
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolten salt construction of stable oxygen vacancies on TiO2 for enhancement of visible light photocatalytic activity†
Yu Zoua,
Kaimeng Yanga,
Qirong Chen*b,
Haitao Wangc and
Xiangfu Meng *a
*a
aDepartment of Chemistry, Capital Normal University, Beijing 100048, China. E-mail: xfmeng@cnu.edu.cn; Tel: +86-10-68902974
bBeijing Center for Physical and Chemical Analysis (BCPCA), Beijing 100089, China. E-mail: qrchen@163.com
cBaxter Healthcare, 12F No. 989, Shanghai 200031, China
First published on 31st October 2018
Abstract
The construction of defects on TiO2 surface has attracted great interest due to their prominent effect on photocatalytic activity. However, most synthesis methods often lead to unstable oxygen vacancies which limits their effect for improvement of visible light photoactivity. In this work, stable oxygen vacancies were successfully introduced in commercial TiO2 (P25) via one-step molten salt (MS) method. Due to the incomplete combination of trifluoroacetic acid (TFA) adsorbed on TiO2 in MS, the lattice oxygen atoms of TiO2 were consumed resulting in the formation of oxygen vacancies both on the surface and in the bulk of TiO2. The optical adsorption edge of oxygen defective TiO2 showed a substantial shift toward to visible light region combining with a color variation from white to dark blue. Meanwhile, the morphology and crystalline phase were also changed because of the presence of oxygen vacancies. As a result, the blue TiO2 with rich oxygen vacancies exhibited a considerably enhanced photocatalytic activity for decomposition of rhodamine B (RhB) and selective oxidation of benzyl alcohol under visible light irradiation.
Introduction
TiO2 has always been one of the most investigated semiconductor materials for dealing with environmental and energy issues.1,2 However, the photocatalytic efficiency of TiO2 is often limited because of its wide band gap and high recombination rate of photogenerated charge carriers.3 Recently, self-doping by Ti3+ and/or oxygen vacancies (Vo) has been proven to be an effective strategy to extend the light absorption of TiO2 into the visible-light region, even the near infrared area.4 Due to the narrowed band gap and enhanced conductivity, defective TiO2 shows high visible light adsorption and suppressed photogenerated carriers recombination.5 Various methods have been adopted to introduce Ti3+ or oxygen vacancies defects in surface or bulk of TiO2, such as high temperature and pressurized hydrogenation,6,7 plasma treatment,8 and vacuum activation,9 etc. Unfortunately, the oxygen defective TiO2 obtained via these approaches are not stable and easily reoxidize in air leading to a decrease of oxygen vacancies concentration.10 Therefore, the synthesis of stable defective TiO2 with high oxygen vacancies concentration still remains a large challenge.In addition to defect engineering, controlling the exposed facets of TiO2 nanocrystals is another efficient method to improve the photogenerated charges separation because the photogenerated electrons favourably transfer from (001) to (101), while holes migrate from (101) to (001).11 It has been reported that oxygen-deficient anatase TiO2 sheets with dominant (001) facets showed an enhanced hydrogen evolution rate in photocatalytic water splitting reactions.12 Recent research demonstrated that the Ti3+/Vo's sites and the co-exposed (101) and (001) facets of the TiO2 nanocrystals could promote CO2 photoreduction activity.13 Even though the synthetic strategies for preparing well-defined (001) facet dominant anatase TiO2 were developed, it is still challenging to one-step construct oxygen deficiency on special crystal facets.
In the present work, we have successfully developed a one-step molten salt (MS) strategy for synthesis of stable Ti3+-self doped blue TiO2 exposed with (001) facets from pristine white TiO2 using trifluoroacetic acid (TFA) as reducing and capping agent simultaneously. Benefitting from the low oxygen partial pressure and oxygen isolation effect of MS, the incomplete combustion of TFA at high temperature will consume the lattice oxygen atoms of TiO2, leading to the formation of oxygen vacancies both on the surface and in the bulk. Furthermore, the releasing HF from decomposition of TFA favours the formation of exposed (001) facets. As a result, stable oxygen defective blue TiO2 exposed with (001) facets was obtained, which showed narrowed band gap and enhanced photocatalytic activity in photodegradation of rhodamine B (RhB) and selective oxidation of benzyl alcohol (BA) under visible light irradiation. These findings demonstrate a facile approach for synchronously constructing oxygen defects and (001) facets in blue TiO2.
Experimental method
Materials and sample preparation
1 g of TiO2 (Degussa, P25) was mixed with different amount of trifluoroacetic acid (Sigma-Aldrich, China) under stirring at room temperature. After drying in ambient, the modified P25 was finely ground with 5.8 g of LiCl–KCl (Sigma-Aldrich, China) eutectic mixture (45/55 wt%) and then the mixture was placed in a crucible and annealed at 400 °C for 3 h. After cooling to room temperature, the solid product was washed using water to remove the eutectic salt and blue color product was obtained after drying at 60 °C overnight. The samples were labelled as P25-MS, B-TiO2-0.1, B-TiO2-0.5, B-TiO2-1.0, and B-TiO2-2.0, according to the usage of TFA for 0 mL, 0.1 mL, 0.5 mL, 1 mL, and 2 mL, respectively.Sample characterization
The UV-vis absorbance spectra were obtained for the dry-pressed disk samples using a Scan UV-vis spectrophotometer (Varian, Cary 500) equipped with an integrating sphere assembly, using BaSO4 as the reflectance sample. The spectra were recorded at room temperature in air within the range 200–800 nm. The electron paramagnetic resonance (EPR) spectra were measured using JEOL JES-FA200 EPR spectrometer at 100 K. Fourier transform infrared (FTIR) spectra were recorded with KBr disks containing the powder sample with the FTIR spectrometer (Nicolet Magna 550). Raman spectra were identified by using a Thermo-scientific DXR spectrometer in the range of 100–1000 cm−1 at 532 nm. The instrument employed for XPS studies was a Perkin-Elmer PHI 5000C ESCA system with Al Kα radiation operated at 250 W. The shift of the binding energy due to relative surface charging was corrected using the C 1s level at 284.6 eV as an internal standard. The morphologies were observed by transmission electron microscopy (FEI Tecnai G20, 200 kV). X-ray diffraction (XRD) patterns were recorded in the range 20–60° (2θ) using D/MAX-2550 diffractometer (Rigaku, Japan) with Cu Kα radiation (λ = 1.5406 Å).Photocatalytic experiments
The photocatalytic activity of each sample was evaluated in terms of the degradation of rhodamine B and selective oxidation of benzyl alcohol. 0.02 g B-TiO2 was added into a 100 mL quartz photo-reactor containing 20 mL of RhB solution (10 mg L−1). The mixture was stirred for 30 min in the dark in order to reach the adsorption–desorption equilibrium. A 350 W Xe lamp equipped with a UV cut-off filters (λ ≥ 420 nm) was used as a visible light source. The lamp was cooled with flowing water in a quartz cylindrical jacket around the lamp and ambient temperature is maintained during the photocatalytic reaction. At the given time intervals, the analytical samples were taken from the mixture and immediately centrifuged, then filtered through a 0.22 μm Millipore filter to remove the photocatalysts. The filtrates were analyzed by recording variations in the absorption in UV-vis spectra of RhB a Cary 100 ultraviolet visible spectrometer. For benzyl alcohol oxidation, 20 mg of B-TiO2 was suspended in 20 mL of toluene with 0.1 mmol of benzyl alcohol. O2 was bubbled through the mixture at a rate of 10 mL min−1. The irradiation condition was identical to degradation of RhB. After the photocatalytic reaction, the products were analyzed using a gas chromatograph system (TRACE 1310GC, Thermo Fisher) with a flame ionization detector (FID).Results and discussion
Molten salt synthesis of defective blue TiO2 (B-TiO2) was carried out in LiCl–KCl eutectic mixture (45/55 wt%) at 400 °C for 2 h using TFA modified commercial TiO2 (Degussa P25) as precursor. Unexpectedly, after cooling to room temperature, blue color solid product was obtained, while it still kept white color without addition of TFA or with small molten salt dosage (Fig. S1†). The solid product was washed using water to remove the eutectic salt and dark blue powder was obtained after drying at 60 °C overnight. The optical response of blue B-TiO2-2.0 and P25 is shown in Fig. 1a. It is clearly seen that B-TiO2-2.0 displays a substantial enhancement of optical absorption in visible light region with a tail extending to NIR region, while commercial P25 only responds to ultraviolet light due to its intrinsic wide band gap. The strong visible light absorption is attributed to the existence of oxygen vacancies or Ti3+ defects, which can induce a continuous donor level of electronic states below the conduction band.14 As seen in the inset of Fig. 1a, the color of TiO2 turned from white to dark blue via molten salt treatment. The band gaps of B-TiO2-2.0 and P25 obtained from Kubelka–Munk transformation of the reflectance spectrum are 2.85 eV and 3.10 eV, respectively. The narrowed band gap for B-TiO2 is due to the raised defects band of electronic states.15 In order to further confirm the defect species in B-TiO2, electron paramagnetic resonance (EPR) is used to detect the paramagnetic species with unpaired electrons either in the surface or bulk regions. As shown in Fig. 1b, pristine P25 exhibits weak EPR signal because of the absence of paramagnetic species. In contrast, a prominent signal at g = 1.93–1.98 appears in B-TiO2, demonstrating the presence of Ti3+ in the bulk.16 The shoulder peak at g = 2.001 confirms the surface oxygen vacancies.17 The formation mechanism of oxygen vacancies or Ti3+ is explained as followed: when TFA modified P25 was calcinated in MS, the thermal decomposition of TFA was suppressed by the oxygen isolation effect of LiCl–KCl eutectic mixture and the incomplete combustion production of TFA, such as CO, consumed the lattice oxygen in TiO2 quickly, leading to the formation of amount of oxygen vacancies on the surface and in the bulk of TiO2. The electrons located at the oxygen vacancies can be transferred to the adjacent Ti4+ to generate the reduced Ti3+ centres, which narrows the band gap and causes strong visible absorption in the long wavelength range. In fact, it was found that the surface of LiCl–KCl eutectic mixture was quite dense when cooling down to room temperature, suggesting that MS prevented the oxygen permeating into the mixture to reoxidize the oxygen defective. Further investigation demonstrated that the relative concentration of surface oxygen vacancies and bulk Ti3+ can be precisely controlled by tuning the annealing temperature and time. A longer annealing time and a higher temperature will lead to increase of the bulk Ti3+ defect concentration and decrease of surface oxygen vacancy concentration (Fig. S2†).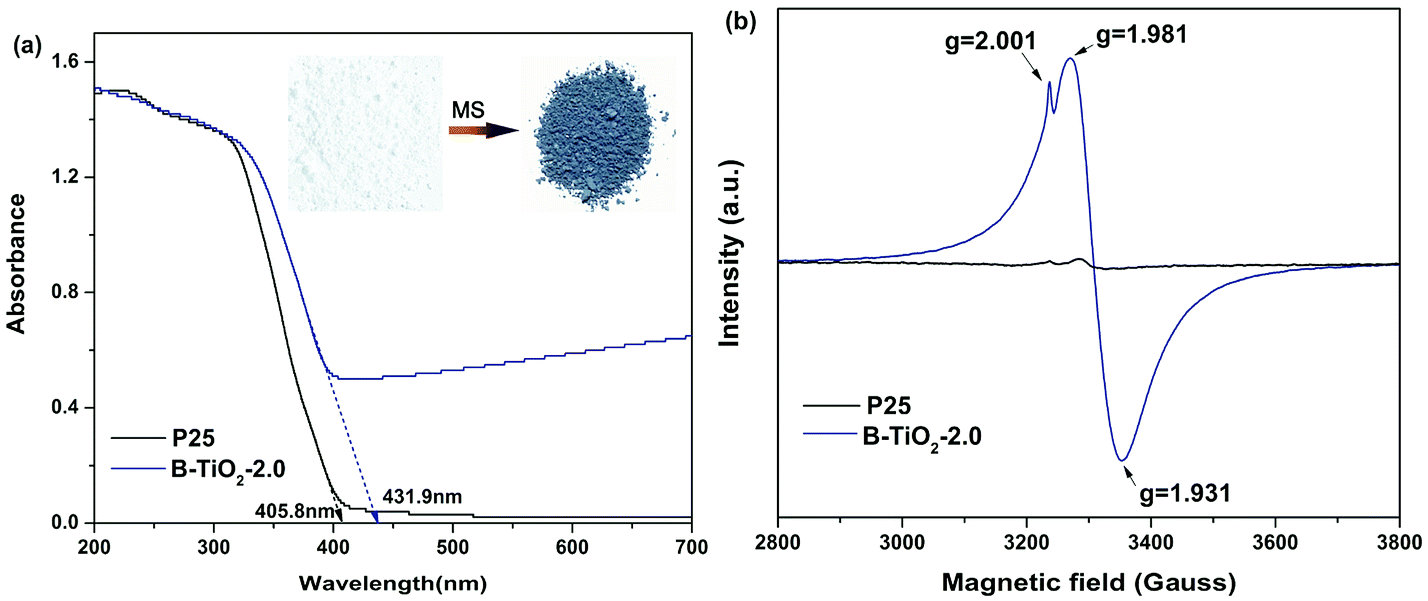 | ||
| Fig. 1 (a) UV-visible diffuse reflectance spectra and (b) X-band EPR spectra of B-TiO2-2.0 and P25. | ||
The surface structure of blue B-TiO2-2.0 is further investigated by Fourier transform infrared (FTIR) spectroscopy, Raman spectra, and X-ray photoelectron spectroscopy (XPS). As shown in Fig. 2a, the FTIR spectrum of B-TiO2-2.0 exhibits stronger –OH stretching bands at 3430 cm−1 and 1643 cm−1 than that of P25, suggesting there are more oxygen vacancies on the surface of B-TiO2-2.0. As reported in the literature,18 the dissociative adsorption of water is more like to bond with oxygen vacancies leading to the formation of more hydroxyl groups. Raman spectra are often used to preliminarily investigate the exerted influence of oxygen deficiency on the geometric structure since the Eg mode in Raman spectra is more sensitive to oxygen defects than the Ti–O stretching mode.19 Fig. 2b shows the Raman spectra of B-TiO2 and P25. It is clear to see that the strongest Eg mode at 142.7 cm−1 is shifted to 143.8 cm−1, accompanying with peak broadening. The blue-shift and peak broadening effect has been observed in several recent studies on hydrogenated TiO2,20 which is attributed to the presence of lattice disorder resulting from phonon confinement or the decreasing of correlation length resulting from localized defects like oxygen vacancy.
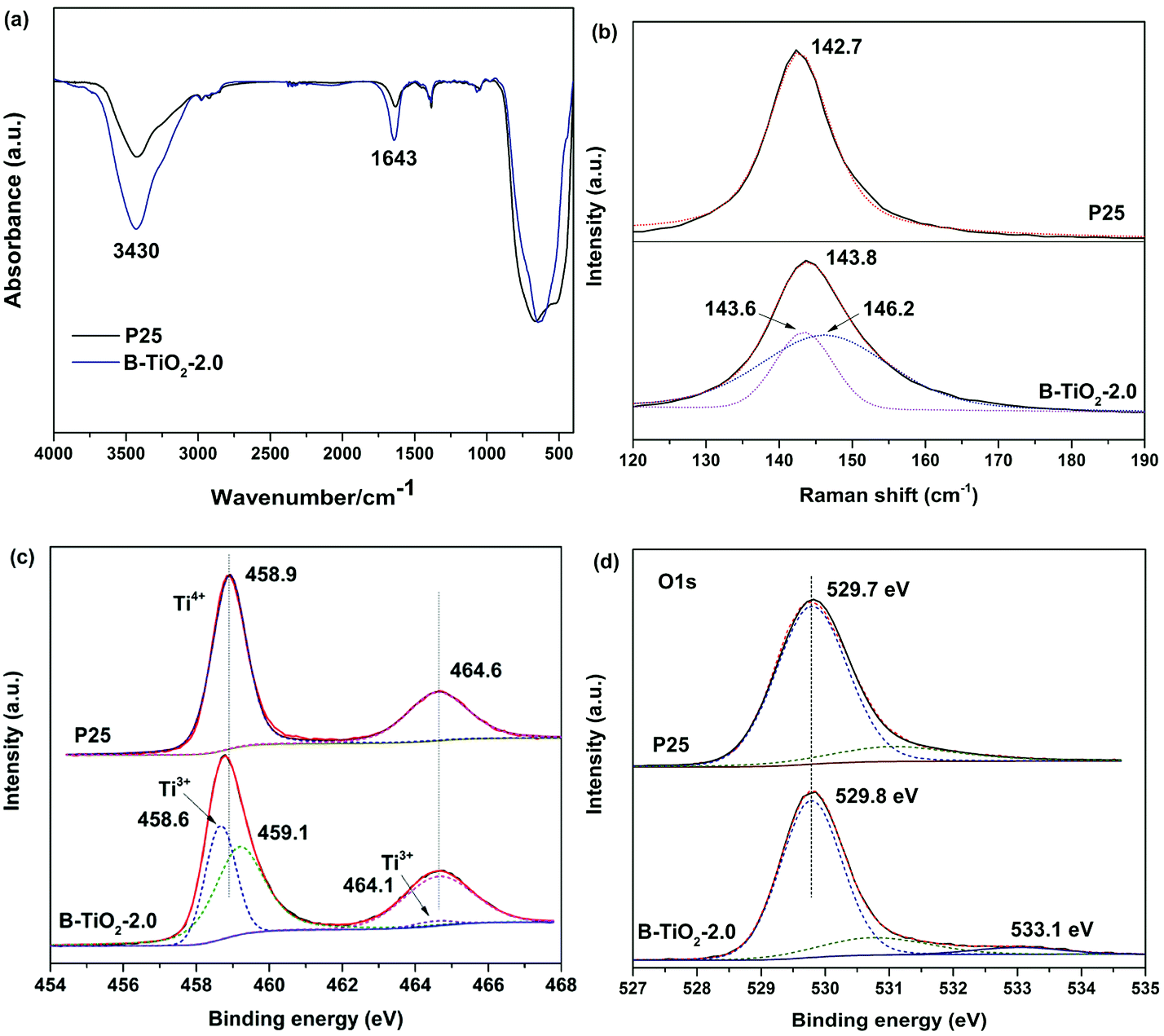 | ||
| Fig. 2 (a) FTIR spectra, (b) Raman spectra, (c) Ti 2p high-resolution XPS spectra and (d) O 1s high-resolution XPS spectra of B-TiO2-2.0 and P25. | ||
XPS was also employed to study the surface chemical states of blue TiO2. Fig. 2c shows the Ti 2p core level spectra of B-TiO2-2.0 and P25. The surface of P25 is dominated by two major peaks at 458.9 and 464.6 eV for typical characteristics of Ti4+. In contrast, two lower energy peaks at 458.6 and 464.1 eV for Ti3+ are observed in B-TiO2-2.0 after Gauss fitting of Ti 2p peaks. This result demonstrates that Ti3+ is formed on the surface or subsurface of blue TiO2 due to the charge compensation of oxygen vacancy. O 1s XPS spectra of both of samples exhibit a main peak at 529.7 eV, corresponding to the lattice oxygen (Ti–O) in TiO2 (Fig. 2d). Intriguingly, a small peak at 533.1 eV assigned to surface hydroxyl groups (Ti–OH) appear in B-TiO2-2.0, which is consistent with FTIR results. Because the oxygen vacancies on the surface are filled by reaction with H2O in the air, oxygen vacancies induce dissociation of water molecules and form two hydroxyl groups via H+ transfer to neighboring lattice oxygen.
The morphology and phase of blue B-TiO2-2.0 are further investigated by transmission electron microscopy (TEM) and X-ray diffraction (XRD). Fig. 3a and b showed the typical images of P25 treated in MS without and with TFA, respectively. Interestingly, truncated bipyramidal TiO2 with exposed (001) facets are observed clearly after treatment with molten salt method. This is probably attributed to the truncation effect of released HF during TFA decomposition, which usually used as capping agent for (001) facets. According to the literature,21 the presence of the molecular form of HF but not F− is essential for formation of the exposed (001) facets of anatase TiO2. In fact, when TFA was replaced by NaF, no (001) facets were observed in the samples (Fig. S3†). It is worth mentioning that the coexistence of (101) facet and (001) facet is beneficial to spatial separation of photogenerated charges due to that the photogenerated holes and electrons prefer to be captured on the (001) and (101) surfaces, respectively.22 High resolution F 1s XPS spectra give the direct evidences of the presence of F in the B-TiO2-2.0 (Fig. 3c). The centroid of the F 1s peak at 685.0 eV is consistent with fluorine bound to the surface of TiO2 (i.e., terminal Ti–F bond).23 Beside the main peak, a shoulder peak at 688.2 eV appears in the B-TiO2-2.0, demonstrating the substitution of F atoms for O atoms in TiO2 lattices.24,25 Recent research work has reported that fluoride anion is efficient for the stabilization of reduced TiO2 because of its tendency to form a strong bond with the surface Ti atoms.26 Moreover, fluorine-occupied surface oxygen vacancies can maintain the intrinsic nature of the defects.27 In our work, the in situ generated HF during TFA decomposition not only induces the formation of (001) facets, but also stabilizes the surface oxygen vacancies by formation of Ti–F–Ti bonds. The blue colors remain stable even after calcination at 300 °C for 2 h in air (Fig. S4†).
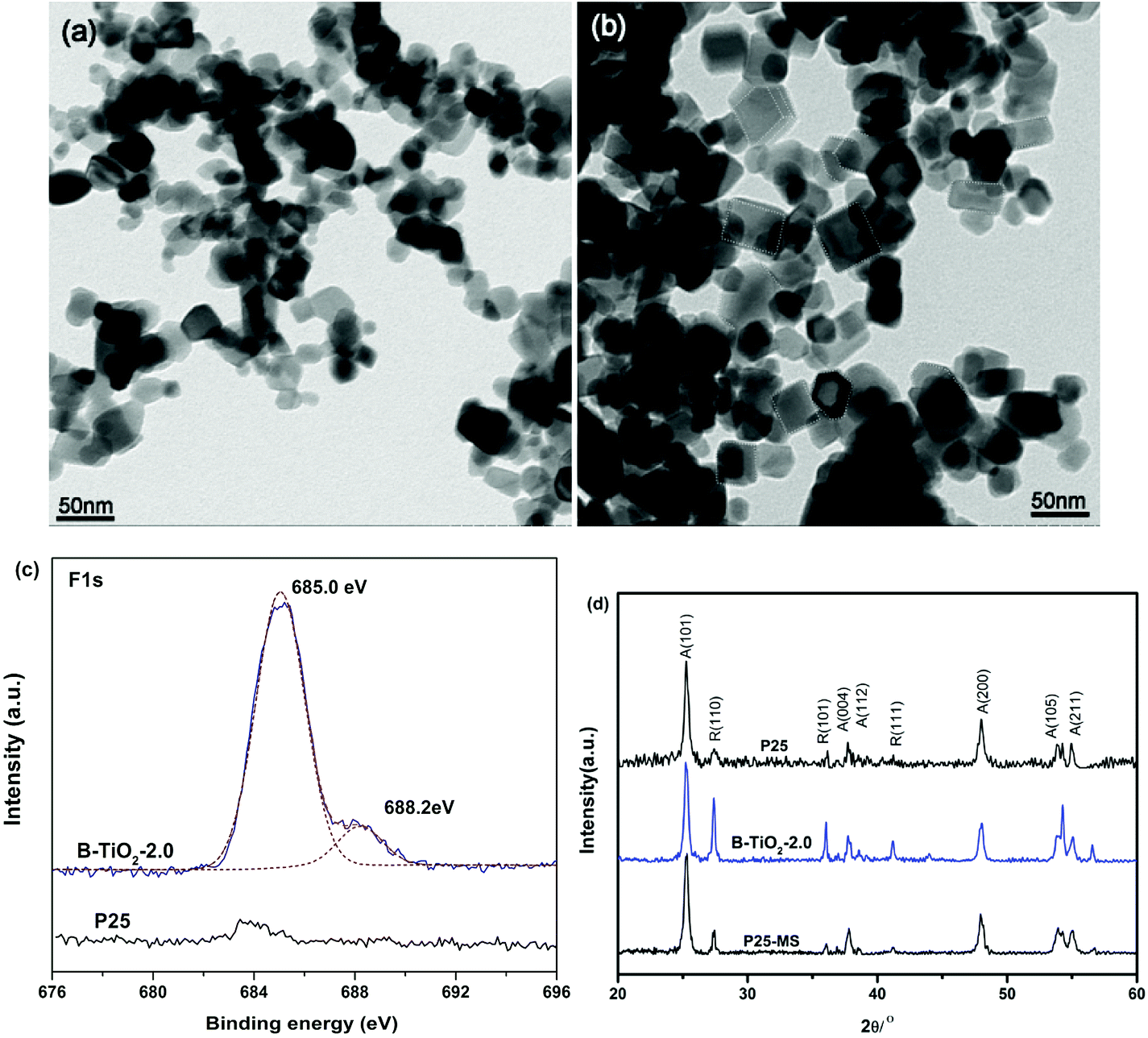 | ||
| Fig. 3 TEM image of P25 annealed in MS without (a) and with (b) TFA, (c) F 1s XPS spectra and (d) XRD patterns. | ||
Beside the morphology variation, the crystalline phase changes of TiO2 during MS process are also analyzed. Fig. 3d displays the XRD patterns of blue B-TiO2-2.0, P25-MS and P25. It is clear to see that the (110) peak intensity of rutile phase increases greatly, suggesting that the relative amount of rutile phase in B-TiO2-2.0 is significantly increased compared with pristine P25 and P25-MS. It has been reported that oxygen vacancies could induce the anatase to rutile phase transformation at relative low temperature.28 Since the phase transformation involves an overall contraction or shrinking of the oxygen structure, the removal of oxygen ions might be expected to accelerate the transformation, which could provide more nucleation centres for rutile phase.29 Interestingly, a less amount of TFA (0.1 mL) could induce higher rutile ratio (Fig. S5†). Further increasing the TFA amount decreases the relative amount of rutile phase because substitution of fluoride ions for an oxygen ion reduces the number of anion vacancies and inhibits the transformation from anatase to rutile phase.30 These findings give a new way to tune the exposed facets percentage and phase composition of TiO2 by controlling the amount of TFA.
Photocatalytic activity of B-TiO2 and P25 is evaluated by photodegradation of RhB and photocatalytic oxidation of benzyl alcohol under visible light irradiation (λ ≥ 420 nm). Fig. 4a shows the typical time depending concentration variation of RhB under visible light irradiation. As expectedly, the pristine P25 shows poor visible light photocatalytic activity due to its wide band gap and weak visible light adsorption. By contrast, B-TiO2 exhibits higher photocatalytic activity, which is nearly 10 times higher than that of P25. Increasing the amount of TFA can further improve the photocatalytic activity. When the amount of TFA is 2 mL, the photocatalytic activity achieves a maximum due to the saturated adsorption of TFA on TiO2 surface. More amount of TFA has slight effect on the improvement of photocatalytic activity (Fig. S6†). It is worth noting that P25-MS shows higher degradation rate than P25, suggesting that MS treatment can improve the photocatalytic activity. Moreover, MS dosages also influence the photocatalytic activity and small amount of molten salt led to a poor visible light photocatalytic activity (Fig. S7†). Fig. 4b shows the photocatalytic conversion of benzyl alcohol. It appears that the conversion rate of benzyl alcohol increases with the amount of TFA, indicating that the photo-oxidation activity is improved greatly. The conversion rate maximum achieves to 58% and the selectivity for all samples is above 99%. Importantly, B-TiO2 has high stability, which can keep the photocatalytic activity even after calcination at 300 °C (Fig. S8†). Valence band (VB) XPS results shows that the valence band maximum (VBM) of B-TiO2-2.0 is downward shifted from 2.34 eV to 2.59 eV after molten salt treatment, indicating that the presence of oxygen vacancies and Ti3+ defect species can substantially shift the VB maximum downwards due to the band-blending effects in the band gap.31 Recent report demonstrated that the reduced TiO2 exhibited worse UV photocatalytic activity than that of pristine TiO2 because the Ti3+ deep and shallow trap states below the conduction band reduced the lifetime of photogenerated charge carriers.32 However, in our present work, full spectrum light irradiation photocatalytic experiment showed that B-TiO2-2.0 also exhibited higher UV photocatalytic activity (Fig. S9†). Combined with the VB XPS and UV-vis diffuse reflectance spectra (DRS) results, the energy band diagram of the samples can be calculated as shown in Fig. 4d. The downward VBM position is beneficial for the enhancement of oxidation capacity of photogenerated holes.
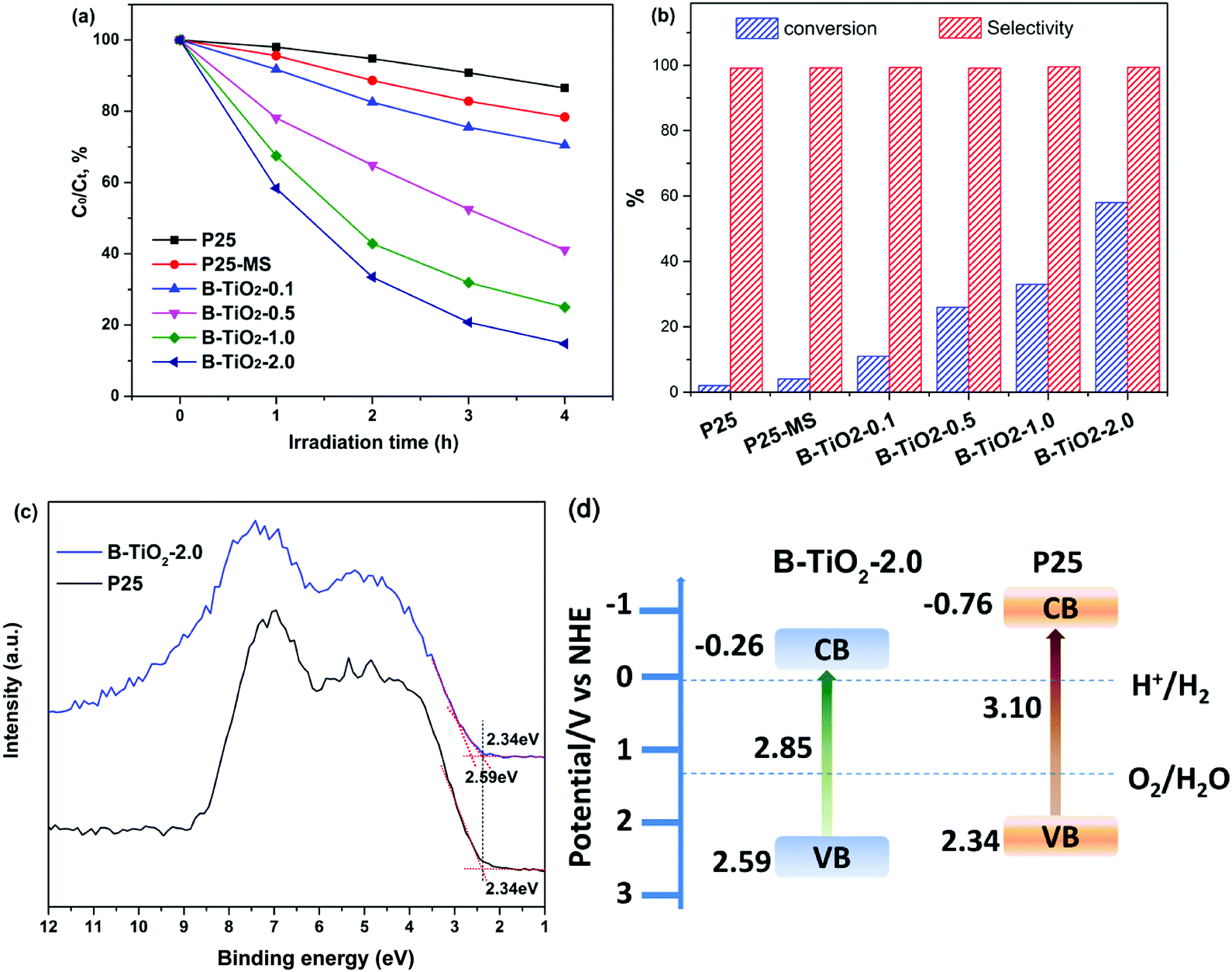 | ||
| Fig. 4 (a) Photodegradation of RhB over different samples under visible light irradiation. (b) Photocatalytic oxidation of benzyl alcohol after irradiation for 7 h. (c) Valence-band XPS spectra. (d) Schematic illustrations of the energy bands. | ||
Based on the above analysis, there are several reasons for that the visible light photocatalytic activity is enhanced. Firstly, the surface oxygen vacancies and bulk Ti3+ constructed via MS method narrows the band gap and increases the visible light adsorption. Secondly, the coexistence of (101) and (001) facets improves the spatial separation of photogenerated charges. Third, the substitution of F for oxygen in lattices is very favorable for the adsorption of O2 so as to promote the photo-oxidation activity.
Conclusions
In summary, we have developed a facile molten salt approach to synthesize reduced TiO2 exposed with (001) facets. The as-prepared blue TiO2−x displays enhanced photocatalytic oxidation activity under visible light irradiation due to the narrowed band gap, which is caused by the existence of oxygen vacancies and bulk Ti3+ defects. The relative concentration of oxygen vacancies and bulk Ti3+ can be tuned by the amount of TFA. The present study demonstrates a general and economic method for narrowing the ban gap and improving the visible light photocatalytic activity, which is also suitable for other semiconductor oxides (e.g. CeO2, WO3, Co3O4, etc.).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the financial support from the National Natural Science Foundation of China (No. 51203094), Capacity Building for Sci-Tech Innovation-Fundamental Scientific Research Funds (Grant No. 025185305000/208) and Beijing Natural Science Foundation and Beijing Academy of Science and Technology (No. L140005).Notes and references
- H. Xu, S. Ouyang, L. Liu, P. Reunchan, N. Umezawa and J. Ye, J. Mater. Chem. A, 2014, 2, 12642–12661 RSC.
- X. Chen and A. Selloni, Chem. Rev., 2014, 114, 9281–9282 CrossRef CAS PubMed.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- X. Pan, M. Yang, X. Fu, N. Zhang and Y. Xu, Nanoscale, 2013, 5, 3601–3614 RSC.
- J. Su, X. Zou and J. S. Chen, RSC Adv., 2014, 4, 13979–13988 RSC.
- G. H. Yin, X. Y. Huang, T. Y. Chen, W. Zhao, Q. Y. Bi, J. Xu, Y. F. Han and F. Q. Huang, ACS Catal., 2018, 8, 1009–1017 CrossRef CAS.
- X. B. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- X. Kong, Y. Xu, Z. Cui, Z. Li, Y. Liang, Z. Gao, S. Zhu and X. Yang, Appl. Catal., B, 2018, 230, 11–17 CrossRef CAS.
- M. Xing, J. Zhang, F. Chen and B. Tian, Chem. Commun., 2011, 47, 4947–4949 RSC.
- W. Z. Fang, F. Dappozze, C. Guillard, Y. Zhou, M. Y. Xing, S. Mishra, S. Daniele and J. L. Zhang, J. Phys. Chem. C, 2017, 121, 17068–17076 CrossRef CAS.
- G. Liu, H. G. Yang, J. Pan, Y. Q. Yang, G. Q. Lu and H.-M. Cheng, Chem. Rev., 2014, 114, 9559–9612 CrossRef CAS PubMed.
- G. Liu, H. G. Yang, X. Wang, L. Cheng, H. Lu, L. Wang, G. Q. Lu and H.-M. Cheng, J. Phys. Chem. C, 2009, 113, 21784–21788 CrossRef CAS.
- L. J. Liu, Y. Q. Jiang, H. L. Zhao, J. T. Chen, J. L. Cheng, K. S. Yang and Y. Li, ACS Catal., 2016, 6, 1097–1108 CrossRef CAS.
- A. Naldoni, M. Allieta, S. Santangelo, M. Marelli, F. Fabbri, S. Cappelli, C. L. Bianchi, R. Psaro and V. Dal Santo, J. Am. Chem. Soc., 2012, 134, 7600–7603 CrossRef CAS PubMed.
- C. Di Valentin, G. Pacchioni and A. Selloni, J. Phys. Chem. C, 2009, 113, 20543–20552 CrossRef CAS.
- B. Pattier, M. Henderson, A. Pöppl, A. Kassiba and A. Gibaud, J. Phys. Chem. B, 2010, 114, 4424–4431 CrossRef CAS PubMed.
- J. Cai, M. Wu, Y. Wang, H. Zhang, M. Meng, Y. Tian, X. Li, J. Zhang, L. Zheng and J. Gong, Chem, 2017, 2, 877–892 CAS.
- O. Bikondoa, C. L. Pang, R. Ithnin, C. A. Muryn, H. Onishi and G. Thornton, Nat. Mater., 2006, 5, 189–192 CrossRef CAS.
- I. D. C. Silva, F. A. Sigoli and I. O. Mazali, J. Phys. Chem. C, 2017, 121, 12928–12935 CrossRef CAS.
- X. Yu, B. Kim and Y. K. Kim, ACS Catal., 2013, 3, 2479–2486 CrossRef CAS; N. Liu, C. Schneider, D. Freitag, U. Venkatesan, V. R. R. Marthala, M. Hartmann, B. Winter, E. Spiecker, A. Osvet, E. M. Zolnhofer, K. Meyer, T. Nakajima, X. Zhou and P. Schmuki, Angew. Chem., Int. Ed., 2014, 53, 14201–14205 CrossRef PubMed.
- H. Zhang, Y. Wang, P. Liu, Y. Han, X. Yao, J. Zou, H. M. Cheng and H. Zhao, ACS Appl. Mater. Interfaces, 2011, 3, 2472–2478 CrossRef CAS PubMed.
- P. Zhou, H. N. Zhang, H. W. Ji, W. H. Ma, C. C. Chen and J. C. Zhao, Chem. Commun., 2017, 53, 787–790 RSC.
- G. S. Wu, J. P. Wang, D. F. Thomas and A. C. Chen, Langmuir, 2008, 24, 3503–3509 CrossRef CAS PubMed.
- J. C. Yu, J. G. Yu, W. K. Ho, Z. T. Jiang and L. Z. Zhang, Chem. Mater., 2002, 14, 3808–3816 CrossRef CAS.
- W. W. Yan, Q. R. Chen, X. F. Meng and B. Wang, Sci. China Mater., 2017, 60, 449–460 CrossRef CAS.
- W. Z. Fang, F. Dappozze, C. Guillard, Y. Zhou, M. Y. Xing, S. Mishra, S. Daniele and J. L. Zhang, J. Phys. Chem. C, 2017, 121, 17068–17076 CrossRef CAS.
- L. Xu, L. Ming and F. Chen, ChemCatChem, 2015, 7, 1797–1800 CrossRef CAS.
- R. Chandana, P. Mohanty, A. C. Pandey and N. C. Mishra, J. Phys. D: Appl. Phys., 2009, 42, 205101 CrossRef.
- R. D. Shannon and J. A. Pask, J. Am. Ceram. Soc., 1965, 48, 391–398 CrossRef CAS.
- K. Lv, B. Cheng, J. Yu and G. Liu, Phys. Chem. Chem. Phys., 2012, 14, 5349–5362 RSC.
- J. Pan, G. Liu, G. M. Lu and H. M. Cheng, Angew. Chem., Int. Ed., 2011, 50, 2133–2137 CrossRef CAS PubMed.
- S. K. Cushing, F. Meng, J. Zhang, B. Ding, C. K. Chen, C.-J. Chen, R.-S. Liu, A. D. Bristow, J. Bright, P. Zheng and N. Wu, ACS Catal., 2017, 7, 1742–1748 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07543c |
| This journal is © The Royal Society of Chemistry 2018 |