
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of double mutations T790M/L858R on conformation and drug-resistant mechanism of epidermal growth factor receptor explored by molecular dynamics simulations†
Fangfang Yana,
Xinguo Liu *a,
Shaolong Zhanga,
Jing Sua,
Qinggang Zhanga and
Jianzhong Chen*b
*a,
Shaolong Zhanga,
Jing Sua,
Qinggang Zhanga and
Jianzhong Chen*b
aSchool of Physics and Electronics, Shandong Normal University, Jinan, 250358, China. E-mail: liuxinguo@sdnu.edu.cn
bSchool of Science, Shandong Jiaotong University, Jinan, 250357, China. E-mail: chenjianzhong1970@163.com; jzchen@sdjtu.edu.cn
First published on 29th November 2018
Abstract
Epidermal growth factor receptor (EGFR) is one of the most promising targets for the treatment of cancers. Double mutations T790M/L858R lead to different degrees of drug resistance toward inhibitors. In this study, molecular dynamics (MD) simulations followed by principal component analysis are performed to study the conformational changes of EGFR induced by T790M/L858R. The results suggest that T790M/L858R cause obvious disturbance of the structural stability of EGFR. Molecular mechanics-Poisson Boltzmann surface area (MM-PBSA) and residue-based free energy decomposition methods are integrated to explore the drug-resistant mechanism of T790M/L858R toward inhibitors. The results show that the decrease in van der Waals interactions of inhibitors with the mutated EFGR relative to the wild-type (WT) one is the main force inducing drug resistance of T790M/L858R toward inhibitors TAK-285, while drug resistance toward W2P and HKI-272 is dominated by the decrease in van der Waals interactions and the increase in polar interactions. We expect that the information obtained from this study can aid rational design of effective drugs to relieve drug resistance of EGFR induced by T790M/L858R.
1 Introduction
The receptor tyrosine kinase (RTK) superfamily is a transmembrane protein in signal transduction and implicated in the regulation of physiological processes of cells, such as cellular differentiation and migration.1 As a member of the RTK family, epidermal growth factor receptor (EGFR) has attracted more attention due to its special role in the development of lung cancer. EGFR is over-expressed in many malignant tumors including lung cancer, breast cancer, liver cancer and so forth.2–4 Previous studies have shown that abnormal expression and activation of EGFR can accelerate the proliferation and metastasis of cancerous cells and promote the angiogenesis and anti-apoptosis of tumors,5,6 which plays an important role in the occurrence and development of tumors. It has been reported that the EGFR gene is of importance for regulating growth of lung cancer, moreover the transitional expression of EGFR is also indicative of advanced lung cancer.7 Therefore, EGFR has been one of the most promising targets for the treatment of cancers.Non-small cell lung cancer (NSCLC) accounts for more than 80% of lung cancer patients. Many studies have shown that the most common mutations of drug resistance in NSCLC are EGFR mutations.8–11 The previous works indicated that mutations in EGFR kinase domains are an early event in development of lung adenocarcinomas and may induce drug resistance toward EGFR inhibitors.12,13 The analysis on tumor samples revealed that T790M mutation always coexists with the somatic mutation L858R.14 The mutation at 858th residue of EGFR kinase domain is the extremely frequent carcinogenic mutation,15,16 which is also regarded as the most common kinase mutation relating with cancers.17,18 The mutation of the leucine at 858th residue of EGFR into the positively charged arginine produced certain drug resistance toward the current inhibitors. The 790th residue of the EGFR kinase is known as the “gatekeeper”, which is an important determinant of inhibitor bindings to EGFR. The binding pocket of EGFR consists of hinge region, C-helix, A- and P-loops (Fig. 1A). It is worth noting that the 790th residue is located in the hinge region and depth of the binding pocket, while that the 858th residue in the A-loop (Fig. 1A and B). The mutation T790M, namely the short side-chain of threonine replaced by the long side-chain of a methionine, generates drug resistance toward the current inhibitors.19,20 In facts, the double mutations T790M/L858R not only retain the characteristics of single mutation, but also have some unique properties.21,22 Although some works were involved in investigating drug resistance of single mutation and double mutations of EGFR, the effect of double mutations T790M/L858R on the conformational changes of EGFR, binding ability and atomic-level binding mechanism are still lacking. Therefore, understanding drug-resistant mechanism of T790M/L858R is essential for rational design of effective drugs targeting EGFR.
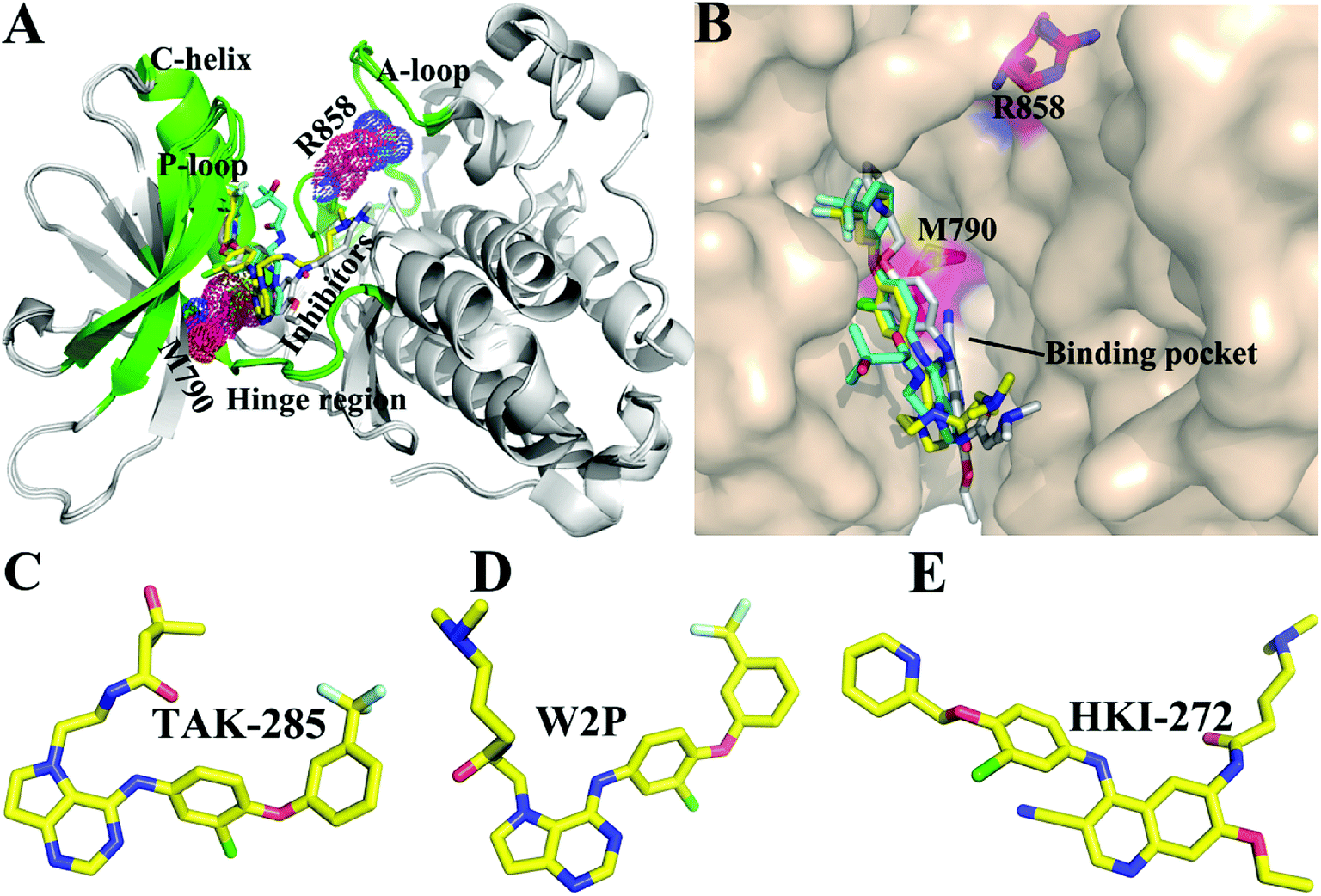 | ||
| Fig. 1 Structures of molecules (A) superimposed structures of EGFR kinase with three ligands are shown in cartoon modes. The main domains of EGFR (hinge region, C-helix, A-loop, and P-loop) are colored with green. (B) Binding pocket of EGFR kinase is displayed in surface modes. Structures of three ligands are shown in stick modes: (C) TAK-285, (D) W2P, and (E) HKI-272. | ||
So far, a variety of inhibitors of EGFR have been developed, among which some are testing in clinical trials, and some have been even applied to treat lung cancer, including gefitinib,23 erlotinib,24 and lapatinib.25 The previous studies demonstrated that different EGFR mutants exhibit distinctive sensitivities to various inhibitors.23,26 In addition, multiple inhibitors have potential safety concerns or side-effect due to their poor selectivity toward other kinases,27,28 which makes treatment of cancers face greater challenges. Thus, it is urgent to delve into drug-resistance mechanisms and the conformational changes of EGFR induced by T790M/L858R. In this work, to obtain theoretical guidance for design of novel inhibitors alleviating drug resistance of the mutated EGFR, we selected three inhibitors TAK-285, W2P, and HKI-272 to probe conformational changes of EGFR and mechanisms of drug resistance induced by T790M/L858R.29 Studies have shown that inhibitors can competitively binding to the APT binding site,30–33 which will lead to the dimerization and reorganization of receptor.34,35 The structures of these investigated inhibitors are shown in Fig. 1C–E. These three inhibitors are the representative derivatives of pyrrolo[3,2-d]pyrimidine, and exhibit significant anti-tumor effects in vivo.36,37 Meanwhile, the work from Tomoyasu Ishikawa et al. has attested that the pyrrolo[3,2-d]pyrimidine scaffold plays a significant role in design of kinase inhibitors. Therefore, it is necessary to further study the role of this kind of inhibitors in bindings to EGFR. With the aim to relieve the side-effect and drug resistance, we performed detailed analyses involving the conformational changes of EGFR and binding mechanisms of inhibitors to the WT/mutant EGFRs.
Molecular dynamics (MD) simulations have been a universal tool to study the conformational changes of proteins and ligand–protein binding mechanisms.38–47 Moreover, MD simulation studies of EGFR have attracted wide attention in recent years.48–50 Several methods have been proposed to calculate binding free energies of inhibitors to proteins, such as free energy perturbation (FEP),51–53 thermodynamics integration (TI)54,55 and molecular mechanics Poisson–Boltzmann/generalized Born surface area (MM-PBSA/GBSA).56–58 Although the FEP and TI can obtain high accuracy in calculating binding free energies of inhibitor–protein complexes, they require a large amount of statistical sampling, which is expensive in computational time.58 On the contrary, MM-PBSA/GBSA methods can not only obtain rational results, but also save more computational resource and time. Furthermore, these two methods have been successfully employed to predict binding free energies of inhibitors to proteins.59–72 Thus, in this work, a combination of MD simulations and MM-PBSA method was applied to probe drug resistant mechanism of T790M/L858R toward inhibitors. In addition, to further investigate the conformational change of EGFR induced by T790M/L858R, principal component analysis (PCA) and dynamics analysis were performed on MD trajectories.73,74 We expect that this work can provide theoretical hints for design of effective inhibitors alleviating drug resistance of the mutated EGFR in future.
2 Materials and methods
2.1 Preparation of simulation system
The crystal structures of T790M/L858R-mutant EGFR associated with three inhibitors were taken from Protein Data Bank (PDB).75 The PDB IDs of 3W2O, 3W2P and 3W2Q separately correspond to the TAK-285/, W2P/ and HKI-272/mutated EGFRs and the 5EDP for the mutated EGFR of the apo state. In order to maintain consistency of atomic coordinates, the WT EGFRs were constructed from their corresponding T790M/L858R complexes by using the leap module in Amber 16.76 Meanwhile, the leap module was also applied to add all missing hydrogen atoms to EGFR. All crystal water molecules were persisted in the initialized models. The protein was simulated using the ff99SB force field77 and the parameters of inhibitors were generated using the general Amber force field (GAFF).78 The PROPKA program was applied to assign the protonation states of residues at PH 7.0.79,80 The AM1-BCC procedure in Amber 16 was employed to assign the atomic charges to inhibitors.81,82 Then, six complexes were immersed into the solvent environment provided by the truncated periodic octahedral box of TIP3P water molecules, keeping a separation margin of 12.0 Å along each dimension from the solute.83 All positively charged systems were neutralized by an appropriate number of Cl− counterions.2.2 Molecular dynamics simulations
Two-stage energy optimization was performed using the steepest descent and conjugate gradient methods to remove several high-energy contacts. Firstly, these compounds were constrained in their initial positions by a harmonic constant of 100 kcal mol−1 Å−2 so as to better optimize the counterions and water molecules. Secondly, energy minimization was carried out on all atoms without restrictions. Then, each system undergoes a slow heating process of 1 ns from 0 K to 300 K and was equilibrated for another 1 ns at the normal temperature 300 K. The chemical bonds involving hydrogen atoms were fixed by the SHAKE algorithm.84 The long-range electrostatic interactions were treated by the particle mesh Ewald (PME) algorithm85,86 and a cutoff of 10 Å was employed to compute electrostatic and van der Waals interactions. After that, 170 ns MD simulations were run on six systems by the PMEMD module in Amber, during which the temperature and pressure were separately controlled at 300 K and 1 atm.2.3 Principal component analysis
In order to investigate the effect of T790M/L858R on dynamical properties of EGFR, PCA based on the dimensional reduction method was performed on the equilibrated MD trajectories.73,87 The eigenvectors and eigenvalues obtained from the diagonalization of the covariance matrix constructed using the atomic coordinates from MD trajectories are used to characterize protein motions. The eigenvectors and the eigenvalues respectively embody the direction and magnitude of the concerted motions. The functionally concerted motions of proteins can be generally described by the first few principal components. In this work, PCA was carried out using the CPPTRAJ module in Amber88 and the porcupine plots for investigating the concerted movements of the systems were plotted using the VMD89 software.2.4 MM-PBSA calculation
MM-PBSA method has been a universal and reliable tool for evaluating binding ability of inhibitors to proteins. Thus, the current study applied MM-PBSA to compute binding free energies (ΔGbind) of three inhibitors to the WT and mutated EGFRs based on 200 conformations taken from the last 100 ns of MD simulations at a time interval of 500 ps. The contribution of the entropy to the binding free energy was also taken into account. All counterions and water molecules were removed from each system during the calculation. In this study, ΔGbind is determined by the following equation:| ΔGbind = ΔEele + ΔEvdw + ΔGpol + ΔGnonpol − TΔS | (1) |
| ΔGnonpol = γ × ΔSASA | (2) |
3 Results and discussion
3.1 Structural stability and flexibility of EGFR
In order to better evaluate dynamic stability of all systems during MD simulation, root-mean-square deviations (RMSDs) of backbone atoms in the WT and T790M/L858R EGFRs with respect to their initial structures were calculated through the entire MD simulations and the results were given in ESI (Fig. S1†). It is observed that the RMSD values of six systems tend to converge after 70 ns of MD simulations, indicating that all systems basically reach equilibrium. The averaged RMSDs of the WT/mutated apo state of EGFRs and EGFRs associated with TAK-285, W2P and HKI-272 are 2.45/2.69 Å, 2.05/2.22 Å, 2.38/2.58 Å and 1.64/2.22 Å, respectively. This result shows that dual mutations T790M/L858R disturb structural stability of EGFR during MD simulations.Root-mean-square fluctuations (RMSFs) of Cα atoms in the WT and mutated EGFRs were computed to further explore influence of T790M/L858R on structural flexibility of EGFR (Fig. 2). As for the WT/mutant apo state of EGFRs (Fig. 2A), the flexibility of residues near 915–930 are enhanced due to T790M/L858R. It is found that T790M/L858R affect the flexibility of EGFRs complexed with three inhibitors (Fig. 2B–D), especially for the regions near residues 750–767, 780–788, 855–877 and 915–930. Except for the slight difference in the RMSF values of residues 750–767 and 915–930 between the WT and mutated EGFRs with HKI-272 (Fig. 2D), the RMSF values of residues 750–767 and 915–930 are increased in the mutated EGFRs with TAK-285 and W2P compared to the WT one (Fig. 2B and C), indicating that the flexibility of these regions is strengthened by T790M/L858R. The structural flexibility of the region near residues 780–788 is obviously increased in the mutated EGFRs with TAK-285 and W2P relative to the WT ones, but that of this region is weakened in the mutated EGFR with HKI-272 relative to the WT one. It is worth noting that the structural fluctuation of the A-loop (residues 855–877) in the mutated EGFR is reduced compared to the WT one, especially for the mutated EGFR complexed with HKI-272.
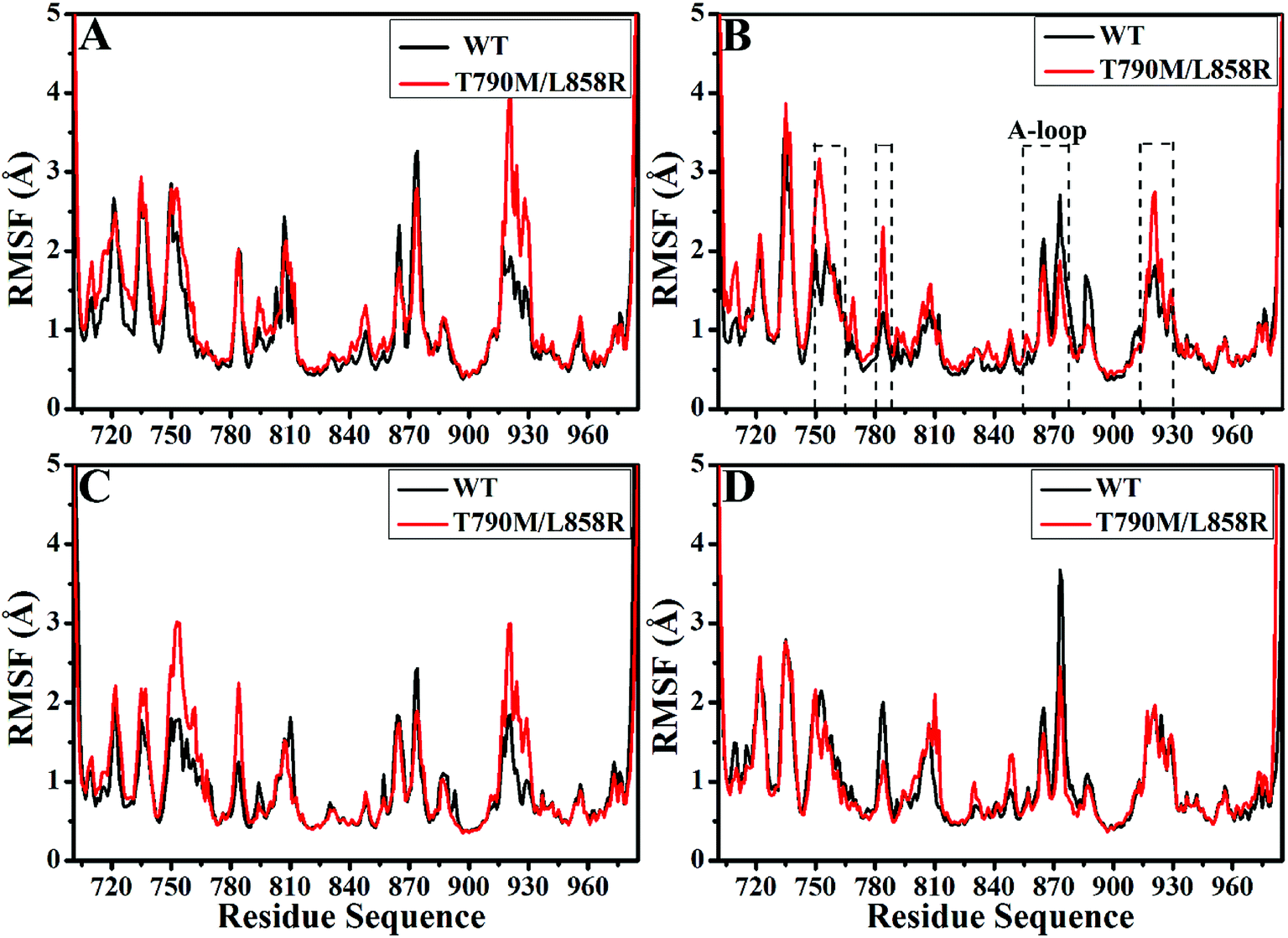 | ||
| Fig. 2 Root-mean square fluctuations (RMSFs) of Cα atoms in the WT and T790M/L858R EGFRs calculated from the equilibrated trajectories. (A), (B), (C) and (D) separately correspond to the WT/mutant apo EGFRs, EGFRs associated with TAK-285, W2P and HKI-272. | ||
3.2 Changes of internal dynamics induced by T790M/L858R
PC analysis has been a universal tool to investigate conformational changes of proteins. In the current work, PC analysis was performed on six systems to explore the changes in internal dynamics of EGFR caused by T790M/L858R. Plots of eigenvalues vs. the corresponding eigenvector indices obtained from the diagonalization of the covariance matrix were depicted in Fig. 3, in which the eigenvalues are usually applied to embody the strength of motions and the eigenvectors are used to reflect the direction of internal motions in EGFR. The first five eigenvalues account for 68%/74%, 57%/69%, 67%/69% and 61%/71% of the total motions for the WT/mutated apo EGFRs, WT/mutated EGFRs with TAK-285, W2P, and HKI-272. The subsequent eigenvalues quickly decrease in amplitude to reach a series of constrained and more localized fluctuations. The first eigenvalues of three mutated EGFRs are much higher than the WT EGFRs, indicating that T790M/L858R strengthen motion of EGFR. This result agrees basically with the previous RMSD analysis. In addition, in order to gain a deeper insight into the changes in the binding pocket of EGFR induced by mutations, the volume of binding pocket of each complex was calculated by utilizing the POVME procedure92 based on the equilibrated MD trajectories (Fig. 4). The averaged volume of binding pocket of the mutated EGFRs is smaller than that of the WT ones, and the decrease in volume may be related to motional difference of residues in the binding pocket caused by T790M/L858R.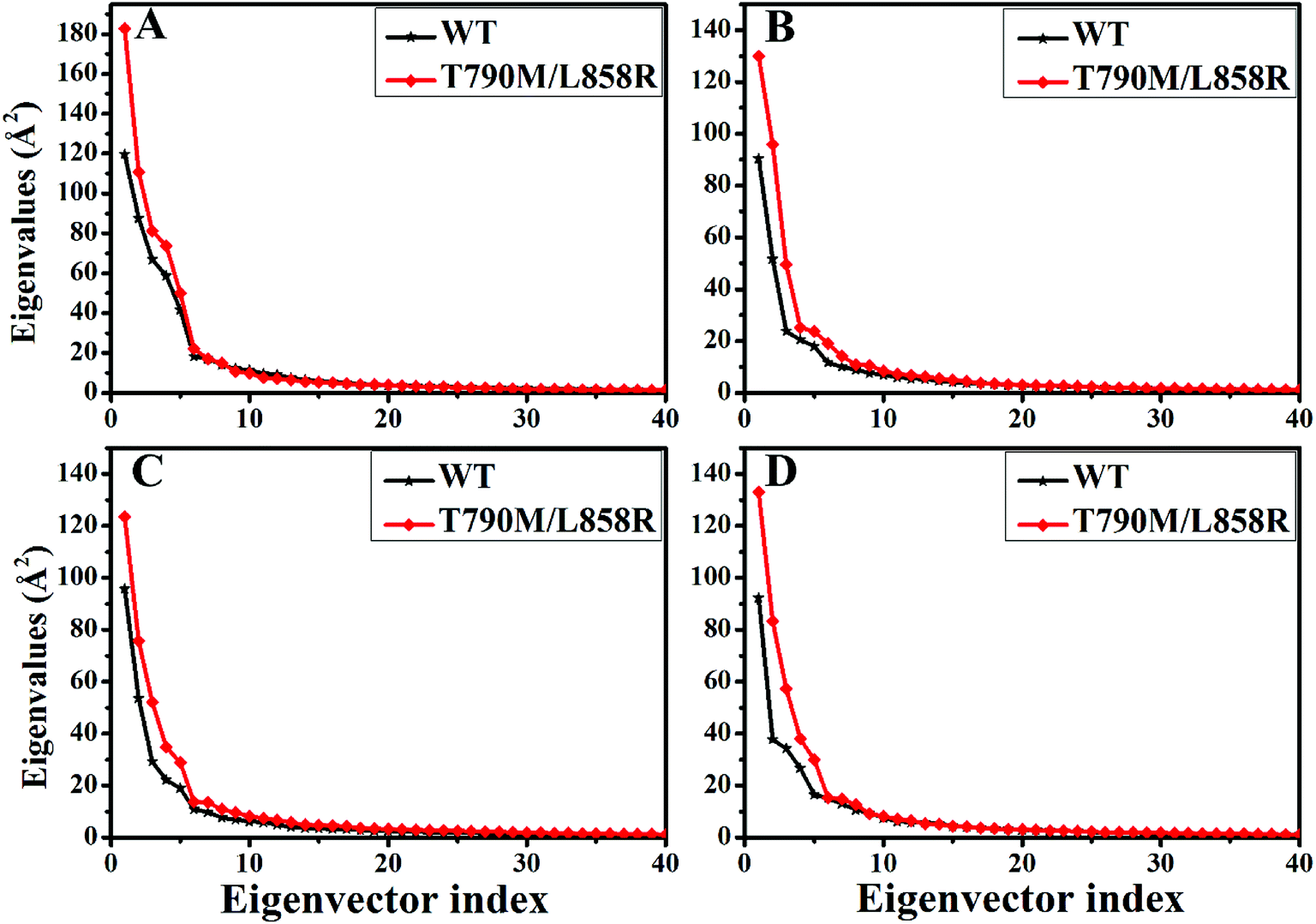 | ||
| Fig. 3 The function of the eigenvalues vs. the eigenvector indices taken from the diagonalization of Cα covariance matrix in the WT and mutated apo EGFRs (A) and EGFRs complexed with inhibitors: (B) TAK-285, (C) W2P and (D) HKI-272. | ||
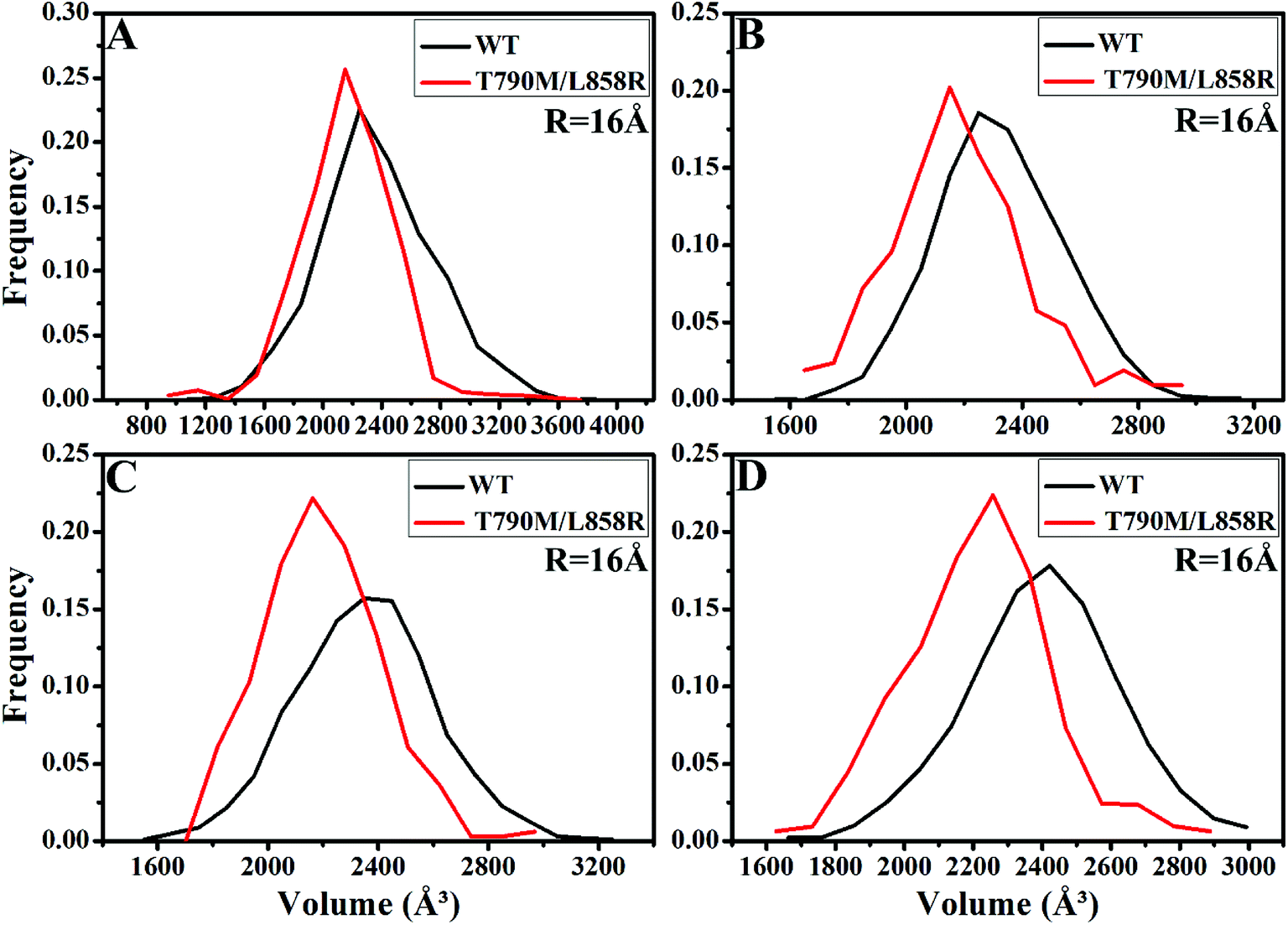 | ||
| Fig. 4 The frequency distribution of the volume of binding pocket in the WT/mutant apo EGFRs (A) and EGFRs complexed with inhibitors: (B) TAK-285, (C) W2P and (D) HKI-272. | ||
To further understand impact of T790M/L858R on internal motions of EGFR, six porcupine plots were depicted in Fig. 5 by using the VMD software. The direction of each arrow characterizes movement direction of Cα atoms, and the length of each arrow embodies motion strength of Cα atoms. As shown in Fig. 5, the concerted motions of domains have changed obviously due to T790M/L858R. Compared with the WT EGFR in its apo state (Fig. 5A1), double mutations make the motion direction and intensity of A-loop change significantly (Fig. 5A2), which may provide great contribution to the decrease of binding pocket. In the WT EGFR complexed with TAK-285 (Fig. 5B1), the P-loop, C-helix and A-loop have outward tendencies of movement. However, double mutations not only change movement strength of the P-loop, but also make the P-loop move downward (Fig. 5B2). In addition, the motion intensity of the C-helix and A-loop is significantly weakened by T790M/L858R, which tends to make the binding pocket become narrower and smaller. Except for the slight movement of the A-loop in the WT EGFR with W2P (Fig. 5C1), the C-helix and P-loop show obvious outward tendency. Although motion strength of the A-loop increased by T790M/L858R (Fig. 5C2), motion strength of the C-helix and P-loop is obviously decreased due to mutations, which is not beneficial to inhibitor bindings. By comparison between the WT and mutated EGFR with HKI-272 (Fig. 5D1 and D2), it is found that T790M/L858R produces smaller effects on movement of the C-helix (752–767), which is in agreement with the previous RMSF analysis (Fig. 2D). Furthermore, T790M/L858R not only weakens movement strength of the P-loop in the mutated EGFR, but also changes motion direction of this loop (Fig. 5D2). The above analysis reasonably explains the reason why the volume of the binding pockets becomes smaller in the mutant systems, meanwhile also proves that T790M/L858R generate significant influence on dynamic behavior of EGFR.
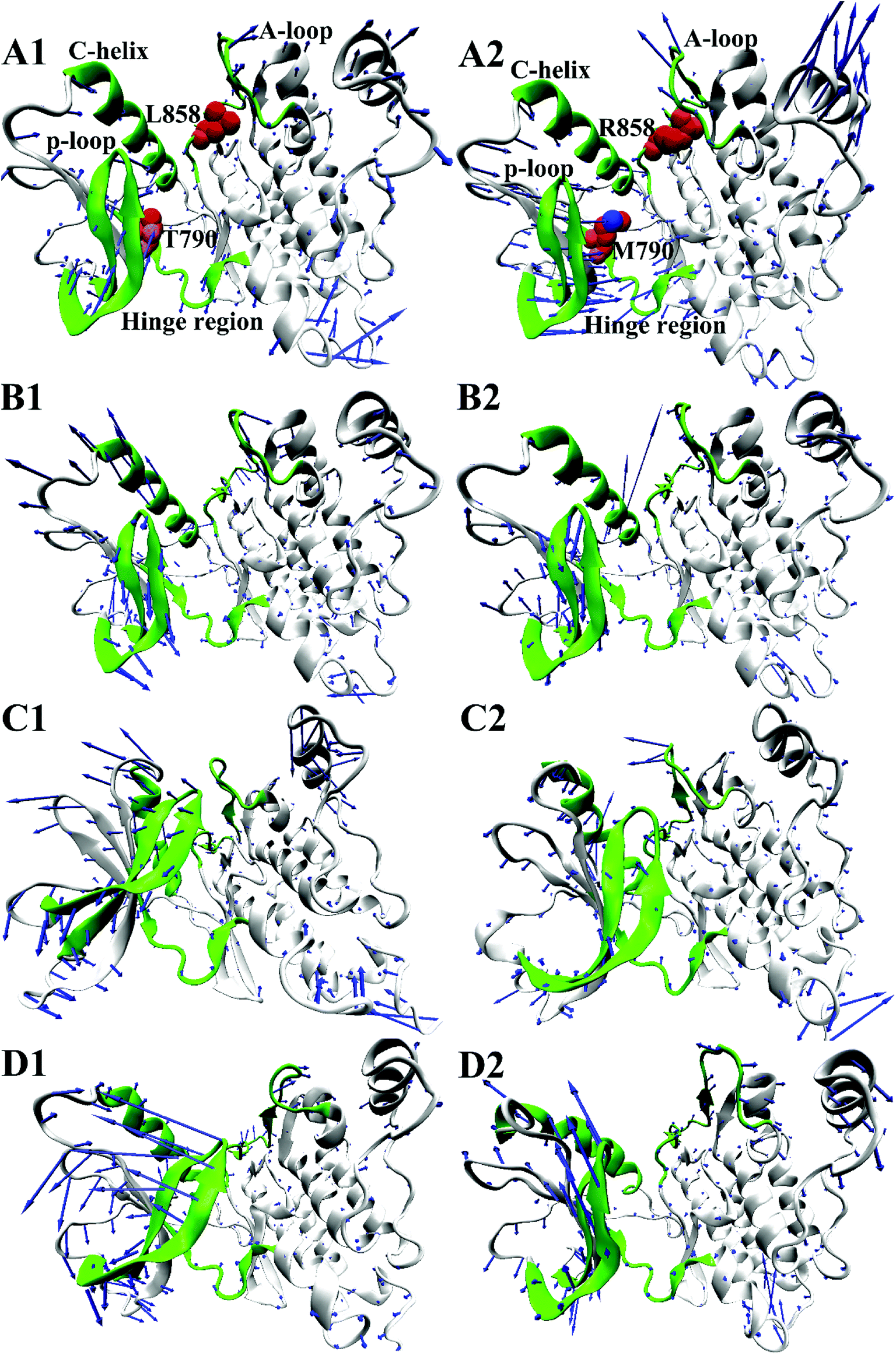 | ||
| Fig. 5 Concerted motions of domains corresponding to the first eigenvector (PC1) obtained from the principal component analysis: (A1/A2), (B1/B2), (C1/C2) and (D1/D2) respectively correspond to the WT/mutated apo EGFRs, EGFRs complexed with inhibitors TAK-285, W2P and HKI-272. | ||
Free energy landscapes can provide useful information for exploring the conformational dynamics of proteins. To further probe the conformational change of EGFR caused by T790M/L858R, free energy landscapes of all investigated systems were constructed by projecting MD trajectories on the first two principal components PC1 and PC2 (Fig. 6 and S2†). Different clusters in pictures represent different conformational states. As shown in Fig. S2,† the WT EGFR in its apo state displays two energy basin, while the WT EGFRs complexed with three inhibitors exhibit single energy basin that is mainly distributed in a conformational subspace (Fig. 6A1–C1). However, T790M/L858R completely break the originally conformational states of EGFR and induce multiple energy basins, which makes EGFR span multiple different conformational subspaces (Fig. 6A2–C2 and S2†). This result suggests that T790M/L858R exerts significant effect on the conformation of EGFR and induce conformational diversity of EGFR.
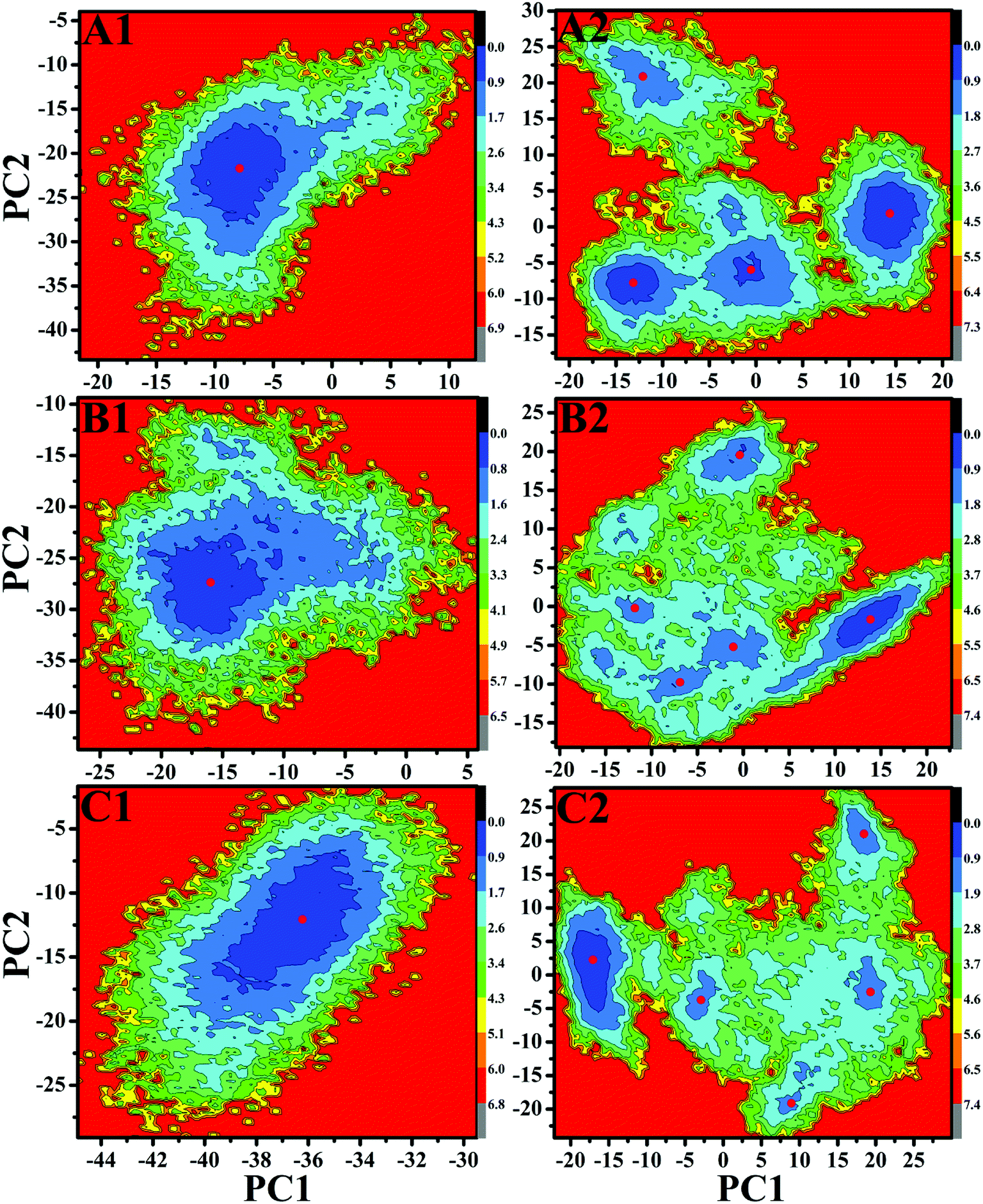 | ||
| Fig. 6 Free energy landscapes of the WT/mutated EGFRs constructed by projecting MD trajectories on the first two principal components PC1 and PC2: (A1/A2), (B1/B2) and (C1/C2) respectively correspond to the WT/mutated EGFRs complexed with inhibitors TAK-285, W2P and HKI-272. | ||
3.3 Binding free energies calculated by MM-PBSA method
The analyses on conformational changes of EGFR suggest that T790M/L858R produce great influence on the conformation of the binding pocket in EGFR, which may further affect binding affinity of inhibitors to EGFR. To assess the effect of T790M/L858R on the binding ability of inhibitors to EGFR, binding free energies of six complexes were computed based on 200 snapshots taken from the equilibrated MD trajectories. In this study, the contribution of entropy changes (−TΔS) to binding free energies was calculated by using the NMA method93 based on 50 snapshots (Table 1). It is noted that the order of the calculated binding free energies is consistent with the experimentally determined one,29 which indicates that the current calculation is theoretically believable.| Energy | TAK-285 | W2P | HKI-272 | |||
|---|---|---|---|---|---|---|
| WT | T790M/L858R | WT | T790M/L858R | WT | T790M/L85R | |
a All values are in kcal mol−1.b ΔGele+pol = ΔEele + ΔGpol.c ΔGbind = ΔEele + ΔEvdw + ΔGpol + ΔGnonpol − TΔS.d The experimental values were derived from the experimental IC50 values using the equation ΔGexp = −TR![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnIC50. In this calculation, the nonlinear regression analysis of the percentage inhibitions was used to compute the IC50 mentioned in ref. 29. lnIC50. In this calculation, the nonlinear regression analysis of the percentage inhibitions was used to compute the IC50 mentioned in ref. 29. |
||||||
| ΔEele | −30.59 ± 4.06 | −20.23 ± 4.42 | −30.16 ± 4.33 | −28.26 ± 4.91 | −23.05 ± 4.12 | −17.86 ± 4.15 |
| ΔEvdw | −62.49 ± 2.64 | −56.14 ± 2.60 | −60.41 ± 2.76 | −55.63 ± 2.28 | −61.71 ± 2.68 | −59.64 ± 2.38 |
| ΔGpol | 60.61 ± 5.07 | 49.58 ± 5.01 | 58.38 ± 4.99 | 57.29 ± 3.62 | 51.04 ± 5.53 | 49.28 ± 4.87 |
| ΔGnonpol | −7.13 ± 0.13 | −6.53 ± 0.31 | −7.34 ± 0.16 | −6.89 ± 0.05 | −7.43 ± 0.33 | −7.50 ± 0.32 |
| bΔGele+pol | 30.02 ± 3.56 | 29.36 ± 3.38 | 28.22 ± 3.15 | 29.03 ± 3.01 | 27.99 ± 3.22 | 31.42 ± 3.83 |
| −TΔS | 22.43 ± 4.15 | 21.76 ± 4.18 | 22.91 ± 4.13 | 21.80 ± 3.79 | 22.93 ± 4.03 | 21.47 ± 3.99 |
| cΔGbind | −17.17 | −11.56 | −16.62 | −11.69 | −18.22 | −14.25 |
| dΔGexp | −11.54 | −6.97 | −11.45 | −7.00 | −11.82 | −9.87 |
To reveal the driving forces behind drug resistance, the individual components of binding free energies were calculated and compared. According to Table 1, although the electrostatic interactions of inhibitors with EGFR provide favorable contributions to inhibitor bindings, this beneficial force is completely screened by the unfavorable polar solvation energies (ΔGpol) to produce the unfavorable polar interactions (ΔGele+pol). Because of T790M/L858R, the polar interaction of TAK-285 with the mutated EGFR is decreased by 0.66 kcal mol−1 relative to the WT one, while polar interactions of W2P and HKI-272 with the mutated EGFR are increased by 0.81 and 3.43 kcal mol−1, respectively. This result suggests that the increase in the polar interactions provides partial contribution for drug resistance of T790M/L858R toward W2P and HKI-272, but not to TAK-285. van der Waals interactions (ΔEvdw) of three inhibitors TAK-285, W2P and HKI-272 with EGFR are favorable for inhibitor bindings. However, these interactions are reduced by 6.35, 4.78 and 2.07 kcal mol−1 in the mutant systems relative to the WT ones, indicating that the decrease in van der Waals interactions also provides partial contribution for drug resistance of T790M/L858R toward three inhibitors. Non-polar interaction (ΔGnonpol) is beneficial to bindings of inhibitors to EGFR. However, the changes in ΔGnonpol caused by T790M/L858R are much smaller than van der Waals interactions, thus non-polar interactions do not provide main contribution to drug resistance. In summary, drug resistance of T790M/L858R in EGFR toward W2P and HKI-272 is mostly dominated by the decrease of van der Waals interactions and the increase in polar interactions in mutated systems, while the main force driving drug resistance of mutations toward TAK-285 mainly comes from the decrease in the van der Waals interactions caused by mutations.
3.4 Drug-resistant mechanisms of mutated EGFR toward inhibitors
To explore the role of separate residues in drug resistance, binding free energies were divided into contributions of individual residues to inhibitor bindings. The residues with relatively significant contributions to binding free energies were shown in Fig. 7. The previous analyses of binding free energies show that the non-polar solvation energies do not provide main contributions to drug resistance, thus we pay more attention to three components including the electrostatic interactions, van der Waals interactions and polar interactions. To further elucidate the drug-resistance mechanism of T790M/L858R toward inhibitors, the separate components of inhibitor–residue interactions were listed in Table S1.† In addition, hydrogen bonding interactions of three inhibitors with EGFR were also analyzed by using the CPPTRAJ program in Amber and the results were shown in Table 2. The geometric positions of inhibitors relative to the key residues of EGFR are depicted in Fig. 8, S3 and S4† by using the lowest energy structure extracted from MD simulations.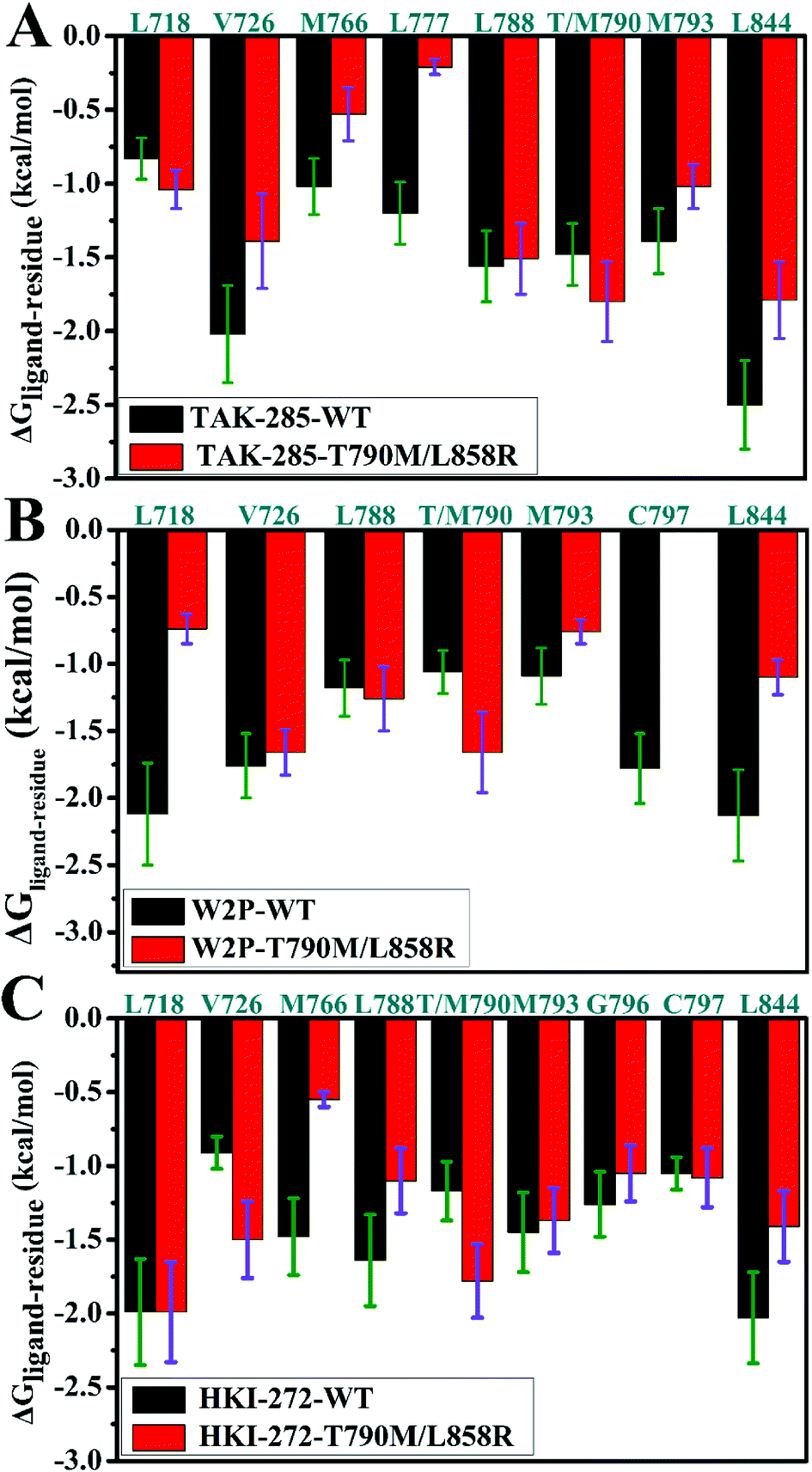 | ||
| Fig. 7 Comparisons of inhibitor–residue interactions between the WT and mutated EGFRs complexed with inhibitors: (A) TAK-285, (B) W2P and (C) HKI-272. | ||
| Compound | aHydrogen bonds | Distance (Å) | Angle (Å) | bOccupancy (%) |
|---|---|---|---|---|
| a Hydrogen bonds are determined by the acceptor⋯donor distance of <3.5 Å (ref. 94) and acceptor⋯H-donor angle of >120°.b Occupancy (%) is defined as the percentage of simulation time that a specific hydrogen bond exists. | ||||
| TAK-285-WT | M793–N–H⋯TAK-285–N | 3.15 | 151.74 | 89.87 |
| TAK-285-T790/L858R | M793–N–H⋯TAK-285–N | 3.11 | 158.15 | 87.19 |
| W2P-WT | M793–N–H⋯W2P–N19 | 3.15 | 154.07 | 92.38 |
| C797–N–H⋯W2P–O33 | 2.91 | 161.44 | 80.30 | |
| W2P-T790M/L858R | M793–N–H⋯W2P–N19 | 3.27 | 152.52 | 81.59 |
| C797–N–H⋯W2P–O33 | 2.90 | 159.64 | 15.69 | |
| HKI-272-WT | M793–N–H⋯HKI-272-NBA | 3.07 | 158.56 | 98.90 |
| C797–N–H⋯W2P-OAG | 2.92 | 155.40 | 59.50 | |
| HKI-272-T790M/L85R | M793–N–H⋯HKI-272-NBA | 3.12 | 152.81 | 90.74 |
| C797–N–H⋯W2P-OAG | 2.94 | 156.02 | 55.49 | |
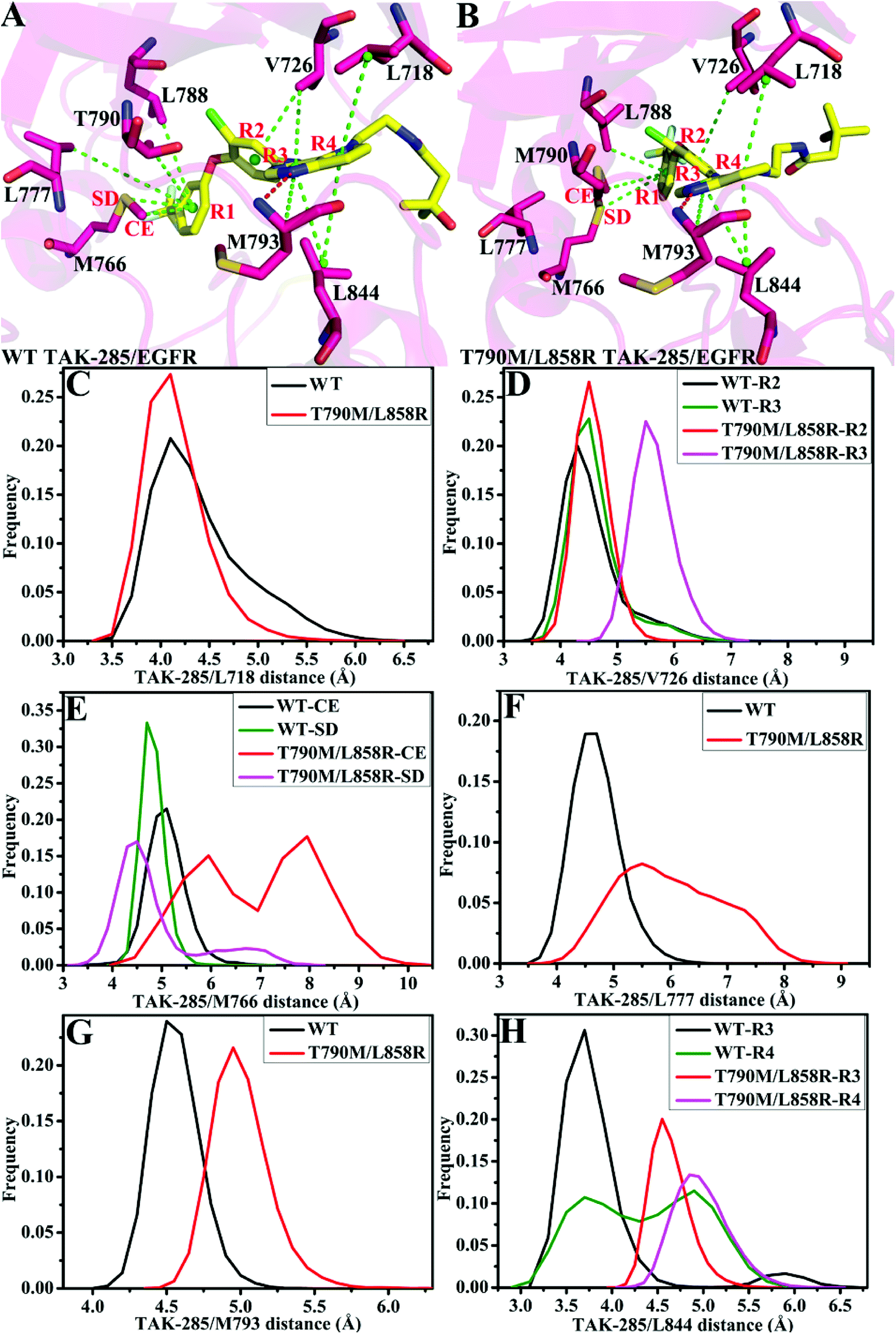 | ||
| Fig. 8 Geometric positions of inhibitor TAK-285 relative to the key residues in the WT and mutated EGFRs. The hydrogen bonds are shown in red line: (A/B) the WT/double mutant EGFRs associated with TAK-285. (C)–(H) are the frequency distribution of distances between TAK-285 and key residues. | ||
As for TAK-285, seven residues V726, M766, L777, L788, T790, M793 and L844 provide important contributions for binding of TAK-285 to the WT EGFR, but in the mutated system six residues L718, V726, L788, M790, M793, and L844 produce strong interactions with TAK-285 (Fig. 7A). Structurally, the alkyl of L788 can form the CH–π interaction with the phenyl ring of TAK-285 in both the WT and mutant systems (Fig. 8A and B), and the difference in this interaction of L788 with TAK-285 is only 0.05 kcal mol−1 between the WT and mutated EGFRs (Fig. 7A). Thus, L788 does not play the role in drug resistance of T790M/L858R toward TAK-285. Except for L788, the energy contributions of the other key residues were significantly changed due to T790M/L858R (Fig. 7A). Structurally, the alkyl group of L718 produces the CH–π interaction with the hydrophobic ring of TAK-285, and the interaction energy of L718 with TAK-285 in the mutated EGFR is strengthened by 0.21 kcal mol−1 compared to the WT one, which owes to the changes in the frequency distribution of the distance between the alkyl group of L718 and the ring of TAK-285 caused by T790M/L858R (Fig. 8C). In addition, the CH–π interaction between the 790th residue of EGFR and TAK-285 is also strengthened by T790M/L858R. The length of the hydrophobic side-chain of M790 in the mutant EGFR is obviously longer than that of T790 in the WT EGFR and shortens the distance of the hydrophobic contact between the 790th residue of EGFR and the phenyl ring of TAK-285, which leads to an increase of 0.32 kcal mol−1 in the CH–π interaction of the 790th residue with TAK-285 in the mutated system relative to the WT one. Thus L718 and the 790th residue do not provide contribution for drug resistance of T790M/L858R on TAK-285 (Fig. 7A). It is also observed that the interactions of TAK-285 with the residues V726, M766, L777, M793 and L844 in the mutated system are respectively decreased by 0.63, 0.49, 0.99, 0.37 and 0.71 kcal mol−1 due to T790M/L858R compared to the WT one, which suggests that V726, M766, L777, M793 and L844 play significant roles in drug resistance of T790M/L858R toward TAK-285. As shown in Table S1,† the energy contributions of V726, M766, L777 and L844 to the bindings mainly originating from van der Waals interactions of these residues with TAK-285 in the WT/mutant EGFRs, while that of M793 to the binding comes from the van der Waals and electrostatic interactions of M793 with TAK-285. The methyl group of V726 in the WT EGFR generates the CH–π interactions with the hydrophobic rings R2 and R3. However, T790M/L858R increase the distance between the rings R2 and R3 of TAK-285 and the methyl of V726, thus the CH–π interactions between them basically disappear in the mutant system (Fig. 8D). According to Fig. 8A, TAK-285 can form the CH–π and S–π interactions with M766 in the WT EGFR, but there is only the S–π interaction formed between TAK-285 and M766 in the mutant EGFR due to the increase in the distance of the CE of M766 away from the hydrophobic R3 of TAK-285 caused by T790M/L858R compared to the WT system (Fig. 8B and E). Structurally, the alkyl of L777 can generate the CH–π interaction with the ring R1 of TAK-285 in the WT EGFR, but this interaction was heavily decreased because of the increase in the distance between L777 and TAK-285 induced by T790M/L858 in the mutated system (Fig. 8A, B and F). Thus, compared to the WT EGFR, the decrease or disappearance of the CH–π interaction of with V726, M766 and L777 with TAK-285 in the mutated one may be the main reasons for drug resistance of T790M/L858 toward TAK-285. M793 can produce hydrogen bonding interactions with TAK-285 (Table 2, Fig. 8A and B), and the difference of electrostatic and polar interactions of M793 with TAK-285 between the WT and mutated EGFRs is very small (Table S1†), which indicates that electrostatic and polar interactions of M793 with TAK-285 are not the main cause of drug resistance. Structurally, the distance of the alkyl in M793 away from the hydrophobic ring R3 of TAK-285 in the mutated EGFR is highly increased by T790M/L858 (Fig. 8G), which heavily weakens the CH–π interaction of M793 with TAK-285 in the mutated EGFR and is partially responsible for drug resistance of T790M/L858 on TAK-285. The interaction of L844 with TAK-285 is mainly contributed by two CH–π interactions between the alkyl group of L844 and two rings (R3 and R4) of TAK-285 in both the WT and mutant EGFRs (Fig. 8A and B). However, the distances of L844 away from R3 and R4 of TAK-285 in the mutated system are extremely increased due to T790M/L858 compared to the WT one (Fig. 8H), which greatly weakens the CH–π interactions between L844 and TAK-285 and provides partial contribution for drug resistance of mutations toward TAK-285.
In the WT EGFR complexed with W2P, the binding attraction of W2P to EGFR mainly comes from seven residues L718, V726, L788, T790, M793, C797 and L844, but in the mutated complex the number of key residues providing significant contributions for the W2P binding is reduced as V726, L788, M790 and L844 due to T790M/L858R (Fig. 7B). The differences in the interactions of W2P with V726 and L788 between the WT and mutated complexes are less than 0.1 kcal mol−1, which shows that these two residues do not offer obvious contributions for drug resistance of T790M/L858R toward W2P. The energy contribution of M790 in the mutated system to binding is increased by 0.6 kcal mol−1 compared to the WT one, and this difference mainly derives from the increase in the van der Waals interactions of M790 with W2P induced by T790M/L858R relative to the WT EGFR (Table S1†). In the WT EGFR, the carbon atom in T790 can produce the CH–π interaction with the hydrophobic ring R1 of W2P. However, in the mutated system, except for the CH–π interaction, the sulfur atom of M790 can form the S–π interaction with the hydrophobic ring R2 due to the increase in the length of the hydrophobic side-chain of M790, which is reflected in the changes in the frequency distribution of the distances between W2P and 790th residues (Fig. S3D†). This result suggests that the 790th residue does not provide contribution for drug resistance T790M/L858R on W2P. As shown in Fig. 7B, the interaction energies of L718, M793, C797 and L844 with W2P in the mutated system are separately reduced by 1.38, 0.33, 1.78 and 1.03 kcal mol−1 due to double mutations compared to the WT one, which indicates that these four residues play significant roles in drug resistance of T790M/L858R toward W2P. The binding attractions of L718 and L844 to W2P are mainly attributed to the van der Waals interactions (Table S1†), while the interaction energies of M793 and C797 with W2P not only come the van der Waals interactions, but also originate from the electrostatic interactions. Hydrophobic residues L718 and L844 can separately generate two CH–π interactions with the hydrophobic rings R3 and R4 of W2P (Fig. S3A†). The increases in the distances of L718 away from the ring R3 of W2P and that of L844 away from the rings R3 and R4 of W2P due to T790M/L858R greatly weaken the CH–π interactions of the alkyls of L718 and L844 with W2P in the mutated system compared to the WT one (Fig. S3C and S3G†), thus the decrease in the van der Waals interaction of L718 and L844 with W2P provides significant contributions for drug resistance of T790M/L858R on W2P. Both M793 and C797 can form the hydrogen bonding interactions with W2P (Fig. S3A and S3B†), and their occupancies are 92.38% and 80.30% in the WT system, respectively. Although T790M/L858R have weak influence on the hydrogen bond M793–N–H⋯W2P–N19, T790M/L858R significantly affect the hydrogen bond C797–N–H⋯W2P–O33 and the occupancy of this hydrogen bond is dropped to 15% in the mutated system (Table 2). The above analyses on hydrogen bonds are basically consistent with the small difference in the electrostatic interaction of M793 with W2P between the WT and mutated EGFRs, as well as a large difference in the electrostatic interaction of C797 with W2P (Table S1†). It can be speculated that the change in hydrogen bonding interaction of C797 with EGFR provides partial contribution to drug resistance of mutations toward W2P. Because of T790M/L858R, the frequency distributions of the distances of M793 and C797 away from W2P has changed (Fig. S3E and S3F†), and the CH–π interaction of M793 with the ring R4 in W2P even disappears, meanwhile, the CH–CH contacts of C797 with W2P is not stable during MD simulations. The van der Waals interactions of W2P with M793 and C797 in the mutated system are separately decreased by 0.32 and 0.37 kcal mol−1 due to T790M/L858R compared to the WT one, which also offers partial contributions for drug resistance of the mutated EGFR toward W2P.
In the WT EGFR complexed with HKI-272, eight residues L718, M766, L788, T790, M793, G796, C797 and L844 generate strong interactions with HIK-271, while in the mutated system eight residues L718, V726, L788, M790, M793, G796, C797 and L844 play key roles in the HKI-272 binding (Fig. 7C). As shown in Fig. 7C, there are very small differences in the energy contribution of L718, M793, G796 and C797 to the HKI-272 binding between the WT and mutated systems, suggesting that although these four residues play important roles in the binding of HKI-272 to EGFR, they do not provide obvious contributions for drug resistance of T790M/L858R toward HKI-272. According to Fig. 7C, T790M/L858R generate significant influence on the interactions of HKI-272 with V726, M766, L788, T790 and L844. The energy contributions of V726 and M790 in the mutated system to the HKI-272 binding are separately strengthened by 0.59 and 0.61 kcal mol−1 compared to the WT one, while the interaction energies of M766, L788 and L844 in the mutant EGFR with HKI-272 are respectively weakened by 0.93, 0.54 and 0.62 kcal mol−1 relative to the WT one. These results indicate that M766, L788 and L844 contribute main force to drug resistance of T790M/L858R toward HKI-272. The sulfur atom of M766 can form the relatively stable S–π interaction with the ring R1 of HKI-272 in the WT system, while this favorable interaction almost disappears in the mutated one due to the increase in the mean distance of the sulfur atom of M766 away from the ring R1 of HKI-272 compared to the WT complex (Fig. S4D†). In addition, the polar interaction of M766 with HKI-272 also shows large difference between the WT and mutant compounds (Table S1†), which suggests that drug resistance of double mutations toward HKI-272 not only comes from the decrease in the van der Waals interaction of HKI-272 with the mutated EGFR relative to the WT one, but also from the slight increase in the polar interaction (Table S1†). Structurally, although L788 can produce the hydrophobic contact with HKI-272 in both the WT and mutant models (Fig. S4A and S4B†), double mutations affect the frequency distribution of the distance of L788 away from the ring R1 (Fig. S4E†). The increase in the distance between L788 and the ring R1 may be the main cause of the decrease in the van der Waals interaction of L788 with HKI-272 in the mutant HKI-272/EGFR complex compared to the WT one. Fig. S4G† displays the frequency distribution of the distance between HKI-272 and L844. It is found that the alkyl group of L844 generates two stable CH–π interactions with the rings R3 and R4 of HKI-272 in the WT system, but L844 can only form a stable hydrophobic contact with R3. Based on the above analysis, it is found that the decrease in the van der Waals interaction of HKI-272 with L788 and L844 in the mutated EGFR relative to the WT one plays a leading role in drug resistance of T790M/L858R toward HKI-272.
Through the above statistical analyses, we can draw reasonable conclusions that the drug resistance of T790M/L858R toward TAK-285 is mainly controlled by five residues (V726, M766, L777, M793 and L844) and that toward W2P come from four residues (L718, M793, C797 and L844), while three residues (M766, L788 and L844) are in an important position in the drug resistance of T790M/L858R toward HKI-272.
4 Conclusions
Investigating the influence of T790M/L858R on the conformational change and drug-resistant mechanism of EGFR is essential for design of efficient inhibitors to relieve drug resistance of the mutated EGFR toward the existing drugs. In the current study, 170 ns traditional MD simulations were performed on the WT and T790M/L858R mutated EGFRs. PC analysis was employed to explore the conformational changes of EGFR induced by T790M/L858R. In addition, MM-PBSA and residue-based free energy decomposition methods were combined to probe drug-resistant mechanisms of the mutated EGFR toward inhibitors.The results from PC analysis indicate that double mutations greatly disturb the structural stability of EGFR, and induce the conformational diversity of EGFR. The calculations of MM-PBSA and residue-based free energy decomposition revealed that drug resistance of EGFR toward TAK-285 and W2P is controlled by the decrease in van der Waals interactions of TAK-285 with the mutated EGFR relative to the WT one, while drug resistance of T790M/L858R toward W2P and HKI-272 is dominated by the decrease in the van der Waals interaction and the increase in the polar interaction of these two inhibitors with the mutated EGFR compared to the mutated one. Therefore, rational optimization of van der Waals interactions of inhibitors with the key residues of EGFR is a key factor in designs of potent inhibitors alleviating drug resistance caused by mutations. This work is expected to provide a theoretical basis for the development of highly effective drugs targeting drug resistance of the mutated EGFR.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China [grant number 11274205], [grant number 11274206], [grant number 11504206]; and major development projects of Shandong Jiaotong University.References
- N. Prenzel, O. M. Fischer, S. Streit, S. Hart and A. Ullrich, Endocr.-Relat. Cancer, 2001, 8, 11–31 CrossRef CAS PubMed.
- F. R. Hirsch, M. Varella-Garcia, P. A. Bunn Jr, M. V. Di Maria, R. Veve, R. M. Bremmes, A. E. Baron, C. Zeng and W. A. Franklin, J. Clin. Oncol., 2003, 21, 3798–3807 CrossRef CAS PubMed.
- C. J. Witton, J. R. Reeves, J. J. Going, T. G. Cooke and J. M. S. Bartlett, J. Pathol., 2003, 200, 290–297 CrossRef CAS PubMed.
- R. A. Walker and S. J. Dearing, Breast Cancer Res. Treat., 1999, 53, 167–176 CrossRef CAS PubMed.
- F. Ciardiello and G. Tortora, N. Engl. J. Med., 2008, 358, 1160–1174 CrossRef CAS PubMed.
- F. R. Hirsch, G. V. Scagliotti, C. J. Langer, M. Varella-Garcia and W. A. Franklin, Lung Cancer, 2003, 41, 29–42 CrossRef.
- V. A. Miller, M. Zakowski, G. J. Riely, W. Pao, M. Ladanyi, A. S. Tsao, A. Sandler, R. Herbst, M. G. Kris and D. H. Johnson, J. Clin. Oncol., 2006, 24, 7003 CrossRef PubMed.
- J. G. Paez, P. A. Jänne, J. C. Lee, S. Tracy, H. Greulich, S. Gabriel, P. Herman, F. J. Kaye, N. Lindeman, T. J. Boggon, K. Naoki, H. Sasaki, Y. Fujii, M. J. Eck, W. R. Sellers, B. E. Johnson and M. Meyerson, Science, 2004, 304, 1497–1500 CrossRef CAS PubMed.
- T. J. Lynch, D. W. Bell, R. Sordella, S. Gurubhagavatula, R. A. Okimoto, B. W. Brannigan, P. L. Harris, S. M. Haserlat, J. G. Supko, F. G. Haluska, D. N. Louis, D. C. Christiani, J. Settleman and D. A. Haber, N. Engl. J. Med., 2004, 350, 2129–2139 CrossRef CAS PubMed.
- W. Pao, V. Miller, M. Zakowski, J. Doherty, K. Politi, I. Sarkaria, B. Singh, R. Heelan, V. Rusch, L. Fulton, E. Mardis, D. Kupfer, R. Wilson, M. Kris and H. Varmus, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13306–13311 CrossRef CAS PubMed.
- B. E. Johnson and P. A. Jänne, Cancer Res., 2005, 65, 7525–7529 CrossRef CAS PubMed.
- X. Tang, H. Shigematsu, B. N. Bekele, J. A. Roth, J. D. Minna, W. K. Hong, A. F. Gazdar and I. I. Wistuba, Cancer Res., 2005, 65, 7568–7572 CrossRef CAS PubMed.
- K. Ohashi, Y. E. Maruvka, F. Michor and W. Pao, J. Clin. Oncol., 2013, 31, 1070–1080 CrossRef CAS PubMed.
- K. P. Chung, J. Y. Shih and C. J. Yu, J. Clin. Oncol., 2010, 28, E701–E703 CrossRef PubMed.
- H. Shigematsu and F. Gazdar Adi, Int. J. Cancer, 2005, 118, 257–262 CrossRef PubMed.
- S. A. Forbes, N. Bindal, S. Bamford, C. Cole, C. Y. Kok, D. Beare, M. M. Jia, R. Shepherd, K. Leung, A. Menzies, J. W. Teague, P. J. Campbell, M. R. Stratton and P. A. Futreal, Nucleic Acids Res., 2011, 39, D945–D950 CrossRef CAS PubMed.
- K. Hashimoto, B. Rogozin Igor and R. Panchenko Anna, Hum. Mutat., 2012, 33, 1566–1575 CrossRef CAS PubMed.
- M. R. Stratton, P. J. Campbell and P. A. Futreal, Nature, 2009, 458, 719–724 CrossRef CAS PubMed.
- S. Kobayashi, T. J. Boggon, T. Dayaram, P. A. Jänne, O. Kocher, M. Meyerson, B. E. Johnson, M. J. Eck, D. G. Tenen and B. Halmos, N. Engl. J. Med., 2005, 352, 786–792 CrossRef CAS PubMed.
- W. Pao, V. A. Miller, K. A. Politi, G. J. Riely, R. Somwar, M. F. Zakowski, M. G. Kris and H. Varmus, PLoS Med., 2005, 2, 225–235 CrossRef CAS.
- L. Sutto and F. L. Gervasio, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10616–10621 CrossRef CAS PubMed.
- B. Liu, B. Bernard and H. Wu Jian, Proteins: Struct., Funct., Genet., 2006, 65, 331–346 CrossRef CAS PubMed.
- C. Yun, T. J. Boggon, Y. Li, M. S. Woo, H. Greulich, M. Meyerson and M. J. Eck, Cancer Cell, 2007, 11, 217–227 CrossRef CAS PubMed.
- J. Stamos, M. X. Sliwkowski and C. Eigenbrot, J. Biol. Chem., 2002, 277, 46265–46272 CrossRef CAS PubMed.
- E. R. Wood, A. T. Truesdale, O. B. McDonald, D. Yuan, A. Hassell, S. H. Dickerson, B. Ellis, C. Pennisi, E. Horne, K. Lackey, K. J. Alligood, D. W. Rusnak, T. M. Gilmer and L. Shewchuk, Cancer Res., 2004, 64, 6652–6659 CrossRef CAS PubMed.
- C. Yun, K. E. Mengwasser, A. V. Toms, M. S. Woo, H. Greulich, K.-K. Wong, M. Meyerson and M. J. Eck, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2070–2075 CrossRef CAS PubMed.
- J. Zhang, P. L. Yang and N. S. Gray, Nat. Rev. Cancer, 2009, 9, 28–39 CrossRef CAS PubMed.
- C. Carmi, E. Galvani, F. Vacondio, S. Rivara, A. Lodola, S. Russo, S. Aiello, F. Bordi, G. Costantino, A. Cavazzoni, R. R. Alfieri, A. Ardizzoni, P. G. Petronini and M. Mor, J. Med. Chem., 2012, 55, 2251–2264 CrossRef CAS PubMed.
- S. Sogabe, Y. Kawakita, S. Igaki, H. Iwata, H. Miki, D. R. Cary, T. Takagi, S. Takagi, Y. Ohta and T. Ishikawa, ACS Med. Chem. Lett., 2013, 4, 201–205 CrossRef CAS PubMed.
- K. Aertgeerts, R. Skene, J. Yano, B. C. Sang, H. Zou, G. Snell, A. Jennings, K. Iwamoto, N. Habuka, A. Hirokawa, T. Ishikawa, T. Tanaka, H. Miki, Y. Ohta and S. Sogabe, J. Biol. Chem., 2011, 286, 18756–18765 CrossRef CAS PubMed.
- E. M. Toloza and T. A. D'Amico, Semin. Thorac. Cardiovasc. Surg., 2005, 17, 199–204 CrossRef PubMed.
- S. M. Thomas and J. R. Grandis, Cancer Treat. Rev., 2004, 30, 255–268 CrossRef CAS PubMed.
- L. Regales, Y. Gong, R. Shen, E. de Stanchina, I. Vivanco, A. Goel, J. A. Koutcher, M. Spassova, O. Ouerfelli, I. K. Mellinghoff, M. F. Zakowski, K. A. Politi and W. Pao, J. Clin. Invest., 2009, 119, 3000–3010 CAS.
- M. A. Lemmon, Exp. Cell Res., 2009, 315, 638–648 CrossRef CAS PubMed.
- M. J. Eck and C.-H. Yun, BBA, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 559–566 CrossRef CAS PubMed.
- Y. Kawakita, H. Banno, T. Ohashi, T. Tamura, T. Yusa, A. Nakayama, H. Miki, H. Iwata, H. Kamiguchi, T. Tanaka, N. Habuka, S. Sogabe, Y. Ohta and T. Ishikawa, J. Med. Chem., 2012, 55, 3975–3991 CrossRef CAS PubMed.
- T. Ishikawa, M. Seto, H. Banno, Y. Kawakita, M. Oorui, T. Taniguchi, Y. Ohta, T. Tamura, A. Nakayama, H. Miki, H. Kamiguchi, T. Tanaka, N. Habuka, S. Sogabe, J. Yano, K. Aertgeerts and K. Kamiyama, J. Med. Chem., 2011, 54, 8030–8050 CrossRef CAS PubMed.
- M. Karplus and J. A. McCammon, Nat. Struct. Mol. Biol., 2002, 9, 646–652 CrossRef CAS PubMed.
- P. Auffinger and E. Westhof, Curr. Opin. Struct. Biol., 1998, 8, 227–236 CrossRef CAS PubMed.
- P. Marimuthu and K. Singaravelu, Biochemistry, 2018, 57, 1249–1261 CrossRef CAS PubMed.
- T. J. Hou, J. M. Wang, Y. Y. Li and W. Wang, J. Chem. Inf. Model., 2011, 51, 69–82 CrossRef CAS PubMed.
- D. F. Shi, Q. F. Bai, S. Y. Zhou, X. W. Liu, H. X. Liu and X. J. Yao, Proteins: Struct., Funct., Genet., 2018, 86, 43–56 CrossRef CAS PubMed.
- M. J. Yang, X. Q. Pang, X. Zhang and K. L. Han, J. Struct. Biol., 2011, 173, 57–66 CrossRef CAS PubMed.
- E. L. Wu, Y. Mei, K. L. Han and J. Z. H. Zhang, Biophys. J., 2007, 92, 4244–4253 CrossRef CAS PubMed.
- C. G. Ji and J. Z. H. Zhang, J. Am. Chem. Soc., 2008, 130, 17129–17133 CrossRef CAS PubMed.
- L. Q. Yang, P. Sang, R. P. Zhang and S. Q. Liu, RSC Adv., 2017, 7, 42094–42104 RSC.
- J. Z. Chen, J. A. Wang, F. B. Lai, W. Wang, L. X. Pang and W. L. Zhu, RSC Adv., 2018, 8, 25456–25467 RSC.
- D. Poger and A. E. Mark, Biochemistry, 2014, 53, 2710–2721 CrossRef CAS PubMed.
- N. F. Endres, R. Das, A. W. Smith, A. Arkhipov, E. Kovacs, Y. Huang, J. G. Pelton, Y. Shan, D. E. Shaw, D. E. Wemmer, J. T. Groves and J. Kuriyan, Cell, 2013, 152, 543–556 CrossRef CAS PubMed.
- A. Arkhipov, Y. Shan, R. Das, N. F. Endres, M. P. Eastwood, D. E. Wemmer, J. Kuriyan and D. E. Shaw, Cell, 2013, 152, 557–569 CrossRef CAS PubMed.
- W. L. Jorgensen and L. L. Thomas, J. Chem. Theory Comput., 2008, 4, 869–876 CrossRef CAS PubMed.
- Y. Kita, T. Arakawa, T. Y. Lin and S. N. Timasheff, Biochemistry, 1994, 33, 15178–15189 CrossRef CAS PubMed.
- X. Y. Jia, M. T. Wang, Y. H. Shao, G. König, B. R. Brooks, J. Z. H. Zhang and Y. Mei, J. Chem. Theory Comput., 2016, 12, 499–511 CrossRef CAS PubMed.
- M. Zacharias, T. P. Straatsma and J. A. McCammon, J. Chem. Phys., 1994, 100, 9025–9031 CrossRef CAS.
- D. L. Beveridge and F. M. DiCapua, Annu. Rev. Biophys. Biophys. Chem., 1989, 18, 431–492 CrossRef CAS PubMed.
- H. Gohlke and D. A. Case, J. Comput. Chem., 2004, 25, 238–250 CrossRef CAS PubMed.
- I. Massova and P. A. Kollman, Perspect. Drug Discovery Des., 2000, 18, 113–135 CrossRef CAS.
- J. M. Wang, P. Morin, W. Wang and P. A. Kollman, J. Am. Chem. Soc., 2001, 123, 5221–5230 CrossRef CAS PubMed.
- L. L. Duan, G. Q. Feng, X. W. Wang, L. Z. Wang and Q. G. Zhang, Phys. Chem. Chem. Phys., 2017, 19, 10140–10152 RSC.
- J. Z. Chen, J. N. Wang and W. L. Zhu, Phys. Chem. Chem. Phys., 2017, 19, 3067–3075 RSC.
- F. F. Yan, X. G. Liu, S. L. Zhang, J. Su, Q. G. Zhang and J. Z. Chen, J. Biomol. Struct. Dyn., 2017 DOI:10.1080/07391102.2017.1394221.
- G. D. Hu, A. J. Ma, X. H. Dou, L. L. Zhao and J. H. Wang, Int. J. Mol. Sci., 2016, 17, 1–15 Search PubMed.
- L. L. Duan, X. Liu and J. Z. H. Zhang, J. Am. Chem. Soc., 2016, 138, 5722–5728 CrossRef CAS PubMed.
- J. Su, X. G. Liu, S. L. Zhang, F. F. Yan, Q. G. Zhang and J. Z. Chen, Chem. Biol. Drug Des., 2018, 91, 828–840 CrossRef CAS PubMed.
- J. Z. Chen, X. Y. Wang, T. Zhu, Q. G. Zhang and J. Z. H. Zhang, J. Chem. Inf. Model., 2015, 55, 1903–1913 CrossRef CAS PubMed.
- T. J. Hou, W. Zhang, D. A. Case and W. Wang, J. Mol. Biol., 2008, 376, 1201–1214 CrossRef CAS PubMed.
- M. D. Disney, I. Yildirim and J. L. Childs-Disney, Org. Biomol. Chem., 2014, 12, 1029–1039 RSC.
- G. D. Hu and J. H. Wang, Eur. J. Med. Chem., 2014, 74, 726–735 CrossRef CAS PubMed.
- H. Y. Sun, Y. Y. Li, M. Y. Shen, S. Tian, L. Xu, P. C. Pan, Y. Guan and T. J. Hou, Phys. Chem. Chem. Phys., 2014, 16, 22035–22045 RSC.
- T. C. Schutt, V. S. Bharadwaj, D. M. Granum and C. M. Maupin, Phys. Chem. Chem. Phys., 2015, 17, 16947–16958 RSC.
- V. S. Bharadwaj, S. Kim, M. T. Guarnieri and M. F. Crowley, Sci. Rep., 2018, 8, 12826 CrossRef PubMed.
- V. S. Bharadwaj, A. M. Dean and C. M. Maupin, J. Am. Chem. Soc., 2013, 135, 12279–12288 CrossRef CAS PubMed.
- S. Wold, K. Esbensen and P. Geladi, Chemom. Intell. Lab. Syst., 1987, 2, 37–52 CrossRef CAS.
- J. Z. Chen, RSC Adv., 2016, 6, 58573–58585 RSC.
- H. Berman, K. Henrick, H. Nakamura and J. L. Markley, Nucleic Acids Res., 2007, 35, D301–D303 CrossRef CAS PubMed.
- D. A. Case, R. M. Betz, D. S. Cerutti, T. E. Cheatham III, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, C. Lin, T. Luchko, R. Luo, B. Madej, D. Mermelstein, K. M. Merz, G. Monard, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. Roitberg, C. Sagui, C. L. Simmerling, W. M. Botello-Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, L. Xiao and P. A. Kollman, AMBER 2016, University of California, San Francisco, 2016 Search PubMed.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins: Struct., Funct., Genet., 2006, 65, 712–725 CrossRef CAS PubMed.
- J. M. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- D. C. Bas, D. M. Rogers and J. H. Jensen, Proteins: Struct., Funct., Genet., 2008, 73, 765–783 CrossRef CAS PubMed.
- H. Li, A. D. Robertson and J. H. Jensen, Proteins: Struct., Funct., Genet., 2005, 61, 704–721 CrossRef CAS PubMed.
- A. Jakalian, B. Jack David and I. Bayly Christopher, J. Comput. Chem., 2002, 23, 1623–1641 CrossRef CAS PubMed.
- A. Jakalian, L. Bush Bruce, B. Jack David and I. Bayly Christopher, J. Comput. Chem., 2000, 21, 132–146 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- T. G. Coleman, H. C. Mesick and R. L. Darby, Ann. Biomed. Eng., 1977, 5, 322–328 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- J. Z. Chen, J. Biomol. Struct. Dyn., 2018, 36, 351–361 CrossRef CAS PubMed.
- D. R. Roe and T. E. Cheatham, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- B. R. Miller, T. D. McGee, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, J. Chem. Theory Comput., 2012, 8, 3314–3321 CrossRef CAS PubMed.
- S. Genheden and U. Ryde, Expert Opin. Drug Discovery, 2015, 10, 449–461 CrossRef CAS PubMed.
- J. D. Durrant, C. A. F. de Oliveira and J. A. McCammon, J. Mol. Graphics Modell., 2011, 29, 773–776 CrossRef CAS PubMed.
- B. S. Xu, H. J. Shen, X. Zhu and G. H. Li, J. Comput. Chem., 2011, 32, 3188–3193 CrossRef CAS PubMed.
- K. Raghavan, M. R. Reddy and M. L. Berkowitz, Langmuir, 1992, 8, 233–240 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06844e |
| This journal is © The Royal Society of Chemistry 2018 |