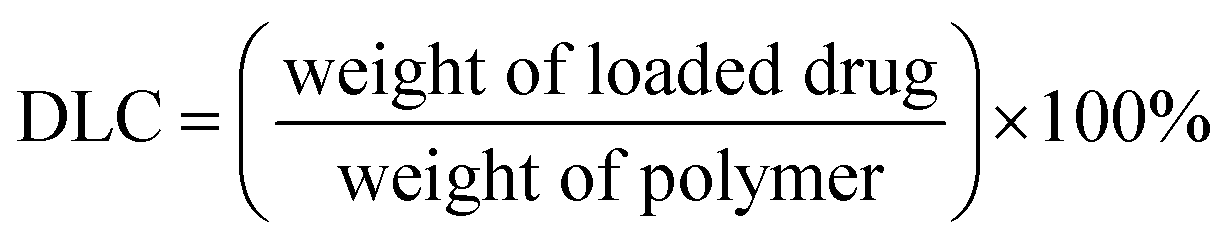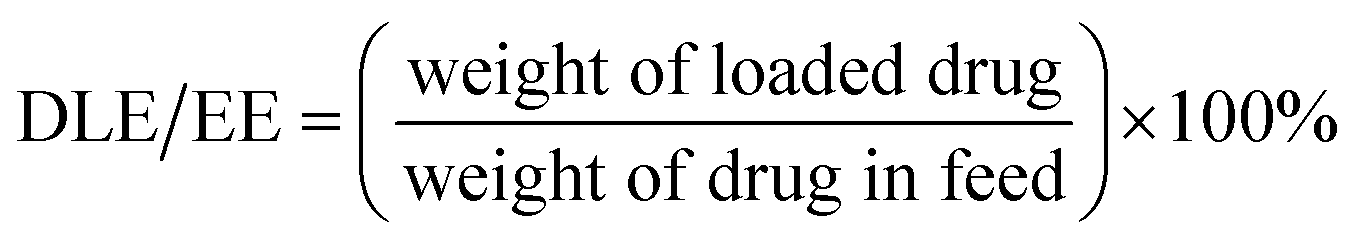DOI:
10.1039/C8RA07060A
(Paper)
RSC Adv., 2018,
8, 37905-37914
Facile synthesis of indole heterocyclic compounds based micellar nano anti-cancer drugs†
Received
23rd August 2018
, Accepted 31st October 2018
First published on 13th November 2018
Abstract
Facile synthesis of micellar “nano” indole heterocyclic anti-cancer compounds is described. The synthesized compounds (11–23) were characterized by UV-VIS, 1H NMR, FT-IR and mass spectroscopy. The binding energies of DNA–compound adducts varied from −20.08 to −23.85 kJ mol−1, and they were stabilized by hydrophobic interactions and H-bonding. The synthesized compounds enter into minor grooves of DNA during adduct formation. The DNA binding constant of compounds 11–23 was 1.00 to 2.00 × 105 M−1. The drug-loading efficiency and drug-loading content in their micellar forms were recorded. Compounds 11, 12, 14 and 19 at a micellar concentration of 670 μL mL−1 displayed excellent anticancer activities against the HepG2/C3A line (25–50%). The potency of nano anticancer drugs was predicted by drug likeness using Lipinski's “rule of five”. Taken together, compounds 11–23 could be used to treat cancers.
1. Introduction
Molecules such as cisplatin, 5-fluorouracil and taxol have been used to treat cancer but cannot cure cancers, especially for late-stage disease. Besides, these molecules show various serious side effects by damaging healthy body cells.1 The most notorious problems with these drugs are a narrow therapeutic range, cytotoxicity and poor solubility.2 Moreover, no single drug can cure different types of cancers.3,4 These challenges can be addressed by developing “nano” anti-cancer drugs because they are cell-specific, targeted drugs that show few side effects.5–15 Hence, different research teams, clinicians and regulatory authorities are looking for effective nano anti-cancer drugs.
Heterocyclic compounds appear to be most effective against various cancers.16,17 Around 60% of the medications used for cancer are based on heterocyclic moieties.18 Among the various heterocyclic moieties, nitrogen-based compounds have been found to be effective against different types of cancers.19 Indoles (Fig. 1) are heterocyclic moieties with a broad range of pharmacological properties, including anti-tumor.20–23 Ahmad et al.24 reported apoptosis of human cancer cells using indoles. Indoles have been shown to inhibit the proliferation of human cancer cells.25–27 Liao et al.28 showed that 2-aryl-3-substituted indoles derivatives were active against the breast cancer cell line MCF-7. Treatment with indoles or their derivatives [e.g., I3C(indole-3-carbinol) and DIM (3,3′-diindolylmethane)] can result in the apoptosis of breast,29 squamous cell carcinoma,30 cholangiocarcinoma,31 colon32 and ovarian tumor cells.33 These indolic heterocycles may be decisive anti-cancer agents for several human cancer cell lines.34 Csipo et al.35 studied the chemosensitization of indole moieties such as I3C and DIM.
 |
| | Fig. 1 Chemical structures of indole-3-carboxaldehyde, indole-2-carboxaldehyde and indole-3-carboxylic acid. | |
Here, we synthesized a heterocyclic series ingrained with indolic moieties, followed by purification and characterization by column chromatography and spectroscopy. DNA binding studies of synthesized molecules were carried out and DNA binding constants calculated. The synthesized molecules were loaded onto micellar “nanocarriers”. The activities of the developed nano anticancer drugs were tested on HepG2(C3A) cell lines. Finally, Lipinski's rule was applied to these synthesized compounds to ascertain if they could become anti-cancer drugs.
2. Experimental
2.1 Chemicals and reagents
Sulfuric acid, potassium hydroxide and formaldehyde of AR grade were purchased from Merck (Mumbai, India), as were LiChrosolv® chloroform, hexane and methanol. Indole-3-carboxaldehyde, 4-nitroaniline, 4-fluroaniline, 4-bromoaniline, 4-chloroaniline, 4-methylaniline, 2-methylaniline and Ct-DNA were purchased from Spectrochem (Mumbai, India). Aluminum silica gel (60 F254) thin-layer plates were procured from Merck (Darmstadt, Germany). The human cancer cell line HepG2/C3A was from National Taiwan University (Taipei City, Taiwan). MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] was obtained from Sigma-Aldrich (Saint Louis, MO, USA). Dulbecco's modified Eagle's medium (DMEM) and antimycotics/antibiotics were purchased from Gibco (Billings, MT, USA). Fetal bovine serum (FBS) was obtained from HyClone (Jülich, Germany). Poly(ε-caprolactone)-block poly(ethylene glycol)-poly(ε-caprolactone), abbreviated as PCL-PEG-PCL (nanocarrier) was obtained from Reinste Nano Ventures (New Delhi, India).
2.2 Instrumentation
Fourier transform infrared (FT-IR) spectroscopy was carried out using a RXIFT spectrometer (LR 64912C; PerkinElmer, Waltham, MA, USA). A UV/VIS spectrometer (T80) from PG Instruments (Lutterworth, UK) was employed. A DPX300 300 MHz system (Bruker, Billerica, MA, USA) was used for NMR studies. A micrOTOF-Q II™ ESI-Qq-TOF mass spectrometer (10262; Bruker) was also used. Melting points were determined using a system developed by Veego Instruments (REC-22038; Mumbai). Deionized water was collected by a Milli-Q (Millipore; Bedford, MA, USA) water-purification system. Synthesized compounds were purified by flash chromatography using a pump (MP-200), UV detector and computer with CHEETAH software.
2.3 Wet chemistry
2.3.1 Syntheses of the condensates of indole (3–10). A clear solution of indole-3-carboxaldehyde (1) (10 mM) was prepared in ethanol/dioxane (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). Equimolar amounts of the corresponding aniline derivatives (2) [4-nitroaniline, 4-fluroaniline, 4-bromoaniline, 4-chloroaniline, 4-methylaniline, 2-methylaniline] were added separately. These were stirred to obtain clear solutions. A few drops of concentrated H2SO4 were added with constant heating under reflux for 4–6 h. The reaction was determined by thin-layer chromatography (TLC) using MeOH:CHCl3 (1:3, v/v) solvent. Solid products started to precipitate and were filtered off after completion of the reaction. They were re-crystallized in an ethanol and dioxane (2:1, v/v) mixture to obtain pure products. The synthesized compounds (3–10) are shown in Scheme 1.
:1, v/v). Equimolar amounts of the corresponding aniline derivatives (2) [4-nitroaniline, 4-fluroaniline, 4-bromoaniline, 4-chloroaniline, 4-methylaniline, 2-methylaniline] were added separately. These were stirred to obtain clear solutions. A few drops of concentrated H2SO4 were added with constant heating under reflux for 4–6 h. The reaction was determined by thin-layer chromatography (TLC) using MeOH:CHCl3 (1:3, v/v) solvent. Solid products started to precipitate and were filtered off after completion of the reaction. They were re-crystallized in an ethanol and dioxane (2:1, v/v) mixture to obtain pure products. The synthesized compounds (3–10) are shown in Scheme 1.
 |
| | Scheme 1 Synthesis of indole derivatives. | |
2.3.2 Syntheses N-substituted indoles (11–23). A solution (15 mL) of indole N-(1H-indol-3-yl methylene)-4-nitroaniline3 (10 mM) was prepared in a mixture of ethanol and dioxane (2:1, v/v). Then, formaldehyde (40%; 1.5 mL) and morpholine (10 mM; 0.087 mL) in ethanol were added to the solution. Then, the mixture was stimulated for 3–4 h and left overnight. The solid was collected by filtration and recrystallized from ethanol and dioxane (2:1, v/v) to yield 11. Compounds 12–23 were synthesized by the same procedure used in the preparation of compound 3. Compounds having Z-geometry of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N were formed because of the peak in IR spectra (i.e., only Z/cis can show a peak in IR spectra). Also, the synthesized compound was stable in biological conditions. The purity of the synthesized compounds was ascertained by column chromatography and flash chromatography.
N were formed because of the peak in IR spectra (i.e., only Z/cis can show a peak in IR spectra). Also, the synthesized compound was stable in biological conditions. The purity of the synthesized compounds was ascertained by column chromatography and flash chromatography.
2.3.3 Encapsulation of synthesized compounds (11–23) in PCL-PEG-PCL nanocarriers. These compounds (3.0 mg each) were dissolved in 1.0 mL of a binary solution of THF:DMSO (1:1, v/v), separately, as was 20 mg of the polymer (PCL60-PEG40-PCL60). The compounds and polymer solutions were mixed in 3.0 mL of double-distilled water and stirred for 3 h. The resultant samples were placed in dialysis bags (molecular weight (MW) cutoff = 8000) and dialyzed for 24 h to obtain micellar solutions, the formation of which was confirmed by UV-VIS spectrometry by determining λmax.
2.4 Docking studies
Docking studies were carried out as per standard protocol.36–38 Heterocyclic molecules containing nitrogen, oxygen and sulfur atom interact with the major and minor grooves of DNA.39 The synthesized compounds contained the same number of nitrogen and sulfur atoms. These heteronuclear compounds tend to interact with DNA, as shown by docking studies theoretically and by DNA binding experimentally. Also, the cancer-causing genes (i.e., oncogenes) are also present on DNA. “DNA intercalation” refers to insertion of molecules into DNA bases and cause frameshift mutations. Our synthesized compounds could act as DNA intercalators.
2.5 DNA binding
Interactions of compounds 11–23 were studied with Ct-DNA (pH 7.4) in a solution of deionized water with tris-(hydroxymethyl)-amino methane buffer (Tris, 10−2 M). Initially, the concentration of freshly prepared Ct-DNA solution was determined by UV-VIS absorption spectrophotometry at 260 nm (ε = 6600 M−1 cm−1) by knowing its absorbance.40 To carry out binding experiments, the absorption spectra of freshly prepared compounds 11–23 at a fixed concentration of 1.6 × 10−4 M were taken separately, and then at different concentrations of DNA (5.15 × 10−5, 3.0 × 10−5, 1.00 × 10−5, 0.9 × 10−5 and 0.7 × 10−5 M). λmax was recorded and the absorbance of the mixture (i.e., with each different solution of DNA and compounds) was also measured. Experiments were repeated five times. The intrinsic DNA binding constant (Kb) was resolved using the Benesi–Hildebrand equation as modified by Wolfe et al.:41| | |
[DNA]/(εa − εf) = [DNA]/(εa − εf) + 1/K(εb − εf)
| (1) |
where the absorption coefficients εa, εf, and εb represent Aobs/[compound], the extinction coefficient for the molecule, and the extinction coefficient for molecule in the fully bound form, respectively. The Kb for dissimilar compounds was ascertained by division of the slopes and intercepts of the plots of [DNA]/(εa − εf) vs. [DNA].
2.6 Determination of drug loading
2.6.2 Anticancer assays. In vitro anticancer profiles of the nano drugs (11–23) were determined reiteratively with a murine microglial cell line (HepG2/C3A) by a cell viability (MTT) assay.43 The principle of the MTT assay is to remove succinic acid from the mitochondria of living cells under the action of hydrogenase and cytochrome C. The tetrazolium ring opens to form purple crystals, which are eluted and have an absorbance peak at 570 nm. The experimental method involved four steps. First, HepG2/C3A cells were seeded in 96-well dishes at 1 × 104/well. Cells were allowed to attach for 24 h and washed several times with phosphate-buffered saline (PBS) and serum-free medium containing micellar solution-encapsulated drugs. Second, the old medium was removed at the end of incubation time, and fresh water containing 0.5 mg mL−1 of MTT added. Third, the medium was cultured for 4 h, the culture medium removed and 100 μL of DMSO was added to the ELISA Reader. Crystals were dissolved by shaking for 5 min, and the absorbance at 570 nm measured. Finally, cell cytotoxicity was calculated using the following equation:
3. Results and discussion
Finally, 13 compounds (11–23) were synthesized (Scheme 1) and purified by flash chromatography. The melting points of synthesized compounds were recorded. The synthesized compounds were characterized by UV-VIS, FT-IR, mass and NMR spectroscopy. Their structural data are described in ESI.†
3.1 Docking studies
To study the in silico properties of compounds 11–23 with DNA, theoretical DNA docking studies were carried out. B-DNA is most common form used due to its wide and deep major grooves, narrow and deep minor grooves, and its pattern of base pairing between the two DNA strands. It gives different hydrogen bond donor/acceptor forms in the major and minor grooves, respectively.
The DNA docking of compounds 11–23 was done with DNA dodecamers d(CGCGAATTCGCG)2 having a (PDB ID: 1BNA) file. The DNA docking results are shown in Fig. SI1–5 (ESI)† and Table 1. Almost all compounds adjusted themselves in a co-crystalized ligand-like appearance in the vicinities of DNA. The binding affinities of compounds 11–23 followed the order 15 > 14 ≈ 13 > 18 ≈ 22 ≈ 12 > 23 ≈ 17 ≈ 20 ≈ 21 > 16 ≈ 11 > 19.
Table 1 Docking parameters of compounds 11–23
| Compound |
Binding energy KJ mol−1 |
No. of H-bonds |
Residues involved in H-bonding (bond length) |
Residues involved in hydrophobic interactions |
| 11 |
−20.50 |
3 |
A: DT′8H:Unk0:O of nitro group (3.55) |
Dt8, Dc9, Dg10, Dg14 & Dc15 |
| B: DG′10H:Unk0:N of CN group (3.43) |
| C: DC′15H:Unko:N of morpholine group (3.48) |
| 12 |
−21.34 |
2 |
A: DG′14H:Unk0:N of CN group (3.40) |
Dt8, Dc9, Dg10, Dc13, Dg14, Dc15 & Dg16 |
| B: DG′10H:Unk0:O of nitro group (3.39) |
| 13 |
−22.59 |
2 |
A: DG′14H:Unk0:O of morpholine group (3.36) |
Dt8, Dc9, Dg10, Dg14, Dc15 & Dg16 |
| B: DG′10H:Unk0:N of CN group (3.42) |
| 14 |
−22.59 |
1 |
A: DG′10H:Unk0:Nof morpholine group (3.51) |
Dt8, Dc9, Dg10, Dc13, Dg14, Dc15 & Dg16 |
| 15 |
−23.85 |
1 |
A: DG′10H:Unk0:Nof morpholine group (3.49) |
Dt8, Dc9, Dg10, Dg14 & Dc15 |
| 16 |
−20.50 |
2 |
A: DC′13H:Unk0:O of methoxy group (3.39) |
Dt8, Dc9, Dg10, Dc13, Dg14, Dc15 & Dg16 |
| B: DG′14H:Unko:O of morpholine group (3.44) |
| 17 |
−20.92 |
1 |
A: DG′10H:Unk0:N of CN group (3.32) |
Dt8, Dc9, Dg10, Dc13, Dg14, Dc15 & Dg16 |
| 18 |
−21.34 |
2 |
A: DG′14H:Unk0:N of CN group (3.43) |
Dt8, Dc9, Dg10, Dc13, Dg14, Dc15 & Dg16 |
| B: DG′10H:Unk0:N of indole group (3.40) |
| 19 |
−20.08 |
2 |
A: DG′10H:Unk0:N of CN group (3.42) |
Dt8, Dc9, Dg10, Dc13, Dg14 & Dc15 |
| B: DC′13H:Unk0:O of nitro group (3.24) |
| 20 |
−20.92 |
2 |
A: DG′10H:Unk0:N of CN group (3.35) |
Dt8, Dc9, Dg10, Dc13, Dg14 & Dc15 |
| B: DG′14H:Unk0:N of indole group (3.46) |
| 21 |
−20.92 |
1 |
A: DC′9H:Unk0:N of indole group (3.57) |
Dt8, Dc9, Dg10, Dc13, Dg14, Dc15 & Dg16 |
| 22 |
−21.34 |
1 |
A: DC′9H:Unk0:N of indole group (3.58) |
Dt8, Dc9, Dg10, Dc13, Dg14 & Dc15 |
| 23 |
−20.92 |
2 |
A: DC′14H:Unk0:N of CN group (3.41) |
Dt8, Dc9, Dg10, Dc13, Dg14 & Dc15 |
| B: DG′10H:Unk0:N of indole group (3.28) |
The number of hydrophobic interactions and H-bonds of compounds 11–23 with DNA are given in Table 1. The hydrogen bonds formed in the DNA-compound adducts were one for 14, 15, 17, 21 and 22, two for 12, 13, 16, 18, 19 and 20, and three for 11 compounds, respectively. During the interaction with DNA, compounds 11–23 adjusted themselves so that their substituted indole, morpholine and N-methylpiperazine rings were within the minor grooves of DNA mostly, whereas Z-geometry CN moieties attached to benzene rings were mainly in the major grooves of DNA. Also, 1–3 H-bonds were formed by this spatial orientation. The residues of DNA involved in H-bonding were guanine, adenine, cytosine and thymine bases (DG'14H, DG'10H, DC'9H, DC'13H, DC'15H, DT'8H) with the amine group of the indole ring, morpholine and N-methylpiperazine rings. Also, cyano and methoxy groups were attached to benzene rings. One hydrogen bond was attached to guanine and cytosine bases. This twisting of rings might have been because of the GC-rich region with a large positive potential responsible for molecular attraction,44 and more repulsion from phosphate groups in the backbone. Also, twisting can lead to more hydrogen bonding. It can be seen in Fig. SI1† that, during the molecular interaction of compound 11 with DNA, it favored the minor grooves of DNA with −20.50 kJ mol−1 of binding energy. Compound 11 formed three hydrogen bonds with the residues (thymine, guanine and cytosine) of DNA with bond lengths of 3.55, 3.43 and 3.48 Å, respectively. With regard to hydrophobic interactions, the nitrogen-containing nucleobases Dt8, Dc9, Dg10, Dg14 and Dc15 were involved. Similarly, compound 19 preferred the minor grooves of DNA and formed two hydrogen bonds with the residues of DNA with a binding affinity of −20.08 kJ mol−1 (ESI†). The residues involved in hydrogen bonding were DG'10H and DC'13H. Besides these interactions, compound 19 (ESI†) also interacted with the nitrogen-containing nucleobases of DNA hydrophobically in a two-dimensional network. The residues involved in these hydrophobic interactions were Dt8, Dc9, Dg10, Dc13, Dg14 and Dc15. It is not useful to discuss the details of docking studies for all compounds as all the compounds interacted with DNA in a similar fashion.
3.2 DNA binding
Interactive study of newly synthesized heterocyclic moieties with DNA is crucial for estimation of their anticancer activity, and to shed light on a viable mechanism of their action. Hence, DNA binding is considered to be the most essential experimental step to measure the activity of anticancer drugs because most anticancer drugs target DNA specifically.45 In general, there are two modes of binding of a drug with DNA: non-covalent or covalent. In covalent binding, a labile ligand is replaced with a nitrogen atom of a DNA base, such as N7 of guanine. In non-covalent binding, there are “electrostatic”, “groove” and “intercalative” types of interactions. Bathochromic and hypochromic shifts revealed the intercalative binding of compounds with DNA. Intercalation enhanced strong stacking interactions between aromatic chromophores and the base pairs of DNA.46 Also, the electrostatic binding of a compound with DNA decreased the π → π* transition energy because the π* orbital of the ligand interacted with DNA base pairs. This led to a lower hypochromic effect with no or negligible bathochromic shift.47 However, covalent bonding led to bathochromism and hyperchromism due to breaking of the DNA structure when a compound interacted with DNA covalently. A compound coordinating with DNA through the N7 position of guanine was indicated by a bathochromic shift. Binding with the outside groove of DNA had a minor or no effect and, rarely, some hyperchromism was shown by groove binding in the UV-VIS spectrum48,49
The purity of DNA in the Ct-DNA stock solution was shown by A260/A280 ≥ 1.80, which indicated a sufficiently DNA protein-free nature.50 With the addition of DNA, slight band shifting was observed at 200–350 nm. The concentration of the stock solution of DNA was determined experimentally by utilizing ε = 6600 M−1 cm−1 at 260 nm. Additional solutions of DNA (3.0, 1.0, 0.9 and 0.7 × 10−4 M) were made from a stock solution (5.15 × 10−5 M). The concentration of the compounds was fixed at 1.6 × 10−4 M. With each different concentration of DNA solution, absorption spectra were recorded.
The DNA binding spectra of the most effective compounds, 11, 21 and 23, are shown in Fig. 2a–c, respectively. Changes such as hypochromism (low to moderate) and slight shifting of bands indicated the interaction of compounds with DNA bands via electrostatic mode due to a decrease of the π → π* transition energy because the π* orbital of the intercalated ligand coupled with the orbital of the base pairs. Graphs of compounds 20 and 17 (ESI†) showed that hyperchromism was because these compounds were covalently bonded with DNA, which was due to breakage of the DNA structure.
 |
| | Fig. 2 (a–c) Absorption spectra of compounds 11, 21 and 23 in the absence and presence of different Ct-DNA concentrations. Arrows indicate the decrease in absorbance upon addition of decreasing concentrations of DNA. Inset: plots of [DNA]/εb vs. [DNA] for the absorption titration of Ct-DNA with compounds. DNA concentrations are (1) 5.15 × 10−5, (2) 3.0 × 10−5, (3) 1.00 × 10−5, (4) 0.9 × 10−5, and (5) 0.7 × 10−5. | |
Graphs of compounds 12, 13, 15, 16, 18 and 19 (ESI†) showed no change in UV-VIS spectra when the different DNA concentrations were added to solutions of the compounds. This confirmed the outer-groove binding of DNA with the compound with low hyperchromism and negligible bathochromism. The Kb was calculated for compounds 11–23 and ranged from 1.0 to 2.0 × 105 M−1, thereby confirming that the synthesized compounds interacted strongly with DNA. Using Microsoft Excel™, regression analysis was carried out for DNA binding studies. The correlation coefficient (R2) was found to be 0.7468–0.9555%. The order of Kb for compounds 11–23 was almost identical. Finally, we summarized that nearly all the synthesized heterocyclic compounds interacted with Ct-DNA through minor grooves, as noted by other scholars.51–53 Overall, the order of binding of differently substituted compounds with DNA was nitro > halogen > methoxy > methyl groups. The binding constants, % hypochromism and wavelength shifts of compounds 11–23 are given in Table 2. Hence, DNA binding studies were comparable with DNA docking studies. Therefore, compounds 11–23 acted as DNA-binding agents.
Table 2 Wavelength shifts, %hypochromism and binding constants of compounds 11–23
| Compound |
λmax (free) |
λmax (bound to DNA) |
Change |
%hypochromisma |
Kbb (M−1) |
| % hypochromicity (H%) = [(Af − Ab)/Af] × 100, where Af and Ab represent the absorbance of free and bound compounds. Binding constants. |
| 11 |
300 nm |
260 nm |
40 |
39.29 |
2.0 × 105 |
| 12 |
261 nm |
262 nm |
1 |
0.72 |
1.0 × 105 |
| 13 |
261 nm |
260 nm |
1 |
0.22 |
1.0 × 105 |
| 14 |
255 nm |
257 nm |
2 |
9.11 |
1.125 × 105 |
| 15 |
255 nm |
255 nm |
0 |
0.34 |
1.125 × 105 |
| 16 |
256 nm |
255 nm |
1 |
3.28 |
1.111 × 105 |
| 17 |
261 nm |
258 nm |
3 |
6.69 |
1.125 × 105 |
| 18 |
262 nm |
264 nm |
2 |
4.53 |
1.111 × 105 |
| 19 |
260 nm |
260 nm |
0 |
0.46 |
1.0 × 105 |
| 20 |
264 nm |
258 nm |
6 |
0.41 |
1.125 × 105 |
| 21 |
260 nm |
261 nm |
1 |
4.17 |
1.0 × 105 |
| 22 |
258 nm |
256 nm |
2 |
5.89 |
1.25 × 105 |
| 23 |
259 nm |
260 nm |
1 |
5.03 |
1.111 × 105 |
3.3 DLC and DLE of drugs encapsulated in micelles
Drug-loaded micelles were prepared by dialysis. All the drugs loaded in micelles showed good results of DLC and DLE (Table 3). DLC ranged from 0.33% to 5.42%. Higher DLC was shown by compound 11 and lower by compound 17. DLE ranged from 3.28% to 54.2%. Compound 11 showed highest DLE and the lowest was shown by compound 17. DLE was relatively good but showing no clear pattern. During dialysis, no precipitate or aggregate was observed. DLC and EE data predicted drug loading into nanocarriers. Also, the encapsulation experiment done with PCL-PEG-PCL nanocarrier showed its efficiency as a nanodrug. Micelle formation is shown in ESI.†
Table 3 DLC and DLE values of compounds 11–23
| Compound |
DLC |
DLE/EE |
| 11 |
5.42% |
54.2% |
| 12 |
2.44% |
24.4% |
| 13 |
4.52% |
45.2% |
| 14 |
3.54% |
35.4% |
| 15 |
0.58% |
5.8% |
| 16 |
3.30% |
33.0% |
| 17 |
0.33% |
3.28% |
| 18 |
4.10% |
41.0% |
| 19 |
1.44% |
14.4% |
| 20 |
4.87% |
48.7% |
| 21 |
3.53% |
35.3% |
| 22 |
4.79% |
47.9% |
| 23 |
3.17% |
31.7% |
3.4 Anticancer profiles of nano drugs
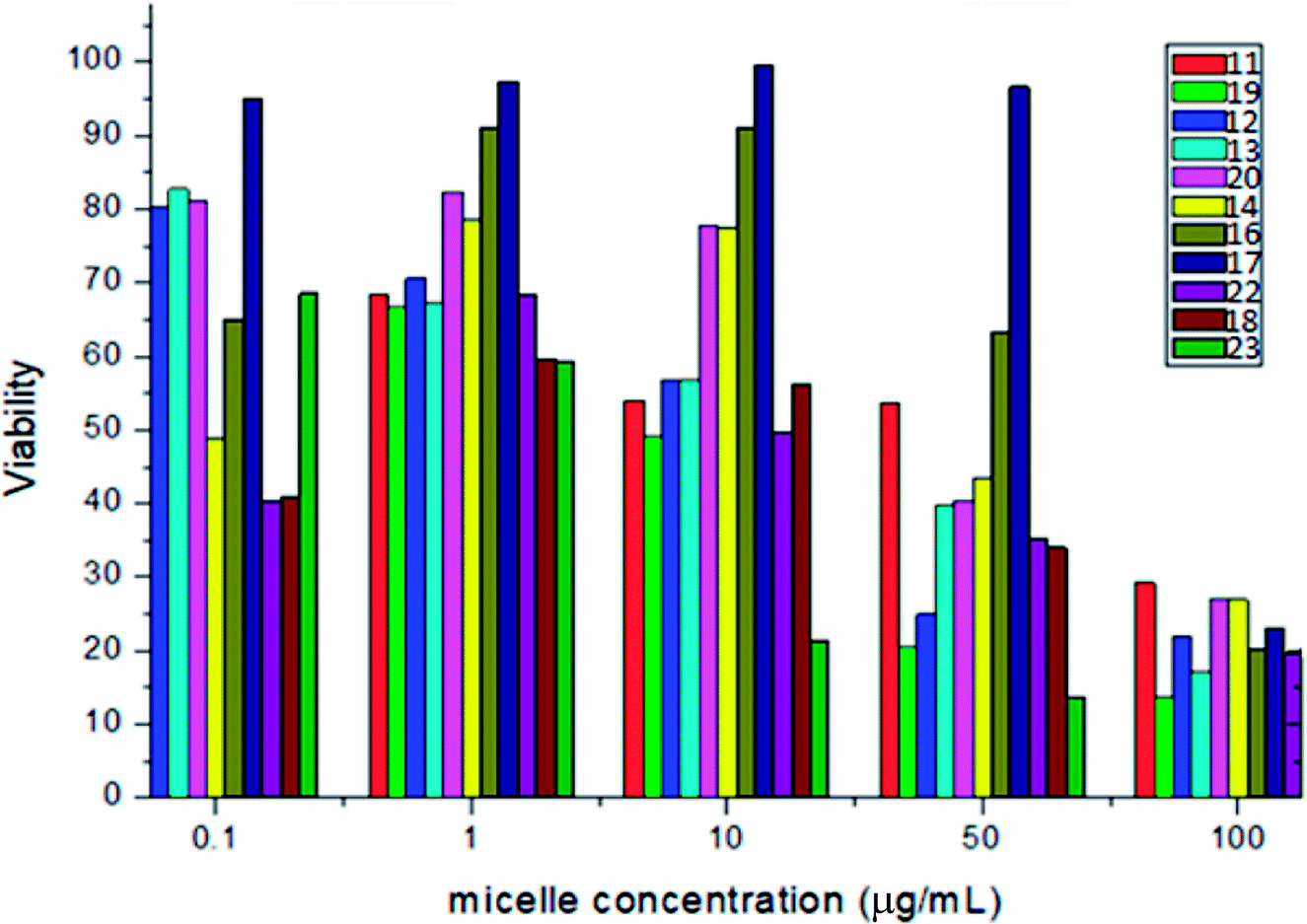
The prepared nano drugs were tested for anticancer activities in terms of % viability on the HepG2/C3A line. The viability of HepG2C3A cells after exposure to compounds 11–23 at varying micellar concentrations (6.7,67, 134, 335, and 670 μL mL−1) are given in Fig. 3 and 4. Fig. 3 reveals that the indole derivatives 11–23 at a micellar concentration of 670 μL mL−1 led to viabilities of HepG2/C3A cells of 25, 35, 28, 48, 32, 30, 39, 50, 40, 45 and 38%. Thus, HepG2/C3A cells displayed low viability upon treatment with heterocyclic moieties 11, 12, 14, 20 and 19, which indicated the good anticancer potential of these synthesized heterocyclic compounds. Fig. 4 shows all the indole moiety derivatives (11–23) caused low viability of HepG2/C3A cells at a micellar concentration of 100 μg mL−1. Also, compound 23 showed low viability at micellar concentrations of 10 and 50 μg mL−1. Therefore, these synthesized compounds interacted differently and had different mechanisms with different cell targets. Also, the half-maximal inhibitory concentration (IC50) was calculated for the micelles formed for compounds 11–23 (Table 4). Compounds 12, 14, 17 and 19 showed IC50 values in the range 1.63–18.70 μg mL−1 at low concentrations. Half of the cells were dead and compound 22 showed a low IC50. Morphologies of HepG2 cells before and after treatment with nanodrugs are shown in Fig. 5a and b: the nano drugs were effective because all cells died after treatment (Fig. 5b).
 |
| | Fig. 3 Percentage viabilities of HepG2/C3A cells upon treatment with compounds 11–23 at micellar concentrations of 6.7, 67, 134, 335 and 670 μL mL−1. | |
 |
| | Fig. 4 Percentage viabilities of HepG2/C3A cells upon treatment with compounds 11–23 at micellar concentrations of 0.1, 1.0, 10, 50 and 100 μg mL−1. | |
Table 4 IC50 values of drugs in micelles in the HepG2/C3A line
| Compounds |
IC50 (μg mL−1) |
| 11 |
3.85 |
| 12 |
2.03 |
| 13 |
3.30 |
| 14 |
1.91 |
| 16 |
5.28 |
| 17 |
3.27 |
| 18 |
3.66 |
| 19 |
1.63 |
| 20 |
4.29 |
| 22 |
18.7 |
| 23 |
7.28 |
 |
| | Fig. 5 Morphology of HepG2 cells (a) before treatment and (b) after treatment with nano drugs. | |
3.5 Drug likeness
In general, the time required to synthesize promising compounds and utilize them is long because their toxicity, pharmacokinetic properties and effects on metabolism must be tested. This leads to considerable burdens on the research-and-development budget of a pharmaceutical company. A number of scientists and researchers are involved to find answers of the above questions. Lipinski's “rule of five” can be used to identify some of the important properties of compounds considered theoretically to be drugs. That is, molecules must have good membrane permeability, MW < 400, logP < 5, number of H-bond donors < 5 and number of H-bond acceptors < 10. For analyses of small drug-like molecules, a filter of logD > 0 and < 3 increases the chances of a compound displaying good intestinal permeability. Moreover, a compound that has >10 hydrogen bond acceptors and 5 hydrogen-bond donors shows poor permeation. Therefore, this rule was applied to compounds 11–23 synthesized according to the procedure described by Veber et al.54 As seen in Table 5, compounds 11–23 followed Lipinski's rule of five.
Table 5 Lipinski's ‘rule-of-five’ data for compounds 11–23
| Compounds |
Mol. wt |
No. of HBA |
No. of HBD |
Mol logP |
Mol logS [log(moles per L)]/(mg L−1) |
Mol PSA (A2) |
Mol vol (A3) |
| 11 |
365.16 |
5 |
1 |
2.74 |
−5.78(0.61) |
61.69 |
352.50 |
| 12 |
397.08 |
3 |
0 |
4.00 |
−6.43(0.15) |
23.43 |
344.16 |
| 13 |
337.16 |
3 |
0 |
3.42 |
−5.80(0.53) |
23.43 |
328.22 |
| 14 |
349.18 |
4 |
0 |
3.12 |
−5.24(2.02) |
30.37 |
354.43 |
| 15 |
333.18 |
3 |
0 |
3.43 |
−5.71(0.65) |
22.73 |
342.94 |
| 16 |
349.18 |
4 |
0 |
3.24 |
−5.54(1.00) |
30.98 |
354.14 |
| 17 |
333.18 |
3 |
0 |
3.55 |
−5.74(0.61) |
23.43 |
343.24 |
| 18 |
353.13 |
3 |
0 |
3.86 |
−6.24(0.20) |
23.43 |
339.50 |
| 19 |
378.19 |
5 |
1 |
2.80 |
−5.39(1.56) |
57.26 |
377.68 |
| 20 |
350.19 |
3 |
0 |
3.47 |
−5.41(1.37) |
19.00 |
353.40 |
| 21 |
362.21 |
4 |
0 |
3.17 |
−4.84(5.18) |
25.93 |
379.61 |
| 22 |
346.22 |
3 |
0 |
3.60 |
−5.35(1.56) |
19.00 |
368.42 |
| 23 |
366.16 |
3 |
0 |
3.92 |
−5.85(0.52) |
19.00 |
364.68 |
4. Conclusions
Thirteen compounds (11–23) were synthesized with good yield and loaded onto micellar nanocarriers. DNA-binding studies indicated that compound–DNA adducts were stabilized mainly by electrostatic attractions, van der Waals forces and hydrogen bonding. The minor grooves of DNA were the areas through which the synthesized compounds interacted with DNA. Hypochromism indicated that the synthesized compounds bound with DNA through electrostatic attraction and intercalations Kb values (1.0 to 2.0 × 105 M−1) indicated the strong binding affinities of compounds 11–23. Good percentages of DLC and DLE were achieved. Cell viability was 25–50% at 670 μL mL−1, which indicated excellent anticancer activities. IC50 values were 1.63–18.70 μg mL−1. Compounds 11–23 followed Lipinski's rule of five. Compounds 11–23 could be used to treat cancers.
Conflicts of interest
There is no conflict of interest for this manuscript.
Acknowledgements
The authors extend their appreciation to the International Scientific Partnership Program ISPP at King Saud University for funding this research work (ISPP # 0037).
References
- K. Saleem, W. A. Wani, A. Haque, A. Malhotra and I. Ali, Nanodrugs: magic bullets in cancer chemotherapy, Top. Anti-Cancer Res., 2013, 58, 437–494 CrossRef.
- M. Pulkkinen, J. Pikkarainen, T. Wirth, T. Tarvainen, V. Haapa-aho, H. Korhonen and K. Järvinen, Three-step tumor targeting of paclitaxel using biotinylated PLA-PEG nanoparticles and avidin–biotin technology: formulation development and in vitro anticancer activity, Eur. J. Pharm. Biopharm., 2008, 70, 66–74 CrossRef CAS PubMed.
- M. Kidwai, R. Venktaramanan, R. Mohan and P. Sapra, Cancer chemotherapy and heterocyclic compounds, Curr. Med. Chem., 2002, 9, 1209–1228 CrossRef CAS PubMed.
- I. Ali, K. Kumerer and H. Y. Aboul-Enein, Mechanistic principles in chiral separations using liquid chromatography and capillary electrophoresis, Chromatographia, 2006, 63, 295–307 CrossRef CAS.
- I. Ali, Nano anti-cancer drugs: pros and cons and future perspectives, Curr. Cancer Drug Targets, 2011, 11, 131–134 CrossRef CAS PubMed.
- I. Ali, S. D. Mukhtar, M. N. Lone, H. S. Ali and H. Y. Aboul-Enein, Recent advances in mesoporous silica and gold based nanovectors in anticancer drug delivery system, Curr. Org. Chem., 2017, 21, 2400–2415 CrossRef CAS.
- I. Ali, M. N. Lone, M. Suhail, S. D. Mukhtar and L. Asnin, Advances in nanocarriers for anticancer drugs delivery, Curr. Med. Chem., 2016, 23, 2159–2187 CrossRef CAS PubMed.
- I. Ali, K. Saleem, H. Y. Aboul-Enein and A. Rather, Social Aspects of Cancer Genesis, Cancer Ther., 2011, 8, 6–14 Search PubMed.
- I. Ali, Nano drugs: Novel agents for cancer chemo-therapy, Curr. Cancer Drug Targets, 2011, 11, 130 CrossRef CAS PubMed.
- I. Ali, K. Saleem, D. Wesselinova and A. Haque, Synthesis, DNA binding, hemolytic, and anti-cancer assays of curcumin I-based ligands and their ruthenium (III) complexes, Med. Chem. Res., 2013, 22, 1386–1398 CrossRef CAS.
- I. Ali and H. Y. Aboul-Enein, Enantioseparation of some clinically used drugs by HPLC using cellulose Tris (3,5-dichlorophenylcarbamate) chiral stationary phase, Biomed. Chromatogr., 2003, 17, 113–117 CrossRef CAS PubMed.
- I. Ali, V. K. Gupta, H. Y. Aboul-Enein and B. sharma, Role of racemization in optically active drugs development, Chirality, 2007, 19, 453–463 CrossRef CAS PubMed.
- H. Y. Aboul-Enein and I. Ali, Determination of tadalafil in pharmaceutical preparation by HPLC using monolithic silica column, Talanta, 2005, 65, 276–280 CrossRef CAS PubMed.
- I. Ali, K. Saleem, I. Hussain and H. Y. Aboul-Enein, Polysaccharides chiral stationary phases in liquid chromatography, Sep. Purif. Rev., 2009, 38, 97–147 CrossRef CAS.
- I. Ali, Z. A. Alothman and M. M. Sanagi, Green synthesis of iron nano-impregnated adsorbent for fast removal of fluoride from water, J. Mol. Liq., 2015, 211, 457–465 CrossRef CAS.
- I. Ali, M. N. Lone, Z. A. Al-Othman, A. Al-Warthan and M. M. Sanagi, Heterocyclic scaffolds: centrality in anticancer drug development, Curr. Drug Targets, 2015, 16, 711–734 CrossRef CAS PubMed.
- H. Y. Aboul-Enein and I. Ali, Comparative study of the enantiomeric resolution of chiral antifungal drugs econazole, miconazole and sulconazole by HPLC on various cellulose chiral columns in normal phase mode, J. Pharm. Biomed. Anal., 2002, 27, 441–446 CrossRef CAS PubMed.
- W. W. Paudler and R. M. Sheets, Recent Developments in Naphthyridine Chemistry, Adv. Heterocycl. Chem., 1983, 33, 147–184 CrossRef CAS.
- S. M. Gomha, N. A. Kheder, M. R. Abdelaziz and Y. N. Mabkhot, A Novel Series of Thiazoles and 1, 3, 4-Thiadiazoles Bearing Thiazole Moiety as Anticancer Agents: Synthesis, Spectral Studies, Biological Evaluation and Structure Activity relationship, ( 2016) Search PubMed.
- I. Ali and H. Y. Aboul-Enein, Enantioseparation of some clinically used drugs by HPLC using cellulose Tris (3, 5-dichlorophenylcarbamate) chiral stationary phase, Biomed. Chromatogr., 2003, 17, 113–117 CrossRef CAS PubMed.
- N. C. Hung, J. M. Lhoste, F. Lavelle, M. C. Bissery and E. Bisagni, Synthesis and antitumor activity of 1-[[(dialkylamino) alkyl] amino]-4-methyl-5H-pyrido [4, 3-b] benzo [e]-and-benzo [g]) indoles. A new class of antineoplastic agents, J. Med. Chem., 1990, 33, 1519–1528 CrossRef.
- H. D. Jain, C. Zhang, S. Zhou, H. Zhou, J. Ma, X. Liu and K. S. Smith, Synthesis and structure–activity relationship studies on tryprostatin A, an inhibitor of breast cancer resistance protein, Bioorg. Med. Chem., 2008, 16, 4626–4651 CrossRef CAS PubMed.
- J. T. Hartmann and L. Kanz, Sunitinib and periodic hair depigmentation due to temporary c-KIT inhibition, Arch. Dermatol., 2008, 144, 1525–1526 Search PubMed.
- A. Ahmad, W. A. Sakr and K. M. Rahman, Mechanisms and therapeutic implications of cell death induction by indole compounds, Cancers, 2011, 3, 2955–2974 CrossRef CAS PubMed.
- H. Y. Aboul-Enein and I. Ali, HPLC enantiomeric resolution of nebivolol on normal and reversed amylose based chiral phases, Pharmazie, 2001, 56, 214–216 CAS.
- I. Ali., H. Y. Aboul-Enein and M. M. Sanagi, Advances in chiral separations by non-aqueous capillary electrophoresis in pharmaceutical and biomedical analysis, Electrophoresis, 2014, 35, 926–936 CrossRef CAS PubMed.
- F. H. Sarkar, Y. Li, Z. Wang and D. Kong, Cellular signaling perturbation by natural products, Cell. Signalling, 2009, 21, 1541–1547 CrossRef CAS PubMed.
- S. Y. Liao, L. Qian, T. F. Miao, H. L. Lu and K. C. Zheng, MFA and docking studies of 2-phenylindole derivatives with anticancer activity, Eur. J. Med. Chem., 2009, 44, 2822–2827 CrossRef CAS PubMed.
- C. Hong, H. A. Kim, G. L. Firestone and L. F. Bjeldanes, 3, 3′-Diindolylmethane (DIM) induces a G1 cell cycle arrest in human breast cancer cells that is accompanied by Sp1-mediated activation of p21WAF1/CIP1 expression, Carcinogenesis, 2002, 23, 1297–1305 CrossRef CAS PubMed.
- Y. Chen, J. Xu, N. Jhala, P. Pawar, Z. B. Zhu, L. Ma and J. M. McDonald, Fas-mediated apoptosis in cholangiocarcinoma cells is enhanced by 3, 3′-diindolylmethane through inhibition of AKT signaling and FLICE-like inhibitory protein, Am. J. Pathol., 2006, 169, 1833–1842 CrossRef CAS PubMed.
- G. Pappa, M. Lichtenberg, R. Iori, J. Barillari, H. Bartsch and C. Gerhäuser, Comparison of growth inhibition profiles and mechanisms of apoptosis induction in human colon cancer cell lines by isothiocyanates and indoles from Brassicaceae, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2006, 599, 76–87 CrossRef CAS PubMed.
- E. J. Kim, S. Y. Park, H. K. Shin, D. Y. Kwon, Y. J. Surh and J. H. Y. Park, Activation of caspase-8 contributes to 3, 3′-diindolylmethane-induced apoptosis in colon cancer cells, J. Nutr., 2007, 137, 31–36 CrossRef CAS PubMed.
- M. Abdelrahim, K. Newman, K. Vanderlaag, I. Samudio and S. Safe, 3, 3′-diindolylmethane (DIM) and its derivatives induce apoptosis in pancreatic cancer cells through endoplasmic reticulum stress-dependent upregulation of DR5, Carcinogenesis, 2005, 27, 717–728 CrossRef PubMed.
- L. J. Yuan, J. B. Liu and X. G. Xiao, Biooxidation of indole and characteristics of the responsible enzymes, Afr. J. Biotechnol., 2011, 10, 19855–19863 CAS.
- I. Csipo, A. H. Montel, J. A. Hobbs, P. A. Morse and Z. Brahmi, Effect of Fas+ and Fas− target cells on the ability of NK cells to repeatedly fragment DNA and trigger lysis via the Fas lytic pathway, Apoptosis, 1998, 3, 105–114 CrossRef CAS PubMed.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function, J. Comput. Chem., 1998, 19, 1639–1662 CrossRef CAS.
- S. Shafia, M. M. Alam, N. Mulakayala, C. Mulakayala, G. Vanaja, A. M. Kalle, R. Pallu and M. S. Alam, Synthesis of novel 2-mercapto benzothiazole, Eur. J. Med. Chem., 2012, 49, 324–333 CrossRef PubMed.
- M. F. Sanner, Python: a programming language for software integration and development, J. Mol. Graphics Modell., 1999, 17, 57–61 CAS.
- G. J. Atwell, J. Y. Fan, K. Tan and W. A. Denny, DNA-Directed Alkylating Agents. 7. Synthesis, DNA Interaction, and Antitumor Activity of Bis(hydroxymethyl)- and Bis(carbamate)-Substituted Pyrrolizines and Imidazoles, J. Med. Chem., 1998, 41, 4744–4754 CrossRef CAS PubMed.
- M. E. Reichmann, S. A. Rice, C. A. Thomas and P. Doty, A further examination of the molecular weight and size of desoxypentose nucleic acid, J. Am. Chem. Soc., 1954, 76, 3047–3053 CrossRef CAS.
- A. Wolfe, G. H. Shimer Jr and T. Meehan, Polycyclic aromatic hydrocarbons physically intercalate into duplex regions of denatured DNA, Biochemistry, 1987, 26, 6392–6396 CrossRef CAS PubMed.
- X. Huang, Y. Xiao and M. Lang, Micelles/sodium-alginate composite gel beads: A new matrix for oral drug delivery of indomethacin, Carbohydr. Polym., 2012, 87, 790–798 CrossRef CAS.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- S. Neidle, DNA minor-groove recognition by small molecules, Nat. Prod. Rep., 2001, 189, 291–309 RSC.
- I. Ali, W. A. Wani, K. Saleem and M. F. Hsieh, Anticancer metallodrugs of glutamic acid sulphonamides: in silico, DNA binding, hemolysis and anticancer studies, RSC Adv., 2014, 4, 29629–29641 RSC.
- I. Ali, A. Haque, K. Saleem and M. F. Hsieh, Curcumin-I Knoevenagel's condensates and their Schiff's bases as anticancer agents: synthesis, pharmacological and simulation studies, Bioorg. Med. Chem., 2013, 21, 3808–3820 CrossRef CAS PubMed.
- R. Indumathy, M. Kanthimathi, T. Weyhermuller and B. U. Nair, Cobalt complexes of terpyridine ligands: crystal structure and nuclease activity, Polyhedron, 2008, 27, 3443–3450 CrossRef CAS.
- P. Zhao, L. C. Xu, J. W. Huang, J. Liu, H. C. Yu, K. C. Zheng and L. N. Ji, Experimental and DFT studies on DNA binding and photocleavage of two cationic porphyrins: effects of the introduction of a carboxyphenyl into pyridinium porphyrin, Spectrochim. Acta, Part A, 2008, 71, 1216–1223 CrossRef PubMed.
- H. Guo, C. Cai, H. Gong and X. Chen, Multi-spectroscopic method study the interaction of anti-inflammatory drug ketoprofen and calf thymus DNA and its analytical application, Spectrochem Acta Part A: Mol and Biomol Spectros, 2011, 79, 92–96 CrossRef CAS PubMed.
- G. Pratviel, J. Bernadou and B. Meunier, DNA and RNA cleavage by metal complexes, Adv. Inorg. Chem., 1998, 45, 251–312 CrossRef CAS.
- I. Ali, A. Haque, K. Saleem and M. F. Hsieh, Bioorganic & Medicinal Chemistry Curcumin-I Knoevenagel's condensates and their Schiff's bases as anticancer agents: synthesis, pharmacological and simulation studies, Bioorg. Med. Chem., 2013, 21, 3808–3820 CrossRef CAS PubMed.
- N. Perin, R. Nhili, M. Cindrić, B. Bertoša, D. Vušak, I. Martin-Kleiner and M. Hranjec, Amino substituted benzimidazo [1, 2-a] quinolines: antiproliferative potency, 3D QSAR study and DNA binding properties, Eur. J. Med. Chem., 2016, 122, 530–545 CrossRef CAS PubMed.
- A. Kung, T. Pieper, R. Wissiack, E. Rosenberg and B. K. Keppler, Hydrolysis of the tumor inhibiting ruthenium (III) complexes HIm trans-[RuCl4 (im) 2] and HInd trans- [RuCl4 (ind) 2] investigated by means of HPCE and HPLC-MS, J. Biol. Inorg Chem., 2001, 6, 292–299 CrossRef CAS PubMed.
- D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, Molecular properties that influence the oral bioavailability of drug candidates, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07060a |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Sofi Danish Mukhtara,
Ming Fa Hsiehc,
Zeid A. Alothmand and
Abdulrahman Alwarthand
*ab,
Sofi Danish Mukhtara,
Ming Fa Hsiehc,
Zeid A. Alothmand and
Abdulrahman Alwarthand