
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of hyperbranched crosslinkers from bio-derived platform molecules for the synthesis of epoxidised soybean oil based thermosets†
Trin Kamjornsupamitr,
Andrew J. Hunt and
Nontipa Supanchaiyamat*
and
Nontipa Supanchaiyamat*
Materials Chemistry Research Center, Department of Chemistry, Center of Excellence for Innovation in Chemistry, Faculty of Science, Khon Kaen University, Khon Kaen 40002, Thailand. E-mail: nontsu@kku.ac.th
First published on 6th November 2018
Abstract
Bio-based carboxyl-terminated hyperbranched crosslinkers have been synthesised by the facile esterification reaction of glycerol with succinic anhydride (Gly-SA). The Gly-SA crosslinking molecules have a large number of terminal carboxyl groups, which can crosslink through the epoxide of epoxidised soybean oil (ESO), making a highly flexible transparent film with excellent oxidative resistance. The effect of different molecular weights of Gly-SA cured ESO on the thermal and mechanical properties of the resulting films was also investigated. This study demonstrated that an increase in the molecular weights of Gly-SA, led to a decrease in the curing rate of mixtures, whilst the glass transition temperature (Tg) of Gly-SA cured ESO increased due to the incorporation of the bulky crosslinker. The use of a Gly-SA crosslinker prepared at 150 °C, resulted in a film (EGS150) with a tensile strength 13 times greater than of the control film, exhibited more than a 220% increase in elongation at break and the Young's modulus quadrupled compared to the value obtained for the control sample. It is noteworthy that the tensile strength and elongation at break improved with increasing Gly-SA chain length, suggesting the pre-polymerised crosslinkers contribute to the enhanced mechanical properties of the materials.
Introduction
The use of bio-based polymers derived from renewable resources have become of interest due to the rapid depletion of crude oil and the potential environmental benefits.1 Plant oils are an attractive feedstock due to their low toxicity, competitive cost, high availability and intrinsic renewability.2–5 The majority of plant oils are utilised within the food industry except for castor and linseed oils, which are used in non-food applications such as lubricants, surface coatings and plastics.6–8 In addition, plant oils have been used for decades in paint formulations and resin applications.9 Plant oils or vegetable oils are fats derived in large volumes from agricultural feedstocks including plants seeds, as triglyceride esters of glycerol and 3 fatty acids.10–14 In 2017/2018, global oil seed production (copra, cottonseed, palm kernel, peanut, rapeseed, soybean and sunflower seed) was estimated to be up to 574 million tonnes, accounting for 345 million tonnes of soybean oil.15Soybean oil is used in various applications such as soaps, paints, coatings, lubricants, and bioplastics.16,17 Typically, soybean oil is composed of palmitic (11%), oleic (23%), linoleic (53%), linolenic (8%) fatty acids, having an average of 4.6 double bonds per triglyceride.9,18,19 The abundance of unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds within the fatty acid constituents of soybean oil facilitate the production of epoxidised soybean oil (ESO).20 ESO can be easily transformed to epoxy thermosets through cationic polymerisation or curing with crosslinking agents such as dicarboxylic acid, anhydrides and diamines.14,21–25 Moreover, ESO derived thermosets materials can be biodegradable in a composting environment.26 Although numerous studies of ESO based thermosets have been reported, the majority of work concerns the use of non-renewable crosslinkers, resulting in low bio-content products.27–29
C double bonds within the fatty acid constituents of soybean oil facilitate the production of epoxidised soybean oil (ESO).20 ESO can be easily transformed to epoxy thermosets through cationic polymerisation or curing with crosslinking agents such as dicarboxylic acid, anhydrides and diamines.14,21–25 Moreover, ESO derived thermosets materials can be biodegradable in a composting environment.26 Although numerous studies of ESO based thermosets have been reported, the majority of work concerns the use of non-renewable crosslinkers, resulting in low bio-content products.27–29
The main challenge in converting ESO thermosets to high performance materials is to overcome the limitation of their short-chain crosslinked structures, which results in a brittle material.30 Long chain dicarboxylic acids have shown promising results when used as crosslinking agents for ESO, providing better flexibility and higher thermal stability as compared to those with shorter chains.14,31 Several studies have developed bio-based crosslinkers for ESO resins to improve properties by providing a larger and stronger structural crosslinkers, such as the preparation of tetrafunctional crosslinkers from gallic acid dicarboxyl terminated oligomeric poly(butylene succinate) (OPBS) and dicarboxyl-terminated polyamide 1010 oligomers (NYL) from castor oil, which can enhance the mechanical properties for ESO-based thermoset materials.32–34 Dai et al. reported a synthesis of polyesters by melt polycondensation of itaconic acid with diols and glycerol, which were used to crosslink acrylated epoxidized soybean oil (AESO). The results indicated that the introduction of the polyesters significantly improved the mechanical properties of the thermosetting resins.35 Other bio-derived crosslinkers including methacrylated eugenol and isosorbide-methacrylate were also utilised as hardeners for AESO,36,37 however such crosslinkers are not fully bio-based and are costly. Therefore, further work is still needed to develop commercially viable sustainable low cost crosslinkers from abundantly available bio-based platform molecules. Glycerol is an abundant co-product of the biodiesel industry and one of the top 12 bio-derived platform molecules.3,38
Herein, carboxylic hyperbranched oligomers (Gly-SA) crosslinking agents have been synthesised via a simple esterification reaction of glycerol (Gly) and succinic anhydride (SA). These bio-derived crosslinkers were utilised in the curing of ESO in order to obtain a fully bio-based thermosetting resin.
Experimental
Materials
Epoxidised soybean oil (ESO) was purchased from Akcros Chemicals as Lankroflex E2307. Glycerol (Gly, 99.5%) was purchased from Carlo erba. Succinic anhydride (SA, 98%) was supplied from Merck. 4-Dimethylaminopyridine (DMAP, 99%) was obtained from Acros Chemicals. All materials were used as received without further purification.Synthesis of carboxyl-terminated hyperbranched oligomers
Carboxyl-terminated crosslinkers with different molecular weights were synthesised by direct esterification of glycerol and succinic anhydride. Four different temperatures; 90 °C, 110 °C, 130 °C and 150 °C of the synthesis temperature were used and the products obtained were named according to their processing temperatures as Gly-SA90, Gly-SA110, Gly-SA130, and Gly-SA150, respectively. The Gly-SA crosslinkers were synthesised according to Scheme 1 from glycerol and succinic anhydride in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 at four different temperatures for 5 hours under a nitrogen atmosphere and the obtained materials were stored at room temperature.
:3 at four different temperatures for 5 hours under a nitrogen atmosphere and the obtained materials were stored at room temperature.
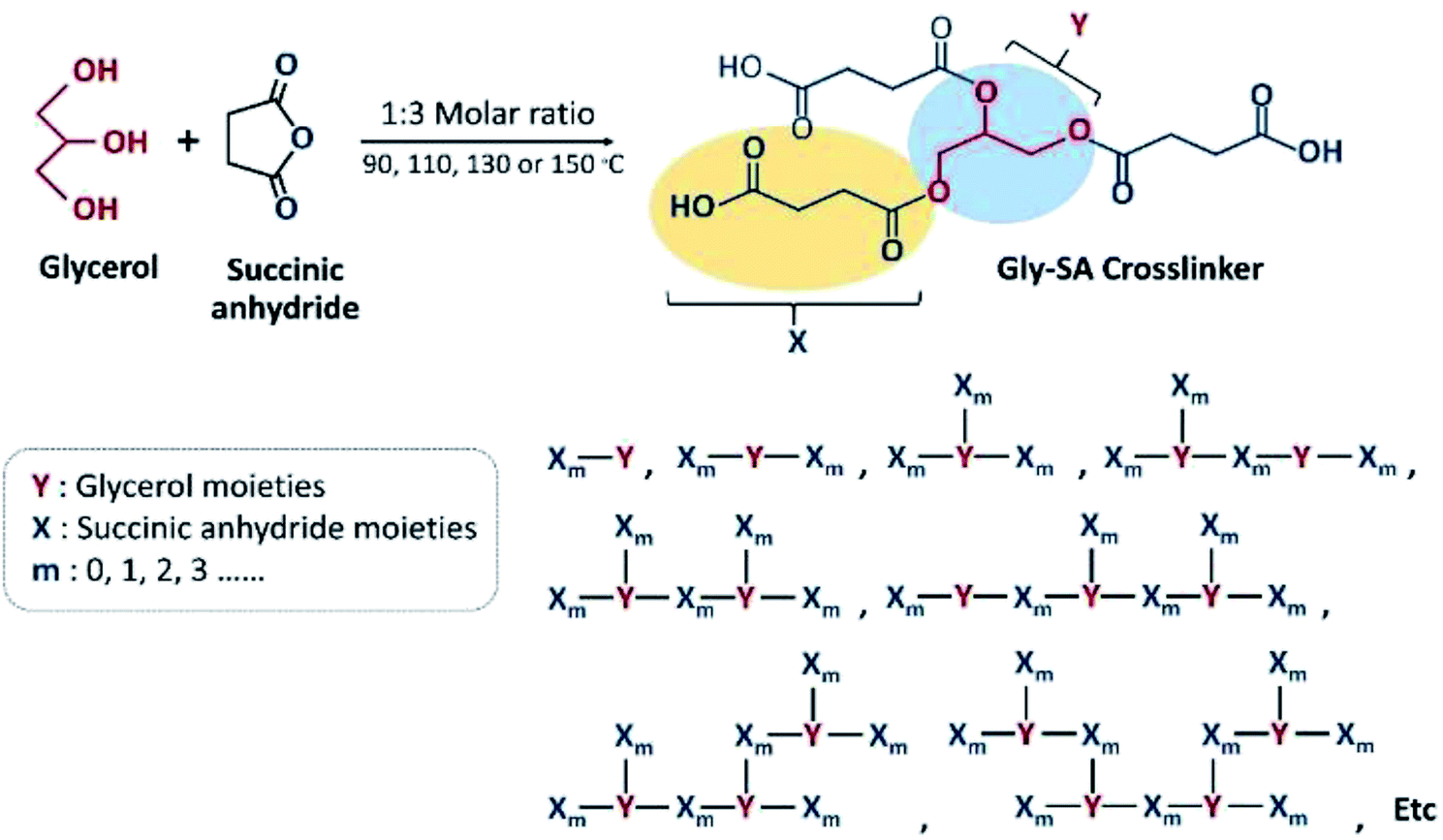 | ||
| Scheme 1 Schematic representation of synthetic route to Gly-SA crosslinker. | ||
NMR of Gly-SA: 1H NMR (DMSO, δ, ppm): 2.4–2.6 (CH2–COOH and CH2–COO–), 3.9–4.2 (–CH2–OCO–), 5.1–5.2 (–CH–O–CO–); 13C NMR (DMSO, δ, ppm): 29.0 (–CH2–COOH & –CH2–COO–), 62.1 (–CH2–OCO–), 69.3 (–CH–O–CO–), 172.2 (–COO–), 173.6 (–COOH).39
Preparation of sample films of Gly-SA cured ESO thermosets
Bio-based thermosetting films were prepared by ring-opening crosslinking reaction of ESO with Gly-SA. Gly-SA was melted at 140 °C for 5 minutes, to this ESO was added to the mixture solution (1:1 weight ratio) and stirred for 15 minutes at 140 °C. Subsequently, DMAP catalyst (0.5% of the total resin weight) was added and the mixture was stirred for a further 2 minutes. The mixture was poured into a silicone tray and cured at 140 °C for 24 hours in order to obtain the thermoset materials (EGS). The plausible cross-linked structure of the EGS resin is presented in Scheme 2.
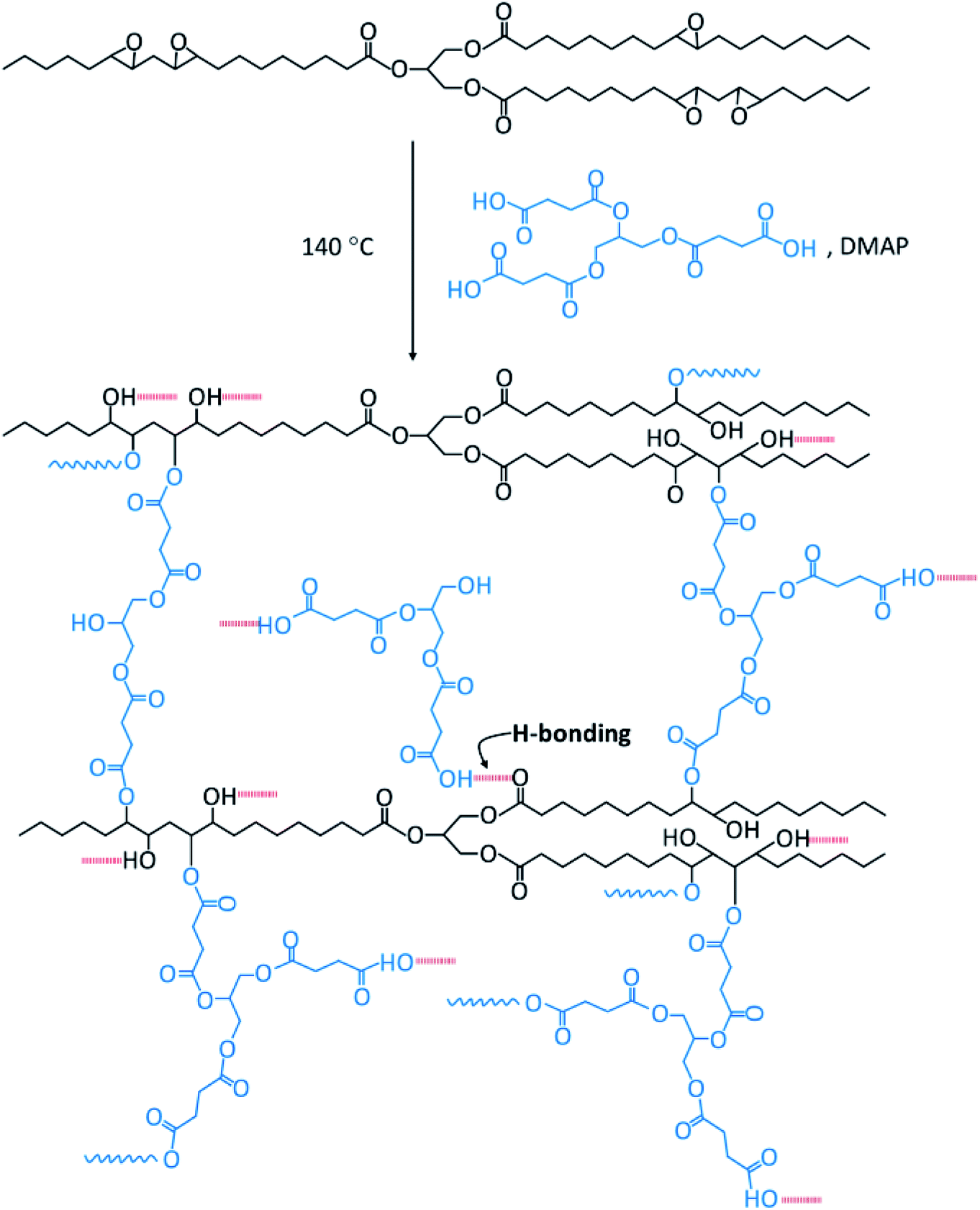 | ||
| Scheme 2 Plausible crosslinked structure for the ESO with Gly-SA. | ||
Characterisation
δ as a function of temperature at a constant frequency were observed.where Wdry,i and Wdry,t are the dry weight (g) before and after treatment in Fenton's reagent, respectively.
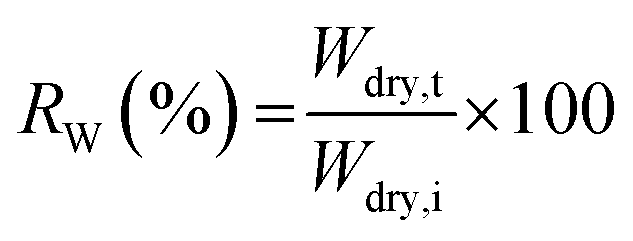
Results and discussion
Synthesis and characterisation of Gly-SA
A schematic representation of the synthesis of Gly-SA crosslinkers and the possibility of the hyperbranched chain is shown in Scheme 1. The obtained products are viscous liquids at room temperature with white to pale yellow colour as shown in Fig. 1. | ||
| Fig. 1 Gly-SA crosslinkers. | ||
Fourier transform infrared (FTIR) spectroscopy was used in attenuated total reflectance (ATR) mode to investigate the reaction between glycerol and succinic anhydride. The FTIR spectra of glycerol, succinic anhydride and Gly-SA are shown in Fig. 2. The disappearance of the anhydride bands at 1775 and 1862 cm−1 confirmed the ring opening reaction of the anhydride. The CO characteristic bands at 1707 and 1730 cm−1 were noted in the spectra of the synthesised crosslinkers, suggesting the presence of carboxylic acid and ester functionalities respectively. Notably, the absorption peak at 1153 cm−1 associated with the C–O stretching vibration indicated the esterification of glycerol and succinic anhydride. FTIR results for Gly-SA90, Gly-SA110 and Gly-SA130 are consistent with the results of Gly-SA150 and are presented in the ESI.†
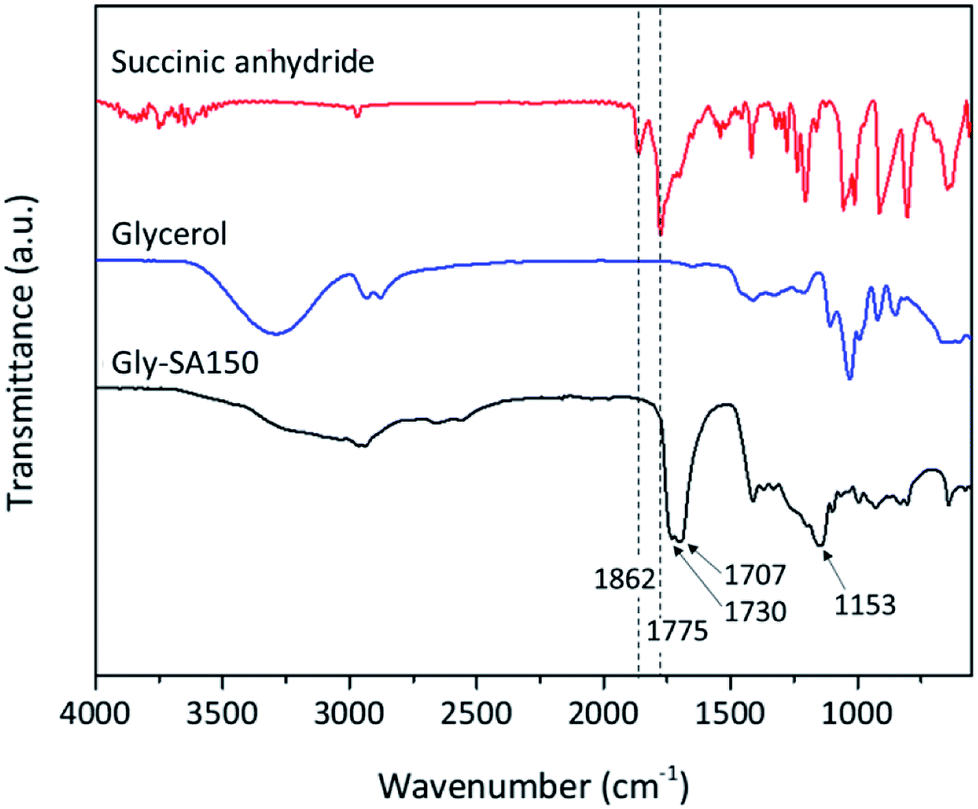 | ||
| Fig. 2 FTIR spectra of glycerol, succinic anhydride and Gly-SA150. | ||
The structures of Gly-SA were further supported by 1H and 13C NMR spectroscopy. The 1H and 13C NMR spectra of Gly-SA90, Gly-SA110, Gly-SA130 and Gly-SA150 are shown in Fig. 3 and 4, respectively. The incorporation of hyperbranched units of the crosslinkers was noted as point (e) in 1H NMR spectra, corresponding to the further esterification reactions of carboxylic acid end groups and hydroxyl group in glycerol molecules (ESI Fig. S2†). The intensity of the peak was found to increase with increasing preparation temperature, indicating the increase in the degree of branching.
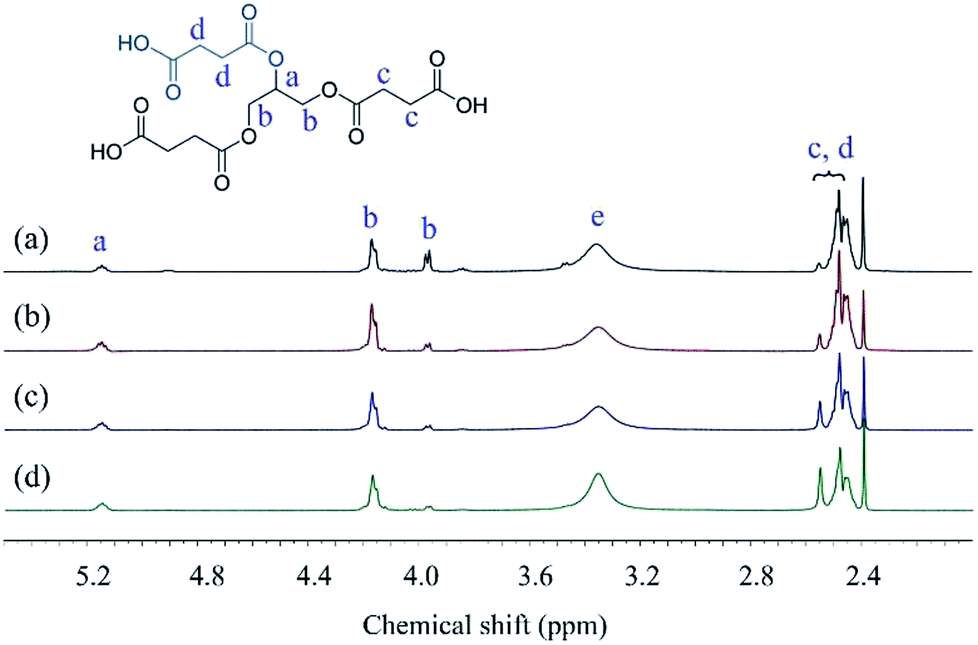 | ||
| Fig. 3 1H NMR spectra of (a) Gly-SA90, (b) Gly-SA110, (c) Gly-SA130 and (d) Gly-SA150. | ||
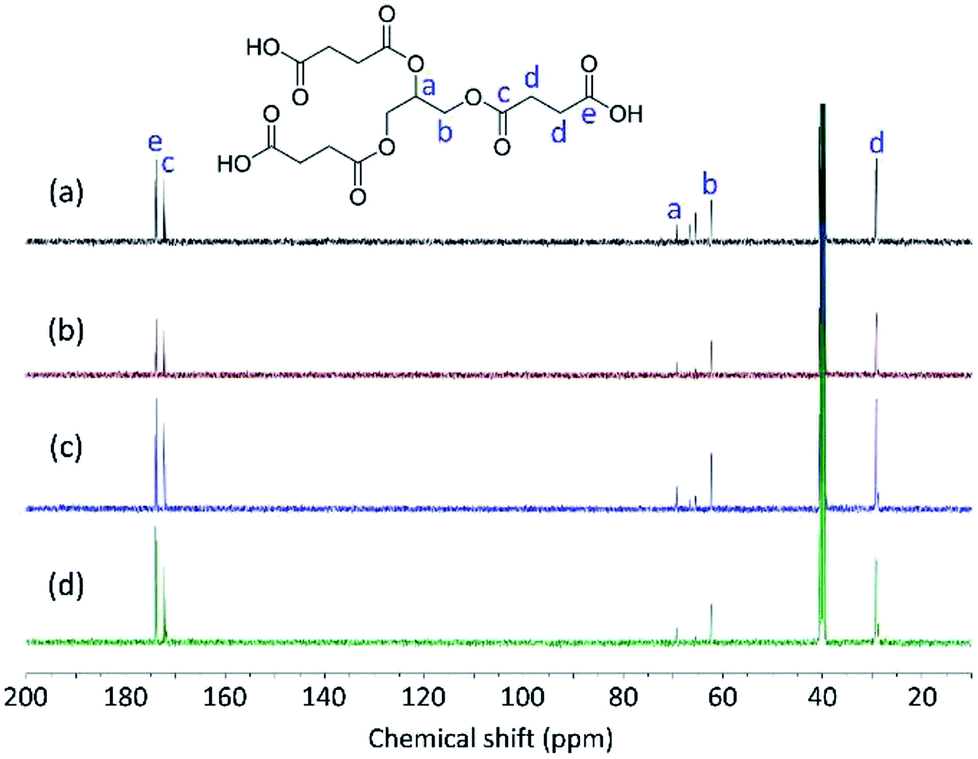 | ||
| Fig. 4 13C NMR spectra of (a) Gly-SA90, (b) Gly-SA110, (c) Gly-SA130 and (d) Gly-SA150. | ||
Molecular weight of Gly-SA crosslinkers
The molecular weights of Gly-SA were determined by gel permeation chromatography (GPC) using THF as the mobile phase. The GPC chromatographs of Gly-SA90, Gly-SA110, Gly-SA130 and Gly-SA150 are shown in Fig. 5.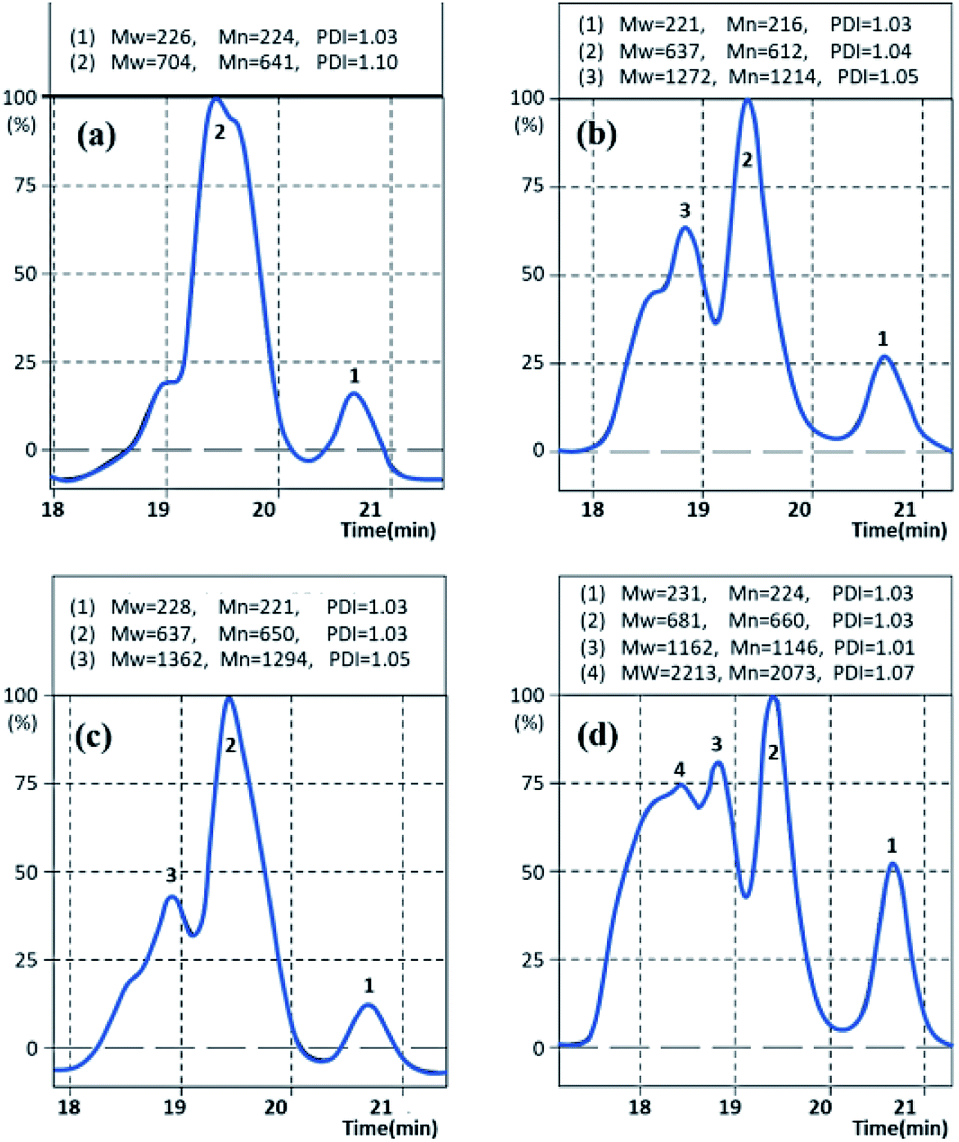 | ||
| Fig. 5 GPC curves of (a) Gly-SA90, (b) Gly-SA110, (c) Gly-SA130 and (d) Gly-SA150. | ||
The peaks of Gly-SA prepared at high temperature were much broader than those of Gly-SA prepared at lower temperature and shifted to shorter retention times owing to the growth of molecular weight. The GPC curves indicated that the products were composed of at least four macromolecular populations and the evolution of the populations with the temperature was clearly seen.
Thermal stability of Gly-SA crosslinkers
TGA and DTG thermograms of Gly-SA crosslinkers were shown in Fig. 6. All Gly-SA crosslinkers exhibited a small loss of water, however a greater loss is observed at preparation temperatures of 130 °C (Gly-SA130) and 150 °C (Gly-SA150), due to the increased degree of esterification. The second weight loss was observed between 160 °C and 330 °C resulting from the degradation of the remaining succinic anhydride and glycerol in the crosslinked mixture.40–42 The weight loss between 330 °C and 430 °C was attributed to the decomposition of the main crosslinker chains with carboxyl-terminated groups.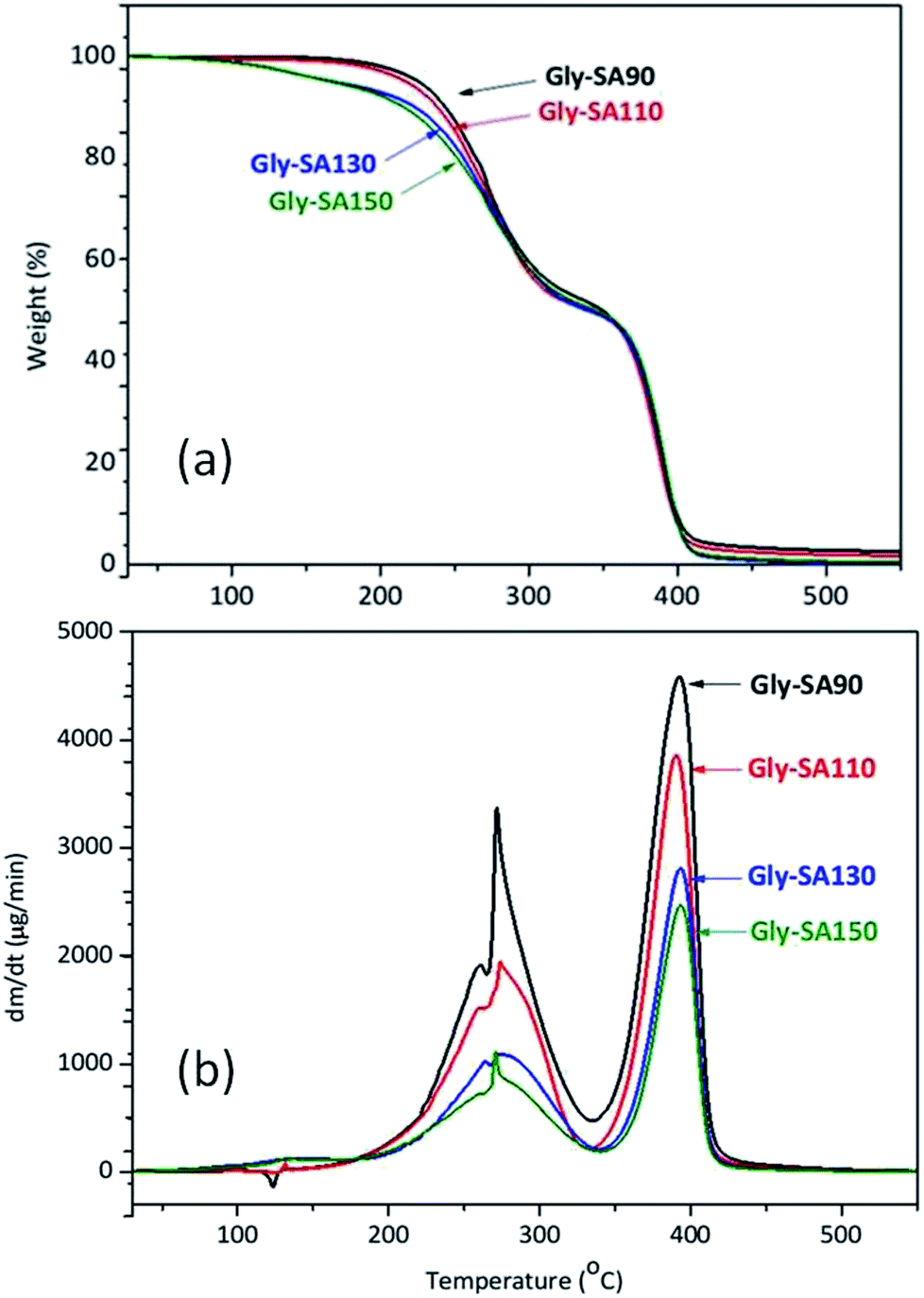 | ||
| Fig. 6 TGA (a) and DTG (b) curves of Gly-SA crosslinkers. | ||
The DTG thermograms (Fig. 6b) revealed that the rate of degradation of Gly-SA crosslinkers increased with decreasing preparation temperature. Furthermore, a shift of the maximum decomposition rate to higher temperature was noted on increasing preparation temperature. This is likely due to the higher molecular weight and greater degree of entanglement of the crosslinkers obtained from higher preparation temperatures.43
Preparation and characterisation of Gly-SA cured ESO thermosets
The synthesised Gly-SA materials were used as crosslinkers for the curing of epoxidised soybean oil (ESO). The envisaged curing reaction of ESO with Gly-SA crosslinkers is the ring opening of epoxide by deprotonated carboxylic acid attacking of carboxyl groups to create a network structure shown in Scheme 2.26 Preparation of films from cured mixtures in silicone moulds at 140 °C produced films with the thickness of 1 mm. The obtained thermoset materials were noted as EGS90, EGS110, EGS130 and EGS150 according to crosslinkers used in the formulations (Gly-SA90, Gly-SA110, Gly-SA130 and Gly-SA150, respectively). Fig. 7 exhibits the ESO based films cured at 4 and 24 hours. | ||
| Fig. 7 Photographs of cured ESO based films (a) ESO homopolymer, (b) EGS90, (c) EGS110, (d) EGS130 and (e) EGS150. | ||
The ESO homopolymer was still not cured after 4 hours. The ESO–Gly-SA films cured for 4 hours showed similar yellow colour as that of ESO homopolymer film, however, those cured for 24 hours showed a darker colour. All films were flexible and transparent as demonstrated in Fig. 8.
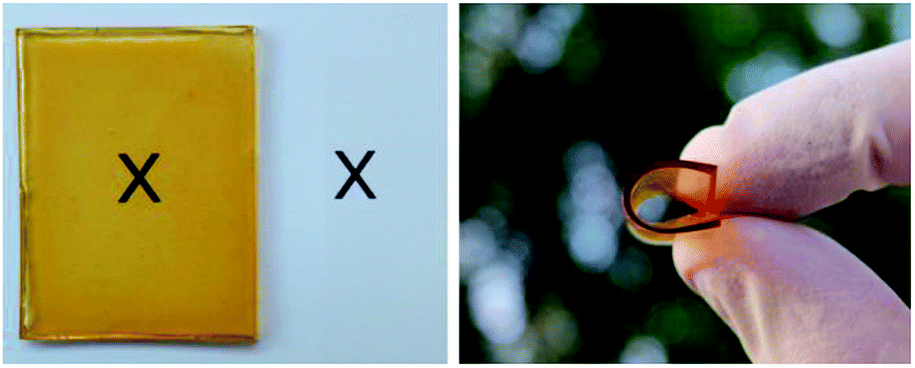 | ||
| Fig. 8 Transparent and flexible EGS150 film. | ||
It is envisaged that at the beginning of the cure process deprotonation of any acid group(s) in crosslinkers occurs. The resulting carboxylate ion then acts as a nucleophile in the ring-opening of the epoxide, leading to alkoxide formation and protonated to form an alcohol. The base catalysts assist the proton transfer in the system. The alcohol functionalities present in the crosslinker and formed in curing can further react with epoxide to create the polymer network. Such reactions are consistent with those previously reported for the cross linking of epoxidised oils with diacids.44 These observations are also backed up with FTIR results.
The crosslinking of ESO and Gly-SA via esterification reaction was studied using ATR-FTIR technique. The FTIR spectra of EGS films are shown in Fig. 9. In the FTIR spectra of all EGS films, the intensity of the band at 830 cm−1 ascribed as the epoxy group decreased due to the ESO ring opening polymerisation reaction by Gly-SA crosslinkers. The band at 3465 cm−1 associating to the hydroxyl functionality was also noted as evidence of epoxide ring opening. A new band at 1003 cm−1 was observed indicating the presence of ether functionality, suggesting possible etherification of the alkoxide group that can subsequently react with other epoxides and contribute to the formation of the crosslinked network.
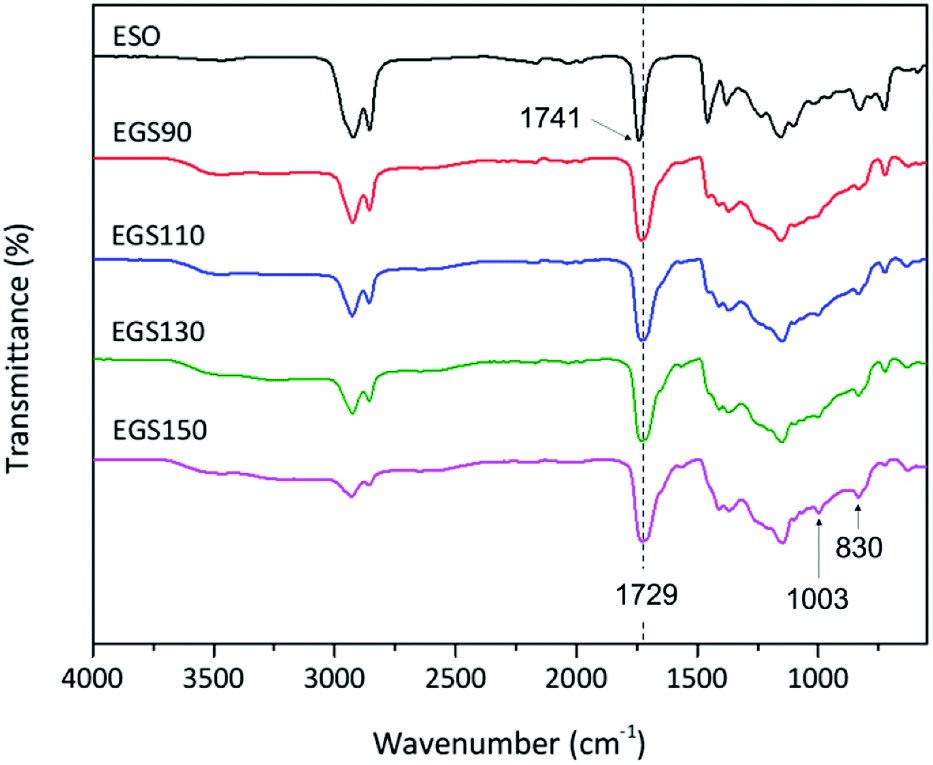 | ||
| Fig. 9 FTIR spectra of ESO and EGS films. | ||
Furthermore, the band at 1711 cm−1 corresponding to the carbonyl group of carboxylic acid observed in Gly-SA crosslinkers was no longer present in the EGS films, providing further evidence of crosslinking through this functionality. This was also confirmed by the shift of the ester band, from 1741 cm−1 for the ESO spectrum to 1729 cm−1 in the thermosets spectra.
Thermal analysis of EGS mixtures and EGS films
Modulated DSC was utilised to study thermal events occurring during the curing process of the bio-based thermosets. An exothermic transition was observed in all mixture samples during the first heating run (Fig. 10).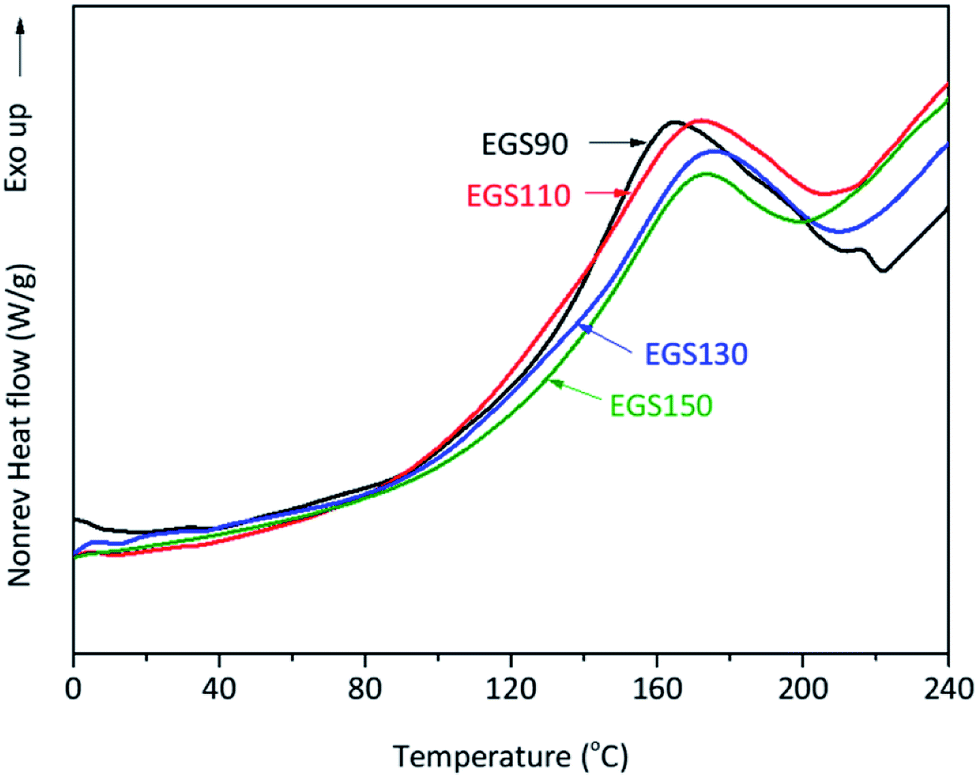 | ||
| Fig. 10 DSC thermograms of crosslinking ESO formed by reaction between ESO and Gly-SA different chain-length. | ||
The analysis showed that the different crosslinkers scarcely affect the peak temperature of the curing process however, the onset temperature tends to rise with increased crosslinker chain length (Table 1). This was anticipated as the steric hindrance can be expected when longer chains are present in the formulation, resulting in retardation of the curing process. The total heat of the reaction of the curing process decreased with increasing chain-length of Gly-SA crosslinkers (Table 1), suggesting less crosslinking occurred. This was probably due to the availability of reactive groups.
| Resin | Total heat of reaction (J g−1) | Onset temperature (°C) | Peak temperature (°C) | Tg (°C) |
|---|---|---|---|---|
| EGS90 | 140.0 | 123.1 | 163.0 | −20.59 |
| EGS110 | 91.0 | 120.4 | 169.1 | −19.44 |
| EGS130 | 83.0 | 129.1 | 171.3 | −18.89 |
| EGS150 | 49.9 | 133.0 | 169.9 | −15.83 |
The glass transition (Tg) of EGS films were analysed by DSC. Fig. 11 shows the second heating scan of EGS films, Tg shifted to higher temperature with increasing chain-length of Gly-SA to −20.59, −19.44, −18.89 and −15.83 °C for EGS90, EGS110, EGS130 and EGS150, respectively, which is explained by the increased molecular weight of Gly-SA.
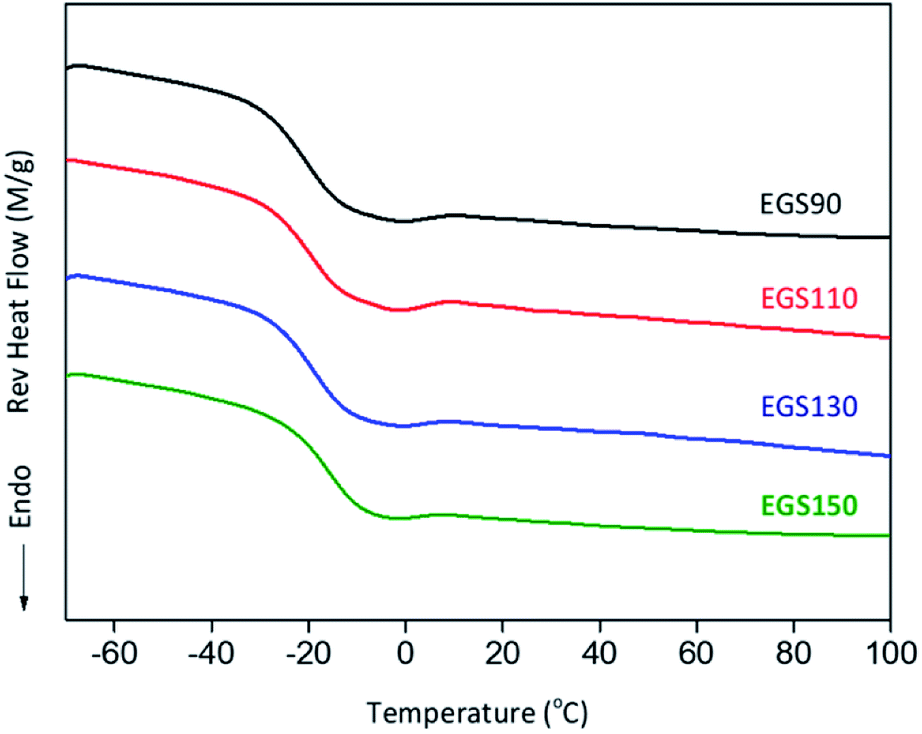 | ||
| Fig. 11 DSC heating scans of different chain-length Gly-SA cured ESO. | ||
The films with crosslinkers prepared from higher temperature showed higher glass transition temperature but highlighted that less crosslinking occurred during the curing process. At higher temperatures Gly-SA crosslinkers could further increase the chain length or even polymerise, leading to higher molecular weights as previously revealed by GPC results. The pre-polymerised crosslinkers contribute to the rise of glass transition in the final thermoset product.
Mechanical properties
The mechanical properties of the sample films were evaluated by tensile testing. Fig. 12 showed the stress–strain curves of the Gly-SA cured ESO (EGS) and cured ESO with DMAP which was used as a control sample. All samples used for tensile testing were cured at 140 °C for 24 hours. The control sample exhibited low mechanical properties with tensile strength (σ) and elongation at break (ε) of 183 ± 7 kPa and 14.1 ± 0.3%, respectively (Table 2). Films comprising of ESO, Gly-SA crosslinkers and DMAP demonstrated higher tensile strength, elongation at break and Young's modulus compared to those of the control. The tensile strength increased to 883 ± 43, 1203 ± 32, 1544 ± 76, and 2323 ± 102 kPa for EGS90, EGS110, EGS130, and EGS150, respectively with the EGS150 film demonstrating a tensile strength which is 13 times higher than that of the control film. The addition of the crosslinkers also enhanced the elongation at break of the materials with EGS150 exhibited more than 220% increase in elongation at break compared to that of the DMAP cured ESO sample. The Young's modulus of the samples with crosslinkers significantly increased to quadruple the value obtained for the control sample. It is noteworthy that the tensile strength and elongation at break increased with increasing Gly-SA chain length, suggesting the pre-polymerised crosslinkers contribute to the enhanced mechanical properties of the materials.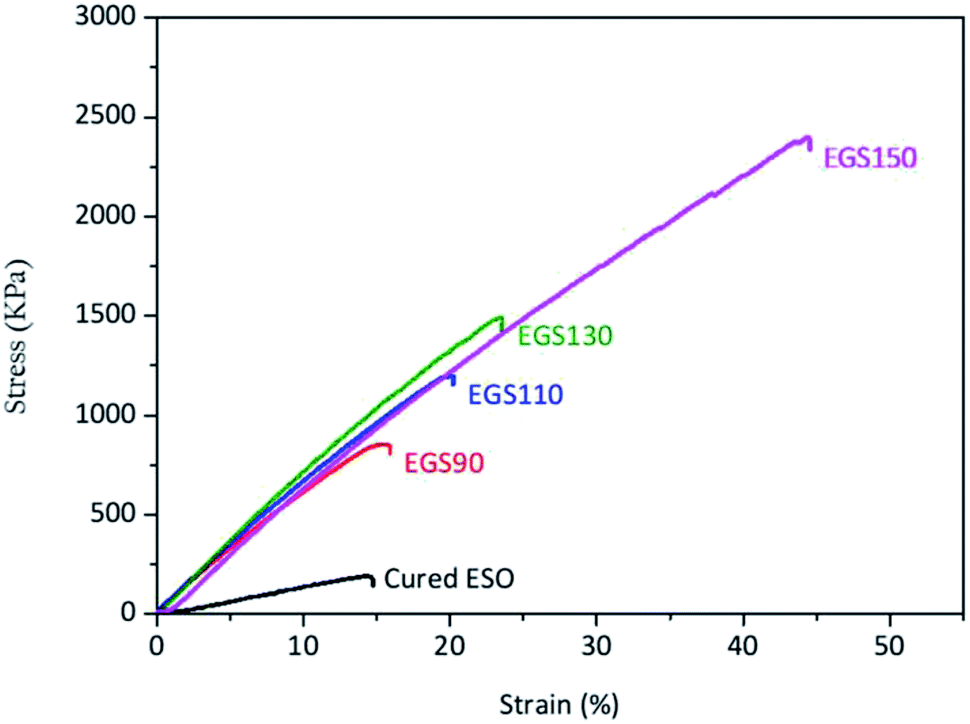 | ||
| Fig. 12 Stress–strain curves of different chain-length Gly-SA cured ESO. | ||
| Properties | Control | EGS90 | EGS110 | EGS130 | EGS150 |
|---|---|---|---|---|---|
| Before ageing | |||||
| Tensile strength (kPa) | 183 ± 7 | 883 ± 43 | 1204 ± 32 | 1544 ± 76 | 2323 ± 102 |
| Elongation at break (%) | 14.1 ± 0.3 | 16.1 ± 0.7 | 20.5 ± 1.1 | 25.6 ± 1.8 | 46.1 ± 2.2 |
| Young's modulus (MPa) | 1.5 ± 0.1 | 6.5 ± 0.0 | 7.1 ± 0.2 | 7.2 ± 0.5 | 6.7 ± 0.4 |
|
|||||
| After ageing | |||||
| Tensile strength (kPa) | 96 ± 8 | 818 ± 88 | 1484 ± 30 | 1679 ± 154 | 1927 ± 65 |
| Elongation at break (%) | 16.5 ± 2.4 | 35.4 ± 1.9 | 65.7 ± 1.2 | 58.9 ± 2.6 | 63.9 ± 3.8 |
| Young's modulus (MPa) | 0.8 ± 0.1 | 3.6 ± 0.5 | 3.4 ± 0.2 | 4.5 ± 0.4 | 4.6 ± 0.5 |
DMA plots between storage modulus (E′) versus temperature, and tanδ versus temperature were provided in Fig. 13. The glass transition temperature (determined from the tanδ peak temperature) shifted to higher temperature with increasing chain-length of Gly-SA (5.0, 5.1, 6.6 and 8.6 °C for EGS90, EGS110, EGS130 and EGS150, respectively). This is in good accordance with DSC results. The increase of the glass transition temperature gives further insight concerning the more dense structure of the thermosets prepared from larger crosslinkers due to the decrease in chain-end concentration.45 Storage moduli of EGS films were significantly higher than that of cured ESO (control sample). It was found that the E′ increased as the molecular weight of the Gly-SA crosslinking agent increased. Storage moduli of all EGS films began to change noticeably at −20 °C. The reduced E′ was due to glassy-to-rubbery state transitions in the sample. The storage moduli at different temperatures were listed in Table 3.
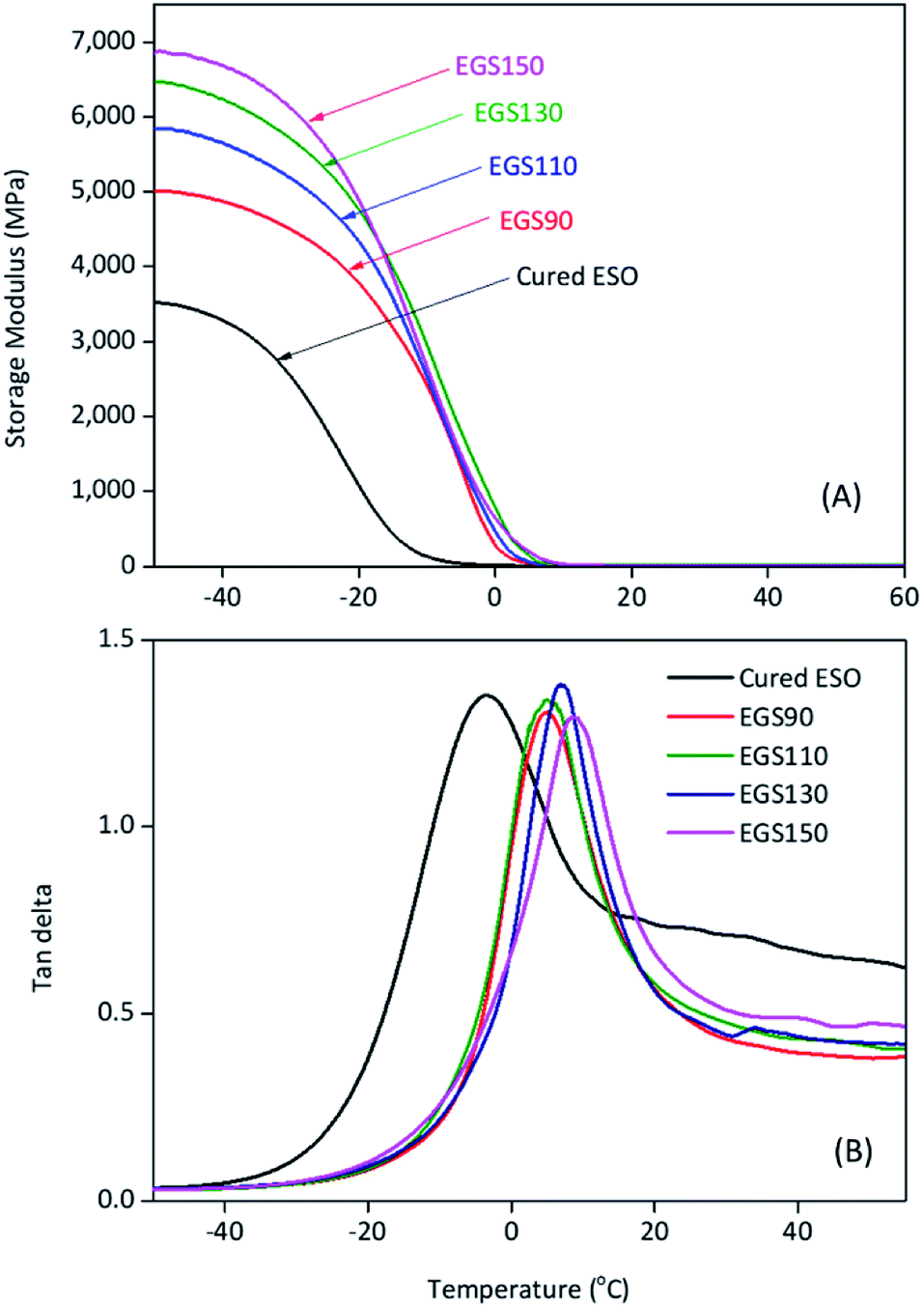 | ||
| Fig. 13 Storage modulus (A) and tanδ (B) curves of different chain-length Gly-SA cured ESO. | ||
δ results of EGS films
| Sample | tanδ peak (°C) |
Storage modulus (MPa) | ||
|---|---|---|---|---|
| −50 °C | −25 °C | 0 °C | ||
| Cured ESO | −3.4 | 3523 | 1864 | 15 |
| EGS90 | 5.0 | 5017 | 4203 | 285 |
| EGS110 | 5.1 | 5843 | 4834 | 459 |
| EGS130 | 6.6 | 6473 | 5311 | 777 |
| EGS150 | 8.6 | 6861 | 5612 | 636 |
Thermal ageing study
The mechanical properties of materials after thermal ageing are reported in Table 2. The Young's modulus clearly decreased after 7 days of ageing in a hot air oven for all samples. The tensile strength also decreased for the control sample, EGS90 and EGS150, however that of EGS110 and EGS130 slightly increased. This suggests that the curing of the formers were completed but the EGS110 and EGS130 may undergo the post-curing process. De Albuquerque et al. also noticed the slight increase in tensile strength after 4 days of thermal ageing, however the fall-off of the tensile strength was noted when the duration of thermal ageing was prolonged.46 The deterioration of the materials was due to oxidative degradation of the polymer. The elongation at break increased after the ageing process, due to the plasticising effect of water in the sample.47Thermal stability of EGS films
The thermal stability of the EGS films were investigated by TGA under nitrogen atmosphere is shown in Fig. 14. The first stage of decomposition of the film was found between 160 and 330 °C, which corresponds to the decomposition of succinic and glycerol remainder in the crosslinkers. These results strongly correlated with TGA data obtained for the crosslinkers, whereas at temperatures from 330 to 490 °C the decomposition of the crosslinked polymer network was observed.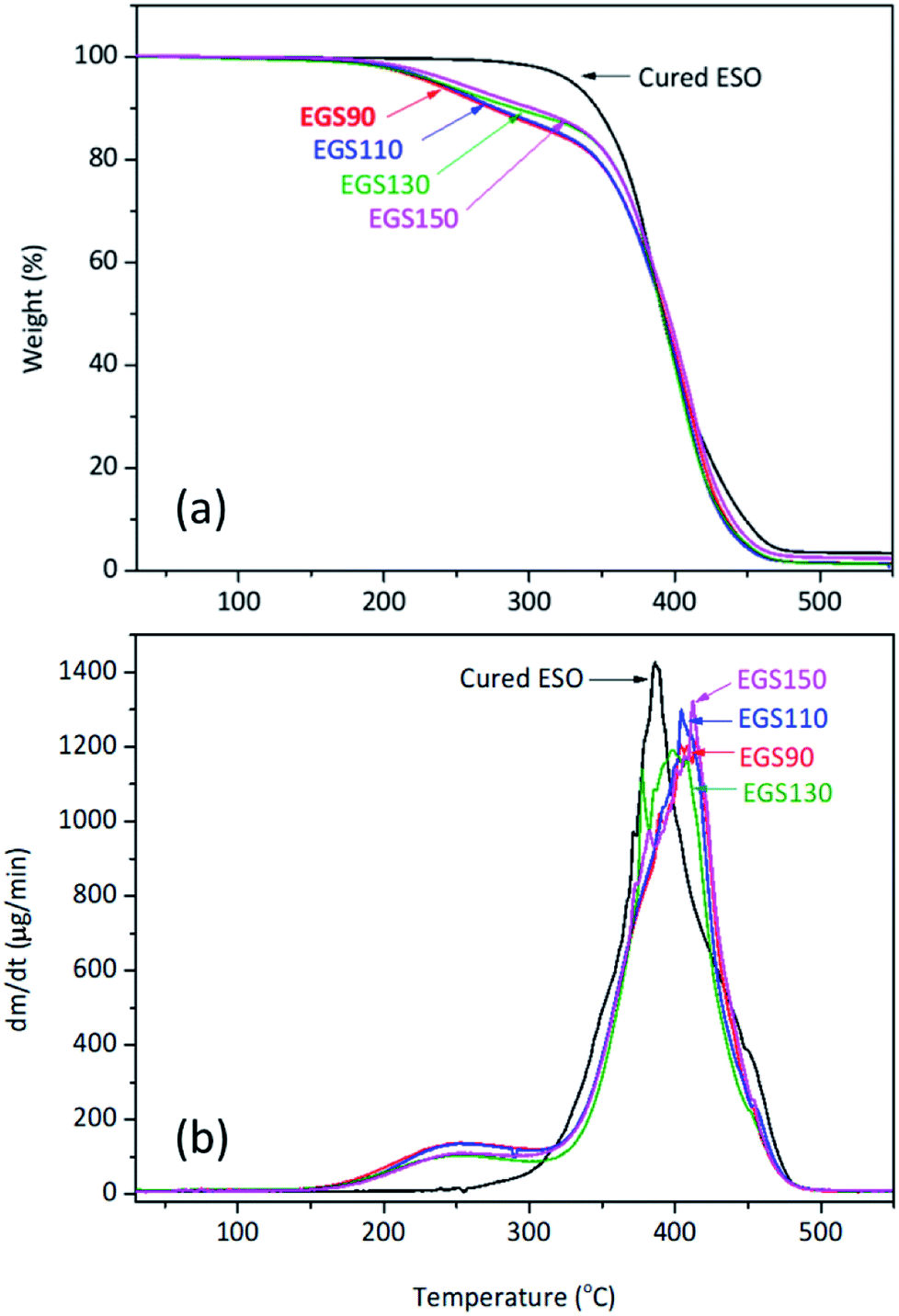 | ||
| Fig. 14 TGA curves of different chain-length Gly-SA cured ESO. | ||
Oxidative stability
During the use of the bio-based thermoset films in applications such as coating, oxygen may diffuse through the film leading to the degradation of thermoset polymer, most of which come from free radical damage. Oxidative stability of EGS films were therefore monitored by treating the films in the Fenton's reagent (2 ppm FeSO4 in 3 wt% H2O2 solution) at 80 °C for 1 h. Fig. 15 demonstrates that all the EGS films exhibited the residual weight (RW) in the region of 90–96%.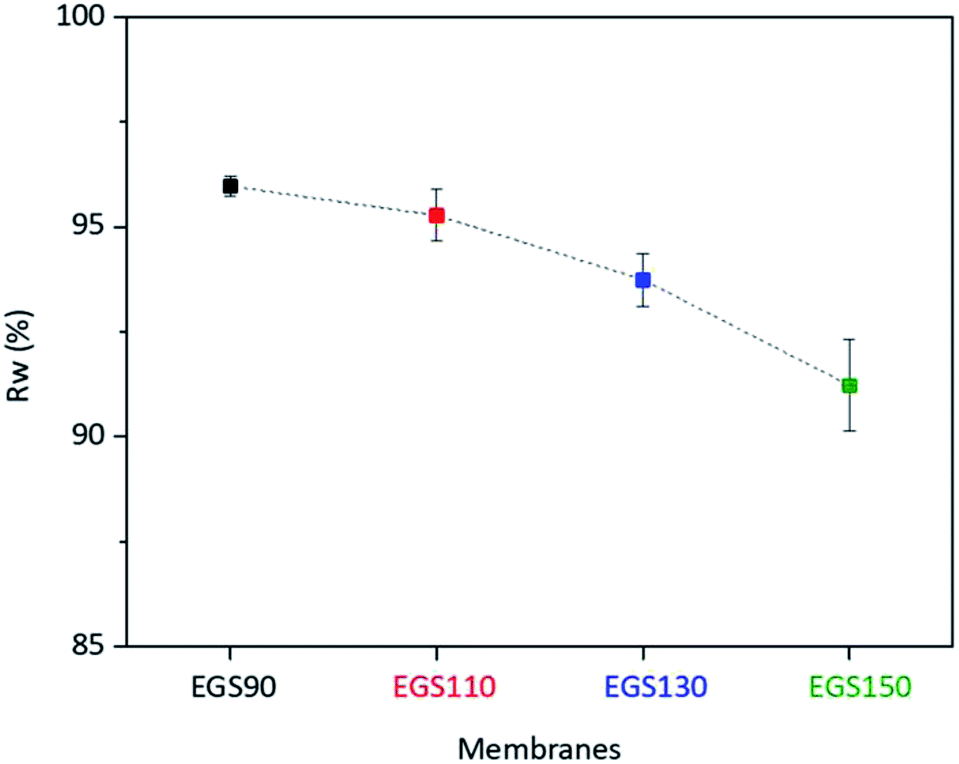 | ||
| Fig. 15 Oxidative stabilities of EGS films. | ||
The EGS film with a long chain crosslinking exhibited slightly lower oxidation stability than one with a short chain cross-linking. This was possibly due to the lower crosslinking density as suggested by lower total heat of reaction obtained from the DSC results. However, all of these bio-based thermosets are considered to demonstrate excellent oxidative resistance with the RW of 96.0%, 95.3%, 93.7% and 91.2% for EGS90, EGS110, EGS130 and EGS150, respectively.
The crosslinkers and thermosets developed in this work offer a number of clear advantages to existing systems reported in the literature including being derived from abundant and low cost bio-derived feedstocks. Other reported crosslinkers are frequently only partially bio-derived, utilising expensive and low volume feedstocks.27–29,36,37 This demonstrates a clear advantage of the EGS films, not only from an environmental standpoint but also from an economical perspective. This study also demonstrated the higher molecular weight crosslinkers, which can be obtained at increased preparation temperatures, led to materials with superior mechanical properties. The fully bio-based materials exhibit promising properties such high thermal stability and excellent oxidative resistance of more than 90%, which is superior compared to other materials previously reported.48,49 Previous literature has demonstrated limited focus on oxidative resistant properties of bio-based thermosets, this further highlights a distinct advantage of this study.
Conclusions
Fully bio-based thermoset materials have been synthesised from epoxidised soybean oil and bio-derived carboxyl-terminated hyperbranched crosslinkers. The crosslinkers were synthesised using the facile esterification reaction of glycerol with succinic anhydride (Gly-SA). The characterisation of the crosslinkers has shown that the molecular weight of the crosslinkers increased as the preparation temperatures increased, suggesting an introduction of hyperbranches to the molecules. FTIR and NMR techniques confirmed the presence of carboxylic acid functionality within the crosslinker molecules which allows the crosslinking with the epoxide of ESO. The obtained thermoset materials demonstrated high flexibility and transparency with excellent oxidative resistance. This study also investigated the effect of different molecular weights of Gly-SA on the thermal and mechanical properties of the thermoset films. DSC analysis revealed a slight decrease of the curing rate when the molecular weights of Gly-SA increase. The incorporation of the bulky of crosslinkers resulted in an increase of the glass transition temperature (Tg). The film produced from Gly-SA crosslinker prepared at 150 °C (EGS150) exhibited a tensile strength 13 times greater than that of the control film. Furthermore a 220% increase in elongation at break and a fourfold increase in Young's modulus compared to the values obtained from the control sample were noted. These results are in good accordance with those obtained from DMA study as the enhancement in Tg and storage moduli were also observed as the molecular weight of the crosslinking agent increased. Moreover these thermoset materials demonstrated excellent oxidative stability, ensuring a potential use of these materials in applications such as coating.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the young researcher development project of Khon Kaen University. The authors would also like to thank Materials Chemistry Research Center and Center of Excellence for Innovation in Chemistry (PERCH-CIC), Faculty of Science, Khon Kaen University.References
- A. Gandini and T. M. Lacerda, Prog. Polym. Sci., 2015, 48, 1–39 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- Y. Lu and R. C. Larock, ChemSusChem, 2009, 2, 136–147 CrossRef CAS PubMed.
- Z. Wang, X. Zhang, R. Wang, H. Kang, B. Qiao, J. Ma, L. Zhang and H. Wang, Macromolecules, 2012, 45, 9010–9019 CrossRef CAS.
- Y. Xia and R. C. Larock, Green Chem., 2010, 12, 1893–1909 RSC.
- B. M. Abdullah and J. Salimon, J. Appl. Sci., 2010, 10, 1545–1553 CrossRef CAS.
- U. Biermann, U. Bornscheuer, M. A. Meier, J. O. Metzger and H. J. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 3854–3871 CrossRef CAS PubMed.
- N. Fox and G. Stachowiak, Tribol. Int., 2007, 40, 1035–1046 CrossRef CAS.
- R. Wool and X. S. Sun, Bio-based polymers and composites, Academic Press, 2011 Search PubMed.
- J. Espana, L. Sánchez-Nacher, T. Boronat, V. Fombuena and R. Balart, J. Am. Oil Chem. Soc., 2012, 89, 2067–2075 CrossRef CAS.
- P. Gogoi, H. Horo, M. Khannam and S. K. Dolui, Ind. Crops Prod., 2015, 76, 346–354 CrossRef CAS.
- T. Tsujimoto, T. Takayama and H. Uyama, Polymers, 2015, 7, 2165–2174 CrossRef CAS.
- T. Tsujimoto and H. Uyama, ACS Sustainable Chem. Eng., 2014, 2, 2057–2062 CrossRef CAS.
- R.-T. Zeng, Y. Wu, Y.-D. Li, M. Wang and J.-B. Zeng, Polym. Test., 2017, 57, 281–287 CrossRef CAS.
- R. Trostle, Global agricultural supply and demand: factors contributing to the recent increase in food commodity prices (rev), DIANE Publishing, 2010 Search PubMed.
- M. Flieger, M. Kantorova, A. Prell, T. Řezanka and J. Votruba, Folia Microbiol., 2003, 48, 27 CrossRef CAS.
- M. S. Lin, C. C. Liu and C. T. Lee, J. Appl. Polym. Sci., 1999, 72, 585–592 CrossRef CAS.
- F. I. Altuna, V. Pettarin and R. J. Williams, Green Chem., 2013, 15, 3360–3366 RSC.
- F. Gunstone, Vegetable oils in food technology: composition, properties and uses, John Wiley & Sons, 2011 Search PubMed.
- B. Cortese, M. H. de Croon and V. Hessel, Ind. Eng. Chem. Res., 2011, 51, 1680–1689 CrossRef.
- S. Kocaman and G. Ahmetli, Prog. Org. Coat., 2016, 97, 53–64 CrossRef CAS.
- C. Öztürk and S. H. Küsefoğlu, J. Appl. Polym. Sci., 2011, 120, 116–123 CrossRef.
- A. Ručigaj, B. Alič, M. Krajnc and U. Šebenik, Eur. Polym. J., 2014, 52, 105–116 CrossRef.
- S. K. Sahoo, S. Mohanty and S. K. Nayak, Chin. J. Polym. Sci., 2015, 33, 137–152 CrossRef CAS.
- J. Xu, Z. Liu, S. Z. Erhan and C. J. Carriere, J. Am. Oil Chem. Soc., 2004, 81, 813–816 CrossRef CAS.
- W. Chow, S. Tan, Z. Ahmad, K. Chia, N. Lau and K. Sudesh, J. Polym. Environ., 2014, 22, 140–147 CrossRef CAS.
- J. D. Espinoza-Perez, B. A. Nerenz, D. M. Haagenson, Z. Chen, C. A. Ulven and D. P. Wiesenborn, Polym. Compos., 2011, 32, 1806–1816 CrossRef CAS.
- S. Tan, Z. Ahmad and W. Chow, Ind. Crops Prod., 2013, 43, 378–385 CrossRef CAS.
- S. Kumar, S. K. Samal, S. Mohanty and S. K. Nayak, Ind. Eng. Chem. Res., 2017, 56, 687–698 CrossRef CAS.
- M. Samper, V. Fombuena, T. Boronat, D. García-Sanoguera and R. Balart, J. Am. Oil Chem. Soc., 2012, 89, 1521–1528 CAS.
- C. Ding, P. S. Shuttleworth, S. Makin, J. H. Clark and A. S. Matharu, Green Chem., 2015, 17, 4000–4008 RSC.
- X.-Y. Jian, X.-P. An, Y.-D. Li, J.-H. Chen, M. Wang and J.-B. Zeng, Macromolecules, 2017, 50, 5729–5738 CrossRef CAS.
- X.-Y. Jian, Y. He, Y.-D. Li, M. Wang and J.-B. Zeng, Chem. Eng. J., 2017, 326, 875–885 CrossRef CAS.
- S. Ma, Y. Jiang, X. Liu, L. Fan and J. Zhu, RSC Adv., 2014, 4, 23036–23042 RSC.
- J. Dai, S. Ma, Y. Wu, L. Han, L. Zhang, J. Zhu and X. Liu, Green Chem., 2015, 17, 2383–2392 RSC.
- K. Liu, S. A. Madbouly and M. R. Kessler, Eur. Polym. J., 2015, 69, 16–28 CrossRef CAS.
- W. Liu, T. Xie and R. Qiu, ACS Sustainable Chem. Eng., 2016, 5, 774–783 CrossRef.
- T. Werpy, G. Petersen, A. Aden, J. Bozell, J. Holladay, J. White, A. Manheim, D. Eliot, L. Lasure and S. Jones, Top value added chemicals from biomass. Volume 1-Results of screening for potential candidates from sugars and synthesis gas, Department of Energy Washington DC, 2004 Search PubMed.
- V. Somisetti, S. Allauddin, R. Narayan and K. Raju, RSC Adv., 2015, 5, 74003–74011 RSC.
- C. Caires, L. Lima, C. Carvalho and M. Ionashiro, Ecletica Quim., 2010, 35, 73–80 CrossRef.
- F. J. Caires, L. Lima, C. Carvalho and M. Ionashiro, Thermochim. Acta, 2010, 500, 6–12 CrossRef CAS.
- A. Gómez-Siurana, A. Marcilla, M. Beltrán, D. Berenguer, I. Martínez-Castellanos and S. Menargues, Thermochim. Acta, 2013, 573, 146–157 CrossRef.
- F. Carrasco, P. Pagès, J. Gámez-Pérez, O. Santana and M. L. Maspoch, Polym. Degrad. Stab., 2010, 95, 116–125 CrossRef CAS.
- N. Supanchaiyamat, P. S. Shuttleworth, A. J. Hunt, J. H. Clark and A. S. Matharu, Green Chem., 2012, 14, 1759–1765 RSC.
- A. Hale, C. W. Macosko and H. E. Bair, Macromolecules, 1991, 24, 2610–2621 CrossRef CAS.
- A. De Albuquerque, K. Joseph, L. H. de Carvalho and J. R. M. d'Almeida, Compos. Sci. Technol., 2000, 60, 833–844 CrossRef CAS.
- N. Esmaeili, F. O. Bakare, M. Skrifvars, S. J. Afshar and D. Åkesson, Cellulose, 2015, 22, 603–613 CrossRef CAS.
- T. Kamjornsupamitr, T. Sangthumchai, S. Youngme and S. Martwiset, Int. J. Hydrogen Energy, 2018, 43(24), 11190–11201 CrossRef CAS.
- W. Liu, S. Wang, M. Xiao, D. Han and Y. Meng, Chem. Commun., 2012, 48, 3415–3417 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07133k |
| This journal is © The Royal Society of Chemistry 2018 |