DOI:
10.1039/C8RA07013J
(Paper)
RSC Adv., 2018,
8, 37606-37617
L-Proline catalyzed one-pot synthesis of polysubstituted pyridine system incorporating benzothiazole moiety via sustainable sonochemical approach†
Received
21st August 2018
, Accepted 18th October 2018
First published on 8th November 2018
Abstract
The efficient, highly convenient synthesis of polysubstituted pyridine derivatives was established via the reaction of N-(benzothiazol-2-yl)-2-cyanoacetamides with an assortment of arylidene malononitriles and arylidene ethyl cyanoacetates in the presence of L-proline as an efficient organocatalyst for this type of ultrasonic-mediated Michael addition. The mechanistic pathway and factors affecting this reaction were also established. The main characteristics of this procedure are high yields, use of a cost-effective catalyst, and easy work-up and purification.
Introduction
Over the last few decades heterocyclic compounds have presented tremendous opportunities in the field of therapeutics and drug design. Many of these compounds play an important role in human life, plants and animals and have now been identified in all aspects of life. Among the heterocyclic compounds, the interesting aza-heterocycles are very important in medicinal chemistry. The presence of nitrogen may modulate the electron distribution inside heterocycles, leading to the modification of both the chemical and physical characteristics of these compounds, which also leads to modification in their biological properties and metabolic pathways.1 One of the most remarkable nitrogen-containing heterocycles is pyridine and its outstanding derivatives since it is the basic moiety in many natural products such as alkaloids.2,3 Moreover, pyridine-containing heterocycles have various biological and pharmacological activities, such as anti-microbial,4–6 anti-viral,7,8 antioxidant,9 anti-diabetic,10 anti-cancer and anti-tumor,11–14 anti-malarial,15,16 anti-inflammatory,17,18 and anti-amoebic activities.19 In addition, some pyridine-containing compounds exhibit analgesic potency20 and enzyme inhibition.21 Furthermore, pyridine ring systems are used in medicine for the treatment of iron overload disease via the chelation effect of these compounds.22 Additionally, benzothiazole is one of the most significant heterocycles, which is the core structure of many pharmaceuticals and natural products.23–25 Recently, the concept of organocatalysis and sonochemical reactions has significantly grown as a result of both the juvenility of the concept and, more importantly, the selectivity and effectiveness of many organocatalytic reactions. Moreover, the sonication process has more inherent advantages compared to conventional heating, such as higher yields, moderate reaction conditions, shorter reaction times and easier work-up.26–28
Consequently, due to the above intriguing characteristics and applications, pyridines and fused pyridine derivatives are notable structures, which have attracted the interest of synthetic organic chemists.29,30 However, although numerous protocols have been developed in the last decade for the synthesis of multisubstituted pyridine derivatives,31–33 the development of highly efficient, convenient methods for the synthesis of polysubstituted pyridines is still a major demand in modern synthetic chemistry. Recently, in our laboratory we directed our effort to the development of efficient protocols for the synthesis of pyridine and fused pyridine derivatives.34–37 As a continuation of our research towards developing these protocols utilizing green methodologies,38–41 herein, we report an efficient, eco-friendly strategy for the synthesis of polysubstituted pyridine derivatives utilizing L-proline as an eco-friendly organocatalyst, which is mediated by ultrasonic irradiation as an efficacious green energy source.42,43
Results and discussion
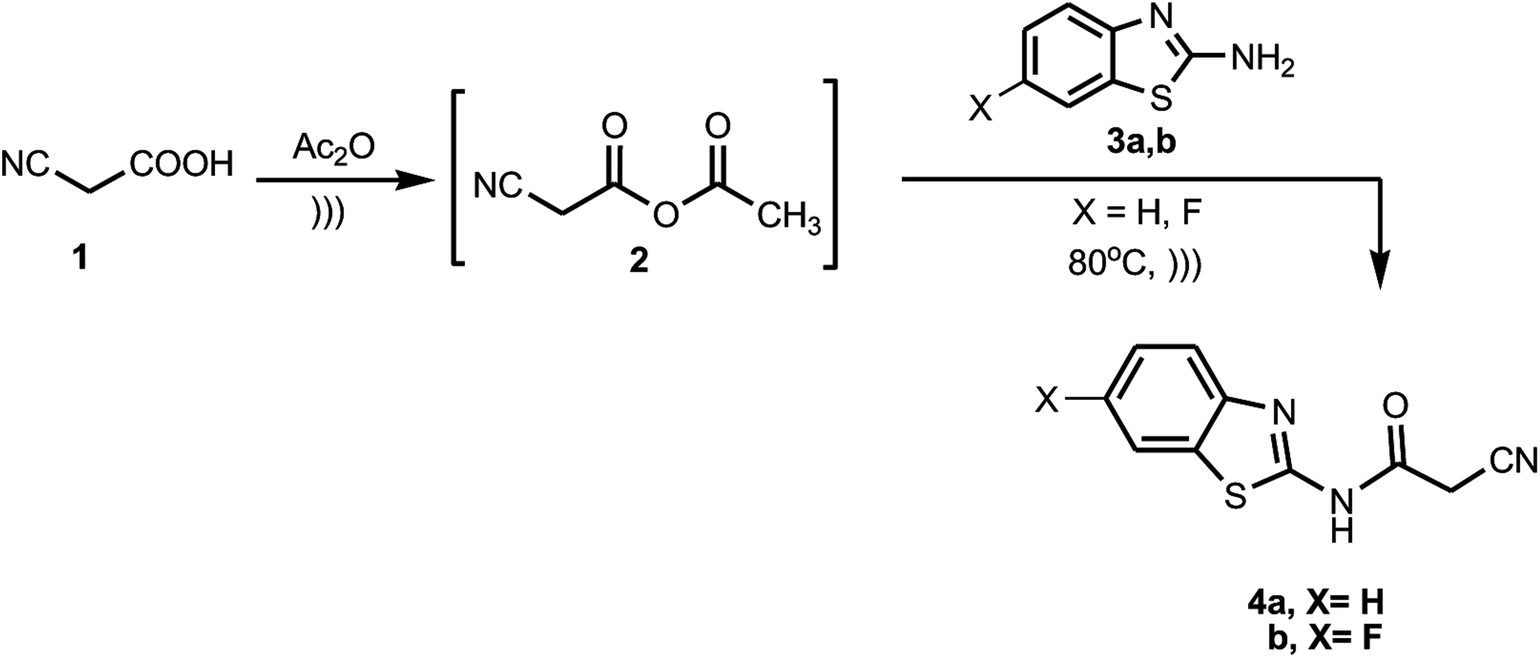
The synthetic strategy of our research to obtain the targeted compounds commenced by preparing benzothiazole cyanoacetamide derivatives 4a and b, which were prepared by the treatment of the 2-amino-benzothiazole derivatives 3a and b with a preheated mixture of cyanoacetic acid (1) in acetic anhydride under sonication (Scheme 1).
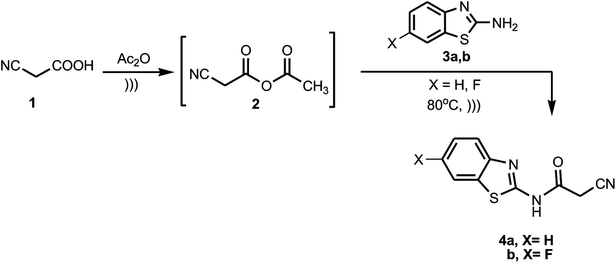 |
| | Scheme 1 Synthesis of benzothiazole cyanoacetamide derivatives 4a and b. | |
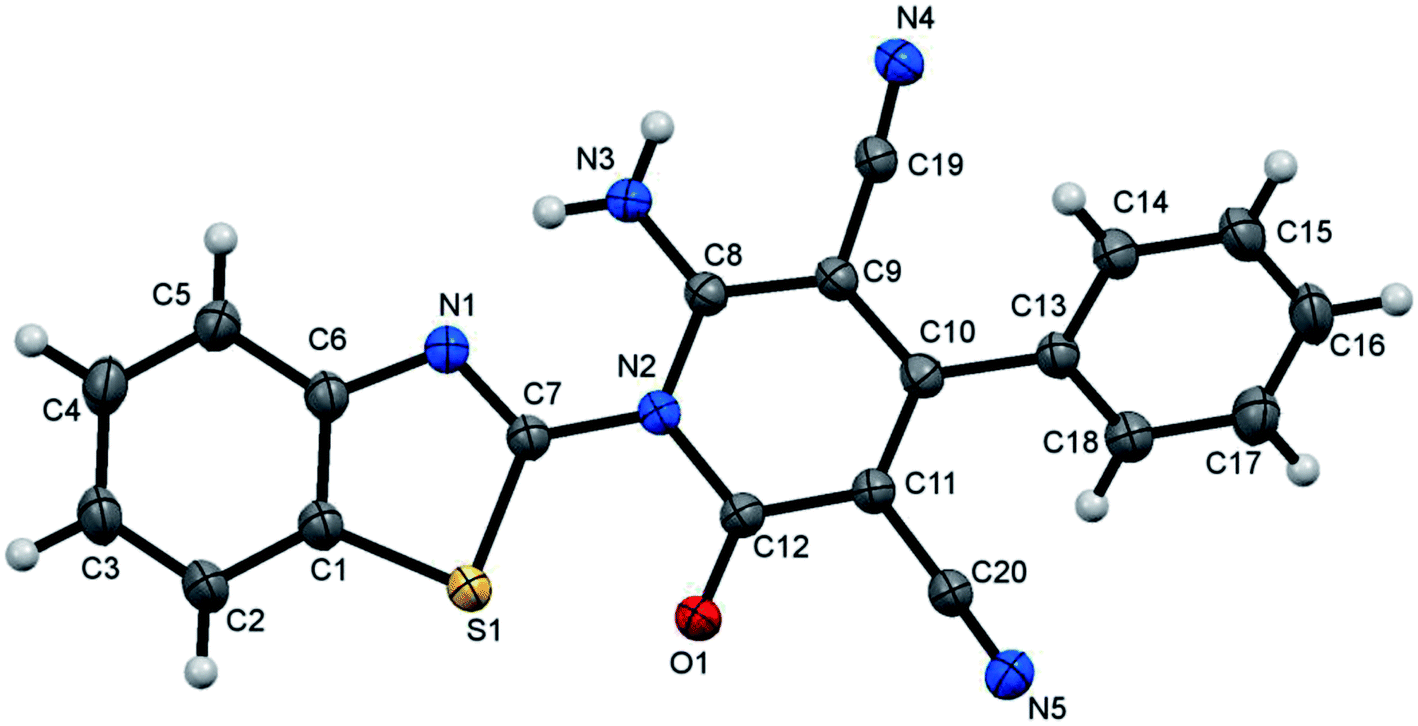
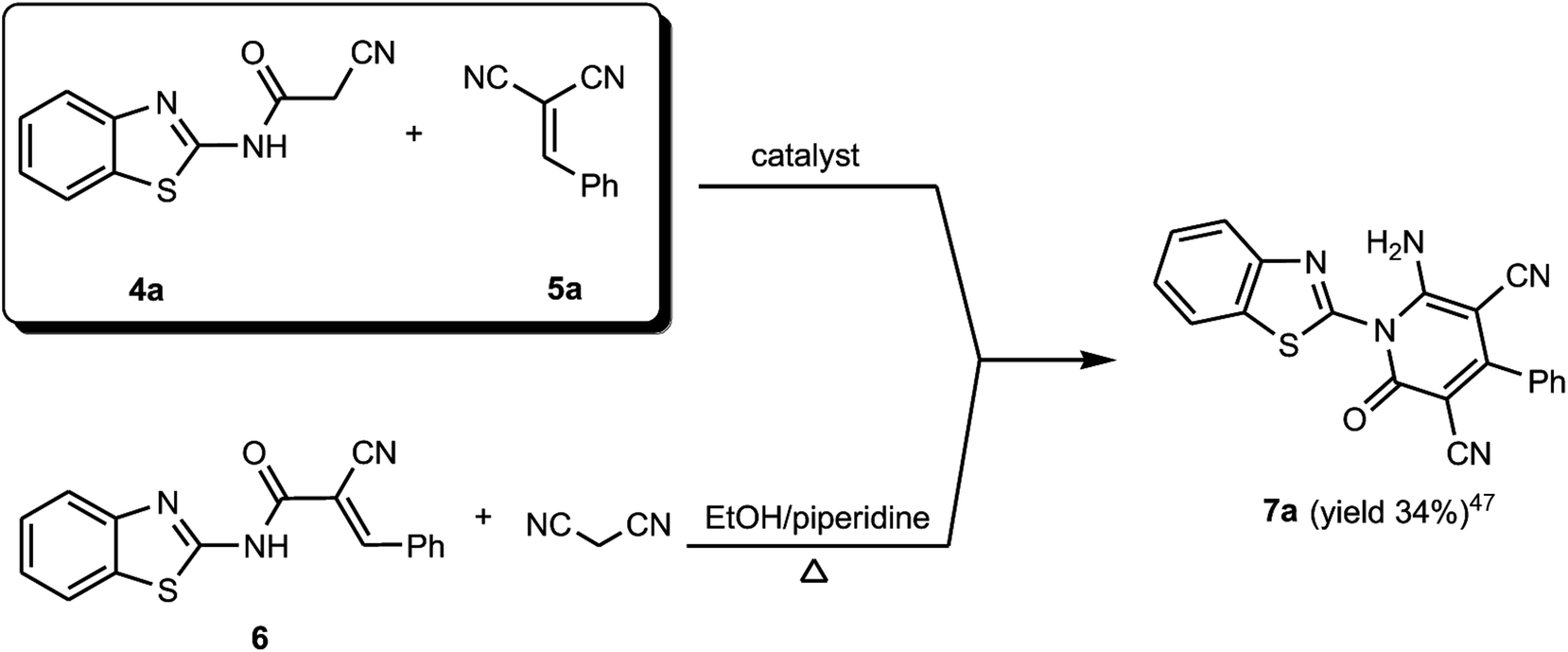
The formed benzothiazole cyanoacetamide derivatives 4a and b represent valuable precursors to be tested for the synthesis of polyfunctional substituted pyridine derivatives. Thus, the active methylene moiety in the N-(benzothiazol-2-yl)-2-cyanoacetamide derivatives 4a and b has high capability to undergo nucleophilic addition reaction with a variety of electrophilic reagents, such as arylidene malononitriles 5a–h via a Michael-type addition reaction.44–46 Thus, in this context of our study, which aimed to establish the best reaction conditions for the synthesis of pyridine derivatives via a green methodology, the reaction between N-(benzothiazol-2-yl)-2-cyanoacetamide (4a) and 2-benzylidenemalononitrile (5a) was tested in the presence of different types of organocatalysts as a model reaction. Firstly, we applied piperidine as catalyst to conduct the cycloaddition reaction between N-(benzothiazol-2-yl)-2-cyanoacetamide (4a) (5 mmol) and 2-benzylidenemalononitrile (5a) (5 mmol) in ethanol (20 mL) under both conventional heating and ultrasonic irradiation to study the effect of the type of energy source, and the results are listed in Table 1. The structure of the obtained product was assigned as 6-amino-1-benzothiazol-2-yl-2-oxo-4-phenyl-1,2-dihydro-pyridine-3,5-dicarbonitrile (7a) based on the data obtained from several analysis tools, such as 1H NMR, 13C NMR, and mass and accurate mass determination. Moreover, this structure was also confirmed by the X-ray single crystal structure determination (Fig. 1 and Scheme 2). It is worth mentioning that the pyridine derivative 7a was obtained earlier by Stetinova et al.47 through a comparable route via the reaction of 3-aryl-N-(2-benzothiazolyl)-2-cyano-2-propenamides (6) with malononitrile in ethanolic solution containing piperidine. However, the main disadvantage of their route is the low yield of the obtained pyridine derivative 7a in comparison with our synthetic procedure, even with the use of piperidine as the catalyst (Scheme 2). As shown in Table 1 (entries 1 and 2), the use of ultrasonic irradiation as the mode of activation for our reaction incredibly increased the reaction yield from 59% (which was obtained using only thermal heating) to 85%. Also, as noted, the reaction time was remarkably minimized from 2 hours (using conventional heating) to 20 minutes (using ultrasonic irradiation). From the results depicted in Table 1, we observed that this cycloaddition reaction could be effectively catalyzed by L-proline Table 1 (entry 6), while piperidine, morpholine, DBU, DBACO, and imidazole are less effective organocatalysts since they afforded lower yields. This may be attributed to the zwitterionic characteristic of L-proline, which makes it act as a bifunctional catalyst, in addition to the H-bonding of proline in the enamine transition state playing an essential role in the reaction outcome. Furthermore, an enhancement was observed for both the reaction rate and yield when the above-mentioned reaction was conducted under ultrasonic irradiation (at 80 °C, 110 W). The use of ultrasound irradiation in chemical reactions in solution, as an alternative energy source, has many advantages, including dramatic improvements in both stoichiometric and catalytic chemical reactions. In some cases, ultrasonic irradiation can increase the reactivity and the reaction rate by nearly a hundred-fold, which proceeds by the generation, growth, and collapse of acoustic bubbles in the reaction mixture. Accordingly, this assists in decreasing the reaction time and significantly enhancing the reaction yield. Then, we tried to use water as a green energy solvent to conduct the above reaction; however, we were unsuccessful mostly because most organic compounds do not dissolve adequately in water, as shown in Table 1 (entries 13 and 14).48 Alternatively, ethanol performed well as the reaction medium since it is a comparatively nonhazardous solvent to the environment, as shown in Table 1 (entry 6).49
Table 1 Optimization of the conditions for the synthesis of 7aa
| Entrya |
Catalyst |
Solvent |
Method |
Time (min) |
Yield (%) |
| Reaction conditions: N-(benzothiazol-2-yl)-2-cyanoacetamide (4a) (5 mmol), 2-benzylidenemalononitrile (5a) (5 mmol), in solvent (20 mL) and catalyst (10 mol%) (in case of US, the temperature was 80 °C at 110 W). NR: no reaction. |
| 1 |
Piperidine |
EtOH |
Heating |
120 |
59 |
| 2 |
Piperidine |
EtOH |
US ))) |
20 |
85 |
| 3 |
Morpholine |
EtOH |
Heating |
120 |
43 |
| 4 |
Morpholine |
EtOH |
US ))) |
20 |
77 |
| 5 |
L-Proline |
EtOH |
Heating |
120 |
74 |
| 6 |
L-Proline |
EtOH |
US ))) |
20 |
95 |
| 7 |
DBU |
EtOH |
Heating |
120 |
30 |
| 8 |
DBU |
EtOH |
US ))) |
20 |
45 |
| 9 |
DABCO |
EtOH |
Heating |
120 |
41 |
| 10 |
DABCO |
EtOH |
US ))) |
20 |
52 |
| 11 |
Imidazole |
EtOH |
Heating |
120 |
35 |
| 12 |
Imidazole |
EtOH |
US ))) |
25 |
56 |
| 13 |
L-Proline |
H2O |
Heating |
120 |
NRb |
| 14 |
L-Proline |
H2O |
US ))) |
60 |
NRb |
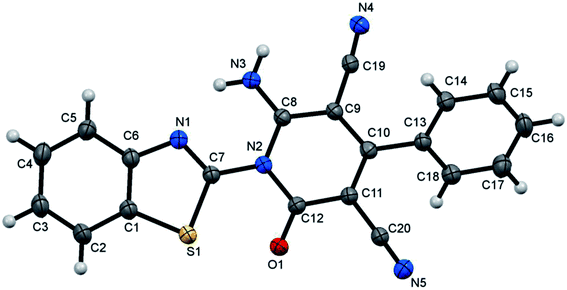 |
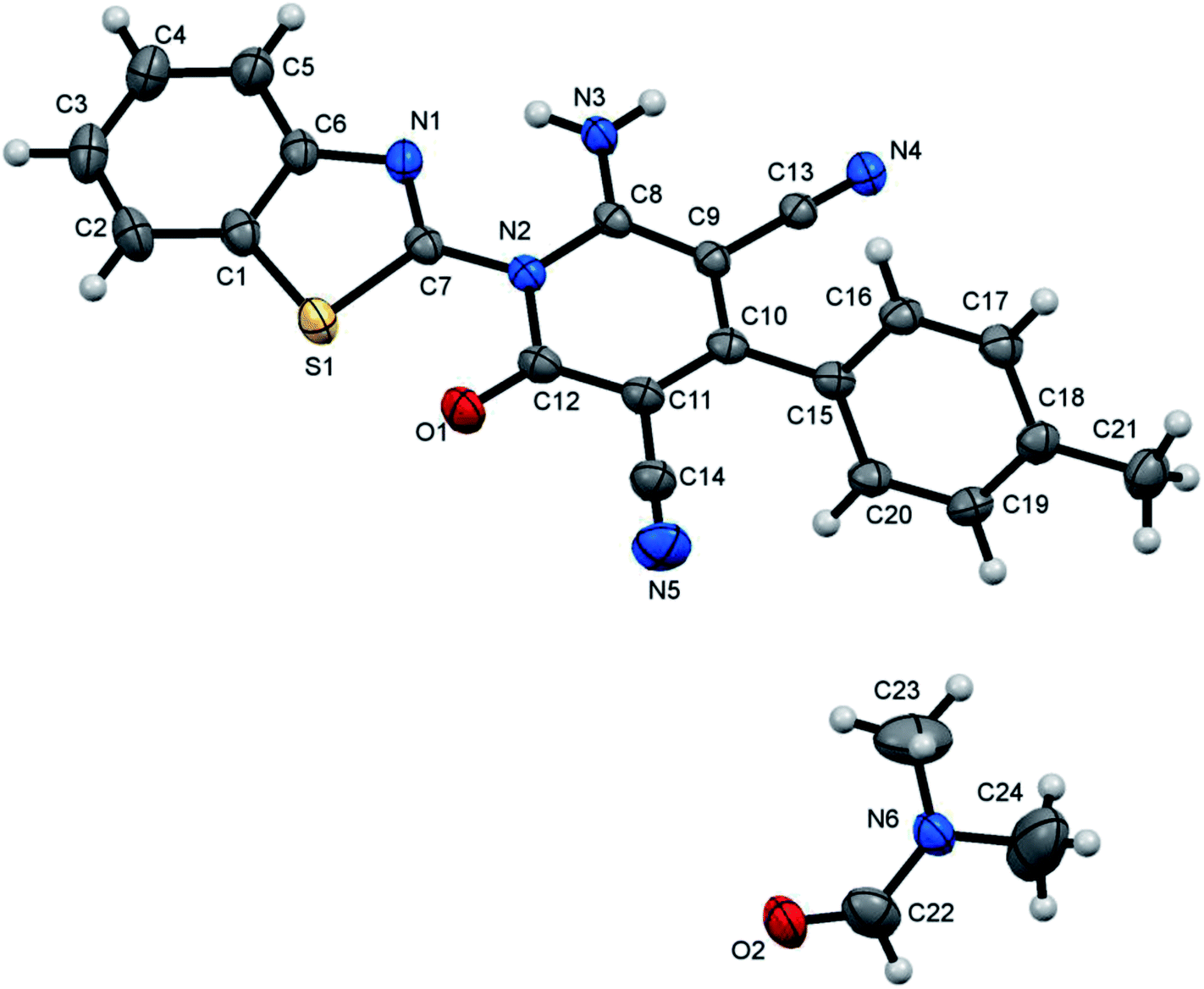
| | Fig. 1 X-ray single crystal structure determined for compound 7a. | |
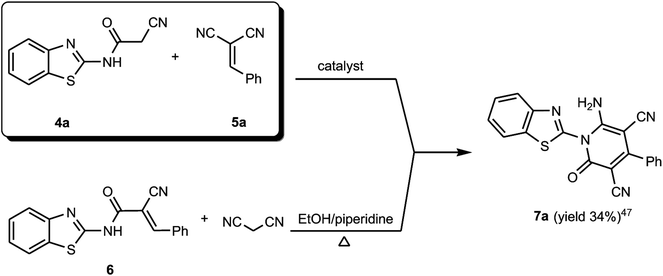 |
| | Scheme 2 Synthesis of benzothiazol-2-yl-pyridine derivative 7a. | |
Then, to estimate the influence of catalyst loading on this procedure, the model reaction (Scheme 2) was conducted by applying different amounts of L-proline. The results depicted in Table 2 confirmed that 10 mol% L-proline is the preferable molar ratio, which afforded the coveted product in excellent yield of 95%, Table 2 (entry 6). When the molar ratio of L-proline was increased more than 10 mol%, the yield diminished, Table 2 (entries 1–5). Moreover, when the loading of the catalyst molar ratio was less than 10 mol%, the desired product was obtained in a slightly lower yield than that with 10 mol% of L-proline, Table 2 (entries 7 and 8).
Table 2 Optimization mol% of L-proline for the synthesis of 7aa
| Entrya |
mol% of L-proline |
Method |
Yield (%) |
| The mixture of N-(benzothiazol-2-yl)-2-cyanoacetamide (4a) (5 mmol) and 2-benzylidenemalononitrile (5a) (5 mmol), in ethanol (20 mL) and L-proline was sonicated at 80 °C, 110 W. |
| 1 |
100 |
US ))) |
27 |
| 2 |
50 |
US ))) |
38 |
| 3 |
30 |
US ))) |
53 |
| 4 |
20 |
US ))) |
69 |
| 5 |
15 |
US ))) |
82 |
| 6 |
10 |
US ))) |
95 |
| 7 |
5 |
US ))) |
93 |
| 8 |
2 |
US ))) |
88 |
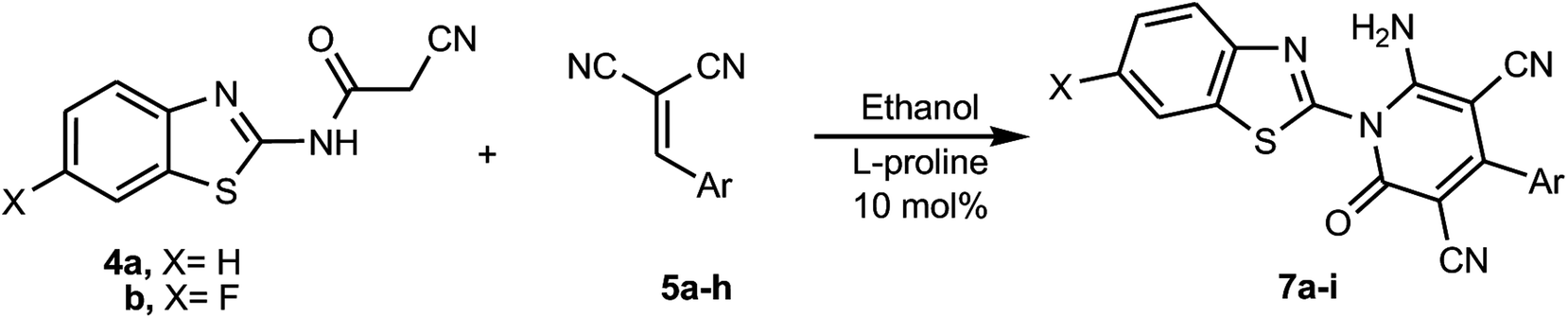
With the optimized green methodology in hand, we investigated the scope of this method for the synthesis of the benzothiazol-2-yl-pyridine derivatives 7a–i. This methodology was found to be applicable for the synthesis of a wide range of pyridone derivatives 7a–i through the reaction of N-(benzothiazol-2-yl)-2-cyanoacetamide (4a) and 2-cyano-N-(6-fluorobenzothiazol-2-yl)acetamide (4b) with an assortment of arylidenemalononitriles 5a–h in ethanol containing 10 mol% of L-proline under ultrasonic irradiation. It afforded the corresponding 6-amino-1-benzothiazol-2-yl-2-oxo-4-arylpyridine-3,5-dicarbonitrile derivatives 7a–i in excellent yields, as summarized in Scheme 3 and Table 3. In a similar manner to that used to characterize 7a, the structures of 7b–i were also determined from their full spectral data, which included 1H and 13C NMR spectra, IR, MS and HRMS and representative examples by X-ray crystallographic analysis for compounds 7b, 7d and 7i, as depicted in Fig. 2–4.
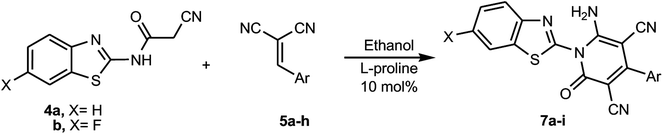 |
| | Scheme 3 Synthesis of 6-amino-1-benzothiazol-2-yl-2-oxo-4-arylpyridine-3,5-dicarbonitrile derivatives 7a–i. | |
Table 3 The reactions of N-(benzothiazol-2-yl)-2-cyanoacetamide derivatives 4 with arylidenemalononitriles 5a
| Entry |
Products |
X |
Ar |
Yield (%)-mp (°C) |
Reported yield (%)-mp (°C) |
| Reaction conditions: the mixture of N-(benzothiazol-2-yl)-2-cyanoacetamide derivatives 4 (5 mmol) and arylidenemalononitriles 5 (5 mmol) in ethanol (20 mL) and L-proline (10 mol%) was sonicated at 80 °C, 110 W for 20 min. |
| 1 |
7a |
H |
C6H5 |
95/300–302 |
34/299–302 |
| 2 |
7b |
H |
4-Me–C6H4 |
91/300–301 |
17/299–301 |
| 3 |
7c |
H |
4-MeO–C6H4 |
92/above 300 |
17/308–311 |
| 4 |
7d |
H |
4-Cl–C6H4 |
87/above 300 |
25/315–317 |
| 5 |
7e |
H |
4-NO2–C6H4 |
90/above 300 |
32/306–309 |
| 6 |
7f |
H |
3-NO2–C6H4 |
89/above 300 |
— |
| 7 |
7g |
H |
2,4-MeO–C6H4 |
96/above 300 |
— |
| 8 |
7h |
H |
C4H3S |
90/above 300 |
— |
| 9 |
7i |
F |
4-Cl–C6H4 |
94/above 300 |
— |
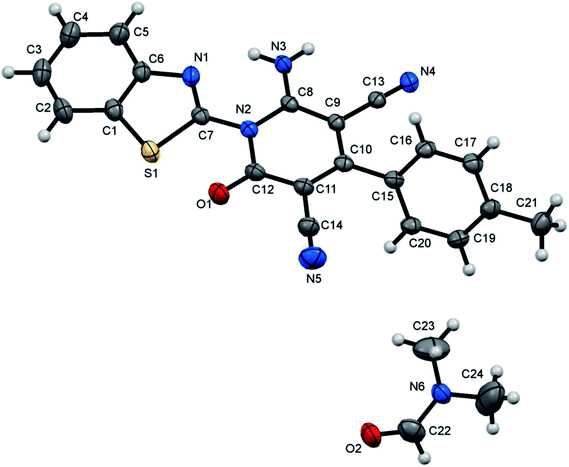 |
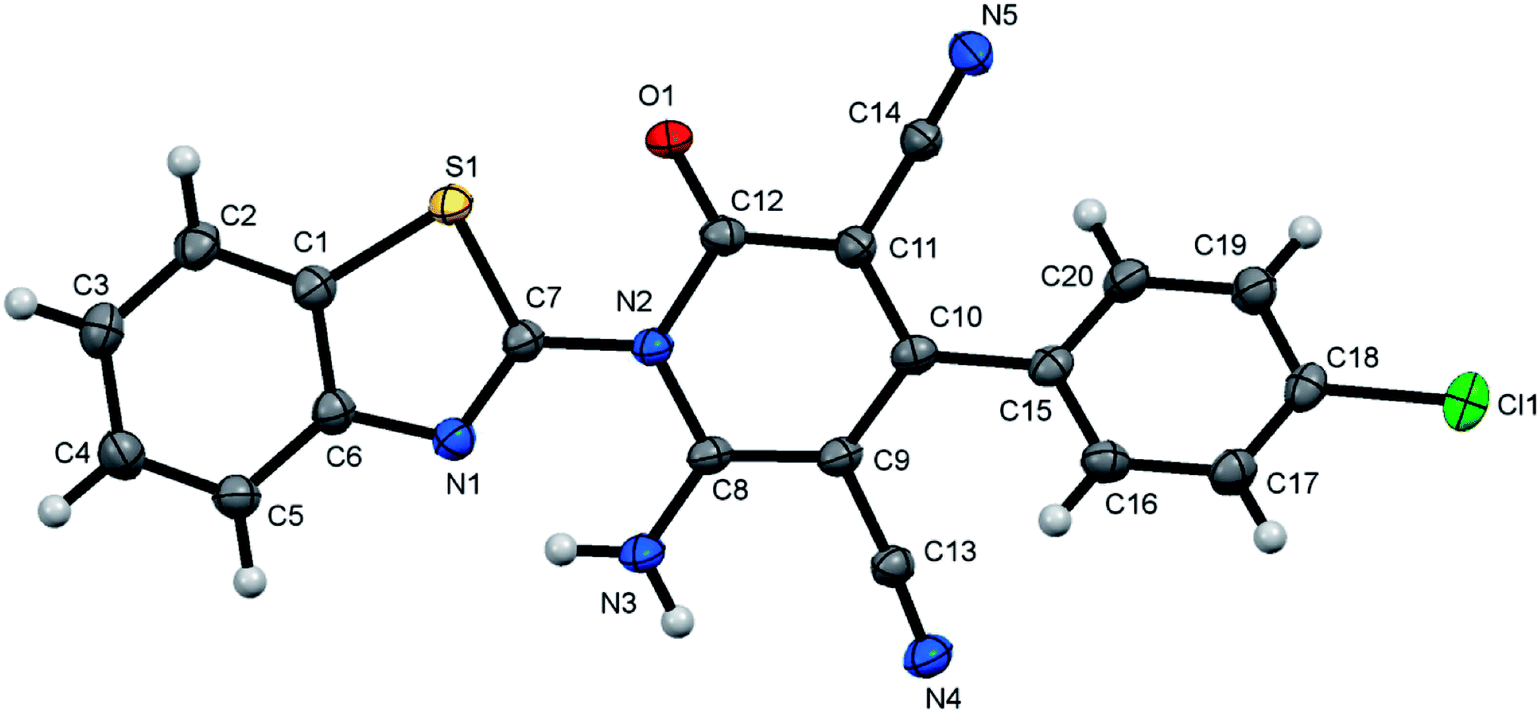
| | Fig. 2 X-ray single crystal structure determined for compound 7b. | |
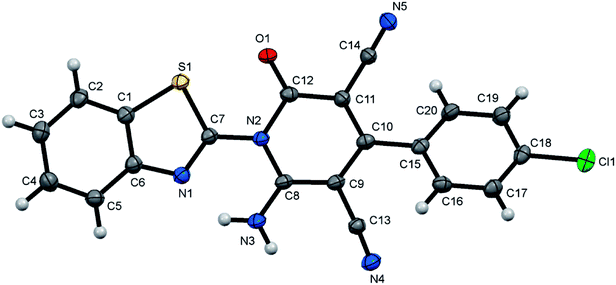 |
| | Fig. 3 X-ray single crystal structure determined for compound 7d. | |
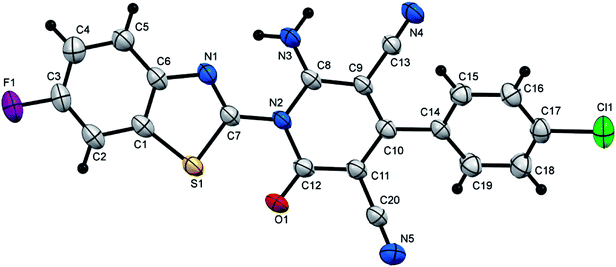 |
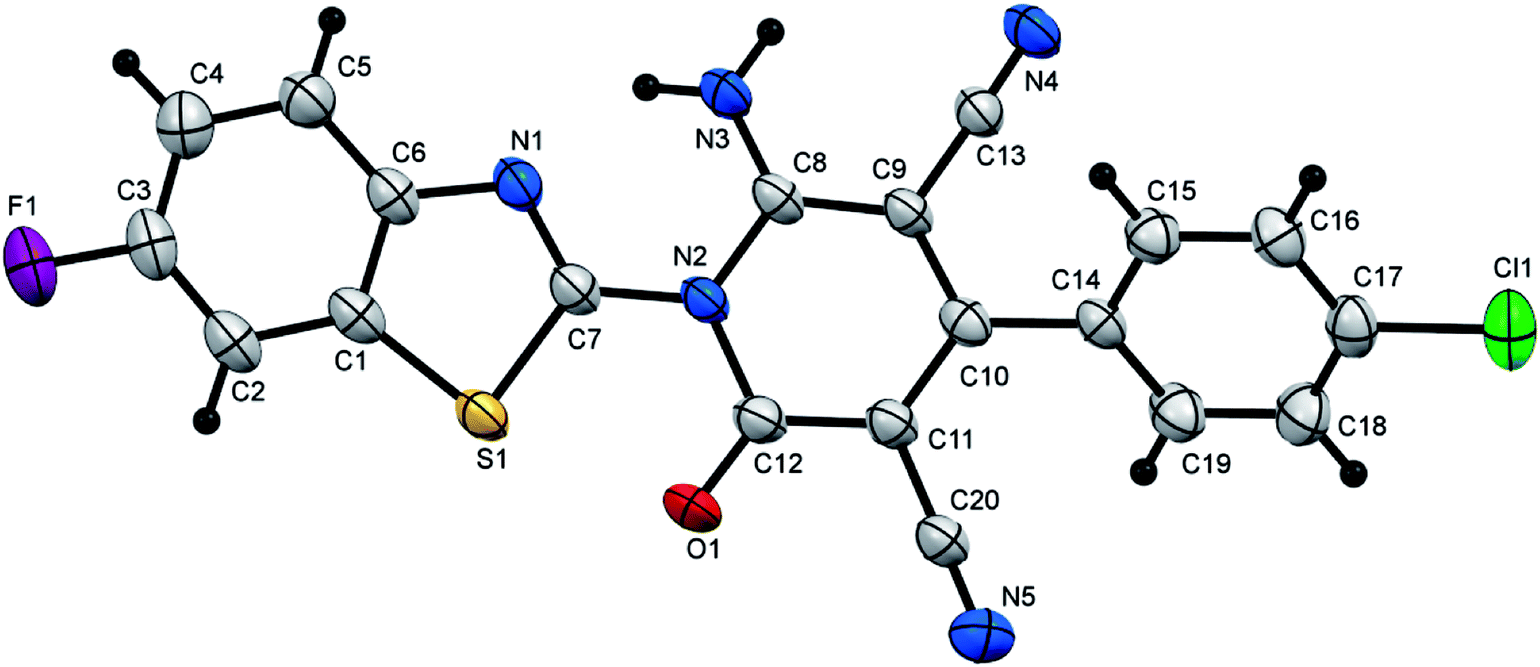
| | Fig. 4 X-ray single crystal structure determined for compound 7i. | |
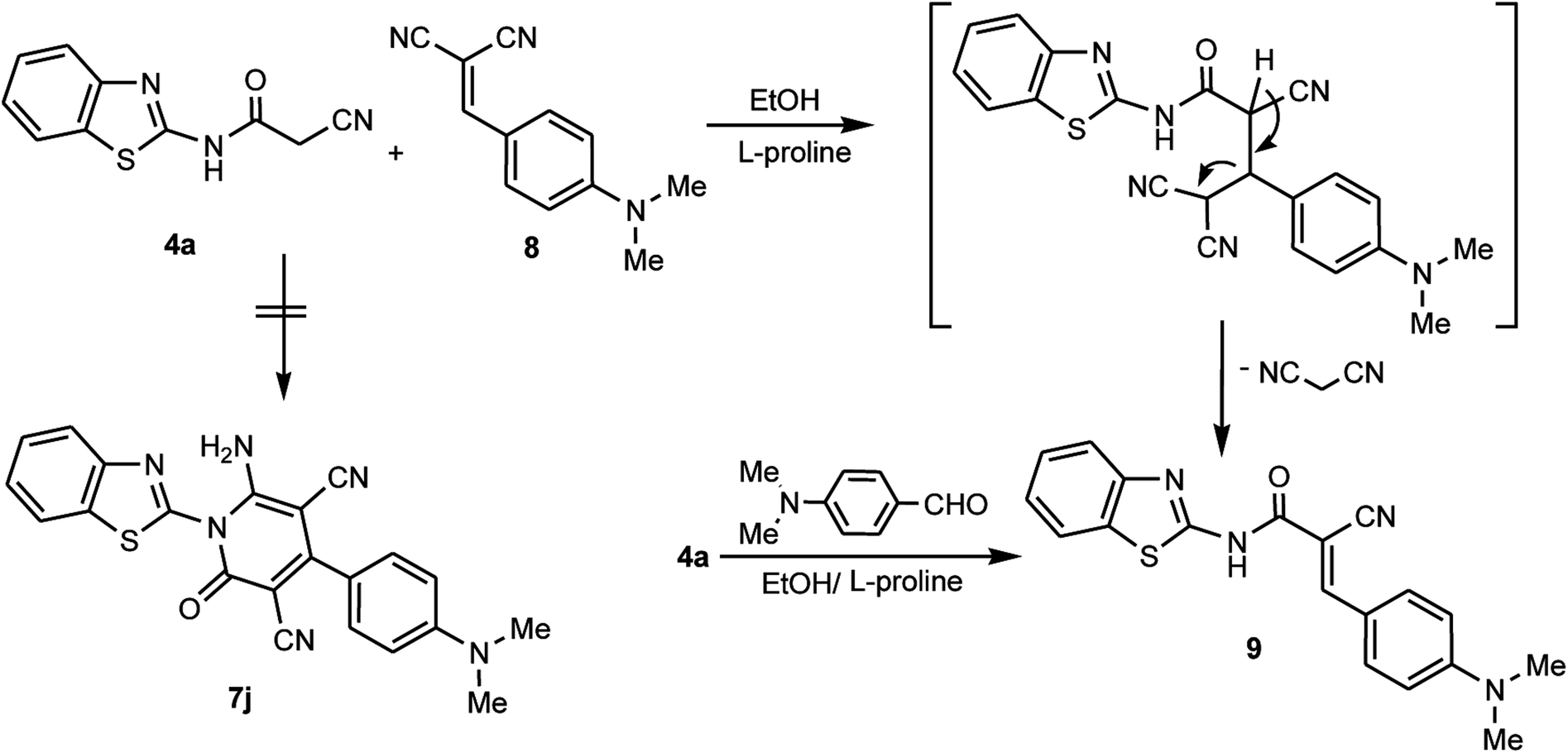
In contrast to the previously noted chemistry, the benzothiazolylcyanoacetamide derivative 4a was reacted with 2-(4-dimethylaminobenzylidene)malononitrile 8 to afford the arylidene derivative 9 and not the corresponding pyridone derivative 7j. This may be attributed to the presence of a para-dimethylamino substituent in the aromatic moiety of the aldehyde, which is a donor-aromatic–acceptor structure. This facilitates the elimination of the malononitrile moiety rather than the cycloaddition of the NH moiety at CN, which is consistent with an earlier study.44 The arylidene derivative 9 could also be obtained through the condensation reaction between the cyanoacetamide derivative 4a and 4-dimethylaminobenzaldehyde in EtOH/L-proline (Scheme 4).
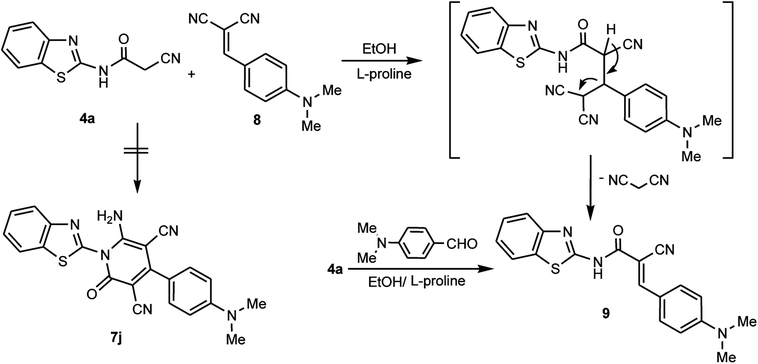 |
| | Scheme 4 Reaction of 4a with 2-(4-dimethylaminobenzylidene)malononitrile 8. | |
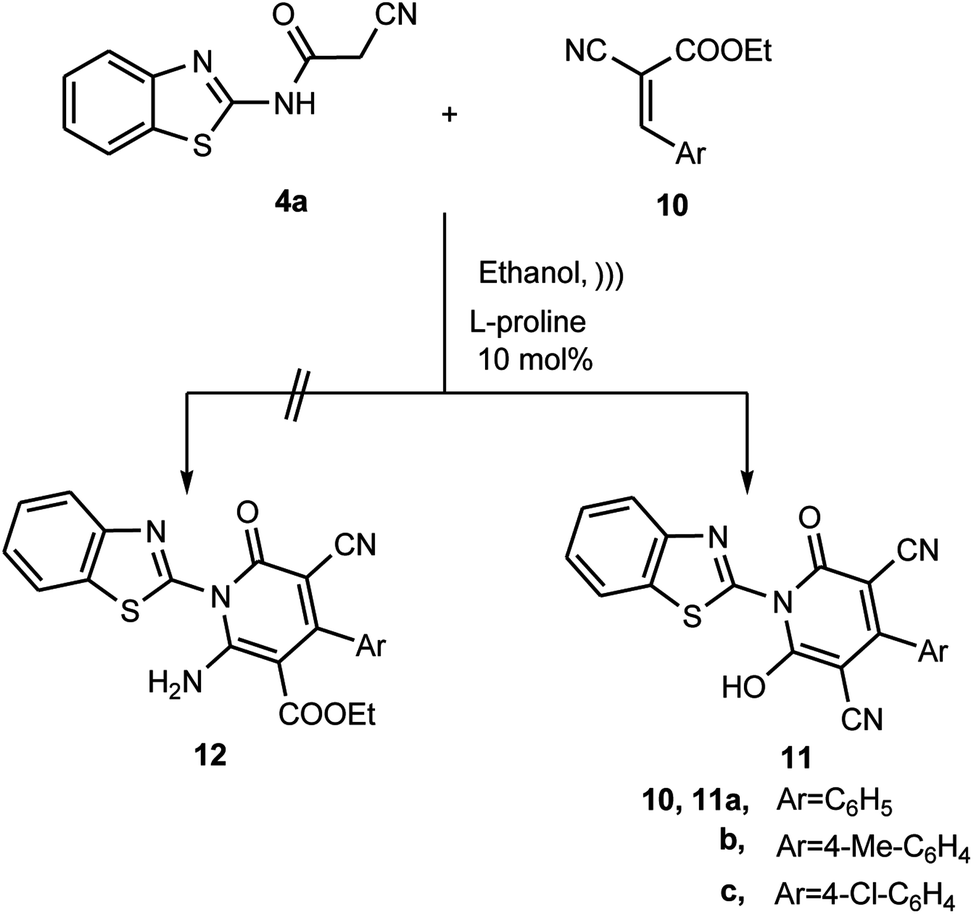
Furthermore, to expand and generalize this L-proline catalyzed Michael-type addition reaction, we investigated the reaction of the N-(benzothiazol-2-yl)-2-cyanoacetamide (4a) and ethyl-2-cyano-3-arylacrylate derivatives 10a–c. The sonication of N-(benzothiazol-2-yl)-2-cyanoacetamide (4a) with ethyl-2-cyano-3-arylacrylate derivatives 10a–c in ethanolic solution containing L-proline afforded the product, where structure 11 was assigned and not 12 based on the different spectrometric analyses (Scheme 5). For example, the 1H NMR spectrum of the reaction product was devoid of the triplet and quartet signal set, which is due to the ethyl ester. Also, it revealed sets of multiplets in the region δ ≈ 7.4–8.7 ppm due to the aromatic protons signals besides the singlet signal at δ ≈ 13.3 ppm, which is due the hydroxyl group proton. In addition, all the data obtained from the IR, MS, HRMS and 13C NMR spectra are consistent with the hydroxypyridone structure 11.
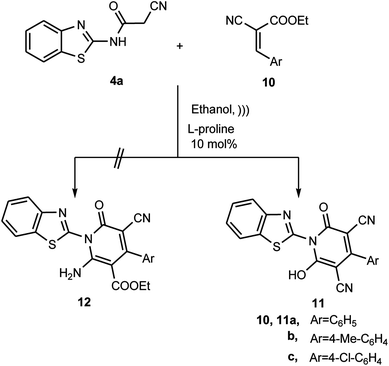 |
| | Scheme 5 Reaction of 4a with ethyl-2-cyano-3-arylacrylates 10a–c. | |
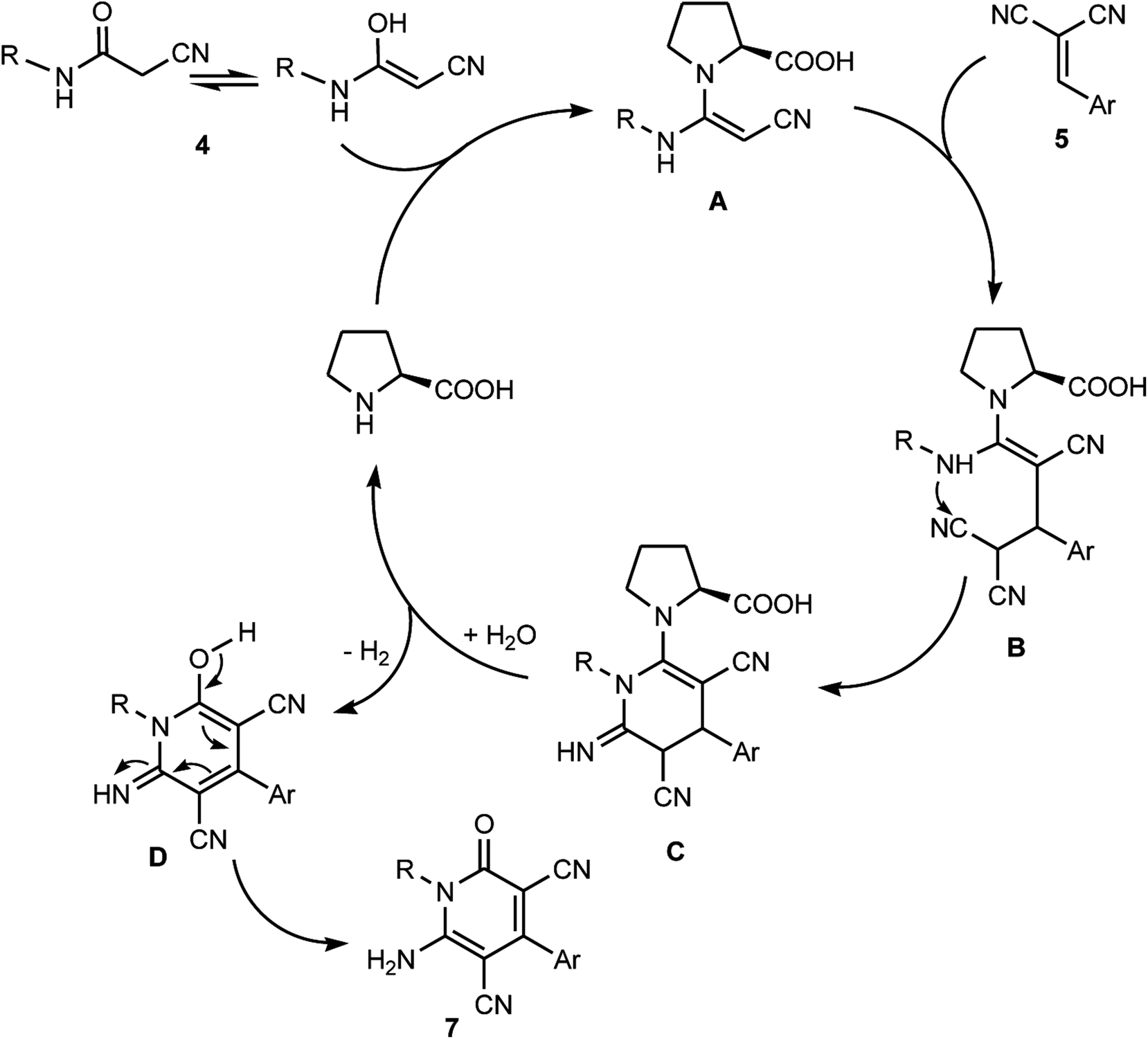
Remarkably, various factors make this Michael addition protocol for the synthesis of multisubstituted pyridone derivatives highly appealing. These include the uncommonly high efficiency of the reaction, which was manifested in the excellent yield of the reaction product in comparison with the reported procedure for the assembly of 7, the employment of ultrasonic irradiation as a clean, sustainable energy source, simple workup procedure and moderate reaction conditions. The plausible mechanistic pathway for the formation of the multisubstituted pyridone derivatives 7 is presented in Scheme 6. It is believed that the enamine adduct A was firstly formed between the cyanoacetamide 4 and L-proline, then the last intermediate underwent nucleophilic addition to 2-arylidenemalononitriles 5a–h to generate the non-isolable intermediate B, which underwent intramolecular cyclization via the attack of the NH moiety at CN to form the pyridine-imine intermediate C. In the presence of water and by losing one hydrogen molecule, the intermediate D was liberated, which eventually rearranged to afford the corresponding pyridone derivatives 7 (Scheme 6).
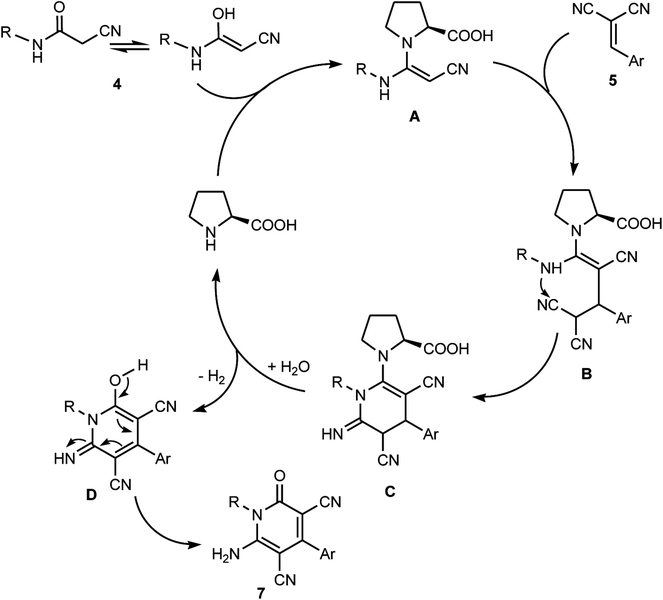 |
| | Scheme 6 Plausible mechanistic pathway for the formation of polysubstituted pyridone derivatives 7. | |
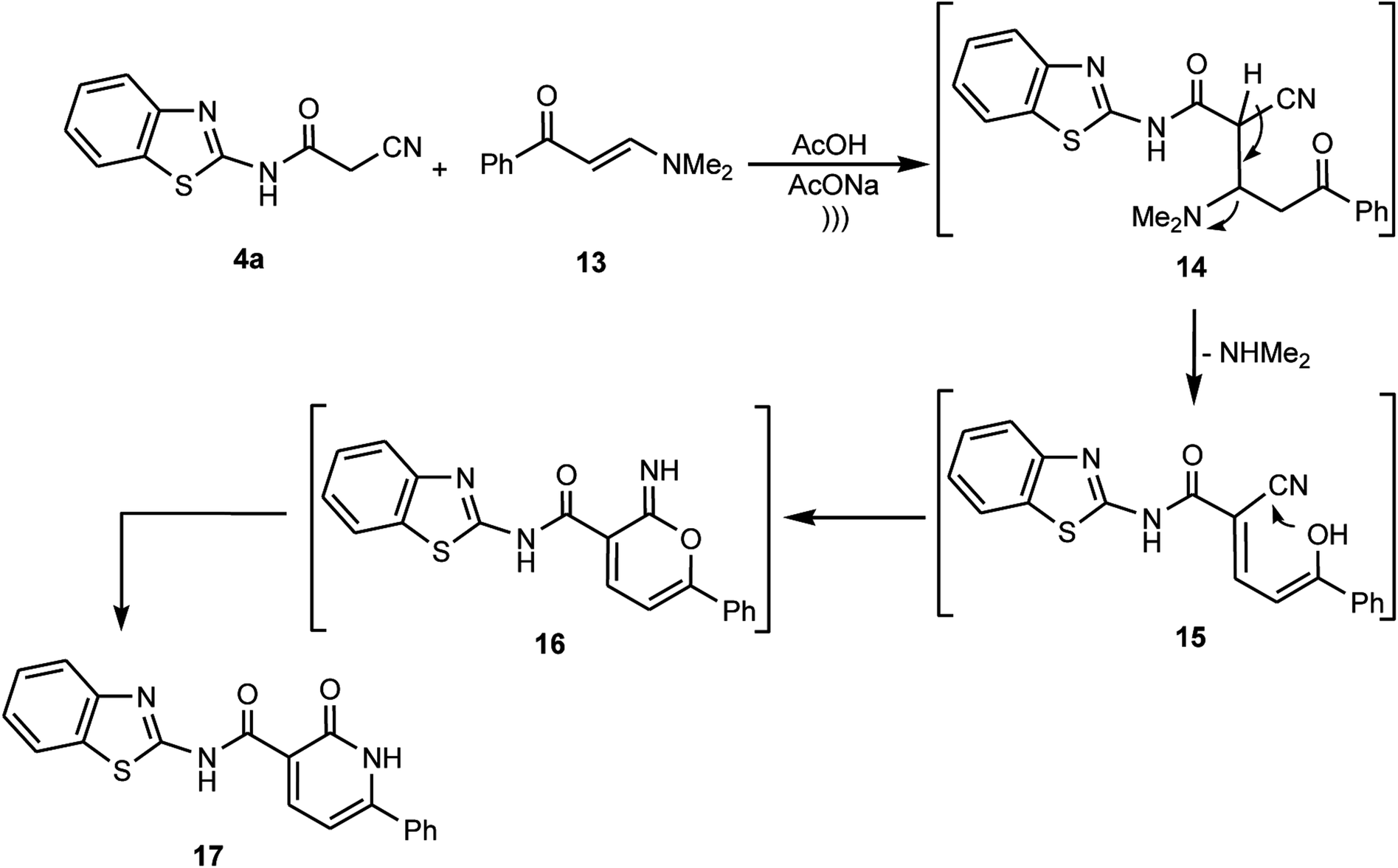
Furthermore, the cyanoacetamide 4a was reacted with the enaminone 13 in a buffered solution of acetic acid containing anhydrous sodium acetate under ultrasonic irradiation to afford a product whose structure was assigned as pyridone 17 based on the obtained results from different spectrometric analyses. For example, the 1H NMR spectrum exhibited two NH signals due to the amide NH and the pyridone NH at δ ≈ 13.22 and 13.59 ppm, respectively, in addition to two doublets at δ ≈ 6.96 and 8.54 ppm for the two adjacent C–H protons in the pyridone ring, plus sets of multiplets in the region δ ≈ 7.96–8.55 ppm, which are due to the aromatic proton signals. Also, MS and HRMS showed the exact molecular weight corresponding the pyridone structure 17. Moreover, the data obtained from the 13C NMR spectra clarify the presence of two signals due to 2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups. Thus, we postulate that cyanoacetamide 4a underwent nucleophilic addition to the enaminone 13 to give the non-isolable adduct 14, which by elimination of the dimethylamine moiety afforded the intermediate 15, which underwent intramolecular cyclization via the attack of the OH moiety at CN to form the pyrane-imine intermediate 16, and eventually rearranged to afford the corresponding pyridone derivative 17 (Scheme 7).
O groups. Thus, we postulate that cyanoacetamide 4a underwent nucleophilic addition to the enaminone 13 to give the non-isolable adduct 14, which by elimination of the dimethylamine moiety afforded the intermediate 15, which underwent intramolecular cyclization via the attack of the OH moiety at CN to form the pyrane-imine intermediate 16, and eventually rearranged to afford the corresponding pyridone derivative 17 (Scheme 7).
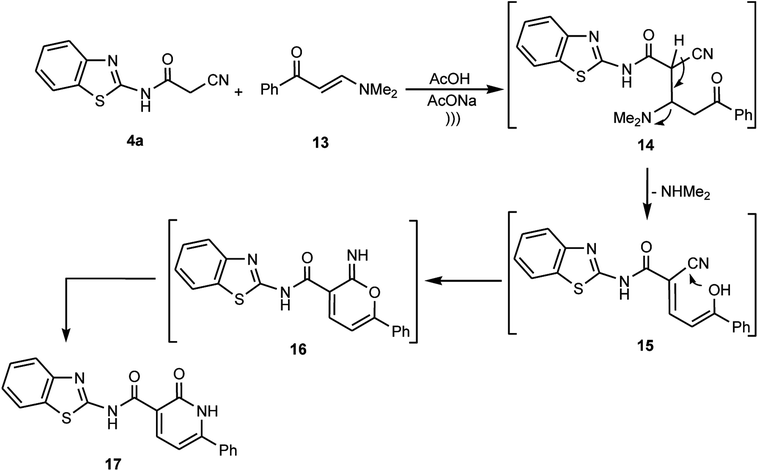 |
| | Scheme 7 Reaction of cyanoacetamide 4a with the enaminone 13. | |
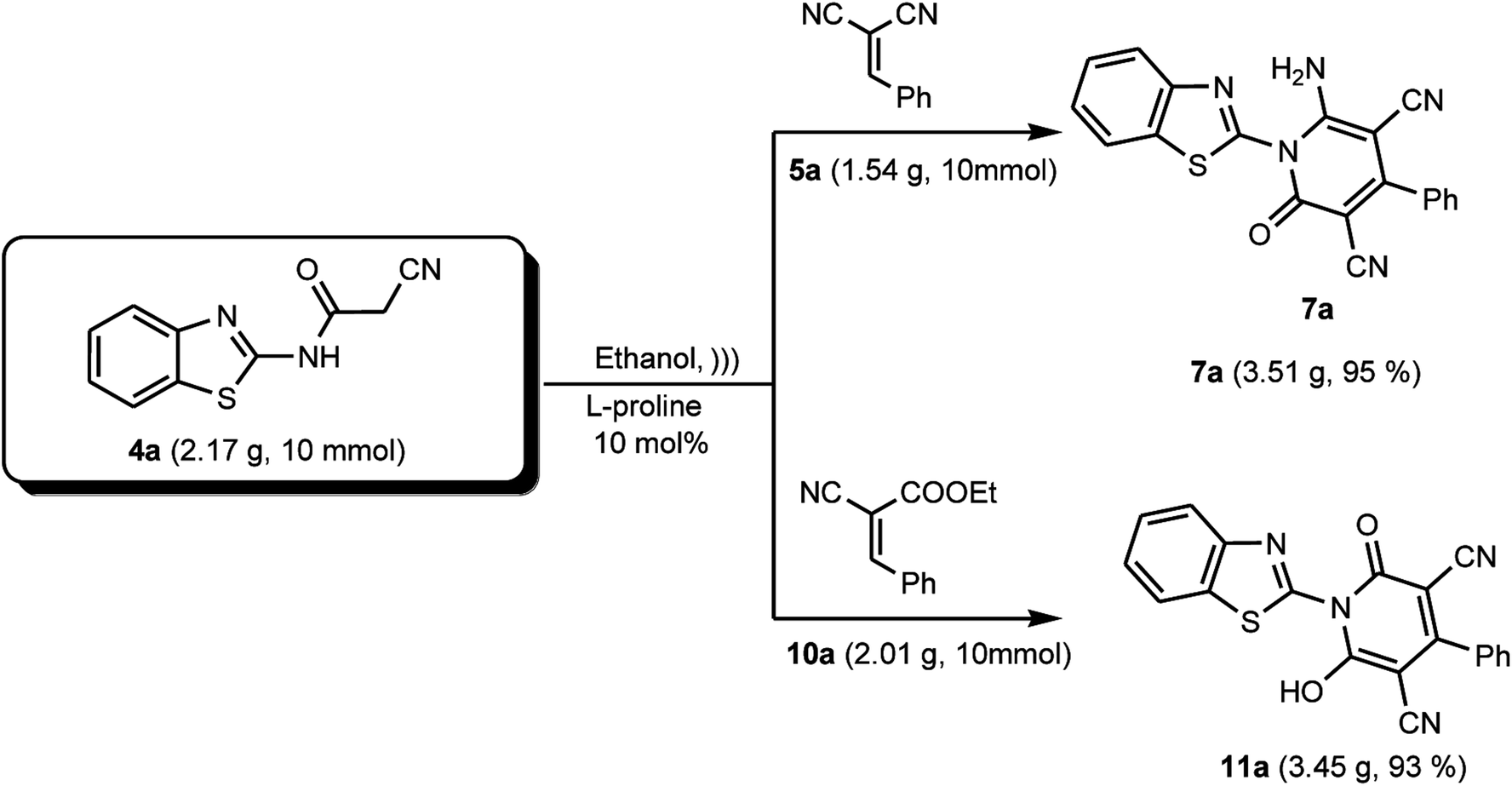
Finally, with the aim to evaluate the scalability of the L-proline catalyzed research protocol reported in this study, we conducted this reaction on a gram-scale under the optimized reaction conditions, which furnished the desired products 7a and 11a, as representative examples, in excellent yields of 95% and 93%, respectively (Scheme 8).
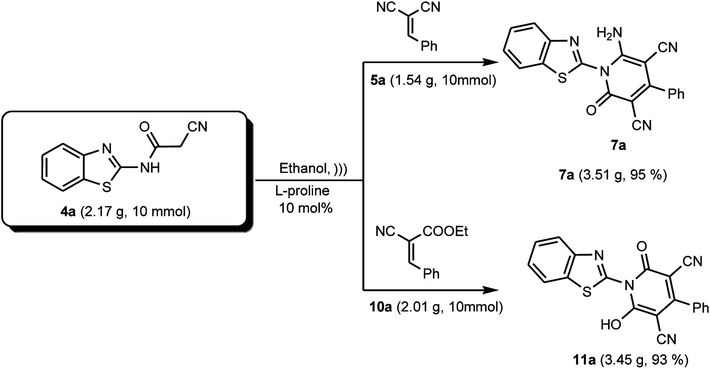 |
| | Scheme 8 Scaled-up synthesis of compounds 7a and 11a. | |
Conclusion
In the present research, L-proline was successfully employed as an environmentally precious organocatalyst for the synthesis of a variety of polysubstituted pyridone systems incorporating the benzothiazole moiety. In combination with the use of ultrasonic irradiation as a sustainable energy source for conducting, this synthesis makes it valuable in comparison with the traditional methods. The current protocol has several unrivaled merits including excellent yields of reactions that may significantly participate in the commercialization of drugs. In addition, it utilizes a cost-effective catalyst and has easy work-up and purification. Also, in terms of efficiency, the use of ultrasonic irradiation for this process proved to be superior.
Experimental
General
Melting points were recorded on a Griffin melting point apparatus and are uncorrected. IR spectra were recorded using KBr disks on a Jasco FT-IR-6300 spectrophotometer. 1H NMR (400 MHz or 600 MHz) and 13C NMR (100 MHz or 150 MHz) spectra were recorded at 25 °C using DMSO-d6 as the solvent with TMS as the internal standard on a Bruker DPX 400 or 600 super-conducting NMR spectrometer. Chemical shifts are reported in ppm. Low-resolution electron impact mass spectra [MS (EI)] and high-resolution electron impact mass spectra [HRMS (EI)] were obtained using a high resolution GC-MS (DFS) thermo spectrometer at 70.1 eV and a magnetic sector mass analyzer. The reactions were followed and the homogeneity of the prepared compounds verified using thin layer chromatography (TLC). X-ray crystal structures were determined using a Rigaku R-AXIS RAPID diffractometer and Bruker X8 Prospector, and the collection of single crystal data was done at room temperature using Cu-Kα radiation. The structures were solved using direct methods and expanded using Fourier techniques. Non-hydrogen atoms were refined anisotropically. The structures were solved and refined using the Bruker SHELXTL software package (structure solution program - SHELXS-97 and refinement program – SHELXL-97).50 Data were corrected for absorption effects using the multi-scan method (SADABS). Sonication was performed in an MKC6, Guyson ultrasonic bath (Model-MKC6, operating frequency 38 kHz ± 10% and an output power of 110 W) with a digital timer (6 s to 100 min) and heater, which allowed the solution to be heated from 20 °C to 80 °C in 1 °C increments. The inner tank dimensions were 150 × 300 × 150 mm (length × width × depth) with a fluid capacity of 6 L. The N-(benzothiazol-2-yl)-2-cyanoacetamide derivatives 4a and b were prepared according to a literature procedure with slight modification.51
General procedure for the preparation of the cyanoacetamide derivatives 4a and b
A solution of cyanoacetic acid (1) (10 mmol) in Ac2O (10 mL) was sonicated in an MKC6, Guyson ultrasonic bath (Model-MKC6, operating frequency 38 kHz ± 10% and output power of 110 W) for about 1 min until the cyanoacetic acid was completely dissolved. Then, the appropriate 2-aminobenzothiazole derivatives 3a and b (10 mmol) were added to the reaction mixture, and sonication was continued for a further 5 min at 80 °C. The reaction mixture was left to cool to room temperature. The solid product formed was collected by filtration and recrystallized from the appropriate solvent and identified as (4a and b, respectively).
N-(Benzothiazol-2-yl)-2-cyanoacetamide (4a)51. Recrystallized from EtOH/dioxane mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as creamy white crystals, yield: 2.15 g (99%), mp 220–221 °C; IR (KBr): 3288 (NH), 2261 (CN), 1688 cm−1 (CO); 1H NMR (DMSO-d6): δ = 4.14 (s, 2H, CH2), 7.29–8.01 (m, 4H, Ar–H) and 12.74 (br, 1H, NH); 13C NMR (DMSO-d6): δ = 27.3 (CH2), 116.0 (CN), 121.5, 122.6, 124.7, 127.1, 132.3, 149.1, 158.4 (Ar–C) and 163.8 (CO); MS (EI): m/z (%) 217 (M+, 31.93), 218 (M+ + 1, 4.6). HRMS (EI): m/z calcd for C10H7N3OS (M+) 217.0304, found 217.0304.
:1) as creamy white crystals, yield: 2.15 g (99%), mp 220–221 °C; IR (KBr): 3288 (NH), 2261 (CN), 1688 cm−1 (CO); 1H NMR (DMSO-d6): δ = 4.14 (s, 2H, CH2), 7.29–8.01 (m, 4H, Ar–H) and 12.74 (br, 1H, NH); 13C NMR (DMSO-d6): δ = 27.3 (CH2), 116.0 (CN), 121.5, 122.6, 124.7, 127.1, 132.3, 149.1, 158.4 (Ar–C) and 163.8 (CO); MS (EI): m/z (%) 217 (M+, 31.93), 218 (M+ + 1, 4.6). HRMS (EI): m/z calcd for C10H7N3OS (M+) 217.0304, found 217.0304.
2-Cyano-N-(6-fluorobenzothiazol-2-yl)acetamide (4b). Recrystallized from EtOH/dioxane mixture (1:1) as yellowish white crystals, yield: 2.3 g (98%), mp 193–194 °C; IR (KBr): 3305 (NH), 2258 (CN), 1685 cm−1 (CO); 1H NMR (DMSO-d6): δ = 4.12 (s, 2H, CH2), 7.28–7.94 (m, 3H, Ar–H) and 12.65 (brs, 1H, NH); 13C NMR (DMSO-d6): δ = 26.38 (CH2), 108.17, 108.35 (d, 2JCF = 27 Hz), 114.34, 114.50 (d, 2JCF = 24 Hz) 115.14 (CN), 121.87, 121.94 (d, 3JCF = 10.5 Hz), 132.76, 132.83 (d, 3JCF = 10.5 Hz), 145.12, 157.56, 158.08, 159.68 (Ar–C) and 162.99 (CO); MS (EI): m/z (%) 235 (M+, 64.99), 236 (M+ + 1, 7.94). HRMS (EI): m/z calcd for C10H6FN3OS (M+) 235.0210, found 235.0210.
General procedure for the preparation of 6-amino-1-(benzothiazol-2-yl)-2-oxo-4-arylpyridine-3,5-dicarbonitrile derivatives 7a–i
Mixtures of cyanoacetamide derivatives 4a and b (5 mmol), arylidene malononitrile 5a–h (5 mmol) and L-proline (0.5 mmol, 10 mol%) in ethanol (20 mL) were sonicated in an MKC6, Guyson ultrasonic bath (Model-MKC6, operating frequency 38 kHz ± 10% and output power of 110 W) for 20 minutes at 80 °C. The reaction was followed by TLC and continued until the starting substrates were completely consumed, then left to cool to room temperature. In each case, the solid product formed after cooling to room temperature was separated by filtration, washed with ethanol, dried and recrystallized from the appropriate solvent to give the pure products 7a–i.
6-Amino-1-(benzothiazol-2-yl)-2-oxo-4-phenyl-1,2-dihydropyridine-3,5-dicarbonitrile(7a)47. Creamy white crystals, yield: 1.75 g (95%), mp 300–302 °C; IR (KBr): ν/cm−1 3321 (NH2), 2210 (CN), 1677 (CO); 1H NMR (DMSO-d6): δ = 7.59–7.65 (m, 7H, Ar–H), 8.12 (d, J = 8.4 Hz, 1H, Ar–H), 8.22 (d, J = 8.4 Hz, 1H, Ar–H) and 8.71 ppm (s, 2H, NH2); 13C NMR (DMSO-d6): δ = 75.84 (C5), 87.68 (C3), 114.53, 114.96, 122.10, 123.23, 125.97, 126.10, 127.39, 128.20, 129.99, 134.21, 136.34, 149.12, 154.09, 156.50, 158.67 and 162.03 ppm (Ar–C and CO); MS (EI): m/z (%) 370 (M+ + 1, 26.84), 369 (M+, 100); HRMS (EI): m/z calcd for C20H11N5OS (M+) 369.0679, found 369.0679. Crystal data, moiety formula: C20H11N5OS, M = 369.40, triclinic, a = 6.43370(10) Å, b = 11.1793(3) Å, c = 12.2318(3) Å, V = 872.02(3) Å3, α = 93.9110(10)°, β = 94.6660(10)°, γ = 94.2320(10)°, space group: P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, Dcalc = 1.407 g cm−3, no. of reflection measured = 14792, θmax = 66.44°, R1 = 0.0347 (CCDC 1858431).52
, Z = 2, Dcalc = 1.407 g cm−3, no. of reflection measured = 14792, θmax = 66.44°, R1 = 0.0347 (CCDC 1858431).52
6-Amino-1-(benzothiazol-2-yl)-2-oxo-4-p-tolyl-1,2-dihydropyridine-3,5-dicarbonitrile (7b)47. Creamy white crystals, yield: 1.74 g (91%), mp 300–301 °C; IR (KBr): ν/cm−1 3319 (NH2), 2224, 2211 (CN), 1674 (CO); 1H NMR (DMSO-d6): δ = 2.42 (s, 3H, CH3), 7.42–7.63 (m, 6H, Ar–H), 8.13 (d, J = 7.2 Hz, 1H, Ar–H), 8.25 (d, J = 7.2 Hz, 1H, Ar–H) and 8.76 ppm (s, 2H, NH2); 13C NMR (DMSO-d6): δ = 20.99 (CH3), 75.83 (C5), 87.51 (C3), 115.37, 115.94, 122.83, 123.69, 126.27, 126.42, 127.88, 129.22, 131.54, 137.35, 140.43, 149.98, 154.29, 156.88, 159.33 and 162.51 ppm (Ar–C and CO); MS (EI): m/z (%) 384 (M+ + 1, 25.97), 383 (M+, 100); HRMS (EI): m/z calcd for C21H13N5OS (M+) 383.0835, found 383.0835. Crystal data, moiety formula: C21H13N5OS, M = 383.43, sum formula: C24H20N6O2S, M = 456.52, triclinic, a = 9.3107(2) Å, b = 10.8553(2) Å, c = 12.3036(3) Å, V = 1117.81(4) Å3, α = 71.3360(10)°, β = 71.5960(10)°, γ = 84.7560(10)°, space group: P, Z = 2, Dcalc = 1.356 g cm−3, no. of reflection measured = 13761, θmax = 66.66°, R1 = 0.0725 (CCDC 1858432).53
6-Amino-1-(benzothiazol-2-yl)-4-(4-methoxyphenyl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (7c)47. Creamy white crystals, yield: 1.84 g (92%), mp above 300 °C; IR (KBr): ν/cm−1 3311 (NH2), 2214 (CN), 1677 (CO); 1H NMR (DMSO-d6): δ = 3.87 (s, 3H, OCH3), 7.16 (d, J = 8.0 Hz, 2H, Ar–H), 7.56–7.65 (m, 4H, Ar–H), 8.13 (d, J = 7.2 Hz, 1H, Ar–H), 8.25 (d, J = 7.2 Hz, 1H, Ar–H) and 8.76 ppm (s, 2H, NH2); 13C NMR (DMSO-d6): δ = 55.37 (OCH3), 75.82 (C5), 87.40 (C3), 114.03, 115.56, 116.14, 122.81, 123.69, 126.29, 126.44, 129.85, 137.32, 149.96, 154.33, 156.89, 159.41, 160.98 and 162.14 ppm (Ar–C and CO); MS (EI): m/z (%) 400 (M+ + 1, 27.12), 399 (M+, 100); HRMS (EI): m/z calcd for C21H13N5O2S (M+) 399.0784, found 399.0784.
6-Amino-1-(benzothiazol-2-yl)-4-(4-chlorophenyl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (7d)47. Creamy white crystals, yield: 1.75 g (87%), mp above 300 °C; IR (KBr): ν/cm−1 3329 (NH2), 2224, 2210 (CN), 1678 (CO); 1H NMR (DMSO-d6): δ = 7.58–7.66 (m, 4H, Ar–H), 7.70 (d, J = 7.6 Hz, 2H, Ar–H), 8.13 (d, J = 7.6 Hz, 1H, Ar–H), 8.26 (d, J = 7.6 Hz, 1H, Ar–H) and 8.82 ppm (s, 2H, NH2); 13C NMR (DMSO-d6): δ = 75.87 (C5), 87.65 (C3), 115.14, 115.70, 122.86, 123.72, 126.30, 126.44, 128.91, 129.87, 133.31, 135.35, 137.36, 150.02, 154.14, 156.85, 159.17 and 161.33 ppm (Ar–C and CO); MS (EI): m/z (%) 405 (M+ + 2, 37.45), 404 (M+ + 1, 51.95), 403 (M+, 100); HRMS (EI): m/z calcd for C20H10ClN5OS (M+) 403.0289, found 403.0288. Crystal data, moiety formula: C20H10ClN5OS, M = 403.84, triclinic, a = 6.4441(8) Å, b = 12.311(2) Å, c = 12.747(2) Å, V = 873.1(2) Å3, α = 61.344(5)°, β = 80.031(6)°, γ = 83.005(6)°, space group: P, Z = 2, Dcalc = 1.536 g cm−3, no. of reflection measured = 8558, θmax = 54.9°, R1 = 0.0349 (CCDC 1858433).54
6-Amino-1-(benzothiazol-2-yl)-4-(4-nitrophenyl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (7e)47. Yellow crystals, yield: 1.86 g (90%), mp above 300 °C; IR (KBr): ν/cm−1 3297 (NH2), 2216 (CN), 1679 (CO); 1H NMR (DMSO-d6): δ = 7.59–7.67 (m, 2H, Ar–H), 7.90 (d, J = 8.0 Hz, 2H, Ar–H), 8.14 (d, J = 7.6 Hz, 1H, Ar–H), 8.26 (d, J = 7.6 Hz, 1H, Ar–H), 8.47 (d, J = 8.0 Hz, 2H, Ar–H) and 8.92 ppm (s, 2H, NH2); 13C NMR (DMSO-d6): δ = 75.83 (C5), 87.69 (C3), 115.00, 115.55, 122.97, 123.84, 124.10, 126.43, 126.56, 129.72, 137.46, 140.83, 148.67, 150.14, 154.11, 156.98, 159.14 and 160.61 ppm (Ar–C and CO); MS (EI): m/z (%) 415 (M+ + 1, 30.02), 414 (M+, 100); HRMS (EI): m/z calcd for C20H10N6O3S (M+) 414.0530, found 414.0530.
6-Amino-1-(benzothiazol-2-yl)-4-(3-nitrophenyl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (7f). Pale yellow crystals, yield: 1.84 g (89%), mp above 300 °C; IR (KBr): ν/cm−1 3328 (NH2), 2209 (CN), 1676 (CO); 1H NMR (DMSO-d6): δ = 7.59–7.66 (m, 2H, Ar–H), 7.94 (t, J = 7.8 Hz, 1H, Ar–H), 8.06 (d, J = 7.8 Hz, 1H, Ar–H), 8.13 (d, J = 7.8 Hz, 1H, Ar–H), 8.24 (d, J = 7.8 Hz, 1H, Ar–H), 8.45–8.48 (m, 2H, Ar–H), and 8.88 ppm (s, 2H, NH2); 13C NMR (DMSO-d6): δ = 76.01 (C5), 87.82 (C3), 114.74, 115.24, 122.59, 122.73, 123.59, 124.99, 126.20, 126.32, 130.58, 134.37, 135.87, 136.99, 147.56, 149.79, 153.93, 156.74, 158.84 and 159.89 ppm (Ar–C and CO); MS (EI): m/z (%) 415 (M+ + 1, 27.34), 414 (M+, 100); HRMS (EI): m/z calcd for C20H10N6O3S (M+) 414.0530, found 414.0531.
6-Amino-1-(benzothiazol-2-yl)-4-(2,4-dimethoxyphenyl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (7g). Yellow crystals, yield: 2.05 g (96%), mp above 300 °C; IR (KBr): ν/cm−1 3308 (NH2), 2216 (CN), 1675 (CO); 1H NMR (DMSO-d6): δ = 3.89 (s, 3H, OCH3), 3.92 (s, 3H, OCH3), 6.71 (s, 1H, Ar–H), 6.75 (d, J = 8.4 Hz, 1H, Ar–H), 7.30 (t, J = 7.8 Hz, 1H, Ar–H), 7.45 (t, J = 7.8 Hz, 1H, Ar–H), 8.04 (d, J = 7.8 Hz, 1H, Ar–H), 8.20–8.25 (m, 2H, Ar–H) and 8.78 ppm (s, 2H, NH2); 13C NMR (DMSO-d6): δ = 55.94, 56.31 (2OCH3), 75.80 (C5), 87.34 (C3), 98.36, 107.70, 113.12, 114.18, 115.24, 116.49, 121.84, 122.98, 123.82, 125.95, 126.40, 130.47, 147.69, 153.81, 161.31, 161.34, 162.68 and 165.87 ppm (Ar–C and CO); MS (EI): m/z (%) 430 (M+ + 1, 30.08), 429 (M+, 100); HRMS (EI): m/z calcd for C22H15N5O3S (M+) 429.0890, found 429.0891.
6-Amino-1-(benzothiazol-2-yl)-2-oxo-4-(thiophen-2-yl)-1,2-dihydropyridine-3,5-dicarbonitrile (7h). Yellowish green crystals, yield: 1.68 g (90%), mp above 300 °C; IR (KBr): ν/cm−1 3307 (NH2), 2213 (CN), 1671 (CO); 1H NMR (DMSO-d6): δ = 7.31–7.61 (m, 4H, Ar–H), 7.99 (d, J = 4.4 Hz, 1H, Ar–H), 8.12 (d, J = 7.6 Hz, 1H, Ar–H), 8.24 (d, J = 7.6 Hz, 1H, Ar–H) and 8.77 ppm (s, 2H, NH2); 13C NMR (DMSO-d6): δ = 75.73 (C5), 87.42 (C3), 115.48, 116.00, 122.83, 123.68, 126.27, 126.42, 127.82, 130.87, 131.20, 133.15, 137.35, 149.92, 154.20, 154.44, 156.97 and 159.27 ppm (Ar–C and CO); MS (EI): m/z (%) 376 (M+ + 1, 27.05), 375 (M+, 100); HRMS (EI): m/z calcd for C18H9N5OS2 (M+) 375.0243, found 375.0242.
6-Amino-4-(4-chlorophenyl)-1-(6-fluorobenzothiazol-2-yl)-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (7i). Creamy white crystals, yield: 1.98 g (94%), mp above 300 °C; IR (KBr): ν/cm−1 3317 (NH2), 2225, 2214 (CN), 1678 (CO); 1H NMR (DMSO-d6): δ = 7.51 (d, J = 8.8 Hz, 1H, Ar–H), 7.61 (d, J = 8.0 Hz, 2H, Ar–H), 7.70 (d, J = 8.0 Hz, 2H, Ar–H), 8.14–8.18 (m, 2H, Ar–H) and 8.81 ppm (s, 2H, NH2); 13C NMR (DMSO-d6): δ = 75.95 (C5), 87.70 (C3), 109.04, 109.31 (d, 2JCF = 27 Hz), 115.11, 115.36 (d, 2JCF = 25 Hz) 115.23, 115.78, 125.31, 125.41 (d, 3JCF = 10.0 Hz), 129.00, 129.94, 133.37, 135.45, 138.73, 138.84 (d, 3JCF = 11.0 Hz), 146.96, 154.16, 156.95, 159.04, 159.25 (Ar–C) and 161.42 ppm (Ar–C and CO); MS (EI): m/z (%) 423 (M+ + 2, 38.12), 422 (M+ + 1, 56.89), 421 (M+, 100); HRMS (EI): m/z calcd for C20H9ClFN5OS (M+) 421.0195, found 421.0196. Crystal data, moiety formula: C20H9ClFN5OS, M = 421.83, triclinic, a = 6.6752(3) Å, b = 11.0006(6) Å, c = 13.2731(8) Å, V = 884.07(8) Å3, α = 67.007(3)°, β = 80.203(3)°, γ = 85.509(3)°, space group: P, Z = 2, Dcalc = 1.585 g cm−3, no. of reflection measured = 12731, θmax = 66.14°, R1 = 0.0465 (CCDC 1858434).55
(E)-N-(Benzothiazol-2-yl)-2-cyano-3-[4-(dimethylamino)phenyl]acrylamide (9)
A mixture of the cyanoacetamide derivative 4a (5 mmol), arylidene malononitrile 8 (5 mmol) or 4-(dimethylamino)benzaldehyde (5 mmol), and L-proline (0.5 mmol, 10 mol%) in ethanol (20 mL) was sonicated in an MKC6, Guyson ultrasonic bath (Model-MKC6, operating frequency 38 kHz ± 10% and output power of 110 W) for 20 minutes at 80 °C. The reaction was monitored by TLC and continued until the starting substrates were completely consumed, then left to cool to room temperature. In each case, the solid product formed after cooling to room temperature was separated by filtration, washed with ethanol, dried and recrystallized from ethanol/dioxane mixture (1:1) to give 9 as the pure product. Orange crystals, yield: 1.69 g (97%), mp above 300 °C; IR (KBr): ν/cm−1 3400 (NH2), 2198 (CN), 1679 (CO); 1H NMR (DMSO-d6): δ = 3.07 (s, 6H, 2CH3), 6.84 (d, J = 8.4 Hz, 2H, Ar–H), 7.32 (t, J = 7.6 Hz, 1H, Ar–H), 7.46 (t, J = 7.6 Hz, 1H, Ar–H), 7.65 (d, J = 7.6 Hz, 1H, Ar–H), 7.93–7.95 (m, 3H, Ar–H), 8.30 (s, 1H, arylidene C–H) and 12.94 ppm (s, 1H, NH); 13C NMR (DMSO-d6): δ = 40.64 (2CH3) 98.23, 112.24, 118.16, 118.24, 119.40, 122.32, 124.11, 126.88, 130.40, 133.77, 144.35, 152.37, 154.15, 162.47 and 166.20 ppm (Ar–C and CO); MS (EI): m/z (%) 349 (M+ + 1, 9.05), 348 (M+, 33); HRMS (EI): m/z calcd for C19H16N4OS (M+) 348.1039, found 348.1039.
General procedure for the preparation of 1-(benzothiazol-2-yl)-6-hyroxy-2-oxo-4-arylpyridine-3,5-dicarbonitrile derivatives 11a–c
Mixtures of the cyanoacetamide derivative 4a (5 mmol), ethyl-2-cyano-3-arylacrylate 10a–c (5 mmol) and L-proline (0.5 mmol, 10 mol%) in ethanol (20 mL) were sonicated in an MKC6, Guyson ultrasonic bath (Model-MKC6, operating frequency 38 kHz ± 10% and an output power of 110 W) for 20 minutes at 80 °C. The reaction was controlled by TLC and continued until the starting substrates were completely consumed, then left to cool to room temperature. In each case, the solid product formed after cooling to room temperature was separated by filtration, washed with ethanol, dried and recrystallized from ethanol to give 11a–c as pure products.
1-(Benzothiazol-2-yl)-6-hydroxy-2-oxo-4-phenyl-1,2-dihydropyridine-3,5-dicarbonitrile (11a). Creamy white crystals, yield: 1.72 g (93%), mp above 300 °C; IR (KBr): ν/cm−1 3159 (OH), 2225 (CN), 1677 (CO); 1H NMR (DMSO-d6): δ = 7.36–7.38 (m, 2H, Ar–H), 7.49–7.55 (m, 5H, Ar–H), 8.07 (d, J = 8.4 Hz, 1H, Ar–H), 8.56 (d, J = 8.4 Hz, 1H, Ar–H) and 13.23 ppm (s, 1H, OH); 13C NMR (DMSO-d6): δ = 89.09 (C5), 102.79 (C3), 115.17 (CN), 118.56, 123.27, 123.32, 126.97, 127.01, 127.44, 127.96, 128.63, 135.78, 136.54, 154.33, 156.56, 161.31 and 165.97 ppm (Ar–C and CO); MS (EI): m/z (%) 371 (M+ + 1, 28.12), 370 (M+, 100); HRMS (EI): m/z calcd for C20H10N4O2S (M+) 370.0519, found 370.0521.
1-(Benzothiazol-2-yl)-6-hydroxy-2-oxo-4-p-tolyl-1,2-dihydropyridine-3,5-dicarbonitrile (11b). Creamy white crystals, yield: 1.84 g (96%), mp above 300 °C; IR (KBr): ν/cm−1 3167 (OH), 2223 (CN), 1672 (CO); 1H NMR (DMSO-d6): δ = 2.44 (s, 3H, CH3), 7.27–7.31 (m, 4H, Ar–H), 7.49–7.54 (m, 2H, Ar–H), 8.01 (d, J = 7.8 Hz, 1H, Ar–H), 8.58 (d, J = 7.8 Hz, 1H, Ar–H) and 12.80 ppm (s, 1H, OH); 13C NMR (DMSO-d6): δ = 20.60 (CH3), 97.72 (C5), 102.78 (C3), 114.70 (CN), 118.29, 122.82, 123.01, 126.66, 126.74, 127.06, 128.15, 133.28, 135.48, 137.75, 153.94, 156.11, 159.48, 161.19 and 165.52 ppm (Ar–C and CO); MS (EI): m/z (%) 385 (M+ + 1, 26.54), 384 (M+, 100); HRMS (EI): m/z calcd for C21H12N4O2S (M+) 384.0675, found 384.0677.
1-(Benzothiazol-2-yl)-4-(4-chlorophenyl)-6-hydroxy-2-oxo-1,2-dihydropyridine-3,5-dicarbonitrile (11c). Creamy white crystals, yield: 1.78 g (88%), mp above 300 °C; IR (KBr): ν/cm−1 3180 (OH), 2221 (CN), 1673 (CO); 1H NMR (DMSO-d6): δ = 7.43 (d, J = 8.4 Hz, 2H, Ar–H), 7.49–7.55 (m, 2H, Ar–H), 7.59 (d, J = 8.4 Hz, 2H, Ar–H), 8.08 (d, J = 8.2 Hz, 1H, Ar–H), 8.58 (d, J = 8.2 Hz, 1H, Ar–H) and 13.31 ppm (s, 1H, OH); 13C NMR (DMSO-d6): δ = 98.11 (C5), 102.83 (C3), 115.13 (CN), 118.63, 123.32, 123.34, 127.11, 127.52, 128.18, 129.08, 133.52, 135.44, 135.72, 154.32, 156.65, 160.06, 162.34 and 166.07 ppm (Ar–C and CO); MS (EI): m/z (%) 406 (M+ + 2, 40.02), 405 (M+ + 1, 46.11), 404 (M+, 100); HRMS (EI): m/z calcd for C20H9ClN4O2S (M+) 404.0129, found 404.0129.
N-(Benzothiazol-2-yl)-2-oxo-6-phenyl-1,2-dihydropyridine-3-carboxamide (17). A solution of cyanoacetamide 4a (1.09 g, 5 mmol) and enaminone 13 (0.9 g, 5 mmol) in AcOH (15 mL) containing anhydrous sodium acetate (1.0 g) was sonicated in an MKC6, Guyson ultrasonic bath (Model-MKC6, operating frequency 38 kHz ± 10% and output power of 110 W) for 40 minutes at 80 °C. The reaction was monitored by TLC and continued until the starting substrates were completely consumed, then left to cool to room temperature. The solid product formed after cooling to room temperature was separated by filtration, washed with ethanol, dried and recrystallized from an ethanol/dioxane (1:2) mixture, to give 17 as orange crystals, yield: 1.35 g (78%), mp above 300 °C; IR (KBr): ν/cm−1 3335, 3284 (2NH), 1681, 1654 (2CO); 1H NMR (DMSO-d6): δ = 6.98 (d, J = 7.2 Hz, 1H, H-5), 7.31 (t, J = 7.2 Hz, 1H, Ar–H), 7.44 (t, J = 7.2 Hz, 1H, Ar–H), 7.54–7.6 (m, 3H, Ar–H), 7.77 (d, J = 7.8 Hz, 1H, Ar–H), 7.85 (d, J = 8.0 Hz, 2H, Ar–H), 7.99 (d, J = 7.8 Hz, 1H, Ar–H), 8.54 (d, J = 7.2 Hz, 1H, H-4), 13.22 (s, 1H, NH) and 13.59 ppm (s, 1H, NH); 13C NMR (DMSO-d6): δ = 106.32, 115.48 (CN), 120.71, 121.81, 123.74, 126.26, 127.79, 129.04, 131.32, 131.74, 131.85, 145.47, 148.69, 152.49, 157.05, 162.15 and 163.54 ppm (Ar–C and CO); MS (EI): m/z (%) 348 (M+ + 1, 9.12), 347 (M+, 38.79), HRMS (EI): m/z calcd for C19H13N3O2S (M+) 347.0723, found 347.0724.
Conflicts of interest
The authors declare that they have no competing interests.
Acknowledgements
The facilities of Analab/SAF by the University of Kuwait through research grants GS01/01, GS01/05, GS01/03, and GS03/08 are gratefully acknowledged.
References
- M. Sainsbury, Heterocyclic chemistry, Royal Society of Chemistry, Cambridge 2001 Search PubMed.
- T. Taniguchi and K. A. Ogasawara, Org. Lett., 2000, 2, 3193–3195 CrossRef CAS PubMed.
- G. D. Henry, Tetrahedron, 2004, 60, 6043–6061 CrossRef CAS.
- I. Gomez, E. Alonso, D. J. Ramon and M. Yus, Tetrahedron, 2000, 56, 4043–4052 CrossRef CAS.
- A. Carreno, C. Zuniga, D. Paez-Hernandez, M. Gacitua, R. Polanco, C. Otero, R. Arratia-Perez and J. A. Fuentes, New J. Chem., 2018, 42, 8851–8863 RSC.
- K. R. Amperayani, K. N. Kumar and U. D. Parimi, Res. Chem. Intermed., 2018, 44, 3549–3564 CrossRef CAS.
- M. Vrabel, M. Hocek, L. Havran, M. Fojta, I. Votruba, B. Klepetařova, R. Pohl, L. Rulišek, L. Zendlova, P. Hobza, I. H. Shih, E. Mabery and R. Mackman, Eur. J. Inorg. Chem., 2007, 2007, 1752–1769 CrossRef.
- J. Fu, S. Karur and K. B. Pfister, PCT Int. Appl., WO 2018073753 A1 20180426, 2018.
- A. Worachartcheewan, S. Prachayasittikul, R. Pingaew, C. Nantasenamat, T. Tantimongcolwat, S. Ruchirawat and V. Prachayasittikul, Med. Chem. Res., 2012, 21, 3514–3522 CrossRef CAS.
- S. Riaz, I. U. Khan, M. Bajda, M. Ashraf, Q. Ain, A. Shaukat, T. Ur Rehman, S. Mutahir, S. Hussain, G. Mustafa and M. Yar, Bioorg. Chem., 2015, 63, 64–71 CrossRef CAS PubMed.
- J. Easmon, G. Pürstinger, K.-S. Thies, G. Heinisch and J. Hofmann, Med. Chem., 2006, 49(21), 6343–6350 CrossRef CAS PubMed.
- G. S. Kumar, Y. Poornachandra, S. K. Gunda, K. R. Reddy, J. Mohmed, K. Shaik, C. G. Kumar and B. Narsaiah, Bioorg. Med. Chem. Lett., 2018, 28, 2328–2337 CrossRef PubMed.
- M. Wei, X. Peng, L. Xing, Y. Dai, R. Huang, M. Geng, A. Zhang, J. Ai and Z. Song, Eur. J. Med. Chem., 2018, 154, 9–28 CrossRef CAS PubMed.
- M. P. Savic, J. J. Ajdukovic, J. J. Plavsa, S. S. Bekic, A. S. Celic, O. R. Klisuric, D. S. Jakimov, E. T. Petri and E. A. Djurendic, Med. Chem. Commun., 2018, 9, 969–981 RSC.
- J. Liu, Z. Wang, A. Levin, T. J. Emge, P. R. Rablen, D. M. Floyd and S. Knapp, J. Org. Chem., 2014, 79, 7593–7599 CrossRef CAS PubMed.
- T. M. Chapman, S. A. Osborne, C. Wallace, K. Birchall, N. Bouloc, H. M. Jones, K. H. Ansell, D. L. Taylor, B. Clough, J. L. Green and A. A. Holder, J. Med. Chem., 2014, 57, 3570–3587 CrossRef CAS PubMed.
- N. S. Gray, T. Zhang, J. Hatcher and S. J. Buhrlage, PCT Int. Appl., WO 2018098367 A1 20180531, 2018.
- S. K. Nechipadappu and D. R. Trivedi, J. Mol. Struct., 2017, 1141, 64–74 CrossRef CAS.
- N. Bharti, M. R. Maurya, F. Naqvi and A. Azam, Bioorg. Med. Chem. Lett., 2000, 10, 2243–2245 CrossRef CAS PubMed.
- S. M. Sondhi, M. Dinodia and A. Kumar, Bioorg. Med. Chem., 2006, 14, 4657–4663 CrossRef CAS PubMed.
- M. Lopez-Martinez, H. Salgado-Zamora, E. R. San-Juan, S. Zamudio, O. Picazo, M. E. Campos and E. B. Naranjo-Rodriguez, Drug Dev. Res., 2010, 71, 371–381 CrossRef CAS.
- T. B. Chaston and D. R. Richardson, J. Biol. Inorg Chem., 2003, 8, 427–438 CrossRef CAS PubMed.
- A. Kamal, M. A. Syed and S. M. Mohammed, Expert Opin. Ther. Pat., 2015, 25, 335–349 CrossRef CAS PubMed.
- R. S. Keri, M. R. Patil, S. A. Patil and S. Budagumpi, Eur. J. Med. Chem., 2015, 89, 207–251 CrossRef CAS PubMed.
- L. L. Bozec and C. J. Moody, Aust. J. Chem., 2009, 62, 639–647 CrossRef.
- C. Palomo, M. Oiarbide and A. Mielgo, Angew. Chem., Int. Ed., 2004, 43, 5442–5444 CrossRef CAS PubMed.
- S. Letort, M. Lejeune, N. Kardos, E. Métay, F. Popowycz, M. Lemaire and M. Draye, Green Chem., 2017, 19, 4583–4590 RSC.
- B. Banerjee, Ultrason. Sonochem., 2017, 35, 15–35 CrossRef CAS PubMed.
- L.-C. Campeau and K. Fagnou, Org. Synth., 2011, 88, 22–32 CrossRef CAS.
- J. Z. Deng, D. V. Paone, A. T. Ginnetti, H. Kurihara, S. D. Dreher, S. A. Weissman, S. R. Stauffer and C. S. Burgey, Org. Lett., 2009, 11, 345–347 CrossRef CAS PubMed.
- M. R. Luzung, J. S. Patel and J. Yin, J. Org. Chem., 2010, 75, 8330–8332 CrossRef CAS PubMed.
- J. R. Manning and H. M. L. Manning, J. Am. Chem. Soc., 2008, 130, 8602–8603 CrossRef CAS PubMed.
- T. J. Donohoe, J. A. Basutto, J. F. Bower and A. Rathi, Org. Lett., 2011, 13, 1036–1039 CrossRef CAS PubMed.
- H. M. Ibrahim and H. Behbehani, Molecules, 2014, 19, 2637–2654 CrossRef PubMed.
- H. Behbehani, H. M. Ibrahim and M. H. Elnagdi, Tetrahedron, 2013, 69, 6176–6184 CrossRef CAS.
- H. Behbehani and H. M. Ibrahim, Tetrahedron, 2013, 69, 10535–10543 CrossRef CAS.
- H. M. Ibrahim, H. Behbehani and M. H. Elnagdi, Chem. Cent. J., 2013, 7, 123 CrossRef PubMed.
- B. Banerjee, Ultrason. Sonochem., 2017, 35, 1–14 CrossRef CAS PubMed; H. Behbehani, H. M. Ibrahim and K. M. Dawood, RSC Adv., 2015, 5, 25642–25649 RSC.
- L. S. da Silveira Pinto and M. V. N. de Souza, Synthesis, 2017, 49, 2555–2561 CrossRef.
- D. Bandyopadhyay, S. Mukherjee, L. C. Turrubiartes and B. K. Banik, Tetrahedron, 2012, 19, 969–973 Search PubMed; H. Behbehani and H. M. Ibrahim, RSC Adv., 2016, 6, 52700–52709 RSC.
- L. Pizzuti, L. A. Piovesan, A. F. C. Flores, F. H. Quina and C. M. P. Pereira, Ultrason. Sonochem., 2009, 16, 728–731 CrossRef CAS PubMed; W. A. A. Arafa and H. M. Ibrahim, RSC Adv., 2018, 8, 10516–10521 RSC.
- M. Vidal, M. García-Arriagada, M. C. Rezende and M. Domínguez, Synthesis, 2016, 48, 4246–4252 CrossRef CAS.
- L. Pizzuti, P. L. G. Martins, B. A. Ribeiro, F. H. Quina, E. Pinto, A. F. C. Flores, D. Venzke and C. M. P. Pereira, Ultrason. Sonochem., 2010, 17, 34–37 CrossRef CAS PubMed.
- H. Behbehani, H. M. Ibrahim, S. Makhseed, M. H. Elnagdi and H. Mahmoud, Eur. J. Med. Chem., 2012, 52, 61–65 CrossRef CAS PubMed; A. Ueda, T. Umeno, M. Doi, K. Akagawa, K. Kudo and M. Tanaka, J. Org. Chem., 2016, 81, 6343–6356 CrossRef PubMed; S. Kim, S. Kang, G. Kim and Y. Lee, J. Org. Chem., 2016, 81, 4048–4057 CrossRef PubMed; A. M. Farag, N. A. Kheder and K. M. Dawood, Int. J. Mod. Org. Chem., 2013, 2, 26–39 Search PubMed; I. V. Dyachenko, V. D. Dyachenko and E. B. Rusanov, Russ. J. Org. Chem., 2007, 43, 83–89 CrossRef; S. Bondock, W. Fadaly and M. A. Metwally, Eur. J. Med. Chem., 2010, 45, 3692–3701 CrossRef PubMed.
- L. Wu, R. Jin, L. Li, X. Hu, T. Cheng and G. Liu, Org. Lett., 2017, 19, 3047–3050 CrossRef CAS PubMed; A. B. Smith III, O. Atasoylu and D. C. Beshore, Synlett, 2009, 16, 2643–2646 CrossRef PubMed; M. Fernández, U. Uria, L. Orbe, J. L. Vicario, E. Reyes and L. Carrillo, J. Org. Chem., 2014, 79, 230–239 CrossRef PubMed.
- Y. N. Mabkhot, N. A. Kheder and A. M. Farag, Heterocycles, 2010, 81, 2369–2376 CrossRef CAS; R. E. Khidre, I. A. M. Radini and D. A. Ibrahim, ARKIVOC, 2016, 5, 301–317 Search PubMed; M. A. Raslan, S. M. Sayed and M. A. Khalil, J. Heterocycl. Chem., 2016, 53, 727–733 CrossRef.
- J. Stetinova, R. Kada, J. Lesko, L. Zalibera, D. Il avsky and A. Bartovic, Collect. Czech. Chem. Commun., 1995, 60, 999–1008 CrossRef CAS.
- A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725–748 CrossRef CAS PubMed.
- C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927–934 RSC.
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef PubMed.
- H. M. Ibrahim, S. Makhseed, R. M. Abdel-Motaleb, A. A. Makhlouf and M. H. Elnagdi, Heterocycles, 2007, 71, 1951–1966 CrossRef CAS.
- Crystallographic data for 7a (ref. CCDC 1858431) can be obtained on request from the director, Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EW, UK.
- Crystallographic data for 7b (ref. CCDC 1858432) can be obtained on request from the director, Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EW, UK.
- Crystallographic data for 7d (ref. CCDC 1858433) can be obtained on request from the director, Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EW, UK.
- Crystallographic data for 7i (ref. CCDC 1858434) can be obtained on request from the director, Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EW, UK.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1858431–1858434. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra07013j |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Wael Abdelgayed Ahmed Arafa
*a,
Wael Abdelgayed Ahmed Arafa