dichloromethane, 90:10) and then via Kugelrohr distillation (106 °C at 2 mm Hg) and finally recrystallization in EtOH to yield the product as white crystals (19.9 g, 60%). 1H NMR (400 MHz, chloroform-d): δ = 7.41 (d, J = 5.4 Hz, 2H), 7.08 (d, J = 5.4, 2H).40:dichloromethane, 95:5), yielding a pale solid (3.86 g, 95%). 1H NMR (400 MHz, chloroform-d): δ = 7.14 (d, J = 5.3 Hz, 2H), 7.02 (d, 5.3 Hz, 2H), 4.20 (t, J = 7.1 Hz, 2H), 1.90–1.80 (m, 2H), 1.37–1.21 (m, 6H), 0.88 (t, J = 7.0 Hz, 3H).40:ethyl acetate, 95:5) and Kugelrohr distillation (70 °C at 2 mm Hg). The combined fractions yielded the product as a white solid (10.70 g, 34%). 1H NMR (400 MHz, chloroform-d): δ = 7.98 (dd, J = 8.8, 0.9 Hz, 1H), 7.85 (dd, J = 7.2, 0.8 Hz, 1H), 7.49 (dd, J = 8.8, 7.2 Hz, 1H).:dichloromethane, 60:40), yielding the product as a red solid (930 mg, quantitative). 1H NMR (400 MHz, chloroform-d): δ = 8.34 (s, 1H), 7.86 (dd, J = 8.7, 0.9 Hz, 1H) 7.84 (dd, J = 7.2, 0.9 Hz, 1H), 7.61 (dd, J = 8.7, 7.2 Hz, 1H), 7.20 (d, J = 5.3 Hz, 1H), 7.02 (d, J = 5.3 Hz, 1H), 4.29 (t, J = 7.0 Hz, 2H), 1.96–1.88 (m, 2H), 1.39–1.23 (m, 6H), 0.87 (t, J = 7.0 Hz, 3H).:dichloromethane, 60:40), yielding the product as a red solid (927 mg, 75%). 1H NMR (400 MHz, chloroform-d): δ = 8.29 (s, 1H), 7.87 (dd, J = 8.7, 0.9 Hz, 1H), 7.82 (dd, J = 7.1, 0.9 Hz, 1H), 7.60 (dd, J = 8.7, 7.2 Hz, 1H), 7.20 (s, 1H), 4.22 (t, J = 7.1 Hz, 2H), 1.95–1.85 (m, 2H), 1.40–1.24 (m, 6H), 0.87 (t, J = 6.4 Hz, 3H); 13C NMR (100 MHz, chloroform-d): δ = 156.3, 152.6, 147.7, 145.4, 137.6, 130.3, 129.1, 124.8, 120.9, 120.0, 119.9, 115.8, 113.3, 72.1, 48.1, 32.1, 31.0, 27.3, 23.2, 14.7; calcd for C20H18IN3S3 [M]+: m/z 522.9708, found: 522.9703.:dichloromethane, 60:40), yielding the product as a red solid (310 mg, 69%). 1H NMR (400 MHz, chloroform-d) δ = 8.17 (s, 1H), 7.77 (dd, J = 8.7, 0.9 Hz, 1H), 7.67 (dd, J = 7.1, 0.9 Hz, 1H), 7.46 (dd, J = 8.7, 7.1 Hz, 1H), 7.11 (s, 1H), 4.10 (t, J = 7.1 Hz, 2H), 3.48 (s, 1H), 1.83 (p, J = 7.1 Hz, 2H), 1.36–1.19 (m, 6H), 0.89–0.80 (m, 3H); 13C NMR (100 MHz, chloroform-d) δ = 155.5, 151.8, 147.1, 143.4, 138.0, 129.6, 128.3, 124.2, 119.4, 119.2, 116.5, 116.1, 114.9, 112.5, 82.1, 78.8, 47.3, 31.4, 30.3, 26.6, 22.5, 14.1; HRMS (ESI): calcd for C22H19N3S3 [M + Na]+: m/z 444.0639, found: 444.0675.
:dichloromethane, 90:10) and then via Kugelrohr distillation (106 °C at 2 mm Hg) and finally recrystallization in EtOH to yield the product as white crystals (19.9 g, 60%). 1H NMR (400 MHz, chloroform-d): δ = 7.41 (d, J = 5.4 Hz, 2H), 7.08 (d, J = 5.4, 2H).40:dichloromethane, 95:5), yielding a pale solid (3.86 g, 95%). 1H NMR (400 MHz, chloroform-d): δ = 7.14 (d, J = 5.3 Hz, 2H), 7.02 (d, 5.3 Hz, 2H), 4.20 (t, J = 7.1 Hz, 2H), 1.90–1.80 (m, 2H), 1.37–1.21 (m, 6H), 0.88 (t, J = 7.0 Hz, 3H).40:ethyl acetate, 95:5) and Kugelrohr distillation (70 °C at 2 mm Hg). The combined fractions yielded the product as a white solid (10.70 g, 34%). 1H NMR (400 MHz, chloroform-d): δ = 7.98 (dd, J = 8.8, 0.9 Hz, 1H), 7.85 (dd, J = 7.2, 0.8 Hz, 1H), 7.49 (dd, J = 8.8, 7.2 Hz, 1H).:dichloromethane, 60:40), yielding the product as a red solid (930 mg, quantitative). 1H NMR (400 MHz, chloroform-d): δ = 8.34 (s, 1H), 7.86 (dd, J = 8.7, 0.9 Hz, 1H) 7.84 (dd, J = 7.2, 0.9 Hz, 1H), 7.61 (dd, J = 8.7, 7.2 Hz, 1H), 7.20 (d, J = 5.3 Hz, 1H), 7.02 (d, J = 5.3 Hz, 1H), 4.29 (t, J = 7.0 Hz, 2H), 1.96–1.88 (m, 2H), 1.39–1.23 (m, 6H), 0.87 (t, J = 7.0 Hz, 3H).:dichloromethane, 60:40), yielding the product as a red solid (927 mg, 75%). 1H NMR (400 MHz, chloroform-d): δ = 8.29 (s, 1H), 7.87 (dd, J = 8.7, 0.9 Hz, 1H), 7.82 (dd, J = 7.1, 0.9 Hz, 1H), 7.60 (dd, J = 8.7, 7.2 Hz, 1H), 7.20 (s, 1H), 4.22 (t, J = 7.1 Hz, 2H), 1.95–1.85 (m, 2H), 1.40–1.24 (m, 6H), 0.87 (t, J = 6.4 Hz, 3H); 13C NMR (100 MHz, chloroform-d): δ = 156.3, 152.6, 147.7, 145.4, 137.6, 130.3, 129.1, 124.8, 120.9, 120.0, 119.9, 115.8, 113.3, 72.1, 48.1, 32.1, 31.0, 27.3, 23.2, 14.7; calcd for C20H18IN3S3 [M]+: m/z 522.9708, found: 522.9703.:dichloromethane, 60:40), yielding the product as a red solid (310 mg, 69%). 1H NMR (400 MHz, chloroform-d) δ = 8.17 (s, 1H), 7.77 (dd, J = 8.7, 0.9 Hz, 1H), 7.67 (dd, J = 7.1, 0.9 Hz, 1H), 7.46 (dd, J = 8.7, 7.1 Hz, 1H), 7.11 (s, 1H), 4.10 (t, J = 7.1 Hz, 2H), 3.48 (s, 1H), 1.83 (p, J = 7.1 Hz, 2H), 1.36–1.19 (m, 6H), 0.89–0.80 (m, 3H); 13C NMR (100 MHz, chloroform-d) δ = 155.5, 151.8, 147.1, 143.4, 138.0, 129.6, 128.3, 124.2, 119.4, 119.2, 116.5, 116.1, 114.9, 112.5, 82.1, 78.8, 47.3, 31.4, 30.3, 26.6, 22.5, 14.1; HRMS (ESI): calcd for C22H19N3S3 [M + Na]+: m/z 444.0639, found: 444.0675.2.3 Diamond growth
150 nm thick B:NCD films were grown on (100)-oriented 10 × 102 mm Si p-type conductive substrates by MWPECVD, with trimethylborane (TMB) as the boron source. Characterization performed on samples grown in the same reactor under similar growth conditions indicated that the B:NCD films have boron concentrations of approximately 5 × 1021cm−3 and a resistivity (Ω) below 5 mΩ cm.41,42
2.4 Diazonium electrografting
The B:NCD thin films were functionalized with 4-iodophenyl and 4-azidophenyl moieties by means of electrochemical diazonium grafting of the respective in situ generated benzenediazonium chlorides with cyclic voltammetry. The electrografting was performed with an Autolab PGSTAT30 potentiostat (Eco Chemie B.V.) and was controlled by the GPES Manager software. A one-compartment electrochemical cell was employed in a three-electrode configuration. Hydrogen-terminated B:NCD was used as the working electrode, a platinum wire as the counter electrode and a Ag/AgCl/KCl(sat) electrode as the reference. All potentials are reported versus this reference electrode at room temperature. Diamond working electrodes were functionalized in two steps by immersing each half of the sample in the in situ generated 4-iodo- or 4-azidobenzenediazonium chloride solution. The electrografting process was performed by cycling the potential between +500 and −600 mV vs. Ag/AgCl for 4-iodoaniline and between +300 and −800 mV vs. Ag/AgCl for 4-azidoaniline at a scan rate of 100 mV s−1 for eight scans.25,36 After grafting, the B:NCD samples were sonicated in MilliQ water and subsequently rinsed with toluene, THF, ethanol, and MilliQ water.2.5 Sonogashira cross-coupling
All Sonogashira cross-coupling reactions were prepared in a glovebox under inert atmosphere. The light-harvesting molecule, palladium catalyst, copper(I) iodide, diamond substrate, base, and solvent were loaded in a tailor made Schlenk vial (Fig. S3†) equipped with a magnetic stirring bar. The Schlenk vial was closed off with a rubber septum, removed from the glovebox and placed in an oil bath at 55 °C for 16 h. After surface functionalization, all B:NCD samples were sonicated in toluene (p.a.), a 10% sodium diethyldithiocarbamate solution, acetone, ethanol, and MilliQ water, before drying under a strong stream of nitrogen. More detailed information on the exact reaction conditions applied can be found in Table 1.| Entry | Catalyst system | Dye | Base | O at% | C at% | B at% | S at% | N at% | Pd at% | Cu at% | I at% |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a General reaction conditions: dry THF (6 mL), base (3 mL), 60 °C, 18 h; TEA = triethylamine, DIPA = diisopropylamine.b Reaction outside of the glovebox. | |||||||||||
| 1 | Pd(PPh3)2Cl2 (5 mol%)/CuI (10 mol%) | (50 μmol) | TEA | 3.6 | 89.8 | 2.9 | 1.1 | 2.0 | 0.1 | — | 0.5 |
| 2 | Pd(PPh3)2Cl2 (5 mol%)/CuI (10 mol%) | (50 μmol) | DIPA | 4.0 | 87.2 | 3.3 | 1.8 | 3.0 | 0.2 | — | 0.5 |
| 3 | Pd(PPh3)2Cl2 (10 mol%)/CuI (20 mol%) | (50 μmol) | DIPA | 3.7 | 88.0 | 3.7 | 1.6 | 2.6 | 0.1 | — | 0.3 |
| 4 | Pd(PPh3)2Cl2 (5 mol%)/CuI (10 mol%) | (100 μmol) | DIPA | 4.0 | 85.9 | 3.8 | 2.3 | 3.5 | 0.2 | — | 0.3 |
| 5b | Pd(PPh3)2Cl2 (5 mol%)/CuI (10 mol%) | (100 μmol) | DIPA | 3.4 | 87.4 | 2.9 | 2.2 | 3.2 | 0.6 | — | 0.3 |
| 6 | — | (50 μmol) | DIPA | 4.3 | 89.5 | 3.4 | 0.2 | 1.7 | — | — | 0.9 |
2.6 Cu(I) catalyzed azide–alkyne cycloaddition
All Cu(I) catalyzed azide–alkyne cycloaddition reactions were prepared in a glovebox under inert atmosphere. The light-harvesting molecule, copper source, ligand, diamond substrate and solvents were loaded in a tailor made Schlenk vial (Fig. S3†) equipped with a magnetic stirring bar. The Schlenk vial was closed off with a rubber septum, removed from the glovebox and placed in an oil bath at 55 °C for 16 h. After surface functionalization, all B:NCD samples were sonicated in toluene (p.a.), a 10% sodium diethyldithiocarbamate solution, acetone, ethanol and MilliQ water, before drying under a strong stream of nitrogen. More detailed information on the exact reaction conditions applied can be found in Table 2.| Entry | Catalyst/ligand | O at% | C at% | B at% | S at% | N at% | Cu at% |
|---|---|---|---|---|---|---|---|
| a General reaction conditions: dye (50 μmol), dry THF (6 mL), dry DMF (3 mL), 50 °C, 18 h; THPTA = tris(3-hydroxypropyltriazolylmethyl)amine, PMDETA = N,N,N′,N′′,N′′-pentamethyldiethylenetriamine. | |||||||
| 1 | CuI (100 mol%)/PMDETA (200 mol%) | 4.1 | 84.6 | 3.0 | 1.9 | 6.4 | — |
| 2 | CuI (10 mol%)/THPTA (20 mol%) | 3.4 | 85.9 | 1.9 | 2.2 | 6.6 | — |
| 3 | CuBr (100 mol%)/PMDETA (200 mol%) | 3.1 | 85.2 | 2.1 | 2.5 | 7.1 | — |
| 4 | — | 4.3 | 89.0 | 2.2 | 0.1 | 4.4 | — |
3. Results and discussion
The covalent functionalization of B:NCD electrodes with alkynylated chromophores was performed in two steps, as illustrated in Scheme 1. For the functionalization via Sonogashira cross-coupling, a iodophenyl layer was first grafted onto the diamond surface via diazonium chemistry. When the CuAAC reaction was employed, an azidophenyl layer was primarily grafted on the surface instead.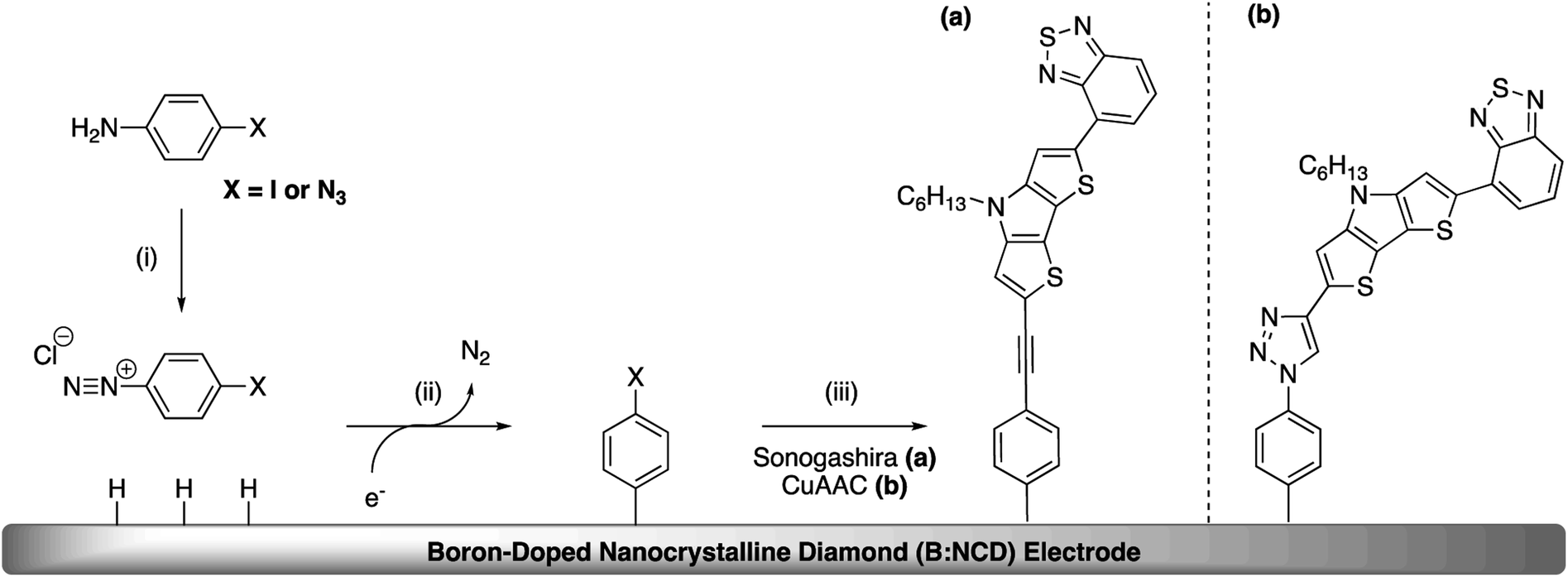 | ||
| Scheme 1 General functionalization strategy of boron-doped nanocrystalline diamond for the design of p-type photoelectrodes.a a(i) In situ diazotization of 4-iodoaniline or 4-azidoaniline; (ii) electrochemical reduction and subsequent grafting of the corresponding diazonium salt; (iii) Sonogashira cross-coupling (a) and Cu(I) catalyzed azide–alkyne cycloaddition (CuAAC) (b) using an alkyne functionalized dithieno[3,2-b:2′,3′-d]pyrrole-2,1,3-benzothiadiazole (alkyne-DTP-BT) chromophore. | ||
3.1 Chromophore synthesis
A light-harvesting donor–acceptor type dye was synthesized to facilitate charge separation of the generated hole and electron after excitation. As we target a high surface coverage, the dye itself should preferentially be rather small, without too long side chains, as this might influence the coupling reactions. Additionally, to obtain a low optical gap chromophore, strong donor and acceptor units have to be combined in a push–pull approach. In this case, the electron rich dithienopyrrole unit was selected as the donor part and combined with a benzothiadiazole acceptor building block.43,44 The synthesis of the alkyne functionalized dithieno[3,2-b:2′,3′-d]pyrrole-2,1,3-benzothiadiazole (alkyne-DTP-BT) chromophore is shown in Scheme 2. Dithienopyrrole 3 was synthesized in two steps via regiospecific lithiation of 3-bromothiophene (1) at −78 °C and subsequent Ullmann coupling, resulting in 3,3′-dibromo-2,2′-bithiophene (2). Afterwards, the bithiophene was fused using a Buchwald–Hartwig amination with hexylamine to generate the donor building block 3, which was then stannylated and coupled to 4-bromo-2,1,3-benzothiadiazole (5) via a Stille cross-coupling. Finally, an alkyne functionality was introduced on the DTP-BT chromophore 6 in 3 steps through iodination with N-iodosuccinimide (NIS),40 Sonogashira cross-coupling with trimethylsilylacetylene and deprotection with tetrabutylammonium fluoride (TBAF). UV-Vis analysis of the newly synthesized dye (9) revealed two strong absorption bands peaking at wavelengths of 335 and 474 nm. As expected, this results in a red dye. The molar extinction coefficient (ε = 18600 M−1 cm−1 at λmax = 474) is slightly lower compared to chromophores previously employed for the sensitization of diamond films.31,34 HOMO (−5.21 eV) and LUMO (−3.29 eV) energy levels were estimated from the oxidation and reduction onsets as measured by cyclic voltammetry. Alternatively, the HOMO energy level can also be estimated by subtracting the optical gap (2.30 eV) from the LUMO energy level, resulting in EHOMO = −5.59 eV.
 | ||
| Scheme 2 Synthetic route toward the alkyne functionalized dithieno[3,2-b:2′,3′-d]pyrrole-2,1,3-benzothiadiazole (alkyne-DTP-BT) chromophore.a a(i) LDA, CuCl2, THF; (ii) t-BuONa, hexylamine, Pd2(dba)3, BINAP, toluene; (iii) n-BuLi, Me3SnCl, THF; (iv) Pd2(dba)3, P(o-tolyl)3, toluene, DMF; (v) NIS, CHCl3, AcOH; (vi) TMS-acetylene, Pd(PPh3)2Cl2, CuI, THF, TEA; (vii) TBAF, THF. | ||
3.2 Primary surface functionalization: diazonium grafting
To immobilize the alkyne-DTP-BT chromophore onto a B:NCD electrode via Sonogashira cross-coupling, an iodine moiety was introduced via electrochemical diazonium grafting of 4-iodoaniline, as reported previously.36 The diazonium salt was generated in situ via diazotization of 4-iodoaniline with an equimolar amount of NaNO2 in HCl (0.5 M). Subsequently, a freshly grown H-terminated diamond thin film was employed as working electrode in a cyclic voltammetry experiment and submerged in the aforementioned solution. The potential was then cycled eight times between +500 and −600 mV (versus a Ag/AgCl reference electrode). Hereby, an electron is ejected from the diamond electrode into the solution, resulting in a reduction of the diazonium salt to the corresponding radical, which can graft onto the surface. Going to more negative potentials can lead to further reduction of the radical to an anion, which is detrimental for the grafting process. The cyclic voltammogram (Fig. 2, top) shows the efficiency of the functionalization protocol as complete passivation of the diamond surface is obtained after one cycle. Actual grafting of the iodophenyl moieties was confirmed by XPS through the detection of high resolution I 3d3/2 and I 3d5/2 peaks at 632 and 622 eV, respectively (Fig. 3, top). XPS analysis of the high resolution iodine peaks revealed that the relative atomic concentration of iodine on the sample surface is about 0.8 at%, which is in agreement with previous results.36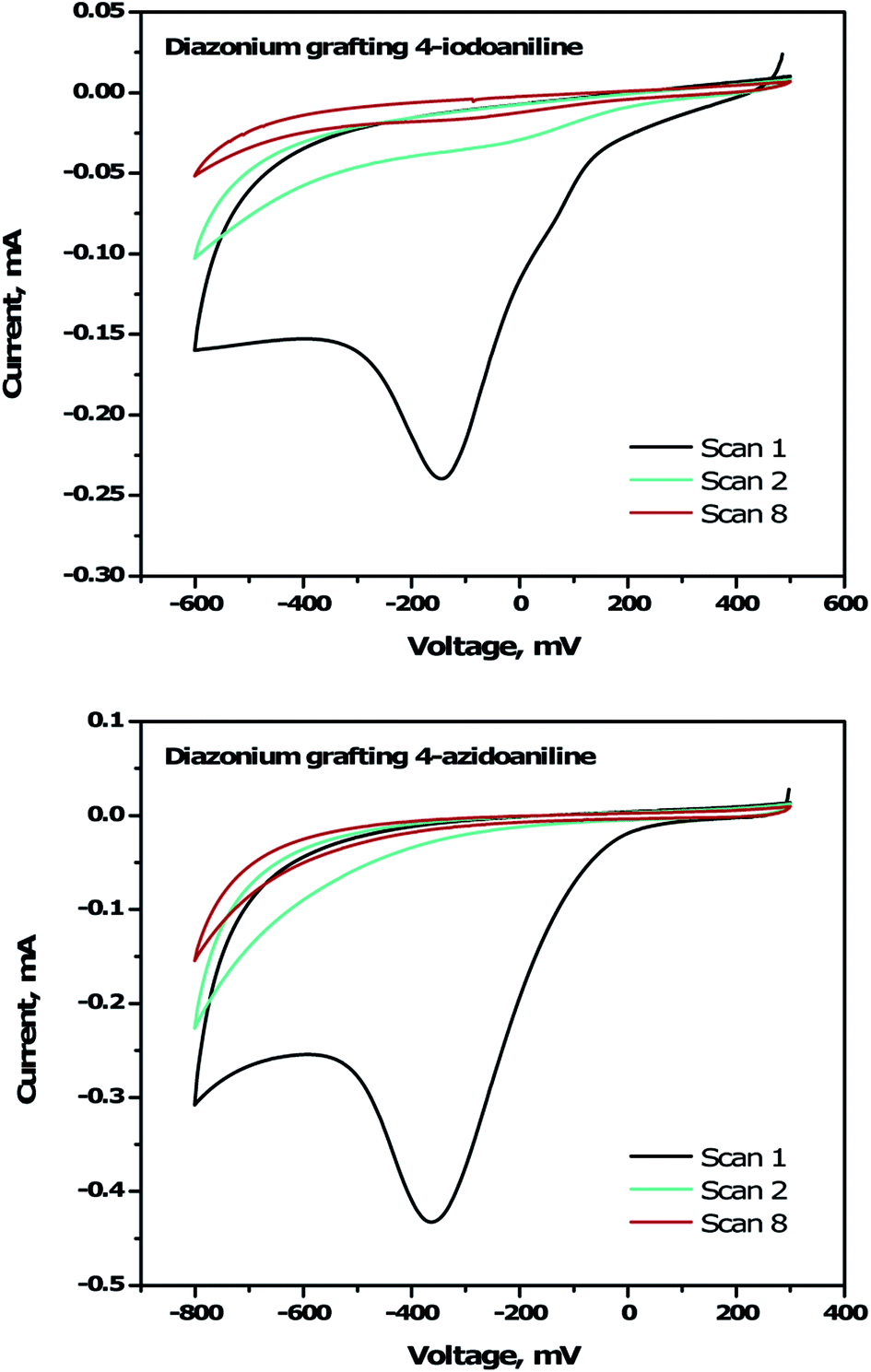 | ||
| Fig. 2 Cyclic voltammograms showing the electrochemical grafting of in situ generated 4-iodobenzenediazonium chloride (top) and 4-azidobenzenediazonium chloride (bottom) on H-terminated B:NCD electrodes at a scan rate of 100 mV s−1 for 8 scans. | ||
 | ||
| Fig. 3 High resolution I 3d and N 1s XPS spectra for iodophenyl (top) and azidophenyl (bottom) functionalized B:NCD electrodes, respectively. The N 1s peak was deconvoluted into three peaks at 399.0, 400.1 and 404.1 eV.45,46 | ||
For the functionalization of diamond via CuAAC chemistry, an azide functional handle is required on the surface. The azide groups were also introduced via electrochemical diazonium grafting, employing 4-azidoaniline hydrochloride. The reductive potential scan was slightly adjusted to ensure optimal functionalization according to a literature protocol,25 by cycling the potential between +300 and −800 mV. Complete passivation of the diamond surface was again obtained after just one scan, as shown in the cyclic voltammogram (Fig. 2, bottom). Grafting of the azidophenyl layer was also confirmed by XPS through the presence of the N 1s signal. Deconvolution of the high resolution N 1s peak (Fig. 3, bottom) revealed three distinct nitrogen species at 399.0, 400.1 and 404.1 eV.45,46 The two signals at the lowest binding energy can be assigned to the more electron rich (outer) azide nitrogen atoms, whereas the signal at higher binding energy comes from the more electron deficient (central) azide nitrogen atom.47 The surface nitrogen content calculated from the high resolution N 1s peak was about 4.8 at%. It should be mentioned that this nitrogen concentration is quite high for nanocrystalline diamond and could be related to multilayer formation (via a radical mechanism and/or azo coupling) during the grafting procedure.17
3.3 Secondary surface functionalization: Sonogashira cross-coupling and CuAAC chemistry
For the development of efficient photocathodes, two parameters are very important, i.e. the amount of dye immobilized on the surface and the contamination thereof, as this can lead to charge trapping of generated holes and electrons. Recently, we demonstrated that the iodine functionalized diamond surface is a suitable platform for covalent attachment of organic molecules through Sonogashira cross-coupling.36 A broad range of reaction conditions were evaluated for the coupling of the 2-ethynylthiophene model system. In all cases, a decent surface coverage was obtained with a relatively low amount of surface contamination by catalyst residues. Accordingly, the optimal catalyst system, Pd(PPh3)2Cl2, was selected for the functionalization of B:NCD with the alkyne-DTP-BT chromophore (Scheme 3). For this specific dye molecule, the reaction conditions were fine-tuned by varying the employed base, catalyst concentration, and chromophore concentration. The degree of surface functionalization and contamination were estimated by the relative atomic concentrations of sulfur, palladium, and copper, as obtained from the XPS spectra (Table 1).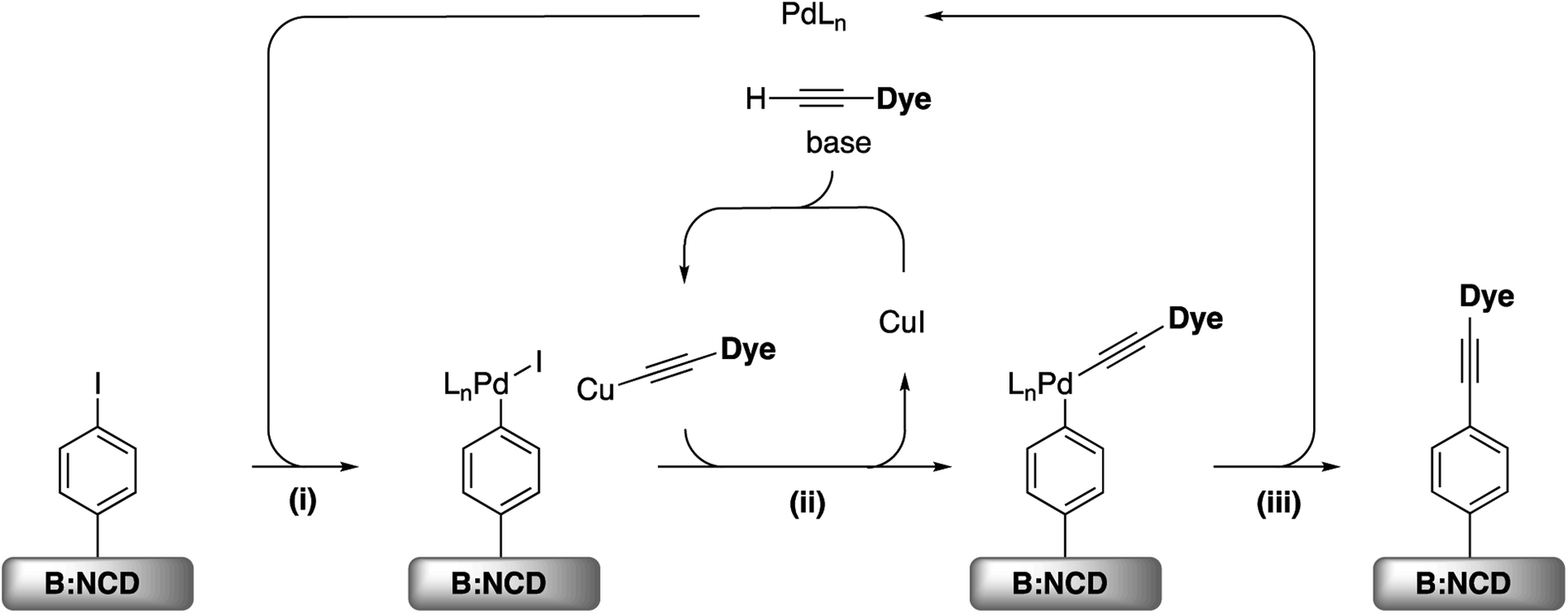 | ||
| Scheme 3 (i) Oxidative addition, (ii) transmetallation, and (iii) reductive elimination for the Sonogashira cross-coupling on iodophenyl functionalized B:NCD surfaces. | ||
Initially (Table 1, entry 1), 50 μmol of the alkyne-DTP-BT dye was dissolved in a mixture of THF and TEA (2:1), with 5 mol% Pd(PPh3)2Cl2 as a catalyst and 10 mol% CuI as co-catalyst. This lead to a surface coverage of 1.1 at% S, while the relative surface concentration of Pd was as low as 0.1 at% and no Cu was detected by XPS. These low amounts of metal residues were obtained by adding an additional step to the rinsing procedure, i.e. sonication in a 10 m/v% solution of sodium diethyldithiocarbamate in MilliQ water. These dithiocarbamates have proven to be especially efficient for the removal of transition metals from organic and aqueous reaction media.48 Additionally, the signal of the B 1s peak was still detected by XPS, which indicates that a relatively thin layer of molecules on the diamond surface is obtained, since the attenuation length of the employed X-rays (1486.6 eV) is approximately 3.5 nm for carbon materials.49 By employing DIPA as a base (Table 1, entry 2), the degree of surface functionalization improved (the S content increased to 1.8 at%), while keeping the amount of metal impurities at a minimum. Doubling the concentration of the catalyst and co-catalyst (Table 1, entry 3) did not significantly influence the outcome of the surface functionalization. On the other hand, doubling the concentration of the dye to 100 μmol (Table 1, entry 4) did increase the relative amount of S on the surface to 2.3 at%, without drastically changing the amount of metal residues. When performing the reaction outside of the glovebox (Table 1, entry 5), a similar surface coverage of sulfur (2.2 at%) was achieved, but the Pd concentration noticeably increased (from 0.2 to 0.6 at%). This could tentatively be attributed to the fact that oxygen can lead to the formation of palladium black, which sticks to the surface, even after thorough rinsing. Finally, a control experiment was performed (Table 1, entry 6) without addition of the catalytic system to confirm actual cross-coupling. A small amount of dye (0.2 at% of sulfur) appears to physisorb on the surface, but this is an order of magnitude lower than in the case of coupling via the Sonogashira reaction.
Apart from the Sonogashira cross-coupling, alkynylated chromophores can also be attached to diamond surfaces by employing CuAAC reactions, often referred to as a ‘click’ chemistry approach (Scheme 4).50 The success of this protocol for diamond functionalization was demonstrated by Szunerits and coworkers by successfully ‘clicking’ both ferrocene and thiophenes onto azide functionalized diamond surfaces with a conversion efficiency of approximately 50%.38,51
 | ||
| Scheme 4 Simplified mechanism for the Cu(I) catalyzed azide–alkyne cycloaddition (CuAAC) on azidophenyl functionalized B:NCD surfaces.52 | ||
A small optimization of the reaction conditions for the CuAAC functionalization was performed. Since the alkyne-DTP-BT dye is not soluble in aqueous media, the traditional catalyst system, containing CuSO4 and sodium ascorbate to reduce copper(II) to copper(I), was replaced with CuI or CuBr. This directly provides the required copper(I) and reduces the amount of reagents that have to be added, which is beneficial to minimize surface fouling. The highest surface coverage based on the relative atomic concentration of sulfur was obtained by employing 50 μmol of the dye in combination with 100 mol% of CuBr and PMDETA as a stabilizing ligand (Table 2, entry 3). This surface coverage of 2.5 at% S is slightly higher than the 2.3% obtained for the Sonogashira cross-coupling. Additionally, no copper residues were detected on the surface by XPS. Based on these results, we can summarize that, in terms of surface functionalization, the CuAAC reaction is slightly more effective than the Sonogashira cross-coupling, and, importantly, no surface contamination was observed at all. Confirmation of the actual CuAAC reaction was again provided by performing a control experiment (Table 2, entry 4), which shows only a small amount of physisorbed dye (0.1 at% S).
The changes on the diamond surface during the two-step functionalization approach for both the Sonogashira cross-coupling and the CuAAC reaction are visualized in Fig. 4. In Fig. 4a, the appearance of the iodine signal after diazonium grafting of 4-iodoaniline can be seen, as well as the appearance of the nitrogen and sulfur signals from the dye after Sonogashira cross-coupling. Based on the ratio of the relative iodine surface concentration before (0.8 at%) and after (0.3 at%) the Sonogashira cross-coupling, the conversion efficiency of the secondary surface functionalization step was calculated to be approximately 63%. Similarly, Fig. 4b shows the grafting of 4-azidoaniline on H-terminated B:NCD films and subsequent ‘clicking’ of the dye. Unfortunately, in this case no approximation of the reaction efficiency could be made since we are not able to distinguish between the nitrogen species in the 1,2,3-triazole linker and the dye. The high resolution S 2p peaks (Fig. 4c and e) look similar for the two coupling strategies and are composed of three main peaks at binding energies of 166.2, 165.4 and 163.9 eV. The two peaks of the deconvoluted high resolution S 2p signal with lowest binding energy are related to the S 2p1/3 (163.9 eV) and S 2p2/3 (165.4 eV) peaks of a S–C bond. These signals arise from the two thiophenes in the dithienopyrrole donor unit.36,38,53 As a result, the peak at 166.2 eV should be attributed to the N–S–N bond in the benzothiadiazole acceptor unit. The very weak peak located at 168.5 eV can possibly be attributed to oxidized sulfur groups (SOx),36 while the peak at 162.0 eV can be referenced to disulfides,54 which could be formed during XPS irradiation of the samples. Contrary to the S 2p signal, XPS analysis did reveal significant differences in the deconvoluted high resolution N 1s signal for substrates functionalized via Sonogashira (Fig. 4d) and CuAAC (Fig. 4f) chemistry. In both cases, a large N 1s peak at a binding energy of 399.9 eV was detected by XPS. This peak can be assigned to the sp2 hybridized nitrogen atoms that are present in the push–pull chromophore. However, when CuAAC chemistry was employed for surface functionalization, an additional peak was detected at 401.1 eV. This peak is typically observed for 1,2,3-triazoles formed after the azide–alkyne cycloaddition reaction.45,46
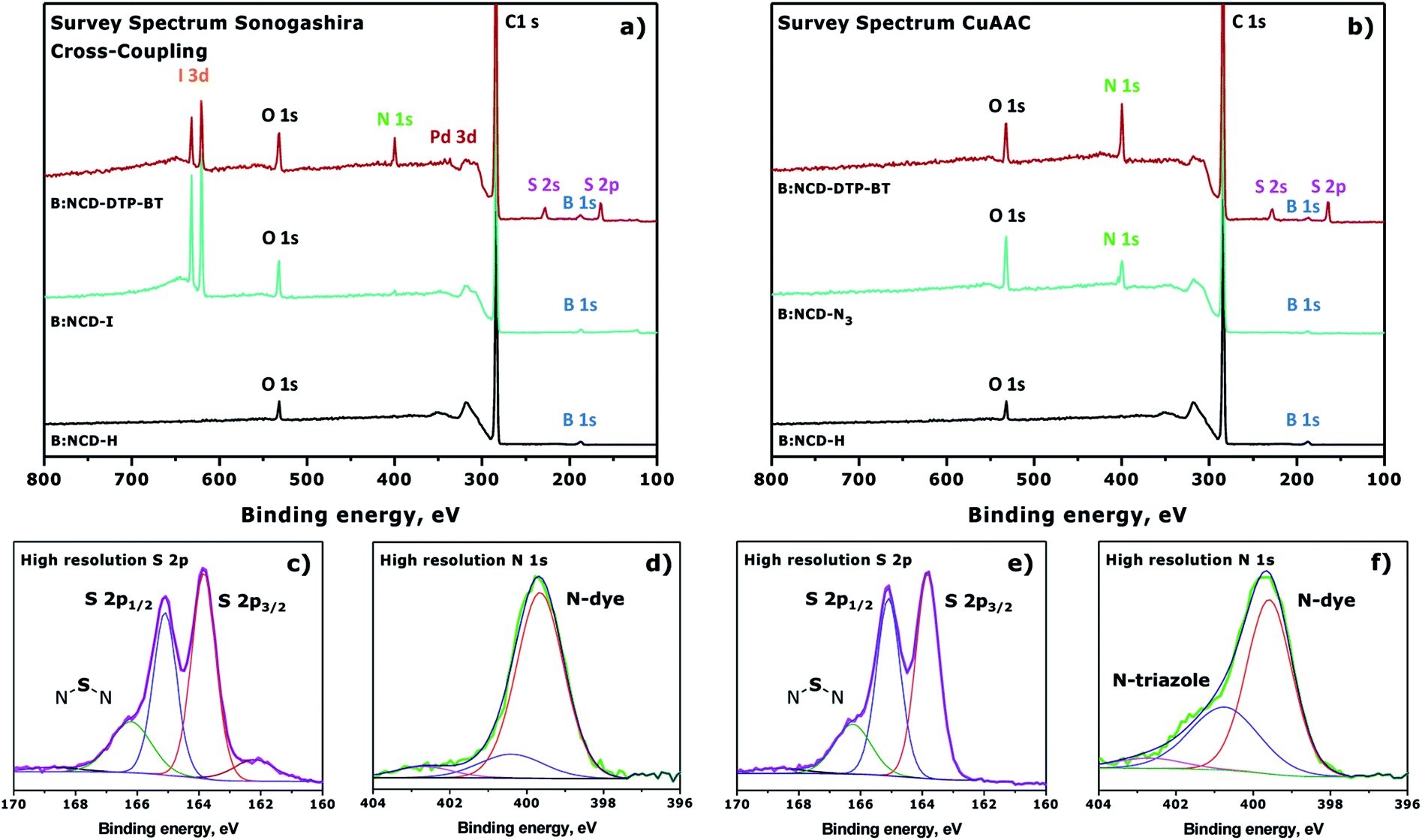 | ||
| Fig. 4 Normalized XPS survey spectra for the surface functionalization of H-terminated B:NCD via (a) Sonogashira cross-coupling (Table 1, entry 4) or (b) CuAAC chemistry (Table 2, entry 2). Deconvoluted high resolution S 2p (c and e) and N 1s (d and f) signals after attachment of the dye via Sonogashira or CuAAC are provided below the XPS survey spectra. | ||
3.4 Surface morphology and Kelvin probe measurements
Kelvin probe measurements have been proven to be highly useful to assess the photovoltage generation on various diamond based opto-electronic devices, from Schottky photodiodes on boron doped diamonds to hybrid systems composed of diamond and organic dyes.55,56 Kelvin probe force microscopy (KPFM) images of H-terminated B:NCD and electrodes functionalized with DTP-BT chromophores via Sonogashira cross-coupling and CuAAC chemistry are shown in Fig. 5. The surface potential of the bare H-B:NCD substrate correlates very well with the surface morphology obtained by AFM (see Fig. S4†). Crystalline grains are well resolvable as their facets and edges have slightly different potentials. Yet the overall potential variations remain within 5 mV RMS. Noticeably, the potential variations remain about the same after the chromophore is grafted to the diamond via CuAAC or Sonogashira chemistry, as shown in the graph in Fig. 5d. However, the surface potential landscape changes significantly: the diamond grains are not resolvable anymore (in spite of some potential fluctuations). This means that the surface potential is homogenized by the chromophore grafting (within 5 mV RMS). It also shows that the surface is well covered by the grafted molecules that create a uniform layer. At the same time, the layer is very thin and does not noticeably affect the surface morphology (as evidenced by Fig. S4†).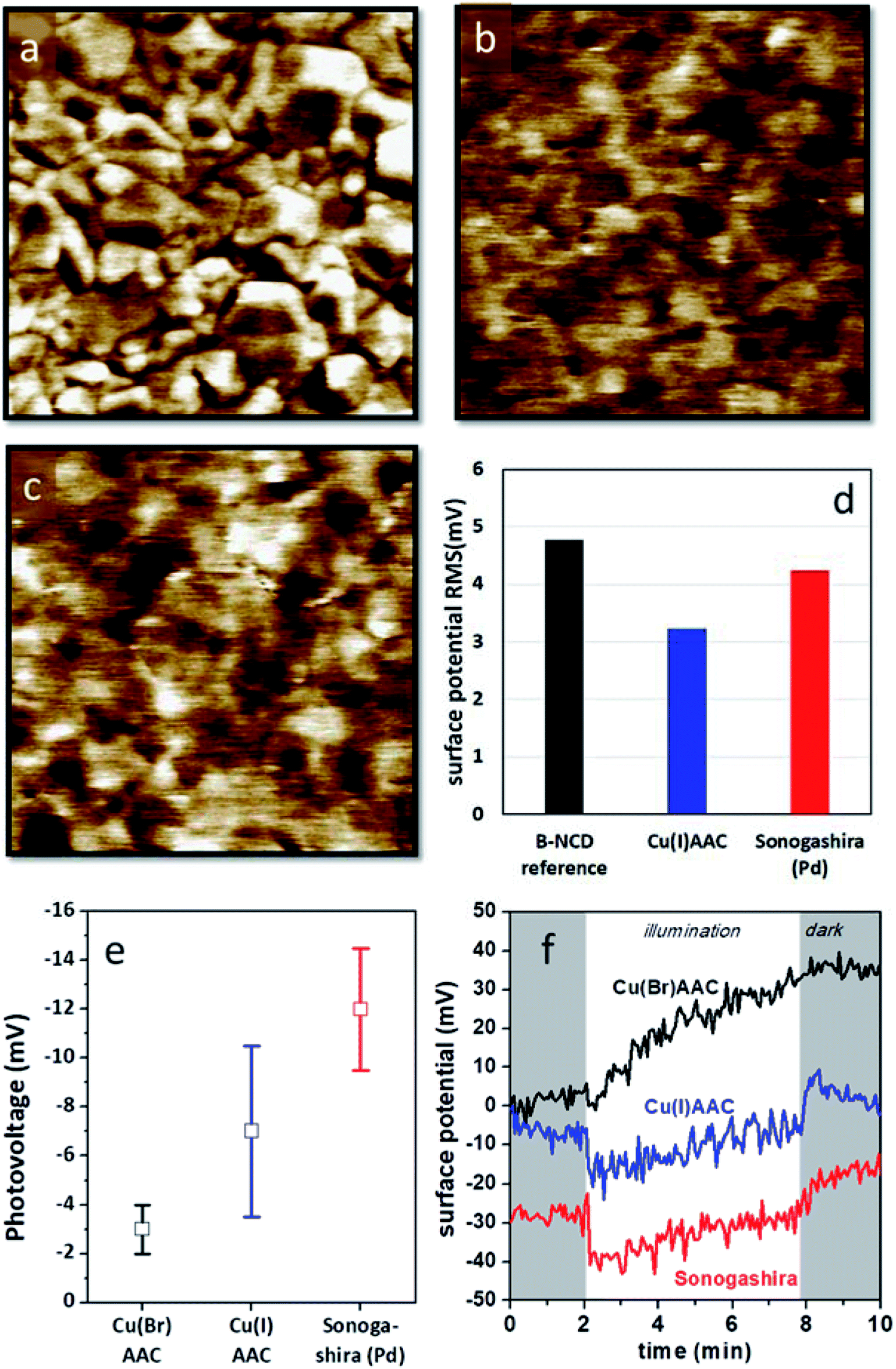 | ||
| Fig. 5 KPFM images of (a) a bare H-terminated B:NCD thin film, (b) a B:NCD film with the chromophore coupled via CuAAC chemistry, and (c) a B:NCD film with the chromophore coupled via Sonogashira. The Z scale is set to 20 mV in all images to visualize subtle potential differences. (d) Surface potential RMS roughness from the KPFM images. (e) Macroscopically measured surface photovoltages and (f) surface potential profiles in time in dark (shaded area) and under illumination (white area) for the three sample types. The potential profiles are stacked along the y-axis for better visibility. For Cu(I)AAC, CuI was used as the catalyst, while for Cu(Br)AAC, CuBr was used instead (according to Table 2). | ||
The photovoltages measured by scanning Kelvin probe (SKP) microscopy on the studied samples are shown in Fig. 5e. One can see that the largest photovoltage was obtained for Sonogashira coupling of the chromophore. It is also noticeable that the lowest photovoltage is obtained for CuAAC (CuBr catalyst) chemistry, which provided the best grafting efficiency in terms of XPS analysis. The photovoltage values were obtained from multiple experiments, and the error bars correspond to the standard deviations of the averaged values. Fig. 5f illustrates the SKP profiles taken in dark and under halogen light illumination on each type of sample. From such profiles, the photovoltage data was extracted. The potential profiles suggest that Sonogashira cross-coupling would provide the most pronounced photoresponse from the given chromophore dye.
3.5 Photoelectrochemical measurements
To complement the photovoltage measurements of the functionalized electrodes, photoelectrochemical measurements were performed using H-terminated, iodophenyl, azidophenyl and DTP-BT functionalized B:NCD thin films. The diamond electrodes were employed as the working electrode in a three-electrode voltammetry setup and they were illuminated with chopped white light in an aqueous electrolyte solution of methyl viologen (MV2+), which functioned as a redox mediator. When performing chronoamperometric measurements (Fig. 6), the electrodes were equilibrated in the dark for a short period before switching the light source on and off for intervals of approximately 10 seconds. Turning on the light source resulted in the generation of a photocurrent, which was fully reversible. Applying a negative bias was shown to positively influence the cathodic photocurrents as a net vectorial electron flow from the cathode to the counter electrode via the redox electrolyte is being generated. Hence, a negative bias of −0.2 V was applied during chronoamperometry.57,58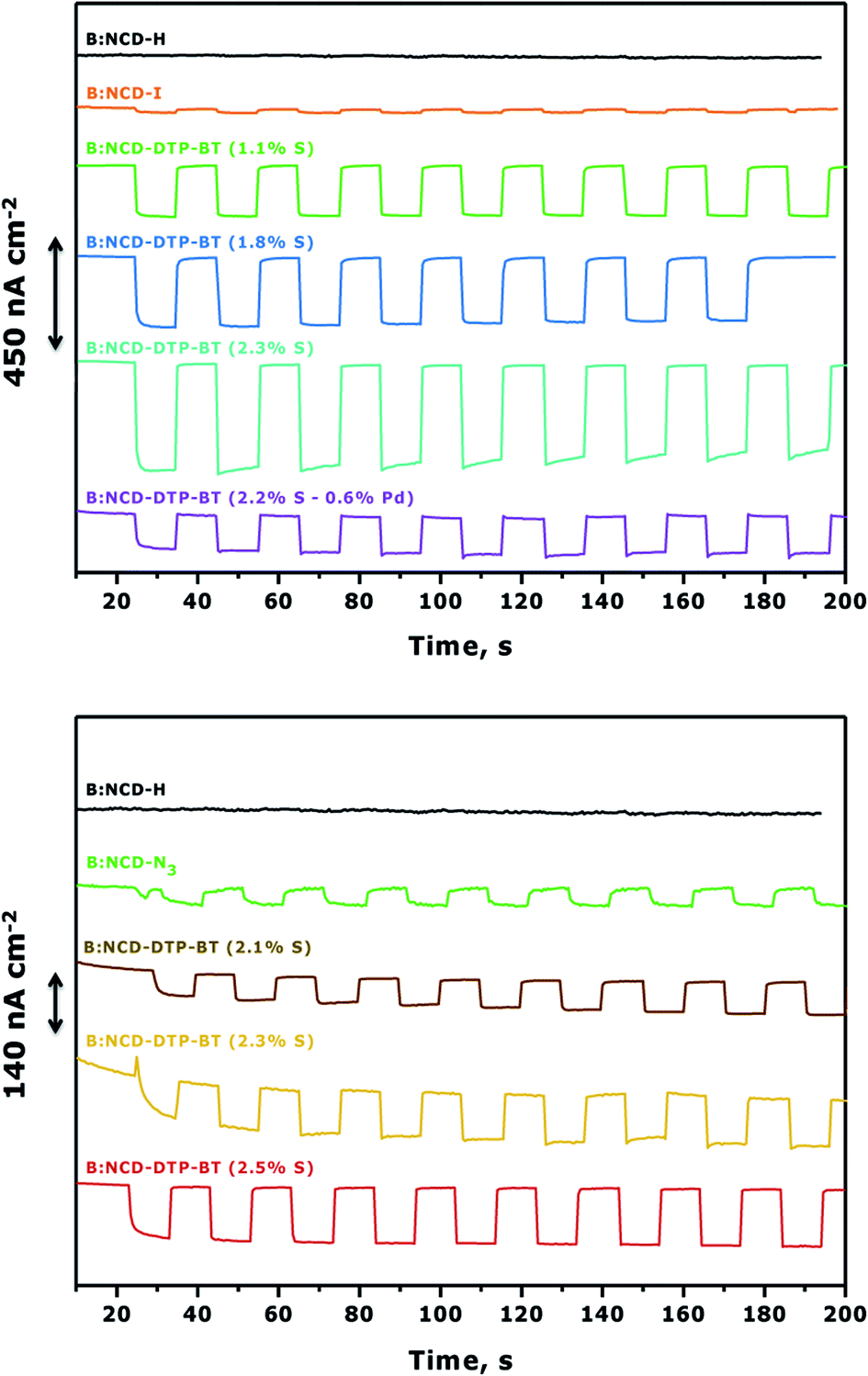 | ||
| Fig. 6 Photocurrent densities (nA cm−2) of H-terminated (B:NCD-H), iodophenyl (B:NCD-I), azidophenyl (B:NCD-N3) and DTP-BT functionalized (B:NCD-DTP-BT) diamond electrodes in a 5 mM methyl viologen solution (in 0.1 M Na2SO4) under white light illumination (90 mW cm−2; simulated AM 1.5G solar spectrum, 10 s 1:1 dark/light interval) at a bias of −0.2 V. Surface functionalization was performed via Sonogashira cross-coupling (top) and CuAAC (bottom). | ||
Fig. 6 (top) clearly shows a good correlation between the relative atomic contractions of S on the surface and the generated photocurrents under white light illumination of 0.9 sun. The diamond electrode that revealed the lowest surface coverage (1.1 at% S) after Sonogashira cross-coupling had the weakest photoresponse of ca. 200 nA cm−2. Upon obtaining a higher surface coverage of 1.8 at% S, the photocurrent increased to 300 nA cm−2, and the highest photocurrent densities (450 nA cm−2) were obtained for the diamond electrode that had the highest amount of dye attached on the surface (2.3 at% S). It should be mentioned that the initial photocurrent decreases over a rather short period of time. However, when illuminating for a few hours, one can see the gradual return of the photocurrent to its original value (Fig. S5†). The higher photocurrents correspond with an increase of the absorption between 350 and 400 nm in the IPCE spectrum (Fig. S6†). This ‘aging-activation’ phenomenon is reported in all long-term studies on diamond-based photocathodes so far, but remains to be fully understood.32,34 Photocurrents were also measured for the sample that contained a higher amount of surface contamination (0.6 at% Pd) (Table 1, entry 5). Despite having a similar surface coverage (2.2 at% S) to the electrode with the highest initial photocurrents (Table 1, entry 4), the generated currents were about three times lower. This gives a first indication that the presence of palladium has a negative effect on the photocurrent. In a control experiment, no photocurrent was obtained upon solar illumination of H-terminated B:NCD, whereas the current for a iodophenyl functionalized electrode was extremely small.
For the functionalization of the diamond electrodes via CuAAC chemistry (Fig. 6, bottom), a similar relation between surface coverage and photocurrent generation was observed. Surprisingly, the observed currents are significantly lower compared to functionalization via Sonogashira, even though the surface coverage is slightly higher and the system is completely devoid of metal contamination (based on XPS). When we compare the functionalization strategies, two major differences are noted. The Sonogashira cross-coupling employs an alkyne linker to attach the dye to the surface and after the reaction residual iodine groups remain on the surface. The CuAAC reaction on the other hand employs a 1,2,3-triazole linker and has remnant azide groups. Since the photocurrents generated by both iodophenyl and azidophenyl functionalized surfaces were negligible, we can assume that the type of residual groups on the surface has a limited influence on the photocurrent. Therefore, the main difference in photocurrent generation is likely related to the employed linker. For now, the exact influence of the alkyne and triazole linkers on the generated photocurrents remains unknown. Possibly, the rigidity (advantageous for orbital overlap) and higher electron density on the alkyne could be more favorable for transporting the generated hole (upon excitation of an electron from the HOMO to the LUMO of the dye) from the chromophore to the hole conducting B:NCD electrode, leading to lower interfacial charge combination and higher currents. Yet, further experiments and computational studies should be performed to confirm this.
To further improve photocurrent generation, a more negative bias of −0.3 V was applied, as shown in Fig. 7. For both the Sonogashira cross-coupling and CuAAC reaction, an increase of the photocurrent was observed from the best performing samples, up to 600 and 240 nA cm−2, respectively. However, for the Sonogashira cross-coupling, the difference in photocurrent between the electrodes with 1.1% S (Fig. 7, top, green curve) and 1.8% S (Fig. 7, top, blue curve) becomes less pronounced. This effect is related to the increasing dark currents at a lower bias as illustrated in the chopped-light polarization curve in Fig. S7.† Additionally, the photocurrent decrease of the electrode with the highest initial photocurrent density (Fig. 7, top, cyan curve) is more pronounced.
 | ||
| Fig. 7 Photocurrent densities (nA cm−2) of DTP-BT functionalized (B:NCD-DTP-BT) diamond electrodes in a 5 mM methyl viologen solution (in 0.1 M Na2SO4) under white light illumination (90 mW cm−2; simulated AM 1.5G solar spectrum, 10 s 1:1 dark/light interval) at a bias of −0.3 V. Surface functionalization was performed via Sonogashira cross-coupling (top) and CuAAC (bottom). | ||
To assess the potential of the Sonogashira cross-coupling for the development of ‘all-carbon’ photovoltaics, we finally compared our results with the functionalization of B:NCD thin films via Suzuki cross-coupling.31 This reaction was previously employed to covalently link Br-BT-Rho and Br-CPDT-Fur dyes to a phenylboronate functionalized diamond surface. Photoelectrochemical measurements resulted in currents of 70 and 150 nA cm−2 under white light illumination with an intensity of 0.15 sun (AM 1.5; 15 mW cm−2) at a bias of −0.2 V, respectively. To facilitate comparison with our results, we assumed a proportional relation between the solar illumination intensity and photocurrent generation for Br-BT-Rho and Br-CPDT-Fur. Doing so, these chromophores should give currents of 420 and 900 nA cm−2 under 0.9 sun illumination (AM 1.5; 90 mW cm−2). Compared to the Br-BT-Rho functionalized electrodes, our system gives similar currents. However, it should be mentioned that the Br-BT-Rho chromophore is more suitable for diamond sensitization due to a better positioning of the HOMO of the dye with respect to the valence band maximum (VBM) of the B:NCD photocathode and its higher molar extinction coefficient (ε = 43200 M−1 cm−1). The Br-CPDT-Rho functionalized electrodes perform even better, as there is a larger driving force for hole injection into the B:NCD electrode and since this dye has an even higher molar extinction coefficient (ε = 73000 M−1 cm−1).31 Despite the less favorable optical properties of the alkyne-DTP-BT dye, photocurrents within the same order of magnitude were obtained for the Sonogashira cross-coupling. This proves the strength of this approach for surface functionalization and indicates that higher photocurrents are within reach.
4. Conclusions
A mono-alkynylated donor–acceptor chromophore was successfully synthesized and covalently anchored onto boron-doped nanocrystalline diamond thin films. The functionalization was achieved through two different two-step functionalization approaches, i.e. electrochemical diazonium grafting and subsequent Sonogashira cross-coupling or CuAAC chemistry. In terms of surface functionalization, the CuAAC reaction was superior, affording a slightly higher chromophore coverage in the absence of surface contamination. However, in terms of photoresponse, the Sonogashira cross-coupling resulted in current densities (450 nA cm−2 at −0.2 V) that are approximately three times higher than those for the CuAAC reaction (140 nA cm−2 at −0.2 V). For the first time we also showed photovoltages generated in such hybrid systems, where again Sonogashira cross-coupling of the chromophore provided the most pronounced effect. Current work focuses on the development of alkynylated chromophores that have higher molar extinction coefficients and more optimal energy level alignment with regards to the diamond photocathode.Conflicts of interest
There are no conflicts of interest.Acknowledgements
The authors thank UHasselt, the Research Foundation – Flanders (FWO Vlaanderen), the Czech Science Foundation projects 13-31783S and 15-01809S, and the European Regional Development Fund project CZ.02.1.01/0.0/0.0/15 003/0000464 for financial support. J. Raymakers and S. Gielen thank the FWO for their PhD fellowships. P. Verstappen is a postdoctoral fellow of the FWO.References
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- K. Kakiage, Y. Aoyama, T. Yano, K. Oya, J. Fujisawa and M. Hanaya, Chem. Commun., 2015, 51, 15894–15897 RSC.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Gratzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- L. Kavan, Curr. Opin. Electrochem., 2017, 2, 88–96 CrossRef CAS.
- C. J. Wood, G. H. Summers and E. A. Gibson, Chem. Commun., 2015, 51, 3915–3918 RSC.
- A. Nattestad, A. J. Mozer, M. K. Fischer, Y. B. Cheng, A. Mishra, P. Bauerle and U. Bach, Nat. Mater., 2010, 9, 31–35 CrossRef CAS PubMed.
- A. Nattestad, I. Perera and L. Spiccia, J. Photochem. Photobiol., C, 2016, 28, 44–71 CrossRef CAS.
- Y. Farre, M. Raissi, A. Fihey, Y. Pellegrin, E. Blart, D. Jacquemin and F. Odobel, ChemSusChem, 2017, 10, 2618–2625 CrossRef CAS PubMed.
- I. R. Perera, T. Daeneke, S. Makuta, Z. Yu, Y. Tachibana, A. Mishra, P. Bauerle, C. A. Ohlin, U. Bach and L. Spiccia, Angew. Chem., Int. Ed. Engl., 2015, 54, 3758–3762 CrossRef CAS PubMed.
- K. Kobashi, K. Nishimura, Y. Kawate and T. Horiuchi, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 4067–4084 CrossRef CAS.
- R. J. Nemanich, J. A. Carlisle, A. Hirata and K. Haenen, MRS Bull., 2014, 39, 490–494 CrossRef CAS.
- J. V. Macpherson, Phys. Chem. Chem. Phys., 2015, 17, 2935–2949 RSC.
- C. H. Y. X. Lim, Y. L. Zhong, S. Janssens, M. Nesladek and K. P. Loh, Adv. Funct. Mater., 2010, 20, 1313–1318 CrossRef CAS.
- J. Stotter, Y. Show, S. Wang and G. Swain, Chem. Mater., 2005, 17, 4880–4888 CrossRef CAS.
- P.-N. Volpe, J. Pernot, P. Muret and F. Omnès, Appl. Phys. Lett., 2009, 94, 092102 CrossRef.
- Y. R. Leroux, H. Fei, J. M. Noel, C. Roux and P. Hapiot, J. Am. Chem. Soc., 2010, 132, 14039–14041 CrossRef CAS PubMed.
- J. Pinson and F. Podvorica, Chem. Soc. Rev., 2005, 34, 429–439 RSC.
- J. Wang, M. A. Firestone, O. Auciello and J. A. Carlisle, Langmuir, 2004, 20, 11450–11456 CrossRef CAS PubMed.
- X. Wang, P. E. Colavita, J. A. Streifer, J. E. Butler and R. J. Hamers, J. Phys. Chem. C, 2010, 114, 4067–4074 CrossRef CAS.
- X. Wang, E. C. Landis, R. Franking and R. J. Hamers, Acc. Chem. Res., 2010, 43, 1205–1215 CrossRef CAS PubMed.
- R. Bogdanowicz, M. Sawczak, P. Niedzialkowski, P. Zieba, B. Finke, J. Ryl, J. Karczewski and T. Ossowski, J. Phys. Chem. C, 2014, 118, 8014–8025 CrossRef CAS.
- T. Nakamura and T. Ohana, Jpn. J. Appl. Phys., 2012, 51, 085201 CrossRef.
- Y. L. Zhong and K. P. Loh, Chem.–Asian J., 2010, 5, 1532–1540 CrossRef CAS PubMed.
- D. Meziane, A. Barras, A. Kromka, J. Houdkova, R. Boukherroub and S. Szunerits, Anal. Chem., 2012, 84, 194–200 CrossRef CAS PubMed.
- W. S. Yeap, M. S. Murib, W. Cuypers, X. Liu, B. van Grinsven, M. Ameloot, M. Fahlman, P. Wagner, W. Maes and K. Haenen, ChemElectroChem, 2014, 1, 1145–1154 CrossRef CAS.
- K. Natsui, T. Yamamoto, M. Akahori and Y. Einaga, ACS Appl. Mater. Interfaces, 2015, 7, 887–894 CrossRef CAS PubMed.
- S. Nakabayashi, N. Ohta and A. Fujishima, Phys. Chem. Chem. Phys., 1999, 1, 3993–3997 RSC.
- Y. L. Zhong, K. P. Loh, A. Midya and Z.-K. Chen, Chem. Mater., 2008, 20, 3137–3144 CrossRef CAS.
- Y. L. Zhong, A. Midya, Z. Ng, Z. K. Chen, M. Daenen, M. Nesladek and K. P. Loh, J. Am. Chem. Soc., 2008, 130, 17218–17219 CrossRef CAS PubMed.
- W. S. Yeap, X. Liu, D. Bevk, A. Pasquarelli, L. Lutsen, M. Fahlman, W. Maes and K. Haenen, ACS Appl. Mater. Interfaces, 2014, 6, 10322–10329 CrossRef CAS PubMed.
- W. S. Yeap, D. Bevk, X. Liu, H. Krysova, A. Pasquarelli, D. Vanderzande, L. Lutsen, L. Kavan, M. Fahlman, W. Maes and K. Haenen, RSC Adv., 2014, 4, 42044–42053 RSC.
- H. Krysova, L. Kavan, Z. V. Zivcova, W. S. Yeap, P. Verstappen, W. Maes, K. Haenen, F. Gao and C. E. Nebel, RSC Adv., 2015, 5, 81069–81077 RSC.
- H. Krysova, Z. Vlckova-Zivcova, J. Barton, V. Petrak, M. Nesladek, P. Cigler and L. Kavan, Phys. Chem. Chem. Phys., 2015, 17, 1165–1172 RSC.
- H. Krysova, J. Barton, V. Petrak, R. Jurok, M. Kuchar, P. Cigler and L. Kavan, Phys. Chem. Chem. Phys., 2016, 18, 16444–16450 RSC.
- R. Chinchilla and C. Najera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- J. Raymakers, A. Artemenko, S. S. Nicley, P. Štenclová, A. Kromka, K. Haenen, W. Maes and B. Rezek, J. Phys. Chem. C, 2017, 121, 23446–23454 CrossRef CAS.
- M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- M. Wang, M. R. Das, M. Li, R. Boukherroub and S. Szunerits, J. Phys. Chem. C, 2009, 113, 17082–17086 CrossRef CAS.
- S. Ghodbane, D. Ballutaud, F. Omnès and C. Agnès, Diamond Relat. Mater., 2010, 19, 630–636 CrossRef CAS.
- A. Gupta, M. M. A. Kelson, V. Armel, A. Bilic and S. V. Bhosale, Tetrahedron, 2014, 70, 2141–2150 CrossRef CAS.
- S. D. Janssens, P. Pobedinskas, J. Vacik, V. Petráková, B. Ruttens, J. D'Haen, M. Nesládek, K. Haenen and P. Wagner, New J. Phys., 2011, 13, 083008 CrossRef.
- S. Drijkoningen, S. D. Janssens, P. Pobedinskas, S. Koizumi, M. K. Van Bael and K. Haenen, Sci. Rep., 2016, 6, 35667 CrossRef CAS PubMed.
- L. E. Polander, L. Pandey, S. Barlow, S. P. Tiwari, C. Risko, B. Kippelen, J.-L. Brédas and S. R. Marder, J. Phys. Chem. C, 2011, 115, 23149–23163 CrossRef CAS.
- H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607–632 CrossRef CAS.
- R. D. Rohde, H. D. Agnew, W. S. Yeo, R. C. Bailey and J. R. Heath, J. Am. Chem. Soc., 2006, 128, 9518–9525 CrossRef CAS PubMed.
- A. C. Gouget-Laemmel, J. Yang, M. A. Lodhi, A. Siriwardena, D. Aureau, R. Boukherroub, J. N. Chazalviel, F. Ozanam and S. Szunerits, J. Phys. Chem. C, 2012, 117, 368–375 CrossRef.
- T. Heinrich, C. H. H. Traulsen, E. Darlatt, S. Richter, J. Poppenberg, N. L. Traulsen, I. Linder, A. Lippitz, P. M. Dietrich, B. Dib, W. E. S. Unger and C. A. Schalley, RSC Adv., 2014, 4, 17694–17702 RSC.
- W. P. Gallagher and A. Vo, Org. Process Res. Dev., 2014, 19, 1369–1373 CrossRef.
- P. J. Cumpson and M. P. Seah, Surf. Interface Anal., 1997, 25, 430–446 CrossRef CAS.
- T. Meinhardt, D. Lang, H. Dill and A. Krueger, Adv. Funct. Mater., 2011, 21, 494–500 CrossRef CAS.
- M. R. Das, M. Wang, S. Szunerits, L. Gengembre and R. Boukherroub, Chem. Commun., 2009, 25, 2753–2755 RSC.
- S. Díez-González, Catal. Sci. Technol., 2011, 1, 166–178 RSC.
- E. O. Sako, H. Kondoh, I. Nakai, A. Nambu, T. Nakamura and T. Ohta, Chem. Phys. Lett., 2005, 413, 267–271 CrossRef CAS.
- X. Chen, Y. Peng and D. Bradshaw, Int. J. Miner. Process., 2013, 125, 129–136 CrossRef CAS.
- J. Čermák, Y. Koide, D. Takeuchi and B. Rezek, J. Appl. Phys., 2014, 115, 053105 CrossRef.
- B. Rezek, J. Čermák, A. Kromka, M. Ledinský and J. Kočka, Diamond Relat. Mater., 2009, 18, 249–252 CrossRef CAS.
- D. Hirayama, K. Takimiya, Y. Aso, T. Otsubo, T. Hasobe, H. Yamada, H. Imahori, S. Fukuzumi and Y. Sakata, J. Am. Chem. Soc., 2002, 124, 532–533 CrossRef CAS PubMed.
- T. Otsubo, Y. Aso and K. Takimiya, J. Mater. Chem., 2002, 12, 2565–2575 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of the alkyne-DTP-BT dye and additional information on the diamond growth, the Schlenk vials applied for diamond functionalization, AFM film morphologies, and additional photoelectrochemical measurements. See DOI: 10.1039/c8ra07545j |
| This journal is © The Royal Society of Chemistry 2018 |