
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMesoporous superacid catalysts for valorisation of refinery naphtha stream†
Sunil Mehla ,
Vamsi Krishsna,
Gandham Sriganesh and
Raman Ravishankar*
,
Vamsi Krishsna,
Gandham Sriganesh and
Raman Ravishankar*
Hindustan Petroleum Green Research and Development Centre, Bangalore, Karnataka, India. E-mail: ramanr@hpcl.in
First published on 2nd October 2018
Abstract
In the current petroleum refining scenario, many refineries end up with surplus naphtha which is either absorbed into the gasoline pool or exported at unattractive prices. Therefore, several options for naphtha valorisation are currently being explored. The usage of Liquified Petroleum Gas (LPG) as a fuel in heating appliances, cooking equipment and automobiles is rapidly increasing. The high specific calorific value, high octane number, clean and efficient combustion of LPG distinguish it as an extremely promising fuel of the future. In the current work, tungstophosphoric acid (TPA) supported on four different mesoporous silica supports were investigated as mesoporous superacids for hydroconversion of refinery naphtha using n-heptane as a model feedstock. The varied levels of interactions of prepared mesoporous silica with tungstophosphoric acid catalysts were observed to have a prominent effect on the strength of the acid sites generated on silica surfaces and as a result affected heptane hydroconversion activity and selectivity of isomerized and cracked products. Interestingly, activity could be tuned towards selective cracking or isomerization-cracking by selection of a suitable topology of mesoporous silica. Hexagonal Mesoporous Silica (HMS) and plugged SBA-15 supported TPA catalysts demonstrated high n-heptane conversion activity and isomerization selectivity whereas KIT-6 and SBA-15 supported TPA catalysts demonstrated high cracking selectivity to LPG.
Introduction
Liquefied Petroleum Gas (LPG) is typically a flammable mixture of propane, n-butane and i-butane. The composition of on spec LPG varies in different countries and the specifications are regulated by respective agencies such as the Gas Processor Association (GPA) and the American Standards for Testing of Materials (ASTM) in the United States and the European LPG Association (AEGPL) in Europe. In addition to C3 and C4 alkanes as major components, LPG typically contains other components in minor quantities depending upon the source of LPG such as methane and ethane, if the source of LPG is an oil well or C3 and C4 olefins if the source of LPG is an oil refinery. LPG finds applications in a variety of industrial processes such as calcination, heating, drying, gloss firing, annealing, melting, blowing, descaling, calendering, baking and many others.1 The use of LPG as automobile fuel, refrigerant, and aerosol propellant is also gaining popularity worldwide.LPG when used as an auto fuel is called Autogas. Autogas is the most widely used alternative fuel in the world. Auto LPG sales increased by 30% basis global average from 2005–06 to 2009–10.2 Vehicles running on Autogas produced 22% lower carbon emissions than petrol, 96% less nitrogen emissions than diesel and 68% less nitrogen emissions than petrol.2 Seven of the ten largest manufacturers in the world produce LPG powered cars and there are 26 million vehicles running on Autogas around the globe.2
In developing countries like India, government is focusing at increasing LPG penetration in rural areas and replacement of kerosene fuel with LPG for its superior and cleaner burning characteristics.1 LPG demand in India increased from 0.41 MMTPA in 1980 to 20 MMTPA in 2015–16. This makes India the fifth largest consumer of LPG in the world and the second biggest importer of LPG after China.3
Approximately 60% of world LPG production is obtained from recovery during extraction of natural gas and oil from the earth. Remaining 40% is obtained from refining of crude oil.3 In petroleum refining industries, LPG is produced from fluid catalytic cracking (FCC), hydrocracking, isomerization and reforming units. However, in all these processes, LPG is not the primary product and instead recovered as a high valued by-product. The fast growth in LPG demand and ever increasing popularity of Autogas and simulated natural gas makes dedicated production of LPG very attractive.
At the same time most refineries have more or less excess of straight run naphtha (SRN) arising out of typical distillation yields from the respective crude oils.4 Refiners have several options to utilize this excess naphtha such as production of solvent grade or food grade and polymer grade hexane, increasing throughput of isomerization and reforming units, direct blending in the gasoline pool with isomerate, reformate and FCC naphtha, co-processing of naphtha in FCC riser or dual riser etc. The issue of availability of excess naphtha and high demand of LPG can be simultaneous solved by development of an appropriate catalyst and process for selective cracking of SRN to LPG.
Catalytic cracking in the absence of hydrogen leads to a high concentration of olefins due to β-scission reactions, which is not useful for LPG (Autogas) production because of LPG specifications set by governing bodies. For example, HD-5 grade of LPG in the United States limits the amount of propylene in LPG to 5 wt%.5 The olefins concentration in LPG is usually limited due to increased emissions of ozone forming toxic compounds and detrimental effect of olefins on engines due to gum formation and deposits.6 Therefore, hydrocracking is an ideal conversion process for production of LPG resulting into only saturated hydrocarbons and absence of olefins in the obtained product. A blend of hydrocracked LPG and fluidized catalytic cracking LPG streams can also be utilized to meet composition specifications.
LPG is typically obtained as a result of sequential cracking of long chain hydrocarbons on strong bronsted acid sites of bi-functional acid catalysts at relatively higher temperatures. Weak and mildly strong bronsted acid sites prefer isomerization reactions.7 Therefore, with proper tuning of bronsted acidity, pore shape and sizes8 several catalysts can potentially be utilized for cracking of SRN to LPG stream such as shape selective zeolites,9 mixed oxides,10 sulfated and tungstated zirconia11 and supported metal oxides.12
Comparatively, stronger acidity, especially Brønsted acidity, was observed for heteropolyacids (HPA) than conventional solid acid catalysts such as acidic oxides and zeolites.13 HPAs are therefore generally more efficient catalysts allowing operation under less severe conditions. The strength of bronsted acid sites and the temperature at which Keggin HPAs lose all acidic protons decreases in the order: H3PW12O40 (465 °C) > H4SiW12O40 (445 °C) > H3PMo12O40 (375 °C) > H4SiMo12O40 (350 °C).14 However, pure HPAs are crystalline materials possessing extremely low surface areas (1–5 m2 g−1) and as a result show low catalytic reactivity in the pure state. Such a shortcoming can be effectively removed by impregnation or incorporation of HPAs on appropriate porous materials with high surface area. Various supports such as silica, alumina, zirconia, carbon etc., have been investigated and silica and carbon were reported as excellent supports for HPAs.14
Ordered Mesoporous Silicas (OMS) have uniform channels, large pore size, high pore volume, high surface area and large number of hydroxyl groups that lead to homogeneous dispersion of active species and high thermal stability.15 These materials are typically modified by incorporation of aluminum,16 titanium17 and zirconium18 etc. depending upon application. Mesoporous silicas are considered as ideal supports for TPA due to the presence of strong interactions with surface hydroxyl groups. Mesoporous silica HPA composites were investigated off late and a strong interaction of HPA with the silica support was reported at low loading levels, whereas bulk properties of HPAs were reported to prevail at higher loadings.19
In 1995 researchers at Michigan State University had reported the synthesis of a mesoporous silica with thick walls using dodecylamine as a neutral template and referred to it as Hexagonal Mesoporous Silica (HMS).20 Santa Barbara Amorphous 15 (SBA-15) is an ordered mesoporous silica with hexagonal pores and was discovered at University of California, Santa Barbara in 1998.21 By 2002 researchers at Utrecht University had found that the mesopores in SBA-15 were curved on a mesoscopic length scale and they developed a modified SBA-15 material, referred to as plugged SBA-15, which demonstrated the presence of micropores along with mesopores.22 Similarly, a three dimensional cubic mesoporous silica, referred as KIT-6 was developed at Korea Advanced Institute of Science and Technology in 2003 using a triblock copolymer and butanol mixture.23 Although, these silicas belong to the same class of porous materials, mesoporous silica, their pore topologies have significant differences and catalysts derived from these structures are expected to have different reactivities.
In this work we have evaluated and rationalized the performance of these four different mesoporous silica and Tungstophosphoric Acid (TPA) composite catalysts for hydroconversion of refinery naphtha to LPG. Tungstophosphoric acid was supported on SBA-15 (2D hexagonal mesoporous silica), plugged SBA-15 (2D hexagonal mesoporous silica with micropores), HMS (disordered mesoporous silica) and KIT-6 (3D cubic mesoporous silica). N-heptane was used a model feed which represents straight run naphtha hydrocarbon and is also typically used as a model compound to study reaction mechanisms.24 The effect of TPA impregnation on ordered mesoporous silicas with different pore shapes and topologies on catalyst properties and respective product selectivities is analysed in the current work.
Materials and methods
1. Catalysts and feedstock
The ordered mesoporous silica supports were prepared as per the procedure reported elsewhere.25 Tetraethylorthosilicate (TEOS) and pluronic-P123 were used for the preparation of SBA-15 under acidic conditions with a typical gel composition of: 1 TEOS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.017 pluronic-P123:9.60 HCl:108 H2O. Similarly plugged SBA-15 was prepared using pluronic P123 and TEOS under acidic conditions with a gel composition of: 1 TEOS:0.01 pluronic-P123:9.60 HCl:108 H2O. KIT-6 was prepared by utilizing n-butanol along with pluronic P123 and TEOS under acidic conditions with a gel composition of: 1 TEOS:0.017 pluronic-P123:1.31 n-butanol:5.20 HCl:194 H2O. HMS was prepared employing dodecylamine and TEOS under acidic conditions with a gel composition of: 1 TEOS:0.33 dodecylamine:15.10 HCl:129 H2O. Hydrothermal treatment was used for all samples at 100 °C for 24 h except HMS. The solid product obtained was filtered, washed, dried at 60 °C for 6 hours and calcined at 550 °C for 1 hour in nitrogen atmosphere followed by 5 hours in air at a heating rate of 1 °C min−1. The final surfactant free materials, viz., SBA-15, plugged SBA-15, KIT-6 and HMS coded as S15, PS15, K6 and HMS, respectively, were directly used for Pt and HPA impregnation.
:0.017 pluronic-P123:9.60 HCl:108 H2O. Similarly plugged SBA-15 was prepared using pluronic P123 and TEOS under acidic conditions with a gel composition of: 1 TEOS:0.01 pluronic-P123:9.60 HCl:108 H2O. KIT-6 was prepared by utilizing n-butanol along with pluronic P123 and TEOS under acidic conditions with a gel composition of: 1 TEOS:0.017 pluronic-P123:1.31 n-butanol:5.20 HCl:194 H2O. HMS was prepared employing dodecylamine and TEOS under acidic conditions with a gel composition of: 1 TEOS:0.33 dodecylamine:15.10 HCl:129 H2O. Hydrothermal treatment was used for all samples at 100 °C for 24 h except HMS. The solid product obtained was filtered, washed, dried at 60 °C for 6 hours and calcined at 550 °C for 1 hour in nitrogen atmosphere followed by 5 hours in air at a heating rate of 1 °C min−1. The final surfactant free materials, viz., SBA-15, plugged SBA-15, KIT-6 and HMS coded as S15, PS15, K6 and HMS, respectively, were directly used for Pt and HPA impregnation.
Tungstophosphoric acid (TPA) was impregnated on different mesoporous silicas using methanol as solvent at a weight loading of 30 wt%. 30 wt% TPA loading was chosen based on previous studies by other researchers. Kozhevnikov reported that TPA in the keggin ion form dominates in loading range of 30 wt% to 50 wt% whereas the lacunary forms of TPA have increased presence at lower loadings.19 We wanted to evaluate the performance of keggin ion and avoid lacunary ions in our catalysts so we have chosen a loading of 30 wt%. On the other hand Siahkali reported that higher loadings than 25 wt% can lead to the formation of large 3D structures and lead to blockage of pores.26 With these reports in mind we have kept TPA loading fixed at 30 wt%. The ingredients were stirred for 3 hours, after which the solvent was removed and the resultant composites were dried at 100 °C overnight. The resulting samples were denoted as TPA@S15, TPA@PS15, TPA@K6 and TPA@HMS in the subsequent sections.
Further, 0.5 wt% Pt was impregnated using hexachloroplatinic acid (H2PtCl6) using methanol as solvent. The ingredients were stirred for 3 hours, after which solvent was removed and the resultant composites were dried at 100 °C for 12 hours. Finally, the catalysts were calcined at 300 °C at a heating rate of 0.2 °C min−1. The resulting samples were denoted as Pt@TPA@S15, Pt@TPA@PS15, Pt@TPA@K6 and Pt@TPA@HMS.
2. Characterization
Powder X-ray diffraction (XRD) patterns of the catalysts were recorded using a Bruker D8 Focus Advance diffractometer equipped with Lynx Eye detector with a Ni-filtered Cu Kα radiation source (λ = 1.5406 Å) in 2θ range of 0.6 to 2 with a step size of 0.01 and step time of 2 s. Fourier transformed infrared spectra (FT-IR) were recorded using a Bruker TENSOR 27 FT-IR spectrometer with 4 cm−1 resolution and 100 scans in mid IR region (4000–600 cm−1) employing KBr pellet technique. Nitrogen adsorption–desorption isotherms were measured at 77 K using Micromeritics 3Flex surface area analyzer, in which catalysts were outgassed at 300 °C for 6 h prior to the sample measurements. Helium was used as the carrier gas. Specific surface area was calculated using BET (Brunauer–Emmett–Teller) equation and pore-size distribution was obtained from desorption branch of the isotherm using BJH (Barrett–Joyner–Halenda) method. Total number of acids sites and acid strengths of the samples were determined by temperature programmed desorption of ammonia (NH3-TPD) using an Altamira AMI-300 equipment. Samples were activated at 300 °C for 1 hour in helium flow. Later, they were cooled and maintained at 100 °C prior to their exposure to ammonia vapour followed by purging with helium for 30 min. The desorption of ammonia was carried out by heating the reactor up to 350 °C at a steady rate of 10 °C min−1.3. Catalyst activity evaluation
n-Heptane conversion was carried out at a reactor temperature of 350 °C at 30 bar in a down flow fixed bed reactor. A catalyst loading of 1.5 g was used and H2/n-heptane molar ratio was maintained at 3.88. Reaction was carried out in an automated fixed bed reactor manufactured by PID Eng. & Tech., Spain. All catalysts were first purged with N2 flow of 50 ml min−1 at 100 °C for 1 hour and subsequently reduced under hydrogen flow of 50 ml min−1 at a pressure of 30 bar and a temperature of 350 °C at a ramp rate of 1 °C min for 3 hours. After reduction, hydrogen (99.9%) flow rate was maintained at 50 ml min−1 and n-heptane feed was pumped into the reactor using a High Performance Liquid Chromatography (HPLC) pump at a Weight Hourly Space Velocity (WHSV) of 2.2 h−1. The liquid products were analysed using a detailed hydrocarbon analyser (DHA) equipped with flame ionization detector (FID) and gas products were analysed by a Refinery Gas Analyser (RGA) equipped with FID and (Thermal Conductivity Detector) TCD detectors. The catalytic activity was reported as n-heptane conversion, isomerization selectivity was calculated as the ratio of sum of all isomer products to n-heptane converted, cracking selectivity was calculated as the ratio of sum of all cracked products to n-heptane converted and LPG selectivity was calculated as ratio of LPG yields to n-heptane converted.Results and discussions
1. Catalyst characterization and analysis
Nitrogen adsorption isotherms for prepared materials were presented in Fig. 1. Typical type-IV isotherms characteristic of mesoporous materials with different hysteresis loop types were obtained for all materials. Initially, the adsorption volume increased at low values of relative pressures attributing to monolayer and multilayer adsorption in micro and mesopores. The onset of hysteresis was observed at a p/po value near 0.64 for S15 and K6 materials. A sudden increase in a narrow range of relative pressures beyond 0.64 for S15 and K6 materials indicated capillary condensation inside tubular pores of uniform size. However, the onset of hysteresis was observed much earlier at p/po values near 0.43 and 0.53 for PS15 and HMS materials, respectively. The adsorption isotherm for PS15 indicated the presence of secondary pores which explained the origin of an early hysteresis. Pore sizes obtained from desorption curve using BJH method and micropores surface area obtained from t-plot of PS15 material also gave evidence for the presence of a secondary pore. Similarly, early hysteresis at low p/po was observed for HMS which could be attributed to the presence of small pores for HMS (57 Å).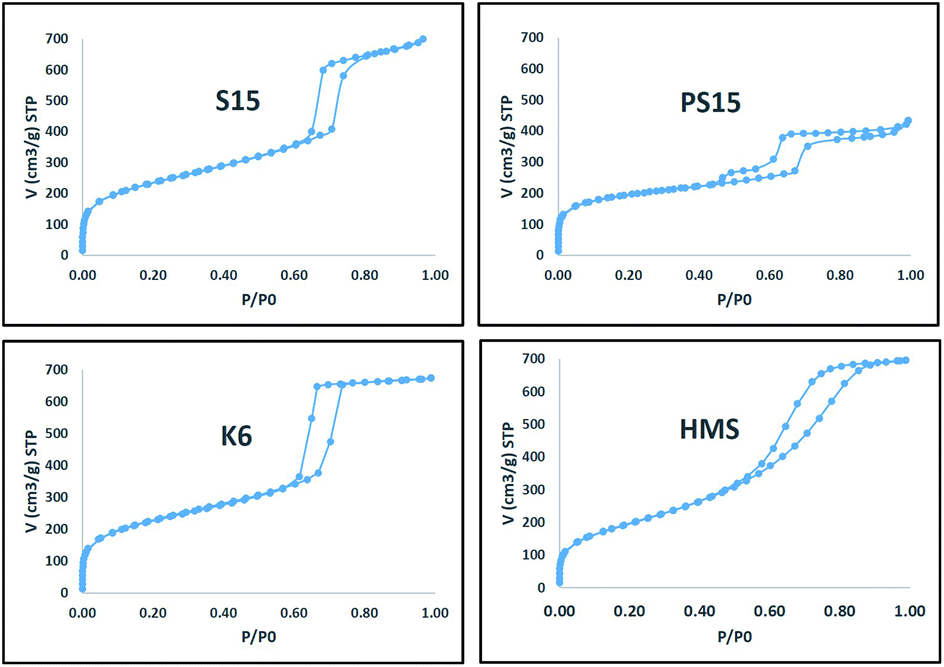 | ||
| Fig. 1 Nitrogen physisorption isotherms obtained for prepared materials. | ||
The pore size distribution plots obtained from desorption curve using BJH method for prepared materials are presented in Fig. 2. S15, K6 and HMS consisted of uniform pores of size 61 Å, 60 Å and 57 Å, respectively. PS15 consisted of a bi-modal pore size distribution of sizes 37 Å and 57 Å. The presence of almost similar pore sizes in all materials facilitated a good comparison of catalysts with respect to catalyst activities and selectivities. As such the observed differences could be directly correlated with differences in TPA mesoporous silica interactions.
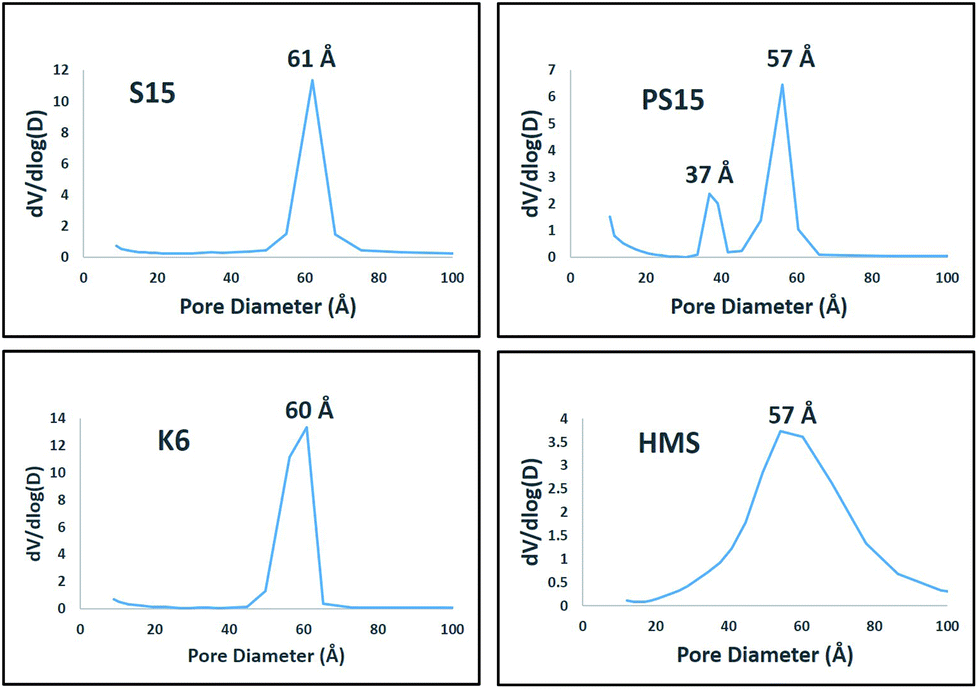 | ||
| Fig. 2 Pore size distribution plots obtained for prepared materials. | ||
The textural properties of catalysts obtained after TPA and platinum impregnation were presented in Table 1. Negligible changes in pore sizes were observed after impregnation indicating the presence of a uniform and fine dispersion of TPA on mesoporous silicas. However, BET specific surface area and micropore volumes were found to decrease owing to a higher TPA loading of 30 wt%. The presence of mesopore, high surface area and pore volume for TPA–OMS composite catalysts indicated the absence of any detrimental effect of TPA and platinum impregnation on mesoporous silicas. The mesoporous topology and structure was completely retained in composite catalysts.
| Material | SBET (m2 g−1) | Smicro (m2 g−1) | Vp (cm3 g−1) | Vmicro (cm3 g−1) | DBJH (nm) | Uptake (μmol g−1) | T (°C) |
|---|---|---|---|---|---|---|---|
| Pt@TPA@S15 | 503 | 8.5 | 0.7 | 0.02 | 6.2 | 267.5 | 197 |
| S15 | 723 | 0 | 1.07 | 0.02 | 6.1 | ||
| Pt@TPA@PS15 | 450 | 123 | 0.43 | 0.06 | 3.8, 5.6 | 206.5 | 215 |
| PS15 | 690 | 203 | 0.63 | 0.09 | 3.7, 5.7 | ||
| Pt@TPA@K6 | 460 | 2.6 | 0.65 | 0.02 | 6.1 | 154.4 | 206 |
| K6 | 754 | 0 | 1.04 | 0.02 | 6.0 | ||
| Pt@TPA@HMS | 397 | 0 | 0.69 | 0 | 5.6 | 189.2 | 190 |
| HMS | 616 | 0 | 1.07 | 0 | 5.7 |
X-ray diffraction (XRD) patterns for prepared materials in the range of 0.5° to 2° are presented in Fig. 3. All materials exhibited well resolved reflections typical of respective mesoporous structures. The XRD patterns for TPA impregnated mesoporous silicas were largely similar to the parent materials with only a slight decrease in intensity owing to the presence of a TPA loading of 30 wt%. Again, XRD characterization corroborates the absence of any detrimental effect of TPA impregnation on the synthesized catalysts, retention of mesoporous structure and uniform and fine dispersion of TPA on mesoporous silicas.
 | ||
| Fig. 3 XRD patterns obtained for prepared materials. | ||
Mesoporous silicas were reported as relatively inert supports for TPA when compared to alumina, zirconia, carbon etc.14 and the Keggin ion was expected to retain its structure when supported on various mesoporous silicas. 31P Magic Angle Spinning (MAS) Nuclear Magnetic Resonance (NMR) Spectroscopy is a useful tool to investigate the integrity of Keggin structure and its interaction with silicas. It was reported that the state of TPA while supported on silica was complex and an interaction between TPA and silica was present indicated by the line broadening in 31P NMR spectrum.27 A peak at −15.1 ppm was associated with bulk TPA, a peak at −14.5 ppm was associated with the interacting form, the amount of which increased with decreasing TPA loadings and a peak at −13.3 ppm was associated with a lacunary or unsaturated species such as PW11, P2W17, P2W18 and P2W21.19 The 31P MAS NMR data for the prepared composite catalysts are presented in Fig. 4. It was observed that the peak corresponding to bulk TPA was absent in all the samples and all mesoporous silicas had different degrees of interactions with TPA giving rise to peaks at −14.09, −14.42, −14.23 and −14.34 ppm for TPA@S15, TPA@PS15, TPA@K6 and TPA@HMS respectively. Kozhevnikov and group had proposed the use of methanol for impregnation to avoid TPA interactions with silica.14 We have observed that all the four TPA impregnated mesoporous silicas prepared by us were strongly interacting with TPA in spite of using methanol as a polar medium for impregnation. It could be concluded from the chemical shifts that the interaction was minimum for PS15 and increased in the order TPA@PS15 < TPA@HMS < TPA@K6 < TPA@S15. In addition, TPA@S15 and TPA@K6 were also observed to show peaks around −13.3 ppm indicating the presence of lacunary or unsaturated species. We concluded that the strong interaction of TPA with silanol groups in TPA@S15 and TPA@K6 catalysts led to agglomeration of keggin ions which after decomposition led to generation of surface lacunary species. Lefebvre and group had observed such lacunary species to be inactive for n-hexane conversion.28 These differences in the structure of TPA supported on mesoporous silicas led to different activities and selectivities for the TPA–OMS composite catalysts.
 | ||
| Fig. 4 31P MAS NMR spectra obtained for prepared materials. | ||
In order to verify the integrity of Keggin ions after TPA and platinum impregnation, FTIR spectra were recorded for the samples and are presented in Fig. 5. All samples indicated four characteristics bands near 1079, 987, 887 and 809 cm−1. These bands could be associated with νas (P–O) in the central PO4 tetrahedron, νas (W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in the exterior WO6 tetrahedron, νas (W–Ob–W) in corner shared octahedral and νas (W–Oc–W) in the edge shared octahedral of the Keggin structure of TPA, respectively. Similar FTIR spectrum for TPA impregnated on silica supports were reported in the literature.27,29,30 Thus, it could be concluded from FTIR spectra that Keggin structure was retained after impregnation in all the materials. Small shifts in bands from ideal values indicated the presence of interaction of TPA with mesoporous silicas. Other bands characteristics of TPA were not observed due to fine dispersion of TPA on silica supports.
O) in the exterior WO6 tetrahedron, νas (W–Ob–W) in corner shared octahedral and νas (W–Oc–W) in the edge shared octahedral of the Keggin structure of TPA, respectively. Similar FTIR spectrum for TPA impregnated on silica supports were reported in the literature.27,29,30 Thus, it could be concluded from FTIR spectra that Keggin structure was retained after impregnation in all the materials. Small shifts in bands from ideal values indicated the presence of interaction of TPA with mesoporous silicas. Other bands characteristics of TPA were not observed due to fine dispersion of TPA on silica supports.
 | ||
| Fig. 5 FTIR spectra obtained for prepared materials. | ||
29Si MAS NMR studies were conducted and the recorded spectra are presented in Fig. 6. A major peak around −110 ppm could be attributed to the presence of Q4 silanol groups (Si-(OSi)4) present in the amorphous walls of mesoporous materials. This peak was observed to be relatively intense for TPA@HMS. The presence of Q3 (XO-Si-(OSi)3) and Q2 ((XO)2-Si-(OSi)2) silanol groups were confirmed by the presence of peaks at ∼−100 ppm and −90 ppm, respectively. The presence of Q3 and Q2 silanol groups indicated the presence of ion-exchanged tungstophosphoric acid Keggin anions and possibly un-exchanged Si–OH groups. The Q2 peaks were clearly evident for TPA@S15 and TPA@K6 catalysts and present as weak shoulders for TPA@PS15 and TPA@HMS catalysts. Since TPA@PS15 and TPA@HMS catalysts also showed the presence of lacunary species we concluded that such lacunary species were bonded with Q2 silanol groups. Other researchers have also reported similar peaks in 29Si MAS NMR studies for different type of silanol groups present in various mesoporous materials.31–34
 | ||
| Fig. 6 29Si MAS NMR spectra obtained over prepared catalysts. | ||
Finally, it was observed that TPA impregnation on the mesoporous silicas led to creation of varying number and strength of acid sites on the composite catalysts. The ammonia uptake and subsequent desorption profile for the samples is provided in Fig. S1† and data obtained for the samples is presented in Table 1. Since TPA may decompose above 350 °C, the TPD data was obtained only till a temperature of 350 °C. A single peak was evident below 350 °C for all catalysts samples and the peak maxima was correlated with the strength of acid sites and total ammonia desorbed was correlated with the number of acid sites. The strength of acid sites was found to increase in the order Pt@TPA@HMS < Pt@TPA@S15 < Pt@TPA@K6 < Pt@TPA@PS15. The number of acid sites obtained from area under the Temperature Programmed Desorption (TPD) curve increased in the following order: Pt@TPA@K6 < Pt@TPA@HMS < Pt@TPA@PS15 < Pt@TPA@S15. It could be concluded that HMS and S15 based composite catalysts are mildly acidic catalysts compared to K6 and PS15 based composite catalysts which possessed relatively stronger acid sites.
2. Catalyst evaluation and discussions
Gas and liquid products obtained from hydroconversion of n-heptane were expressed in percentage yields in Table 2. The order of increase of gas make on the catalysts was Pt@TPA@HMS < Pt@TPA@S15 < Pt@TPA@K6 < Pt@TPA@PS15. Thus gas yields were directly related to the strength of acid sites on the prepared catalyst, which followed the same trend. The liquid yields obtained, therefore, follow the exact reverse trend. It was interesting to note here that the concentration of methane and ethane in the gas products were low and the gas was predominantly composed of C3 and C4 hydrocarbons which form the LPG pool. Similarly, most of the liquid was composed of C7 hydrocarbons and only a small percentage of C5 and C6 hydrocarbons were present.| Catalyst | Pt@TPA@K6 | Pt@TPA@HMS | Pt@TPA@PS15 | Pt@TPA@S15 |
|---|---|---|---|---|
| Gas yield (%) | 33.22 | 28.51 | 34.77 | 30.41 |
| Liq. yield (%) | 66.78 | 71.49 | 65.23 | 69.59 |
| C1 | 0.10 | 0.05 | 0.40 | 0.07 |
| C2 | 0.27 | 0.13 | 0.44 | 0.21 |
| C3 | 19.83 | 16.58 | 20.56 | 17.63 |
| Total C4 | 13.02 | 11.75 | 13.37 | 12.50 |
| Total C5 | 1.94 | 0.69 | 1.85 | 0.78 |
| Total C6 | 2.53 | 1.52 | 2.61 | 1.64 |
| Total C7 | 62.31 | 69.28 | 60.77 | 67.17 |
| Total | 100.00 | 100.00 | 100.00 | 100.00 |
The conversion and selectivities observed for hydrocracking of n-heptane towards C1–C6 range hydrocarbons were presented in Table 3. Dry gas was referred to as the total of methane and ethane. Heptane conversion was observed to decrease in the order of Pt@TPA@PS15 (54.49%) > Pt@TPA@HMS (51.95%) > Pt@TPA@K6 (42.86%) > Pt@TPA@S15 (41.34%). Based on the above data it was rationalized that the presence of lacunary species, which were reported to be inactive for hydrocarbon conversion by Lefebvre and group,28 led to inferior n-heptane conversion over Pt@TPA@K6 and Pt@TPA@S15 catalysts compared to Pt@TPA@PS15 and Pt@TPA@HMS catalysts. The strength of interaction of TPA with silanol groups of the mesoporous silicas, studied using 31P MAS NMR, was the least in the case of TPA@PS15 material, pointing towards generation of strong bronsted acid sites similar to unsupported TPA where as in the case of TPA@HMS the generated acid sites were weaker in strength due to relatively stronger interaction with TPA. These strength of acid sites were also reflected in NH3-TPD studies and found to be lower on Pt@TPA@HMS (190 °C), resulting in high selectivity towards isomerization cracking, compared to Pt@TPA@PS15 (215 °C) which demonstrated higher selectivity to cracking among these two catalysts.
| Catalyst | Pt@TPA@K6 | Pt@TPA@HMS | Pt@TPA@PS15 | Pt@TPA@S15 |
|---|---|---|---|---|
| Conversion % | 42.86 | 51.95 | 54.49 | 41.34 |
| Isomer sel. (%) | 11.59 | 40.46 | 27.42 | 20.02 |
| Cracking sel. (%) | 87.94 | 59.13 | 72.01 | 79.43 |
| Dry gas sel. (%) | 0.86 | 0.34 | 1.54 | 0.69 |
| LPG sel. (%) | 76.64 | 54.53 | 62.27 | 72.88 |
| C1 cracked sel. (%) | 0.23 | 0.10 | 0.73 | 0.17 |
| C2 cracked sel. (%) | 0.63 | 0.24 | 0.81 | 0.52 |
| C3 cracked sel. (%) | 46.26 | 31.92 | 37.73 | 42.65 |
| C4 cracked sel. (%) | 30.38 | 22.62 | 24.54 | 30.24 |
| C5 cracked sel. (%) | 4.53 | 1.33 | 3.40 | 1.89 |
| C6 cracked sel. (%) | 5.90 | 2.93 | 4.80 | 3.97 |
Lefebvre and group had associated the peak around −14.5 ppm in 31P NMR to the interaction of silanol groups of silica (–SiOH) with the proton of Keggin ion leading to the formation of (–SiOH2+) (H2PW12O40−) ionic pairs.35 Since the strength of O–H bonds in silanol groups vary in mesoporous silicas of different topologies, the trends in chemical shifts observed in 31P NMR are representative of the strength of Keggin ion silica interactions and the trends can be used to rationalize differences in catalyst activities. We conclude that TPA@PS15 and TPA@HMS samples possess more acidic protons in the Keggin form of supported Keggin ion paired with silanol groups, which catalyse the hydroconversion of n-heptane. On the other hand the performance of Pt@TPA@S15 and Pt@TPA@K6 was affected due to the presence of lacunary species. Still the strength of acid sites originating from keggin ion on these catalysts were observed to be higher on Pt@TPA@K6 catalyst (206 °C) compared to Pt@TPA@S15 catalyst (197 °C). As a result Pt@TPA@K6 catalyst demonstrated higher cracking selectivity and interestingly the highest among all catalysts. Again, the acidity of protons could be correlated with TPA–silanol interactions and correlated with 31P MAS NMR studies where Pt@TPA@K6 catalyst had presented a weaker interaction with TPA compared to Pt@TPA@S15.
From Table 3, it could also be observed that Pt@TPA@HMS followed by Pt@TPA@PS15 demonstrated higher selectivity towards isomerization compared to the other catalysts. On the other hand, a hydrocracking selectivity as high as 87.94% was observed for Pt@TPA@K6 and 79.43% for Pt@TPA@S15 catalyst. It is known in the literature that weak and mild acid sites are suitable for hydroisomerization, whereas strong acid sites lead to further cracking of the isomerized products formed initially.7 The high acid strength of TPA protons led to predominant cracking of n-heptane on all the samples and the cracking selectivity varied from 59.13% to 87.94%. Since the large pores of mesoporous silica presented no steric hindrance, a major percentage of this cracking selectivity was observed towards LPG. The cracking selectivities could be correlated to the acidic properties of the catalysts. Highest dry gas yields were obtained for Pt@TPA@PS15 > Pt@TPA@K6 > Pt@TPA@S15 and Pt@TPA@HMS which was also the order of decreasing strength of acid sites obtained over the composite catalysts. The liquid products obtained consisted of unreacted n-heptane, isomerized heptane and cracked hexane and pentane hydrocarbons. However, there were no cyclic (naphthenic) and aromatic products observed for any samples in any carbon range (C5, C6 or C7). Such a product distribution is ideal for the hydrocracking objective for the production of LPG. Because the unreacted heptane and isomerized heptane can be recycled back to the feed stream and cracked again to result into a very high selectivity towards LPG.
In Table 4, the isomerization selectivities for the composite catalysts are presented. Pt@TPA@HMS was found to exhibit the highest selectivity (40.46%) towards isomerization and the highest ratio (0.63) of multi branched isomers to mono branched isomers in the C7 range. Multibranched isomers are highly desired for their high octane numbers. A complete distribution of isomers obtained in C4, C5 and C6 range is given as Table S1† and C7 hydrocarbons is given as Table S2 in ESI,† respectively. In C7 isomers 2,2,3-trimethylbutane was observed as the highest octane number isomer and 2-methylhexane, 3-methylhexane and 2-ethylhexane isomers were obtained in high yields according to thermodynamic distribution. Multi/mono ratio can be used to compare the degree of isomerization obtained over catalysts and the ratio decreased in the order: Pt@TPA@HMS > Pt@TPA@S15 > Pt@TPA@K6 >Pt@TPA@PS15. The same order was observed for iC4/nC4 ratio over the prepared catalysts.
| Catalyst | Pt@TPA@K6 | Pt@TPA@HMS | Pt@TPA@PS15 | Pt@TPA@S15 |
|---|---|---|---|---|
| Conversion (%) | 42.86 | 51.95 | 54.49 | 41.34 |
| Isomer sel. (%) | 11.59 | 40.46 | 27.42 | 20.02 |
| Cracking sel. (%) | 87.94 | 59.13 | 72.01 | 79.43 |
| Mono branched (%) | 8.12 | 24.89 | 19.56 | 12.90 |
| Multi branched (%) | 3.47 | 15.57 | 7.85 | 7.12 |
| Multi/mono | 0.43 | 0.63 | 0.40 | 0.55 |
| iC4/nC4 | 5.08 | 6.39 | 4.24 | 6.14 |
| iC5/nC5 | 2.08 | 3.60 | 2.25 | 3.88 |
| iC6/nC6 | 1.96 | 1.71 | 1.95 | 2.34 |
| C3/C4 | 1.52 | 1.41 | 1.54 | 1.41 |
| DG/iC4 | 3.40 | 1.76 | 7.76 | 2.65 |
However, the order of isomer ratios was observed to be significantly different for C5 and C6 range hydrocarbons. These trends can be rationalised by consideration of the presence of lacunary species in Pt@TPA@K6 and Pt@TPA@S15 catalysts. Pt@TPA@HMS showed higher iC6/nC6 and iC5/nC5 ratios compared to Pt@TPA@PS15 because of weaker strength of acid sites favouring isomerisation. Similarly, among Pt@TPA@K6 and Pt@TPA@S15, where the activity was affected by presence of lacunary species, Pt@TPA@S15 was observed to demonstrate better iC6/nC6 and iC5/nC5 ratios because of weaker strength of acid sites favouring isomerization. C6 and C5 isomer distribution followed typical thermodynamic distribution as observed in isomerization reactions. The C3/C4 and DG/iC4 ratios give an insight into the governing mechanisms and could be directly correlated with the acidity data. Protolytic cracking was prevalent on Pt@TPA@PS15 followed by Pt@TPA@K6 composite catalyst with high strength of acid sites. Similarly, the propane selectivity was slightly higher on these catalysts compared to the other catalysts.
Conclusions
Four mesoporous superacid composite catalysts prepared by impregnation of tungstophosphoric acid Keggin ion on KIT-6, HMS, SBA-15 and plugged SBA-15 mesoporous silica supports were tested for hydroconversion of model feed n-heptane. 30 wt% TPA was finely dispersed on all the supports and did not lead to significant reduction in pore sizes and crystallization of bulk TPA phase. TPA Keggin ion was therefore present as an ionic pair linked to the silanol groups of mesoporous silicas. Presence of lacunary species in minor amounts was observed for SBA-15 and KIT-6 based catalysts possibly resulting from strong interactions of KIT-6 and SBA-15 with TPA Keggin ion leading to agglomeration of keggin ions and generation of surface lacunary species after decomposition. These species reduced n-heptane hydroconversion activity. Pt@TPA@PS15 demonstrated the weakest interaction with TPA and led to generation of strongest acid, high n-heptane conversion and selectivity towards isomerization cracking. Relatively weaker interactions of HMS with TPA Keggin ion led to generation of mild acidity of Pt@TPA@HMS, high n-heptane conversion and high isomerization selectivity. All mesoporous superacid catalysts, however, demonstrated hydrocracking as the predominant reaction due to inherent high acid strength of tungstophosphoric acid Keggin ion. Protolytic cracking to different extents was observed on all the catalysts. However no cyclic or aromatic compounds were observed for any catalysts providing an opportunity for recycle of liquid products for increasing cracking yields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work was funded by Hindustan Petroleum Green R&D Centre, Hindustan Petroleum Corporation Limited (HPCL) under project code CD/2012/001 and SM/VK sincerely thank the management for the support and funding. Mr Raghav Krishna and Dr C. S. K. Raju are acknowledged for the product analysis using DHA and catalyst characterization.Notes and references
- A. Thosar, WLPGA Sub-Continent Regional Summit, 2017 Search PubMed.
- L. Yanis and P. Jim, unpublished work.
- A. Abbott, C. Pieron, R. Pecilunas, J. Rockall, N. Xydas, D. Tyler, E. Assous, D. Tyler, L. Poret and M. Kelly, WLPGA Annual Report, 2015 Search PubMed.
- J. M. Ferreira Coelho and A. Szklo, Energy Strategy Reviews, 2015, 6, 80–91 CrossRef.
- A. Boretti, Fuel Process. Technol., 2017, 161, 41–51 CrossRef CAS.
- M. Hajbabaei, G. Karavalakis, J. W. Miller, M. Villela, K. H. Xu and T. D. Durbin, Fuel, 2013, 107, 671–679 CrossRef CAS.
- H. Deldari, Appl. Catal., A, 2005, 293, 1–10 CrossRef CAS.
- S. Mehla, K. R. Krishnamurthy, B. Viswanathan, M. John, Y. Niwate, S. A. Kishore Kumar, S. M. Pai and B. L. Newalkar, Microporous Mesoporous Mater., 2013, 177, 120–126 CrossRef CAS.
- A. Corma, V. G. Alfaro and A. V. Orchilles, Appl. Catal., A, 1999, 245–254 CrossRef CAS.
- R. G. Tailleur and J. B. Platin, J. Catal., 2008, 255, 79–93 CrossRef CAS.
- M. Busto, L. A. Dosso, C. R. Vera and J. M. Grau, Fuel Process. Technol., 2012, 104, 128–135 CrossRef CAS.
- H. Al-Kandari, F. Al-Kharafi and A. Katrib, Catal. Commun., 2008, 9, 847–852 CrossRef CAS.
- M. Misono, I. Ono, G. Koyano and A. Aoshima, Pure Appl. Chem., 2000, 72, 1305–1311 CAS.
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171–198 CrossRef CAS PubMed.
- G. J. d. A. A. Soler-Illia, C. Sanchez, B. Lebeau and J. Patarin, Chem. Rev., 2002, 102, 4093–4138 CrossRef PubMed.
- A. Vinu, V. Murugesan, W. Böhlmann and M. Hartmann, J. Phys. Chem. B, 2004, 108, 11496–11505 CrossRef CAS.
- R. Srivastava, D. Srinivas and P. Ratnasamy, J. Catal., 2005, 233, 1–15 CrossRef CAS.
- S.-Y. Chen, L.-Y. Jang and S. Cheng, Chem. Mater., 2004, 16, 4174–4180 CrossRef CAS.
- I. V. Kozhevnikov, K. R. Kloetstra, A. Sinnema, H. W. Zandbergen and H. v. Bekkum, J. Mol. Catal. A: Chem., 1996, 114, 287–298 CrossRef CAS.
- P. T. Tanev and T. J. Pinnavaia, Science, 1995, 267, 865–867 CrossRef CAS PubMed.
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- A. H. Janssen, P. Van Der Voort, A. J. Koster and K. P. de Jong, Chem. Commun., 2002, 1632–1633, 10.1039/B204943K.
- F. Kleitz, S. Hei Choi and R. Ryoo, Chem. Commun., 2003, 2136–2137, 10.1039/B306504A.
- A. Corma and A. V. Orchillés, Microporous Mesoporous Mater., 2000, 35–36, 21–30 CrossRef CAS.
- P. Selvam and V. Krishna, Chem.–Eur. J., 2017, 23, 1604–1612 CrossRef PubMed.
- A. Ghanbari-Siahkali, A. Philippou, J. Dwyer and M. W. Anderson, Appl. Catal., A, 2000, 192, 57–69 CrossRef CAS.
- X. Yang, J. A. Wang, L. Chen, S. P. R. Sebastian and A. M. Robledo, Catal. Commun., 2012, 28, 202–206 CrossRef CAS.
- A. Oulmekki and F. Lefebvre, React. Kinet. Catal. Lett., 1992, 48, 601–606 CrossRef CAS.
- D. Jin, Z. Liu, J. Zheng, W. Hua, J. Chen, K. Zhu and X. Zhou, RSC Adv., 2016, 6, 32523–32533 RSC.
- P. Yang, S. Zhou, Y. Du, J. Li and J. Lei, RSC Adv., 2016, 6, 53860–53866 RSC.
- J. Trébosc, J. W. Wiench, S. Huh, V. S. Y. Lin and M. Pruski, J. Am. Chem. Soc., 2005, 127, 3057–3068 CrossRef PubMed.
- S. Lei, Z. Yan and H. Heyong, Chem. Lett., 2001, 30, 1164–1165 CrossRef.
- M. Kar, B. Malvi, A. Das, S. Panneri and S. S. Gupta, J. Mater. Chem., 2011, 21, 6690–6697 RSC.
- C. Jo, K. Kim and R. Ryoo, Microporous Mesoporous Mater., 2009, 124, 45–51 CrossRef CAS.
- V. Dufaud, F. Lefebvre, G. P. Niccolai and M. Aouine, J. Mater. Chem., 2009, 19, 1142–1150 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07024e |
| This journal is © The Royal Society of Chemistry 2018 |