
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel multifunctional Ag and Sr2+ co-doped TiO2@rGO ternary nanocomposite with enhanced p-nitrophenol degradation, and bactericidal and hydrogen evolution activity†
Xueyu Wei *ab,
Jiashun Cao*a and
Fang Fanga
*ab,
Jiashun Cao*a and
Fang Fanga
aCollege of Environment, Hohai University, Nanjing – 210098, P. R. China. E-mail: wxyu1027@126.com; cjsprof@163.com
bSchool of Civil Engineering and Architecture, Anhui Polytechnic University, Wuhu – 241000, P. R. China
First published on 12th September 2018
Abstract
In the present study, a novel multifunctional Sr2+/Ag–TiO2@rGO ternary hybrid photocatalyst was prepared via facile sol–gel and hydrothermal methods. The prepared catalyst was well characterized by UV-vis, XRD, Raman, HRTEM and XPS. The synthesized composite was utilised for p-NP degradation, E. coli disinfection and H2 generation under visible light. The Sr2+/Ag–TiO2@rGO catalyst showed enhanced photocatalytic H2 evolution rate (64.3 μmol h−1) compared with Ag–TiO2@rGO (30.1 μmol h−1) and TiO2 (no activity). Nearly complete degradation of 15 mg l−1 p-NP was achieved over Sr2+/Ag–TiO2@rGO after 3 h, while only 66% and 5% was achieved by Ag–TiO2@rGO and TiO2 respectively. Furthermore, TEM analysis was carried out on Escherichia coli (E. coli) before and after visible light irradiation to understand the inactivation mechanism and DNA analysis indicated no fragmentation during inactivation. Radical quantification experiments and ESR analysis suggested that ·OH and O2˙− were the main ROS in the degradation and disinfection processes. The superior photocatalytic H2 evolution rate of Sr2+/Ag–TiO2@rGO was attributed to the synergetic effect between the Ag, Sr2+ and TiO2 components on the rGO surface. The localized SPR effect of Ag induced visible light generated charge carriers into the conduction band of the TiO2 and Sr2+ which further transfer to the rGO for the reduction of H+ ions into H2. The results suggest that Sr2+/Ag–TiO2@rGO structures could not only induce separation and migration efficiency of charge carries, but also improve charge collection efficiency for enhanced catalytic activity. Thus, we believe that this work could provide new insights into multifunctional nanomaterials for applications in solar photocatalytic degradation of harmful organics and pathogenic bacteria with clean energy generation during wastewater treatment.
Introduction
Wastewater comprises various pollutants such as toxic chemicals, pathogenic bacteria and viruses. Although, different strategies have been employed to eliminate these pollutants, it still remains a great challenge to eliminate hazardous chemicals and disease-causing microorganisms employing a single material. This objective could be solved by means of preparing new multifunctional composite materials that are capable of degrading chemical pollutants and eliminating common pathogenic bacteria effectively with simultaneous generation of clean energy (hydrogen) via an eco-friendly process.For many years, nitro aromatic compounds have been considered as one of the most refractory and carcinogenic pollutants in waste water due to their high stability and solubility.1 The reduction of p-nitrophenol (p-NP) to p-aminophenol (p-AP) is more amicable way, since p-AP is an important intermediate compound for the manufacture of analgesic and antipyretic drugs, photographic films, corrosion inhibitor, anticorrosion-lubricant, and hair-dyeing agents.2 Semiconductor photocatalysts imposes a great potential to resolve the serious world energy crisis and several environmental issues induced by extensive consumption of non-renewable fossil fuels and organic chemicals.3 It constitutes a green technology to produce hydrogen and oxygen by breaking down water through harnessing solar light energy under ambient conditions.4,5 Photocatalysis is an efficient technology for addressing with environmental pollution, because of its high efficiency and eco-friendly and pollution free catalytically generated by products.6,7 Titanium dioxide is one of the most potential and intensively used oxides for several environmental remediation, water splitting and organic synthesis applications.8 It has several advantages over other semiconductors for instance, low cost, chemical stability, high catalytic activity, and biocompatibility.8,9 However, due to its wideband gap (Eg ranging from ca. 3.0 to ca. 3.2 eV), its poor response to light in the visible region, and the fast recombination of the photogenerated electron–hole pairs, which drastically render its catalytic applications.10,11 Different metals like Pt, Pd, and Au on different kind of supports have been used as photocatalysts, among them in terms of cost and resources abundance, Ag could be a good replacement due to its high catalytic performance and low cost. Amongst many TiO2 composites, Ag/TiO2 has been widely studied in degradation of nitroaromatic compounds, however the degradation efficiency was low.12,13
Doping of transition elements into TiO2 lattice, by physical and chemical processes, have been found to be useful approaches to obtain visible light sensitized catalysts.14 However, these metal dopants have to be used in small quantities to avoid the recombination of photo-generated electrons and holes, which is bolstered by the large amount of these dopants.15 Thus, low concentration co-doping of cations and anions could be an effective solution to enhance the visible light absorption efficiency as well as to reduce the recombination of the photo-generated charges. Co-doping with two different atoms into TiO2/ZnO matrices has attracted significant attention since they can exhibit higher photo-catalytic activity and unique characteristics compared to doping them with a single element.16–19
Furthermore, enhanced TiO2 based nanostructures can also be designed by means of coupling with an ideal sink of photogenerated electrons to overcome the problem of recombination during photocatalysis. For this, graphene is an ideal choice to accept and shuttle electrons during photocatalysis due to its excellent properties. Graphene is a single-layer, two-dimensional carbonaceous material that has attracted much attention because of its fascinating electronic, mechanical, thermal and optical characteristics.20–22 Due to its remarkable nature, graphene has been widely applied in various areas, such as nanoelectronics, chemical and biochemical sensing, solar cells, especially photocatalysis.23,24 As a result of high specific surface area, excellent transparency, superior electron mobility, and high chemical stability, graphene can be used as an ideal high performance candidate for photocatalyst carrier or promoter.25 Hence graphene based photocatalysts have been considering great research interests for their potential in photocatalysis. For instance, Divya et al. synthesised TiO2/ZnO/RGO/Ag ternary composite through microwave method which showed enhanced activity in degradation of rhodamine B.26 In another study, Biswas et al. prepared TiO2–graphene quantum dot multifunctional material with superior photocatalytic and biofilm eradication properties.27 Moreover, few more studies are also available on the synthesis of single metal cation doped TiO2, graphene based composites but main focus was to examine their performance in hydrogen evolution rather the degradation of organic compounds, however the efficiency was low.28,29
To the best of our knowledge, very limited studies have been reported on the photocatalysis of composite materials consisting of TiO2, graphene and single transition metal cation. Thus, the integration of TiO2, graphene and dual transition metal into nanocomposites is expected to possess enhanced photocatalytic properties compared to single cation doped and pure TiO2. In the present study novel Sr2+/Ag–TiO2@rGO (SAT@rGO) ternary hybrid photocatalyst was prepared via facile sol–gel and hydrothermal methods. Sr2+/Ag–TiO2@rGO catalyst showed enhanced performance in degradation of p-NP, disinfection of E. coli and in hydrogen evolution activity compare to Ag–TiO2@rGO (AT@rGO). We expected our work could give a new train of thought on exploration of graphene-based nanocomposites for various environmental applications.
Materials and methods
Materials
Graphite powder, H2SO4, HNO3, KMnO4, HCl, isopropanol and ethanol were obtained from Alibaba Chemicals. Titanium(IV) isopropoxide, Tween 20, AgNO3, Sr(NO3)2 polyethylene glycol (PEG, 300) were obtained from Sigma.Preparation of SAT@rGO composite
TiO2 nanoparticles are prepared using the following method; a mixture of 5 mL of titanium(IV) isopropoxide in 50 mL isopropanol was added drop wise to 200 mL of distilled water maintained at pH 1.5 while the solution was continuously stirred. This TiO2 sol was dried at 100 °C for 24 h, and then calcined at 450 °C for 4 h to obtain the nanoparticles. Ag/TiO2 (AT) and Sr2+/Ag–TiO2 (SAT) nanoparticles were prepared by the above method with few modifications. For AT nanoparticles preparation, required amount of aqueous solutions of AgNO3, while for SAT nanoparticles required amounts of aqueous solutions of both AgNO3 and Sr(NO3)2 were added drop wise to the TiO2 sol with continuous stirring for 45 min. A small aliquot of distilled water, 0.05 M hydrazine hydrate and 5 mL of tween 20 were added to all the above solutions with continuous stirring for additional 30 min. The resultant sol was sonicated at 80 MHz for 90 min and then dried at 100 °C in a hot air oven for 24 h to get the dry gel. The gel was then calcinated at 450 °C to obtain required nanoparticle powders.Graphene oxide (GO) was prepared from the graphite powder (flake size of <50 m) according to the modified Hummers method.30 Further, preparation of SAT@rGO ternary composite was carried out by a facile hydrothermal treatment which was proposed to provide an intrinsic contact between TiO2 and rGO better than UV photo-reduction and chemical reduction methods.31 Thus hydrothermal method was employed in the present study. Briefly, 100 mg of GO aqueous solution (1 mg mL−1) was exfoliated through sonication for 2 h till to get a stable suspension. To this, 0.90 g of above prepared AT or SAT aqueous suspension (10 mL) and 1 mL of PEG was added drop wise under vigorous stirring until homogenous mixture was obtained. Further, 30 mL of ethanol was added to the above mixture and continued stirring for 2 h, and then transferred to a 100 mL stainless steel Teflon lined autoclave maintained at 180 °C for 12 h. The obtained powder was washed with water and alcohol, and then dried at 80 °C for 6 h to obtain the final composite powder.
Characterization of nanocomposite
Powder XRD crystallogram was recorded using X-ray BRUKER D8 Advance X-ray diffractometer with Cu Kα source (λ = 1.5406 Å). JEOL JEM 2100 high resolution transmission electron microscope (HRTEM) was used for imaging, SAED pattern and energy dispersive X-ray pattern with an accelerating voltage of 200 KV at different magnification. Raman spectroscopy analysis was carried out using Thermo Scientific DXR2. Diffuse reflectance spectra were recorded using JASCO V-670 UV-Vis spectrophotometer. The photoluminescence (PL) spectra were obtained using HITACHI F-7000 fluorescence spectrophotometer. XPS data was acquired using Kratos Axis Ultra 165 spectrometer with a monochromated Al Kα X-ray source (hα = 1486.6 eV).Photocatalytic degradation of p-NP
The photocatalytic performance of the synthesised composites was investigated by visible light degradation of p-nitrophenol (p-NP). A 250 W xenon lamp was used as a light source and the light density was adjusted to 100 mW cm−2. In the degradation experiment 20 mg of the photocatalysts were dispersed in 100 mL of 15 ppm p-NP solutions. The suspensions were then stirred in the dark for 30 min to reach an adsorption/desorption equilibrium. Subsequently the solutions were then irradiated with visible-light while stirring (Fig. S3†). A small aliquot were extracted out at regular intervals and centrifuged to remove the catalyst. The collected p-NP solution was eventually analyzed by using a high performance liquid chromatography (Shimadzu, Japan) equipped with Eclipse XDBC18 (4.6 × 150 mm, 5 μm) reverse phase column. The UV detector wavelength was 265 nm, while the mobile phase consisted of methanol (50%) and acetonitrile + acetic acid (50% + 1.6 mL L−1) fed at a flow rate of 1 mL min−1 for 20 minutes.Bactericidal performance
A Gram negative E. coli bacterium was utilised for photocatalytic disinfection tests at log phase comprising of 108 cfu mL−1 incubated in nutrient broth. The disinfection tests were carried out with a 250 W Xe lamp with cut-off filter as visible light source (Fig. S3†). During the disinfection tests SAT@rGO photocatalyst (5 mg) was stirred with bacterial suspension in saline solution (10 mL, 0.9% NaCl at pH 7.0) and exposed to visible light. Small volume (100 μL) from the test suspension samples were taken at regular time intervals and spread on freshly prepared agar plates, which were further incubated at 37 °C for 24 h. Standard plate count method was used to determine viable number of cells as cfu mL−1. Furthermore, TEM analysis of untreated and treated bacterial samples was carried out by placing 10 μL of each cell dispersion on TEM grids and air-dried before examination using the TEM (JEOL JEM-2100).DNA analysis
The DNA of the bacterial samples was extracted with OMEGA bacteria DNA Kit (D3350-01) from the bacterial suspension solution at different time intervals. Electrophoresis analysis was performed to separate and visualize DNA fragment in a 0.8% agarose gel at 130 V for 25 min. Genomic DNA was stained with the gel stain.Photocatalytic hydrogen evolution
The photocatalytic hydrogen evolution experiment was carried out in a quartz reactor with a magnetic stirrer. Typically, 5 mg of the photocatalysts were dispersed through magnetic stirring in 100 mL of water containing 10 mL triethanolamine (sacrificial agent). A 250 W xenon lamp (∼100 mW cm−2) was utilized as light source. Before irradiation the suspension was stirred in dark for 1 h to obtain adsorption/desorption isotherm. Then the solution was irradiated with visible light for 3 h, the hydrogen product from the aqueous solution was analysed by an offline gas chromatography (Shimadzu GC-2014) equipped with a thermal conductivity detector (TCD) with a molecular sieve (5 Å) column, at 70 °C, using N2 as the carrier gas. Control and the blank (without catalyst) experiments were carried out under identical conditions for comparison.Results and discussion
Characterization of nanocomposites
The crystalline phase of synthesised TiO2, TiO2@rGO, AT@rGO and SAT@rGO ternary composites were analysed by X-ray diffraction as shown in Fig. 1a. All the XRD patterns display majorly the distinctive TiO2 diffraction peaks. In all the samples, anatase phase was confirmed by the 2θ peaks at 25.3°, 37.8°, 47.8°, 53.9°, 54.9°, 62.6°, 69.0°, 70.2° and 75.0° which can be indexed to (101), (004), (200), (105), (211), (204), (116), (220) and (215) crystal planes respectively. Furthermore, either a typical diffraction peak of rGO and doped metal nanoparticles (Ag and Sr) or a peak shift is noticed. These results could be ascribed to that the main characteristic peak of rGO at around 24.5° could be shielded by the main anatase peak at 25.2°, similar kind of results also reported in the literature which also confirms the formation of rGO.32,33 The XRD pattern of the prepared GO shown peaks at 10.8° and 42.2° corresponding to (001) and (100) reflections, while rGO exhibited its typical diffraction peak at 24.8° corresponding to (002) plane respectively (Fig. S1†). These results suggested that the addition of dopant metals did not develop new crystal planes or changes in preferential orientations of TiO2 due to lower amount and relatively lower diffraction intensity of silver, strontium and rGO. | ||
| Fig. 1 X-ray diffraction pattern (a) and Raman spectra of as prepared composites (inset in (b) shows D and G bands of rGO). | ||
The Raman spectra of the prepared TiO2 composites show a strong Raman band at 144 cm−1, which is ascribed to the Eg mode of anatase TiO2 (Fig. 1b). Other bands observed at 397 cm−1, 516 cm−1, and 638 cm−1 were associated with the B1g, A1g, and Eg modes respectively. In AT and SAT composites, all the Raman bands of the TiO2 were observed, while no Raman active band from Ag and Sr was observed due lower doping levels. Furthermore, the B1g, A1g, and Eg modes mode of TiO2 were observed to shift due to the interaction of the metal atoms in TiO2 with the rGO layers. In addition, the AT@rGO and SAT@rGO composites shown D and G bands (shown in inset) of GO at 1347 cm−1 and 1604 cm−1 confirms the presence of rGO in the prepared composites.
The morphology of the synthesized nanocomposite (SAT and SAT@rGO) photocatalysts were characterized by TEM and images are shown in Fig. 2. As shown in Fig. 2a the SAT nanoparticles shows spherical morphology with small agglomeration in the range of 20–40 nm, while Fig. 2c indicates the SAT@rGO composite in which the particles are also in the range of 20–45 nm. HRTEM images (Fig. 2b and d) of the composites indicated the TiO2 with lattice spacing of 0.35 nm corresponds to the (101) plane. From these results, it is evident that SAT nanoparticles were finely embedded in thin layer of graphene sheets. Therefore, a good contact and interaction between SAT nanoparticles and graphene achieved by the proposed sol–gel and hydrothermal process is believed to be favourable in transferring the photogenerated electrons into SAT@rGO composite system during photocatalytic degradation of p-NP and for hydrogen evolution as well.
 | ||
| Fig. 2 TEM images of as prepared SAT (a and b) and SAT@rGO (c and d) composite. | ||
XPS analysis was carried out to ensure the chemical composition and chemical state of the SAT@rGO composite. The apparent XPS scans were obtained for C 1s, Ag 3d, Sr 3d, Ti 2p and O 1s levels (Fig. 3) to investigate the composition of the synthesised composite. In the relevant high-resolution XPS spectrum of C 1s core level the main peak was observed at 284.6 eV (C–C), which corresponds to the adscititious carbon adsorbed on the surface of the composite, while the peak at 285.6 eV corresponds to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. The relevant peak at 288.6 eV (CO) indicates the presence of Ti–O–C bond at the interface of the composite photocatalyst (Fig. 3b).29,33 Ag at 3d core levels (Fig. 3c) shows that the Ag 3d5/2 and Ag 3d3/2 binding energies at 368.0 eV and 374.1 eV were in good agreement with the bulk metallic silver.34,35 The Sr 3d core level (Fig. 3d) peak was observed at 133.7 eV 135.4 eV was in complete agreement with the literature.36,37 The binding energies of Ti 2p (Fig. 3e), the photoelectron peaks at 458.5 and 464.3 eV were representative of Ti 2p3/2 and Ti 2p1/2 core levels respectively.35,36 The O 1s peak at 531.5 eV is attributed to the significant hydroxyl groups on the surface of GO. Furthermore, the O 1s peak of TiO2/graphene at around 530.1 eV may be assigned to O in Ti–O–C bond38 (Fig. 3f). These results further confirm the strong bondage between TiO2 and GO by the proposed hydrothermal method.
C bonds. The relevant peak at 288.6 eV (CO) indicates the presence of Ti–O–C bond at the interface of the composite photocatalyst (Fig. 3b).29,33 Ag at 3d core levels (Fig. 3c) shows that the Ag 3d5/2 and Ag 3d3/2 binding energies at 368.0 eV and 374.1 eV were in good agreement with the bulk metallic silver.34,35 The Sr 3d core level (Fig. 3d) peak was observed at 133.7 eV 135.4 eV was in complete agreement with the literature.36,37 The binding energies of Ti 2p (Fig. 3e), the photoelectron peaks at 458.5 and 464.3 eV were representative of Ti 2p3/2 and Ti 2p1/2 core levels respectively.35,36 The O 1s peak at 531.5 eV is attributed to the significant hydroxyl groups on the surface of GO. Furthermore, the O 1s peak of TiO2/graphene at around 530.1 eV may be assigned to O in Ti–O–C bond38 (Fig. 3f). These results further confirm the strong bondage between TiO2 and GO by the proposed hydrothermal method.
 | ||
| Fig. 3 XPS survey spectrum of SAT@rGO (a), high resolution XPS spectra of C 1s of graphene oxide (b) Ag 3d (c), Sr 3d (d) Ti 2p (e) and O 1s (f). | ||
UV-Vis absorption spectra of the prepared composites were shown in Fig. S2(a).† From the results it indicates that the absorption edge shows a red shift from 340 nm to 390 nm for AT@rGO while it shows a maximum red shift from 340 nm to 400 nm in SAT@rGO. This shift is happened due to decrease in band gap by introducing silver and strontium into TiO2 matrix. Furthermore, from the figure it can be seen that the addition of silver into TiO2 showed a abroad absorption band between 400 nm and 600 nm, which could be attributed to the SPR effect of silver nanoparticles.28,39 The red shifted spectrum shows a possible evidence for good interaction between rGO, TiO2, Ag and Sr species. Hence, the observed red shift behaviour clearly justifies the change in the light absorption characteristics, exhibited by the co-doped TiO2@rGO materials, from UV to visible light region. The inference plot of hν vs. (αhν)2 was utilized to get optical energy band gap values (Eg) as shown in Fig. S2(b).† It showed in the figure that the energy band gap values were reduced to 2.87 eV for and SAT@rGO nanoparticles. It was worth noticed that the doping of TiO2 with two transition metals into its lattice and good interaction with graphene could be accompanied with a decrease in the energy band gap and an increase in the wavelength (red shift). These results indicated that SAT@rGO nanoparticles have greater possibility to exhibit a higher photo–catalytic activity in the visible region.
Visible light degradation of p-NP
The visible light photocatalytic performances of the synthesised composites were evaluated by using p-NP as a representative pollutant for the degradation. The UV-vis absorption spectra of p-NP (Fig. 4a) solution at different time intervals with SAT@rGO composite indicated that the absorption band of p-NP at 402 nm was obviously decreased with increase in irradiation time and disappeared after 3 h. Fig. 4b shows the relative concentrations of p-NP remaining in the solution as a function of the irradiation time with different photocatalysts. The degradation reaction kinetics of p-NP can be described by a Langmuir–Hinshelwood model (eqn (1)) which illustrated that the reactions took place at a solid–liquid interface. The degradation of p-NP with visible-light irradiation revealed pseudo first-order kinetics with regression coefficients (R2) ≥0.90. The HPLC analysis of p-NP at initial and after 3 h of degradation further confirms the competition of mineralisation (Fig. S4†). Among the various tested photocatalysts, SAT@rGO composite exhibited the best performance with rate constant k = 0.503 min−1| ln[Co/Ct] = Kt | (1) |
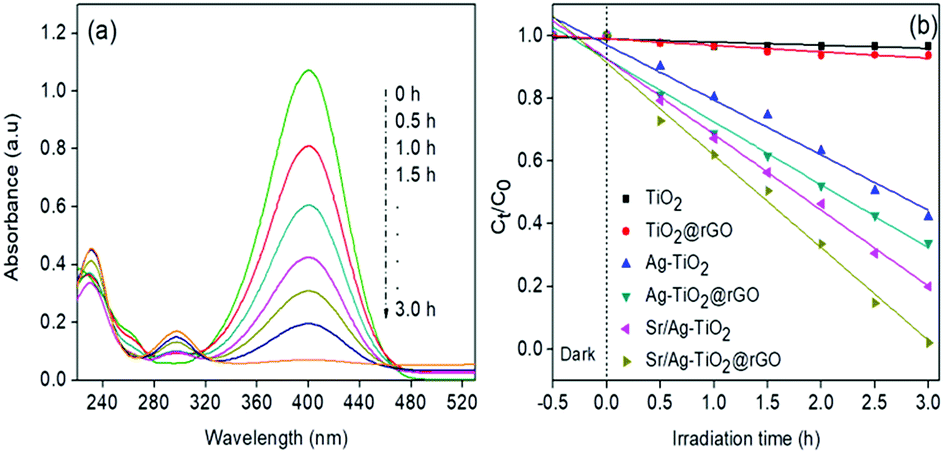 | ||
| Fig. 4 (a) UV-vis absorption spectra of p-NP at different time intervals with SAT@rGO (b) kinetics of p-NP degradation with synthesised graphene composites. | ||
The effectiveness of a catalyst also depends on its stability after prolonged use. Thus, the stability of the most effective photocatalyst (SAT@rGO) was studied by performing recycle tests up to four cycles. The activity of the photocatalyst under visible light did not change significantly and remained above 95% of its original activity (Fig. S5a†). Further to confirm the phase changes in the crystal structure of the synthesised nanorods. XRD spectra were obtained after 4 successful cycles of reuse, indicating that no significant changes in the crystal nature reveals that Sr/Ag–TiO2@rGO composite is an active and stable visible-light driven photocatalyst (Fig. S5b†).
Bactericidal activity against E. coli
Pure TiO2, AT and SAT were tested in order to investigate the effect of transition metal doping on the visible light bactericidal activity. Experiments were carried out in light and dark in order to understand the effect of visible light irradiation during disinfection. From the Fig. 5 it shows that AT@rGO and SAT@rGO nanoparticles have some minimal effect on bacterial survival in the dark, this is due to the presence of Ag or Ag+ and penetration of nanoparticles into the cell membrane, leading to the cell death. AT@rGO nanoparticles showed 26% while SAT@rGO showed 39% of inhibition in the dark after 5 h of incubation. Bare TiO2 and control samples in the presence of visible light have not shown any effect on bacterial survival. Furthermore, nearly 71% of inhibition was found in the case of AT@rGO, while 96% of inhibition has been observed for SAT@rGO nanoparticles after 5 h of incubation.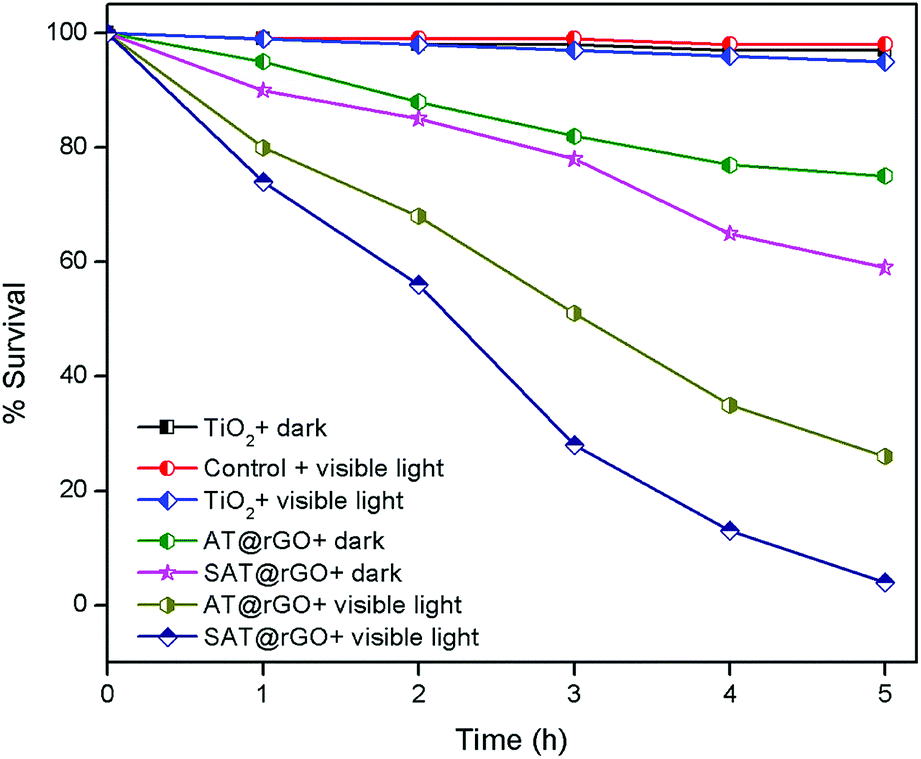 | ||
| Fig. 5 Percentage survival of E. coli with prepared nanoparticles as a function of time. | ||
The enhanced bactericidal activity of both AT@rGO and SAT@rGO composites under visible light irradiation could be attributed to the synergistic effect of the nanoparticles. Further, visible light irradiation induces photoexcitation of the catalyst which leads to the production of reactive oxygen species (ROS) ·OH and O2˙− radicals. These radicals initially damage the surfaces of the bacterial cells with an attack of ROS, before breakage of the cell membranes occur at weak points. A relatively high rate of bactericidal activity was found in the case of SAT@rGO nanoparticles which could be due to the co-doping of two metals leads to high photoexcitation and ·OH and O2˙− radicals production. Our results are in good agreement with some previous reports.40,41
To further understand the morphological changes in the bacterial membrane during photocatalytic disinfection process, TEM analysis was carried out after 5 h of irradiation by SAT@rGO composite. In the control sample, the undamaged cell wall membranes of E. coli bacteria indicating cells were healthy (Fig. 6c). However after 5 h of visible light irradiation in the presence of the catalyst it shown complete damage in the cell wall membrane (Fig. 6d). These results show that the cell membrane ruptured during the photooxidation process by ·OH and O2˙− radicals. Further to identify that the bacterial inactivation was a result of DNA damage, we carried out experiments to examine the effects using SAT@rGO by gel electrophoresis and showed in Fig. S6.† It indicates that the DNA extracted from E. coli suspension did not undergo fragmentation during the photocatalysis process, this results reveals that the disinfection is mainly due to the membrane damage as expected in our results of bacterial disinfection.
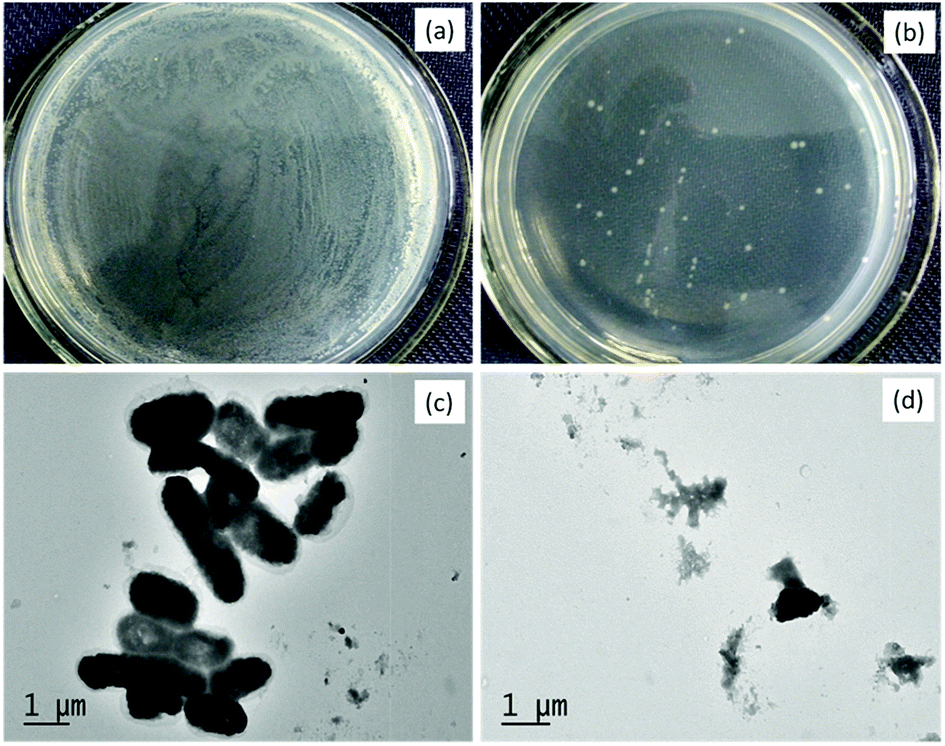 | ||
| Fig. 6 Photographs of E. coli cells before (a) and after (b) irradiation; TEM images of E. coli cells before (c) and after (d) irradiation. | ||
The ESR technique is used to further confirm the reaction mechanism and detect the active species involved in the photodegradation of p-NP and disinfection of E. coli over SAT@rGO under visible light. The characteristic signals of DMPO-·OH and DMPO-·O2− species could be confirmed based on the standard spectra of ·OH (AN = 14.8 G and AH = 14.8 G; g = 2.0032) and ·O2− (AN = 13.2 G and AH = 11.2 G; g = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 032) simulated by Bruker WinEPR SimFonia programme (Fig. 7a and b).42–44 The above results confirms that ·OH, O2˙− and photogenerated holes are the main active species during the photocatalytic process.
032) simulated by Bruker WinEPR SimFonia programme (Fig. 7a and b).42–44 The above results confirms that ·OH, O2˙− and photogenerated holes are the main active species during the photocatalytic process.
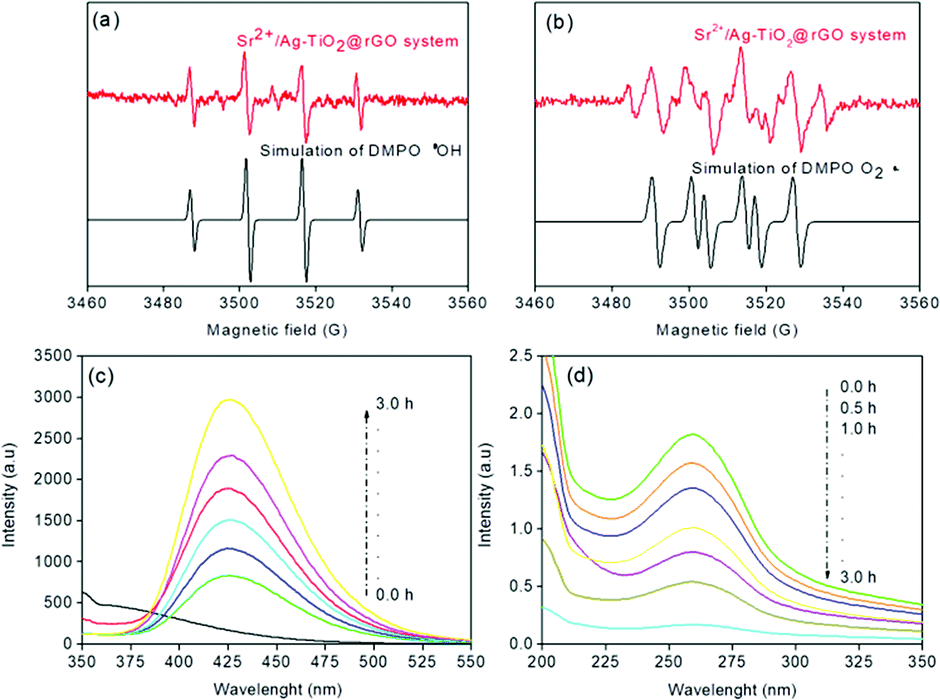 | ||
| Fig. 7 ESR signals of ·OH (a) and O2˙− (b) at ambient temperature, (c) fluorescence spectra of a TAOH, (d) spectra of NBT transformation using SAT@rGO under visible light irradiation. | ||
Further experiments have been carried out to support the mechanism and analyze that O2˙− and ·OH radicals are the main species for the effective disinfection of E. coli and p-NP degradation over SAT@rGO composite. For quantification of ·OH radicals, the terephthalic acid photoluminescence probing technique (TA-PL) was employed since TA could readily react with ·OH radicals to from a highly fluorescent 2-hydroxyterephthalic acid (TAOH) (Scheme S1†).45,46 The highest PL intensity was observed over SAT@rGO composite as expected (Fig. 7c). This should be attributed to the easy charge transfer, which results in the improvement of holes on the VB of SAT@rGO to produce more ·OH, resulting in the highest PL peak of SAT@rGO with respect to time. The quantification of O2˙− production have been carried out thorough the transformation of NBT (Scheme S2†) during the photocatalytic reaction.47 Fig. 7d showed the spectra of the transformation percentage of NBT at different time intervals. From the figure it can be seen that SAT@rGO composite shown much higher transformation percentage of NBT. After the photoexcitation of the catalyst, most of electrons generated are remained in the conduction band to react with O2 to generate more O2˙−. These results demonstrated that due to smaller energy band gap the photogenerated electrons in valance band can easily transfer to the conduction band of SAT@rGO, resulting in more effective charge separation and reducing the probability of recombination of electron–hole pairs, which finally leads to that more photogenerated electrons can react with O2 to produce O2˙− and take part in decomposition of p-NP and disinfection of E. coli.
Photocatalytic H2 production
The as-prepared graphene composites have also been evaluated for photocatalytic water splitting hydrogen generation. From Fig. 8, it shows that SAT@rGO composite shown as high as 187.9 μmol g−1 after 3 h of visible light irradiation with an average production rate 0.643 mmol g−1 h−1 with triethanolamine as sacrificial reagent. Whereas the hydrogen generation rate of AT@rGO was only about 79.8 μmol g−1 after 3 h with 0.310 mmol g−1 h−1, while no significant generation of hydrogen with pure TiO2 and TiO2@rGO composites under visible light irradiation. The results demonstrate that the photocatalytic activity of TiO2 could be greatly improved by addition of silver, strontium nanospecies and rGO. The enhanced visible light photocatalytic performance in H2 production over the SAT@rGO composite could also be attributed to the unique microstructures and synergistic effect of two metals and excellent electron transference capacity of rGO. | ||
| Fig. 8 (a) Hydrogen evolution as a function of time and (b) hydrogen production rate of various composites under visible light irradiation. | ||
Photocatalytic reaction mechanism
The photocatalytic reaction mechanism of the prepared SAT@rGO composite under visible light irradiation is illustrated in Fig. 9. The Ag Fermi energy level is lower to that of conduction band of TiO2. The enhanced visible light activity could to ascribe to the formation of hetero-junction electric field among the TiO2, silver and strontium atoms. Furthermore, SPR photo induced energetic electrons of Ag and electron transfer induced by doped Sr2+ present in the material will diffuse through TiO2/rGO into the conduction band of TiO2 at ultrafast speed. Further energized electrons in the conduction band of TiO2 can be transferred to the rGO sheet since the potential energy of graphene/rGO is lower than conduction band of TiO2 and slightly higher than the potential of H+/H2.48 Thus, the enhanced photocatalytic activity of the SAT@rGO composite could also be attributed to the synergistic effect of dopant metals and improved visible light absorption due to SPR effect of silver and formation of chemical bonding between TiO2 and rGO during hydrothermal process. | ||
| Fig. 9 Proposed photocatalytic degradation and disinfection mechanism over SAT@rGO composite. | ||
Conclusions
In conclusion, SAT@rGO nanocomposite was successfully prepared by sol–gel, hydrothermal methods and photoreduction methods. In this ternary system, TiO2 nanoparticles were doped with two metal atoms (Ag and Sr2+) in the lattice and further this was overspread on the surface of graphene nanosheets uniformly. Visible light degradation of p-nitrophenol and disinfection of E. coli experiments proved that surface plasmon resonance (SPR) phenomenon and synergistic effect of dopant metals had an effect on photoreaction enhancement. The hydrogen evolution rate of SAT@rGO was much higher than AT@rGO and TiO2@rGO. The enhanced photocatalytic performance can be attributed to the presence of rGO, dopant metals and SPR effect, with longer lifetime of photo-generated electron–hole pairs and faster interfacial charge transfer rate. This work proposes that the visible light photo activity of the graphene based ternary nanocomposites is superior to the binary catalysts. We believe that this work could give new insights on investigation of multifunctional graphene based nanocomposites for several environmental applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Natural Science Key Foundation of Education Committee of Anhui Province (No. KJ2016A063 and No. KJ2017A119), the Fundamental Research Funds for the Central Universities (No. 2016B44014), the Natural Science Foundation of China (No. 51502106), the Anhui Provincial Natural Science Foundation (No. 1808085ME133).References
- M. A. J. Harrison, S. Barra, D. Borghesi, D. Vioneb, C. Arsenec and R. I. Olariu, Atmos. Environ., 2005, 39, 231–248 CrossRef.
- S. Saha, A. Pal, S. Kundu, S. Basu and T. Pal, Langmuir, 2010, 26, 2885–2893 CrossRef PubMed.
- X. B. Chen, C. Li, M. Gratzel, R. Kostecki and S. S. Mao, Chem. Soc. Rev., 2012, 41, 7909–7937 RSC.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef PubMed.
- N. S. Lewis, Science, 2007, 315, 798–801 CrossRef PubMed.
- S. I. Nikitenko, T. Chave, C. Cau, H. P. Brau and V. Flaud, ACS Catal., 2015, 5, 4790–4795 CrossRef.
- B. Hu, F. Cai, T. Chen, M. Fan, C. Song, X. Yan and W. Shi, ACS Appl. Mater. Interfaces, 2015, 7, 18247–18256 CrossRef PubMed.
- S. Ananthakumar, J. Ramkumar and S. M. Babu, Renewable Sustainable Energy Rev., 2016, 57, 1307–1321 CrossRef.
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798–801 CrossRef PubMed.
- M. Kapilashrami, Y. Zhang, Y. S. Liu, A. Hagfeldt and J. Guo, Chem. Rev., 2014, 114, 9662–9707 CrossRef PubMed.
- S. A. Ansari, M. M. Khan, M. O. Ansari and M. H. Cho, New J. Chem., 2016, 40, 3000–3009 RSC.
- H. J. Zhang, X. Y. Li and G. H. Chen, J. Mater. Chem., 2009, 19, 8223–8231 RSC.
- X. Wang, Z. Zhao, D. Ou, B. Tu, D. Cui, X. Wei and M. Cheng, Appl. Surf. Sci., 2016, 385, 445–452 CrossRef.
- U. G. Akpan and B. H. Hameed, Appl. Surf. Sci., 2010, 375, 1–11 Search PubMed.
- V. Iliev, D. Tomova and S. Rakovsky, Desalination, 2010, 260, 101–106 CrossRef.
- Z. Q. He, X. Xu, S. Song, L. Xie, J. J. Tu, J. M. Chen and B. Yan, J. Phys. Chem. C, 2008, 112, 16431–16437 CrossRef.
- H. R. Pouretedal, A. Norozi, M. H. Keshavarz and A. Semnani, J. Hazard. Mater., 2009, 162, 674–681 CrossRef PubMed.
- Y. Y. Chen, Y. B. Xie, J. Yang, H. B. Cao, H. Liu and Y. Zhang, Sci. China: Chem., 2013, 56, 1783–1789 CrossRef.
- J. Xiao, Y. Xie, H. Cao, F. Nawaz, S. Zhang and Y. Wang, J. Photochem. Photobiol., A, 2016, 315, 59–66 CrossRef.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef PubMed.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef PubMed.
- H. L. Guo, X. F. Wang, Q. Y. Qian, F. B. Wang and X. H. Xia, ACS Nano, 2009, 3, 2653–2659 CrossRef PubMed.
- H. Zhang, X. J. Lv, Y. M. Li, Y. Wang and J. H. Li, ACS Nano, 2010, 4, 380–386 CrossRef PubMed.
- X. Li, X. Wang, L. Zhang, S. Lee and H. Dai, Science, 2008, 319, 1229–1232 CrossRef PubMed.
- Y. Sun, Q. Wu and G. Shi, Energy Environ. Sci., 2011, 4, 1113–1132 RSC.
- K. S. Divya, M. M. Xavier, P. V. Vandana, V. N. Reethu and S. Mathew, New J. Chem., 2017, 41, 6445–6454 RSC.
- A. Biswas, G. Salunke, P. Khandelwal, R. Das and P. Poddar, New J. Chem., 2017, 41, 2642–2657 RSC.
- Y. Yang, E. Liu, H. Dai, L. Kang, H. Wu, J. Fan, X. Hu and H. Liu, Int. J. Hydrogen Energy, 2014, 39, 7664–7671 CrossRef.
- N. R. Khalid, E. Ahmed, M. Ahmad, N. A. Niaz, M. Ramzan, M. Shakil, T. Iqbal and A. Majid, Ceram. Interfaces, 2016, 42, 18257–18263 CrossRef.
- H. A. Becerril, J. Mao, Z. Liu, R. M. Stoltenberg, Z. Bao and Y. Chen, ACS Nano, 2008, 2, 463–470 CrossRef PubMed.
- W. Fan, Q. Lai, Q. Zhang and Y. Wang, J. Phys. Chem. C, 2011, 115, 10694–10701 CrossRef.
- X. Pan, Y. Zhao, S. Liu, C. L. Korzeniewski, S. Wang and Z. Fan, ACS Appl. Mater. Interfaces, 2012, 4, 3944–3950 CrossRef PubMed.
- H. M. El-Bery, Y. Matsushita and A. Abdel-moneim, Appl. Surf. Sci., 2017, 423, 185–196 CrossRef.
- E. Sumesh, M. S. Bootharaju, Anshup and T. Pradeep, J. Hazard. Mater., 2011, 189, 450–457 CrossRef PubMed.
- Y. Lai, H. Zhang, K. Xie, D. Gong, Y. Tang, L. Sun, C. Lin and Z. Chen, New J. Chem., 2010, 34, 1335–1340 RSC.
- H. A. Hamedani, N. K. Allam, H. Garmestani and M. A. El-Sayed, J. Phys. Chem. C, 2011, 115, 13480–13486 CrossRef.
- S. Sood, A. Umar, S. K. Mehta, A. S. K. Sinha and S. K. Kansal, Ceram. Int., 2015, 41, 3533–3540 CrossRef.
- W. S. Wang, D. H. Wang, W. G. Qu, L. Q. Lu and A. W. Xu, J. Phys. Chem. C, 2012, 116, 19893–19901 CrossRef.
- E. Vasilaki, I. Georgaki, D. Vernardou, M. Vamvakaki and N. Katsarakis, Appl. Surf. Sci., 2015, 353, 865–872 CrossRef.
- M. Li, M. E. Noriega-Trevino, N. Nino-Martinez, C. Marambio-Jones, J. Wang, R. Damoiseaux, F. Ruiz and E. M. V. Hoek, Environ. Sci. Technol., 2011, 45, 8989–8995 CrossRef PubMed.
- X. Wang and T. T. Lim, Water Res., 2013, 47, 4148–4158 CrossRef PubMed.
- B. Jiang, X. Wang, Y. Liu, Z. Wang, J. Zheng and M. Wu, J. Hazard. Mater., 2016, 304, 457–466 CrossRef PubMed.
- D. R. Duling, J. Magn. Reson., Ser. B, 1994, 104, 105–110 CrossRef PubMed.
- J. Zou, J. Ma, L. Chen, X. Li, Y. Guan, P. Xie and C. Pan, Environ. Sci. Technol., 2013, 47, 11685–11691 CrossRef PubMed.
- K. I. Ishibashi, A. Fujishima, T. Watanabe and K. Hashimoto, Electrochem. Commun., 2000, 2, 207–210 CrossRef.
- N. Arunkumar and R. Vijayaraghavan, RSC Adv., 2014, 4, 65223–65231 RSC.
- L. Ye, J. Liu, Z. Jiang, T. Peng and L. Zan, Appl. Catal., B, 2013, 142–143, 1–7 Search PubMed.
- Q. Xiang, J. Wu and M. Jaroniec, Nanoscale, 2011, 3, 3670–3678 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06813e |
| This journal is © The Royal Society of Chemistry 2018 |