DOI:
10.1039/C8RA06589F
(Paper)
RSC Adv., 2018,
8, 34625-34633
Preparation of a novel injectable in situ-gelling nanoparticle with applications in controlled protein release and cancer cell entrapment
Received
5th August 2018
, Accepted 2nd October 2018
First published on 9th October 2018
Abstract
Temperature sensitive injectable hydrogels have been used as drug/protein carriers for a variety of pharmaceutical applications. Oligo(ethylene glycol) methacrylate (OEGMA) monomers with varying ethylene oxide chain lengths have been used for the synthesis of in situ forming hydrogel. In this study, a new series of thermally induced gelling hydrogel nanoparticles (PMOA hydrogel nanoparticles) was developed by copolymerization with di(ethylene glycol) methyl ether methacrylate (MEO2MA), poly(ethylene glycol) methyl ether methacrylate (300 g mol−1, OEGMA300), and acrylic acid (AAc). The effects of acrylic acid content on the physical, chemical, and biological properties of the nanoparticle-based hydrogels were investigated. Due to its high electrostatic properties, addition of AAc increases LCST as well as gelation temperature. Further, using Cy5-labelled bovine serum albumin and erythropoietin (Epo) as model drugs, studies have shown that the thermogelling hydrogels have the ability to tune the release rate of these proteins in vitro. Finally, the ability of Epo releasing hydrogels to recruit prostate cancer cells was assessed in vivo. Overall, our results support that this new series of thermally induced gelling systems can be used as protein control releasing vehicles and cancer cell traps.
1. Introduction
Three-dimensional (3D) injectable hydrogel scaffolds have been widely used in cell therapy and tissue regeneration based on their unique characteristics, including controlled porosity, high water content, and mimicking of the microenvironment of natural extracellular matrices.1,2 Although the 3D porous scaffolds can be either formed in situ or preformed, the injectable in situ forming scaffolds offer many advantages over preformed ones. Specifically, cells and bioactive molecules can be readily incorporated into the in situ forming matrix by simply mixing prior to solidification. In addition, in situ forming gels can be implanted via a needle injection unlike implantation of preformed scaffolds that often require costly surgical procedures with a risk of complications.3 Last but not least, in situ forming scaffolds, but not preformed gel, can easily fill irregularly-shaped defects which are associated with different injuries and trauma.3,4 Consequently, there has been an increasing focus on the development of injectable in situ-forming systems for biomedical applications in recent years. Based on their gelation mechanisms, in situ forming hydrogel scaffolds can be categorized into chemically- and physically-crosslinked scaffolds.5 Chemically-crosslinked hydrogel scaffolds can be formed by either in situ polymerization or crosslinking reactions between the components. However, the toxicity and reactive nature of chemical reagents used in scaffold fabrication may adversely affect the survival and bioactivity of seeded cells and bioactive molecules.6 To overcome such shortcomings, in situ physical-crosslinking mechanisms in response to certain environmental changes such as pH, temperature, ion concentration, solvent and light, have gained in popularity.7–12
Among all in situ gelling systems, thermally induced gelling mechanism is the most common method to produce in situ forming scaffolds. Typically, these gel systems can flow at room temperature but solidify immediately and form 3D scaffolds when brought to body temperature (37 °C). There are two benefits of using thermally induced gel in biomedical applications. Firstly, toxic reagents and crosslinkers are not required for the production of many thermally induced gelling systems. Secondly, thermally induced gelling system can be made of a wide variety of biomaterials including natural polymers and their derivatives, synthetic polymers and polypeptides.8–16 While many of these thermally induced gelling systems were composed of linear or branched polymers, thermosensitive nano-/micro-size polymer particles have recently been used as building blocks for in situ forming hydrogel scaffolds. Compared to these linear/branched polymer-based thermogelling systems, the particle-based ones have several advantages including the reduced viscosity and improved mechanical properties at the same concentration. Furthermore, the building blocks (particles) can be employed as carriers of growth factors or bioactive molecules to deliver them in a controlled manner for guiding differentiation of stem cells.13,17,18 Among these particle-based thermogelling systems, poly(N-isopropylacrylamide) (PNIPAM)-based microgels are the most studied and have been widely explored as in situ forming scaffolds for use in tissue engineering.13,17,19–26 PNIPAM polymer has a lower critical solution temperature (LCST) at near to physiological temperature therefore, PNIPAM-based microgels can be used for reversible cell adhesion or detachment and for triggered release of therapeutics.24,25 However, there are limitations for long-term application in biotechnology.27 The monomer of PNIPAM, N-isopropylacrylamide (NIPAM) is carcinogenic as well as the byproducts are neurotoxic, perhaps generated by hydrolysis of PNIPAM.18 To overcome these concerns, a series of thermosensitive polymers or nanoparticles based on oligo(ethylene glycol) methacrylate (OEGMA) with different ethylene glycol chain lengths have been developed.28–31 By copolymerization of OEGMA monomers with different length of ethylene glycol chain, the as-prepared nanoparticles have a wide range of LCST, similar to those of PNIPAM or PEG-based hydrogels.28–31 For instance, a copolymer hydrogel based on di(ethylene glycol) methacrylate (MEO2MA, n = 2) and OEGMA475 (mw = 475 g mol−1, n = 8, 9) has a volume phase transition temperature (VPTT) between 23 °C and 90 °C.32 These results support the overall hypothesis of this work that thermally induced gelling system can be made of OEGMA particles.
Herein, we fabricated a series of new injectable in situ gelling hydrogel with three monomers, di(ethylene glycol) methacrylate (MEO2MA), OEGMA (mw = 300 g mol−1), and acrylic acid (AAc). The P(MEO2MA-co-OEGMA300-co-AAc) (PMOA) hydrogel has physiochemical and biological properties similar to PNIPAM and PEG. Specifically, we have synthesized three hydrogel nanoparticles with different content of acrylic acid (0, 1, and 3 mol% of AAc.) The physiochemical and biological properties of the three hydrogels were assessed in vitro. Finally, the capability of this new thermally induced gelling system to release proteins and recruit circulating cancer cells was assessed in vivo.
2. Materials and methods
2.1. Materials
Di(ethylene glycol) methyl ether methacrylate (MEO2MA), poly(ethylene glycol) methyl ether methacrylate (300 g mol−1, OEGMA300), acrylic acid (AAc), ethylene glycol dimethacrylate (EGDMA), and ammonium persulfate (APS) were purchased from Sigma-Aldrich (St Louis, Missouri). Sodium dodecyl sulfate (SDS) was obtained from Bio-Rad (Hercules, CA). Milli-Q grade deionized water was used through all experiments.
2.2. Synthesis of P(MEO2MA-co-OEGMA300-co-AAc) (PMOA) hydrogel nanoparticles
A series of hydrogel nanoparticles were synthesized via a free radical precipitation polymerization method.18 In brief, MEO2MA, OEGMA300, AAc, SDS, EGDMA and DI water were mixed in a 500 mL round bottle flask. The mixture was heated to 70 °C in a water bath under nitrogen purging. After 30 min, APS solution was added to the mixture to initiate polymerization. The reaction was allowed to proceed for 6 hours with magnetic stirring at 70 °C under N2 atmosphere. Here three batches of nanoparticles with different AAc contents (0, 1, and 3 mol%) were prepared (Table 1), and defined as PMOA0, PMOA1, and PMOA3, respectively. The above-prepared nanoparticle dispersions were purified with exhaustive dialysis (cutoff: 10 kDa) against deionized water for one week. The purified nanoparticles were concentrated and collected using a centrifuge, and then stored in a refrigerator for further use.
Table 1 Monomer composition in feed (moles). Where the total moles of three monomers are 0.026 moles, SDS is as a detergent and APS is as an initiator
| Samples |
MEO2MA (mol) |
OEGMA300 (mol) |
AAc (mol) |
EGDMA (mol) |
SDS (mol) |
APS (mol) |
| PMOA0 |
0.02314 |
0.0026 |
0 |
0.00026 |
0.000139 |
0.000438 |
| PMOA1 |
0.02288 |
0.0026 |
0.00026 |
0.00026 |
0.000139 |
0.000438 |
| PMOA3 |
0.02236 |
0.0026 |
0.00078 |
0.00026 |
0.000139 |
0.000438 |
2.3. Size, polydispersity, zeta potential, and morphology of the particle
The size, polydispersity, and zeta potential of the hydrogel nanoparticles were determined using a ZetaPALS dynamic light scattering (DLS) detector (Brookhaven Instruments, Holtsville, NY).33 The samples at a concentration of 1 mg mL−1 in DI water were prepared, and sizes of these nanoparticles were determined at 24 and 37 °C, respectively. Furthermore, to observe morphology of the nanoparticles, Scanning Electron Microscope (SEM) was employed as described earlier.34 Briefly, a drop of the diluted particle dispersion was placed onto a glass slide cover adhered to a SEM specimen holder with the conductive tape, and then dried at ambient temperature. After sputter-coating with silver, SEM images were recorded by a Hitachi S-4800 II FE Scanning Electron Microscope.34
2.4. Conductivity measurement
Using a FP30 Conductivity meter (Mettle Toledo, Columbus, OH), carboxyl group content of the nanoparticles was determined quantitatively following a published method.35 1.0 mL of HCl solution (0.01 M) was added into a 20 mL of the nanoparticle dispersion (30 mg mL−1), followed by stirring for 20 min. Conductivity measurement was conducted using NaOH aqueous solution (0.01 M) as a titrant. The COOH contents of these nanoparticles were calculated based on the following formulation: COOH (μmole mg−1) = C × V/W, where C and V are the concentration and used volume of NaOH solution, respectively. W is the solid content of the nanoparticles.
2.5. Turbidity test
To determine the phase transition temperature of the nanoparticles, the turbidity of the nanoparticles in DI water and phosphate buffered saline (PBS, 43.5 mM ionic strength) was respectively measured using a Beckman DU640 UV-visible spectrophotometer (Fullerton, CA).36 Briefly, 2.0 mL of the nanoparticle dispersion (1.0 mg mL−1) was prepared in a UV cuvette, and covered with a lid to keep water from evaporation during the experiments. The cuvette was placed in a water bath equipped with a temperature controller and incubated for one minute after the predetermined temperature was reached. The transmittance of the dispersion at various temperature points was recorded at 550 nm.
2.6. Viscosity test
The viscosity of the nanoparticle dispersions as a function of temperature was measured using an HR-2 Discovery Hybrid Rheometer (TA Instruments).18,37 Two flat parallel plates (25 cm in diameter) were used, and the distance between two plates was adjusted to 0.6 mm. During the experiments, a constant stress of 2 Pa and a frequency of 0.1 Hz was applied. To measure viscosity, 0.6 mL of nanoparticle dispersions (60 mg mL−1 in PBS) was loaded on the plate, viscosity of the samples was recorded with temperature increasing at a rate of 2 °C min−1 from 15 to 40 °C.
2.7. Water loss
The water retention of the hydrogels was determined using an established method.38 Briefly, 1.0 mL of the nanoparticle dispersion (60 mg mL−1 in PBS) was added to an Eppendorf tube, total mass (Wo) of the tube and the sample was recorded. The tube was then transferred into a water bath and incubated at 37 °C for 4 hours. The lost water from the hydrogels during the incubation was discarded carefully from the top, and total mass (Wi) of tube and hydrogel was weighted. The weight percentage of lost water was defined as: (Wo − Wi)/Wo × 100%. To observe pore structures of the hydrogels, at the end of water loss experiments, the thermally gelling hydrogels were quickly frozen in liquid nitrogen and dried in vacuo. The cross sections of the hydrogels were observed under an SEM as described above.
2.8. In vitro release of protein
Bovine serum albumin (BSA) and erythropoietin (Epo) was used as a model protein drug and labelled with Cy®5 dye (Lumiprobe Co., Hunt Valley, MD) following the manufacturer's protocol. The release kinetics of Cy®5-labeled BSA/Epo was then determined as described previously.18 Briefly, 1.0 mL of nanoparticle dispersion (60 mg mL−1 in PBS) was mixed with 1.0 mg of Cy®5-labeled BSA/Epo at room temperature and the mixture was heated to 37 °C to allow for gelation. Then, 2.0 mL of pre-heated PBS media (37 °C) was added onto the top of the hydrogel. The sample was quickly put into an incubator at 37 °C under gentle shaking. At different time intervals, 150 μL of the supernatants were taken and added into wells of a 96-wells plate. Fluorescence intensities were recorded using a microplate reader (Spectra Max Gemini EM XPS, Molecular Devices, San Jose, CA) at an excitation of 640 nm and an emission of 700 nm.
2.9. In vitro cell cytotoxicity
Cytotoxicity of the thermally-gelling hydrogels to cells was conducted according to the publication.39 Briefly, 0.54 mL of DMEM was added into the thermally-gelling hydrogels and incubated at 37 °C in a cell culture incubator. After culture for 3 days, 0.06 mL was taken out as the conditioned media for the cell viability assay. NIH 3T3 fibroblasts were plated at a density of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well in a 48-well plate and cultured in DMEM media containing 20% of the above-collected conditioned media. Cell viability was then characterized using a modified Alamar Blue dye assay.40
000 cells per well in a 48-well plate and cultured in DMEM media containing 20% of the above-collected conditioned media. Cell viability was then characterized using a modified Alamar Blue dye assay.40
2.10. In vivo biocompatibility and cancer cell trap
Animal subcutaneous implantation model was used to determine the tissue compatibility of thermally-gelling hydrogel as described previously.41 In brief, a 100 μL of the pre-gel nanoparticle dispersion (60 mg ml−1) and saline as control was subcutaneously injected in dorsally in a Balb/C mouse (25 g body weight) obtained from The Jackson Laboratory (Bar Harbor, ME). After implantation for 24 hours, the mouse was sacrificed and the implants and the surrounding tissues were isolated, and then sectioned for H&E staining. Additionally, a subcutaneously implanted cancer trap mouse model was used to assess the ability of chemokine-releasing thermal gelling hydrogel for recruiting prostate cancer cells.42 2.0 μL of Epo (10 units in PBS) was mixed with 50 μL of the thermally-gelling nanoparticle dispersion (80 mg mL−1 in PBS), and then was injected into both sides of the back of mice via 18-gauge needle. After 12 hours, intravascular injection of prostate cancer cells (DAB2IP-knockdown PC3 cells, gift from Dr Jer-Tsong Hsieh at the University of Texas Southwestern Medical Center at Dallas) was administered into mice. The cells were labelled with Vybrant™ DiD Cell-Labeling Solution (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's protocol before injection. In vivo cell recruitment to the sites of hydrogel implants was monitored using Kodak In Vivo Imaging System FX Pro (Carestream Health Inc., New Haven, CT, USA) as described previously.42 At the end of the study, implants and surrounding tissues were frozen sectioned and analyzed using H&E and immunohistochemistry as previously described.43 Images were taken utilizing a Leica fluorescence microscope (Leica Microsystem GmbH, Wetzlar, Germany) combined with a Retiga-EXi CCD camera (QImaging, Surrey, BC, Canada). Cell number was calibrated using ImageJ software. This study was performed in strict accordance with the AAALAC guidelines – Guide for the care and use of laboratory animals (NRC 2011) and was approved by Animal Care and Use Committee of the University of Texas at Arlington.
2.11. Statistical analysis
All the data were evaluated using two-tailed Student t-test and presented as mean ± standard deviation. Statistical analyses of all data were performed by a Student t-test. The results showed significance when p value < 0.05. All tests were conducted in triplicate for statistical analysis.
3. Results & discussion
3.1. Synthesis and characterization of hydrogel nanoparticles
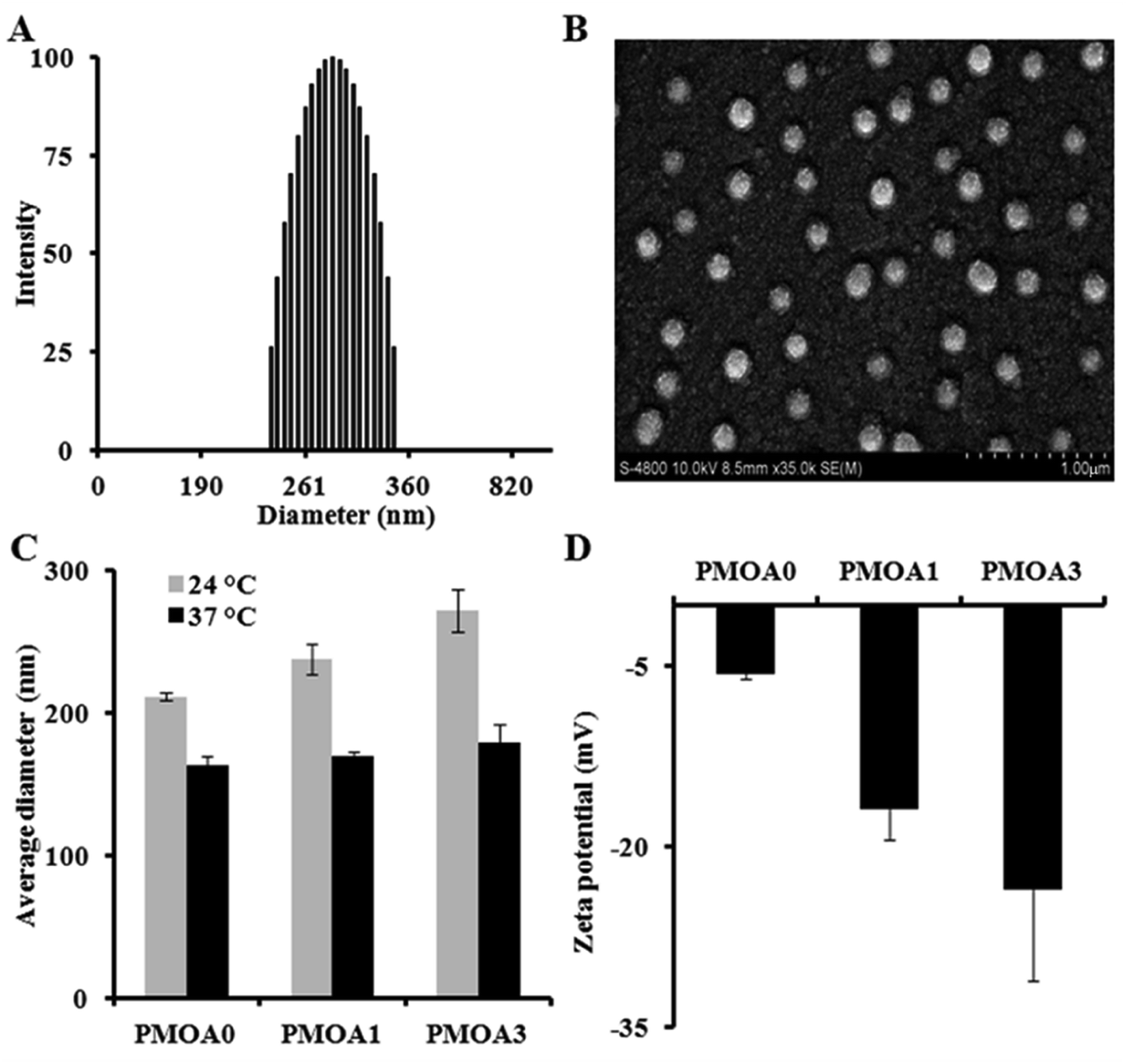
Three of hydrogel nanoparticles with various AAc contents were synthesized via free radical polymerization (Table 1). All three as-prepared particles are highly mono-dispersed. The polydispersity index of the PMOA3 nanoparticles is 0.005 with narrow size distribution of the nanoparticles at room temperature (Fig. 1A). The finding is in line with the observation of SEM (Fig. 1B). Slight reduction of the particle size in SEM images is due to the particle dehydration during SEM sample preparation. From Fig. 1C, one can observe the dependence of size on temperature for all three nanoparticles. The average diameter for the PMOA0 is 211.4 nm at 24 °C but reduces to 163.3 nm at 37 °C. Similar observation has been obtained on the other two samples. The average diameter is 237.9 nm and 271.8 nm at 24 °C while it is 169.2 nm, and 179.2 nm at 37 °C for PMOA1 and PMOA3, respectively. Furthermore, the average size of the hydrogel nanoparticles increases from 211.4 to 237.9 and 271.8 nm at room temperature with increasing of AAc from 0 to 1 and 3%, respectively. This is because more AAc introduces more charged density into the hydrogel nanoparticles, resulting in greater swelling of the particles. Conductivity titration study reveals that the content of carboxyl groups for PMOA1 and PMOA3 are 9.628 and 23.1 nmole mg−1, respectively. The observation can be further confirmed by zeta potential of the hydrogel nanoparticles (Fig. 1D). The zeta potential of the PMOA0, PMOA1 and PMOA3 is −5.69 mV, −16.8 mV to −23.57 mV, respectively. These observations are in good agreement with early publications.44,45
 |
| | Fig. 1 (A) Dynamic Light Scattering (DLS) measurement demonstrating polydispersity of PMOA3 nanoparticle at 24 °C. (B) Scanning Electron Microscope (SEM) image illustrating that the particles are in spherical shape. (C) DLS measurement at both 24 and 37 °C demonstrating an increase in particle size with the increase of acrylic acid content. (D) DLS measurement demonstrating a decrease in zeta potential with the increase of acrylic acid. | |
3.2. Turbidity test and thermally-triggered gelation
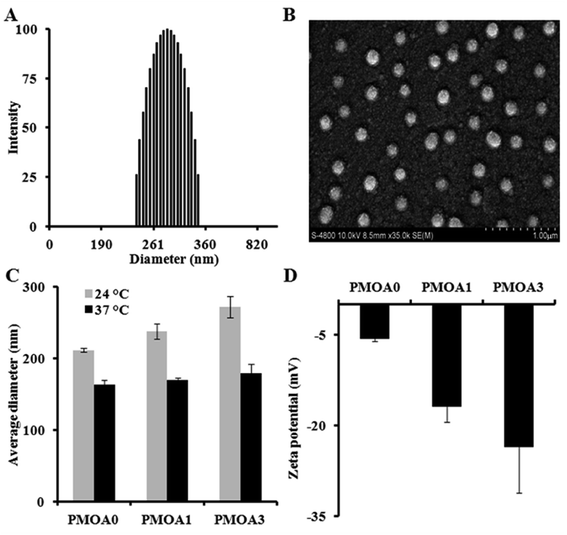
To determine phase transition temperature of the hydrogel nanoparticles, turbidity of the diluted hydrogel nanoparticles (1.0 mg mL−1 in DI water and PBS) at different temperatures was determined using a UV-visible spectrophotometer. The results show that all three nanoparticles exhibit temperature-sensitive property (Fig. 2A). For PMOA0 and PMOA1, transmittance of the dispersions decreases gradually with increasing temperature. There is a sharp drop in the transmittance when heated to 28 °C and 30 °C (defined as phase transition temperature), respectively.46 On the other hand, onset of the phase transition temperature for PMOA3 cannot be reached even when the temperature is raised up to 40 °C. The increase in the phase transition temperature with increasing AAc contents can be explained as follows. More AAc contributes to high ionic strength (as confirmed in Fig. 1D). In addition, the repulsive force between these negatively charged carboxyl groups keeps the polymer chains from aggregation.44 Similar observations are also documented in several recent publications.44,45
 |
| | Fig. 2 (A) Turbidity measurement of thermogelling nanoparticles (1.0 mg mL−1 in deionized water) demonstrating an increase in transition temperature with the increase of acrylic acid. Hydrogel nanoparticles dispersed in PBS (ionic strength: 43.5 mM) have lower transition temperatures than with deionized water. (B) Inversed particle sample (PMOA3) in a test tube illustrating that the hydrogel nanoparticles (60 mg mL−1) form gel at physiological temperature. (C) Rheometry measurement of thermogelling nanoparticles (60 mg mL−1) demonstrating the viscosity as a function of temperature. | |
The phase transition temperatures of these nanoparticles at physiological media (PBS, pH: 7.4 and ionic strength: 43.5 mM) were also investigated (Fig. 2A). One can observe that the phase transition temperature of all three hydrogel nanoparticles shifts to a lower temperature. Furthermore, the presence of salts initiates the nanoparticle's flocculation when temperature is heated above the phase transition temperature. Occurrence of the phenomenon is because the ions in PBS can disturb the closest hydration shells between water and particles, leading to aggregation of the hydrogel particles when heating. On the contrary, all three nanoparticles are highly colloidally stable in DI water at the studied temperature range. Interestingly, all nanoparticles with concentration of 60 mg mL−1 exhibit a thermogelling property in PBS media. At room temperature (24 °C), the nanoparticle dispersions can flow freely. However, the dispersions become physically gelled and cannot escape from the inverted tube when temperature reaches to 37 °C (Fig. 2B). This thermally-triggered gelation may be explained as follow: poly[oligo(ethylene glycol) methacrylate] segments of the nanoparticles turns from hydrophilic into hydrophobic beyond the phase transition temperature, leading to the hydrophobic interaction among the particles to form a physical network in presence of salt as shown in recent studies.24,25 The thermosensitive hydrogels, PMOA0, PMOA 1 and PMOA3, can revert to sol state by cooling down. This temperature-dependent phase change can be carried out indefinitely at least in vitro. Furthermore, the viscosities of the dispersions were measured with increasing temperature. In concurrence with an earlier study, we find a sharp increase in viscosity when the dispersion is heated above the gelation temperature, and the gelation temperature increases with increasing AAc contents (Fig. 2C).25
3.3. Water loss and microstructure of the hydrogel
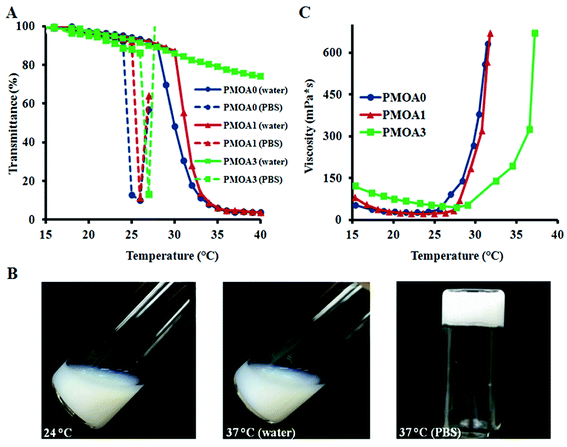
Since the syneresis of hydrogels has adverse effects for applications of the injectable hydrogel scaffold in tissue engineering and drug delivery,47 the water loss of the thermogelling macroscopic hydrogels were carried out, and the results are presented in Fig. 3A. Without AAc (PMOA0), approximately 48.4 wt% of water is expelled from the hydrogel after incubated at 37 °C for 4 hours. As expected, addition of AAc has significant impact on water loss. Water loss for PMOA1 (1% AAc) and PMOA3 (3% AAc) were measured to be 32.4 and 7.6 wt%, respectively. The phenomena is in good agreement with recent observations that associate improved hydrogel charges with increased AAc content which in turn, attributes to enlarged pore size, reduced hydrogel shrinkage and augmented water retention of the hydrogel.21,48 SEM was further used to investigate microstructure of these hydrogels (Fig. 3B–D). An interconnected porous structure can be observed in all three hydrogels. This structure is very critical for hydrogel scaffolds because it allows cells to migrate, and nutrients and waste products to exchange between the scaffolds and surrounding media.49 The presence of AAc makes the pore size of PMOA1 and PMOA3 larger than that of PMOA0 (5.5, 12.3, and 32.1 μm for PMOA0, PMOA1 and PMOA3, respectively). These results are consistent with those of water loss investigation.
 |
| | Fig. 3 (A) Measurement of water loss demonstrating a decrease in water loss with the increase of acrylic acid. 1.0 mL of hydrogel nanoparticles (60 mg mL−1) were incubated at 37 °C for 4 hours, and then the released water was collected from the top of the gels and measured the weight of the lost water. SEM images illustrating porosity of PMOA0 (B), PMOA1 (C), and PMOA3 (D) nanoparticles matrix. | |
3.4. In vitro release of protein
The slow release properties of hydrogels were first characterized using Cy®5-labeled BSA. We find that all three hydrogels show a burst release at first 1 hour but BSA released from PMOA0 (∼43%) is faster than that of PMOA1(∼39%) and PMOA3 (∼31%). The burst release may create the chemokine gradient for initiating cell migration, while subsequent slow release of BSA maintains the gradient for maximal and accumulative recruitment of the cells. After that, BSA gradually released out from the matrix of these hydrogels. PMOA0 released 50% BSA in 11 hours. On the other hand, the release of 50% BSA from PMOA1 and PMOA3 took longer duration for 17 and 23 hours, respectively (Fig. 4A). With regard to the release of Cy®5-labeled Epo, a similar release profile can be observed (Fig. 4B) although Cy®5-labeled Epo shows a faster release than BSA. The faster release of Epo than BSA may be caused by their size difference. Molecular weights of Epo and BSA are 30 and 66.5 kDa, respectively. It is well established that smaller size of protein has faster release rate.50 The surface charge of protein has also been shown to influence protein release from hydrogel nanoparticles.51 However, surface charge of proteins play insignificant role in our findings, since both proteins have a similar isoelectric point (around 4.5).
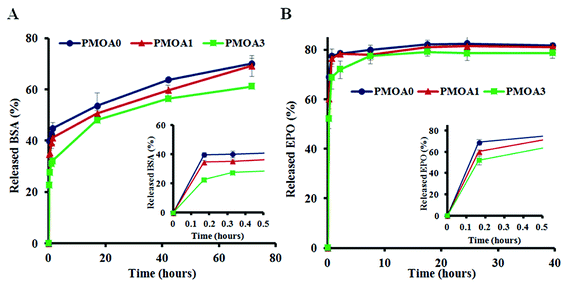 |
| | Fig. 4 In vitro release tests of proteins. (A) Release profiles of Cy®5-labeled BSA and (B) release profiles of Cy®5-labeled Epo. | |
3.5. Hydrogel's responses to cell in vitro and tissue in vivo
To evaluate toxicity of the hydrogels to cell in vitro, the conditioned media of the hydrogels were collected at the defined time points, and the toxicity of them to 3T3 fibroblast cells was estimated using an Alamar Blue assay (Fig. 5A). After 3 days, PMOA3 exhibits the least toxic response among these hydrogels although all three hydrogels exhibit minimum toxicity to 3T3 fibroblasts. Combining the results with the protein release in vitro, PMOA3 was chosen as a protein delivery scaffold in vivo. However, prior to doing that, response of the PMOA3 hydrogel to tissue needs to be further evaluated in vivo. Here a mouse subcutaneous implant model was employed, and the poly(lactic-co-glycolic acid) (PLGA) particle served as a control.41 As expected, PLGA implant triggers more foreign body reactions in the surrounding area of the implant than the PMOA3 gel (Fig. 5B). Quantitative analysis further shows that PMOA3 hydrogel triggers significant lesser inflammatory cell accumulation (∼4×) than the PLGA implants (Fig. 5B). The histological evidence supports that PMOA3 hydrogels possess good tissue compatibility. These results are in consistent with previous finding that poly(oligo(ethylene glycol)) nanoparticles and their derived products have similar tissue responses as the FDA-approved poly(ethylene glycol) polymer.18
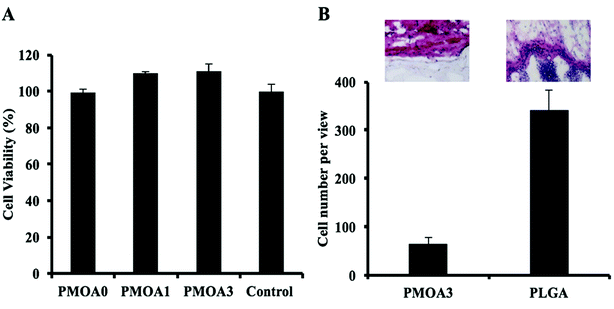 |
| | Fig. 5 In vitro analysis of hydrogel-associated cell and tissue toxicity. (A) In vitro toxicity of the thermogelling gels to cells. (B) H&E staining and cell quantification of tissue surrounding PMOA3 and PLGA implants. | |
3.6. In vivo cancer cell trap
To investigate the capability of thermally induced gelling system as a cancer trap implant, Epo-loaded PMOA3 nanoparticles or Epo alone were implanted in the subcutaneous cavity via 18-gauge needle. After 12 hours, mice were administered IV injection of DiD-labeled prostate cancer cells (PC3-KD cells). After cancer cell implantation, migratory of the cancer cells were imaged after 2 days and 4 days, respectively. Based on NIR imaging, the Epo-loaded PMOA3 implant recruited significantly more PC3-KD cells than Epo alone (Fig. 6A). Total intensity of PC3-KD cells from the implanted site was 3.14 × 106 at Day 2 and 3.66 × 106 at Day 4 (Fig. 6B). Previous studies have demonstrated that Epo receptor is highly expressed in many different cancer cells such as breast, head-and-neck tumors, colon, lung, prostate and melanoma, and Epo-releasing scaffolds prompts more melanoma cell migration.42,52–54 In addition, our previous study has demonstrated that similar hydrogel has controlled protein release properties.18 Equally important, using an in vivo cancer cell trapping mouse model, Epo-releasing PMOA hydrogel is able to recruit cancer cells to the implant site effectively. This injectable, thermogelling PMOA hydrogel may be used to create Epo gradient for recruiting and trapping circulating cancer cells to reduce or delay cancer metastasis.
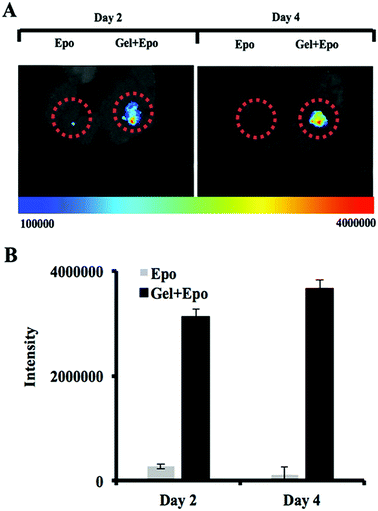 |
| | Fig. 6 The ability of chemokine-releasing thermogelling nanoparticles to recruit prostate cancer cells in mice. (A) Whole body imaging and (B) fluorescent intensity measurement of NIR-labeled prostate cancer cells at the implant site of Epo and Epo-releasing thermogelling nanoparticles in animals. | |
4. Conclusions
In short, an injectable, thermogelling poly(oligo(ethylene glycol))-based nanoparticles are successfully prepared. Both in vitro and in vivo testing confirm that the PMOA hydrogels made of these nanoparticles induces minimal toxicity to cells and tissue responses. By simply mixing the protein with the hydrogel nanoparticle at room temperature, the mixture can easily be injected into the body and turn into the physical hydrogel at the body temperature, serving as a depot for controlled release of proteins. In vivo cancer cell migration experiment shows a dramatically reduction of metastasis by tuning the release of Epo. In general, this study suggests that this PMOA hydrogel nanoparticle has a great potential in protein therapy and drug delivery.
Conflicts of interest
Drs Zhou and Tang have a potential research conflict of interest due to a financial interest with Progenitec Inc. A management plan has been created to preserve objectivity in research in accordance with UTA policy.
Acknowledgements
The authors appreciate Ms. Minsyuan Huang's effort at the beginning of this investigation and Amirali Nojoomi and Dr Kyungsuk Yum's helps for viscosity measurement. This work was partially supported by a grant from Wilson Charitable Foundation Trust and an Idea Development Award No. W81XWH-14-0289 from the Defense Health Program through the Department of Defense Peer Reviewed Prostate Cancer Research Program. Opinions, interpretations, conclusions and recommendations were those of the authors and are not necessarily endorsed by the Department of Defense.
References
- F. J. O'brien, Mater. Today, 2011, 14, 88–95 CrossRef.
- Q. Hou, A. Paul and K. M. Shakesheff, J. Mater. Chem., 2004, 14, 1915–1923 RSC.
- J. D. Kretlow, L. Klouda and A. G. Mikos, Adv. Drug Delivery Rev., 2007, 59, 263–273 CrossRef CAS PubMed.
- J.-A. Yang, J. Yeom, B. W. Hwang, A. S. Hoffman and S. K. Hahn, Prog. Polym. Sci., 2014, 39, 1973–1986 CrossRef CAS.
- B. Choi, X. J. Loh, A. Tan, C. K. Loh, E. Ye, M. K. Joo and B. Jeong, in In Situ Gelling Polymers, Springer, 2015, pp. 5–35 Search PubMed.
- W. E. Hennink and C. F. van Nostrum, Adv. Drug Delivery Rev., 2012, 64, 223–236 CrossRef.
- X. Yang, G. Zhang and D. Zhang, J. Mater. Chem., 2012, 22, 38–50 RSC.
- J. Yan, K. Liu, W. Li, H. Shi and A. Zhang, Macromolecules, 2016, 49, 510–517 CrossRef CAS.
- T. Vo, S. Shah, S. Lu, A. Tatara, E. Lee, T. Roh, Y. Tabata and A. Mikos, Biomaterials, 2016, 83, 1–11 CrossRef CAS PubMed.
- H. Liu, Y. Xiao, H. Xu, Y. Guan, J. Zhang and M. Lang, Chem. Commun., 2015, 51, 10174–10177 RSC.
- E. J. Kim, J. S. Choi, J. S. Kim, Y. C. Choi and Y. W. Cho, Biomacromolecules, 2015, 17, 4–11 CrossRef PubMed.
- Y. Cheng, C. He, J. Ding, C. Xiao, X. Zhuang and X. Chen, Biomaterials, 2013, 34, 10338–10347 CrossRef CAS PubMed.
- H. J. Moon, M. H. Park, M. K. Joo and B. Jeong, Chem. Soc. Rev., 2012, 41, 4860–4883 RSC.
- E. Y. Kang, H. J. Moon, M. K. Joo and B. Jeong, Biomacromolecules, 2012, 13, 1750–1757 CrossRef CAS PubMed.
- C. He, S. W. Kim and D. S. Lee, J. Controlled Release, 2008, 127, 189–207 CrossRef CAS PubMed.
- A. Alexander, J. Khan, S. Saraf and S. Saraf, J. Controlled Release, 2013, 172, 715–729 CrossRef CAS PubMed.
- J. Zhou, G. Wang, L. Zou, L. Tang, M. Marquez and Z. Hu, Biomacromolecules, 2007, 9, 142–148 CrossRef PubMed.
- T. Cai, P. D. Hu, M. Sun, J. Zhou, Y.-T. Tsai, D. Baker and L. Tang, Nanomedicine, 2012, 8, 1301–1308 CrossRef CAS PubMed.
- D. Wang, D. Cheng, Y. Guan and Y. Zhang, Biomacromolecules, 2011, 12, 578–584 CrossRef CAS PubMed.
- Z. Shen, A. Mellati, J. Bi, H. Zhang and S. Dai, RSC Adv., 2014, 4, 29146–29156 RSC.
- Y. Li, Z. Ye, L. Shen, Y. Xu, A. Zhu, P. Wu and Z. An, Macromolecules, 2016, 49, 3038–3048 CrossRef CAS.
- Y. Guan and Y. Zhang, Soft Matter, 2011, 7, 6375–6384 RSC.
- J. Gu, Y. Zhao, Y. Guan and Y. Zhang, Colloids Surf., B, 2015, 136, 1139–1147 CrossRef CAS PubMed.
- T. Gan, Y. Zhang and Y. Guan, Biomacromolecules, 2009, 10, 1410–1415 CrossRef CAS PubMed.
- T. Gan, Y. Guan and Y. Zhang, J. Mater. Chem., 2010, 20, 5937–5944 RSC.
- M. Burek, S. Waśkiewicz, A. Lalik, S. Student, T. Bieg and I. Wandzik, Biomater. Sci., 2017, 5, 234–246 RSC.
- J. Madsen and S. P. Armes, Soft Matter, 2012, 8, 592–605 RSC.
- J.-H. Ryu, R. T. Chacko, S. Jiwpanich, S. Bickerton, R. P. Babu and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 17227–17235 CrossRef CAS PubMed.
- L. Chambre, A. Degirmenci, R. Sanyal and A. Sanyal, Bioconjugate Chem., 2018, 29, 1885–1896 CrossRef CAS PubMed.
- T. Cai, M. Marquez and Z. Hu, Langmuir, 2007, 23, 8663–8666 CrossRef CAS PubMed.
- B. Aktan, L. Chambre, R. Sanyal and A. Sanyal, Biomacromolecules, 2017, 18, 490–497 CrossRef CAS PubMed.
- J.-F. Lutz, K. Weichenhan, Ö. Akdemir and A. Hoth, Macromolecules, 2007, 40, 2503–2508 CrossRef CAS.
- J. Zhou, Y.-T. Tsai, H. Weng, D. W. Baker and L. Tang, Biomaterials, 2011, 32, 9383–9390 CrossRef CAS PubMed.
- S. Y. An, M.-P. N. Bui, Y. J. Nam, K. N. Han, C. A. Li, J. Choo, E. K. Lee, S. Katoh, Y. Kumada and G. H. Seong, J. Colloid Interface Sci., 2009, 331, 98–103 CrossRef CAS PubMed.
- Z. Liu, Z. Ding, K. Yao, J. Tao, G. Du, Q. Lu, X. Wang, F. Gong and X. Chen, J. Magn. Magn. Mater., 2003, 265, 98–105 CrossRef CAS.
- M. M. Good, T. I. Montoya, H. Shi, J. Zhou, Y. Huang, L. Tang, J. F. Acevedo and R. A. Word, PLoS One, 2017, 12, e0186268 CrossRef PubMed.
- A. Nojoomi, E. Tamjid, A. Simchi, S. Bonakdar and P. Stroeve, Int. J. Polym. Mater. Polym. Biomater., 2017, 66, 105–114 CrossRef CAS.
- M. Şen and O. Güven, Eur. Polym. J., 2002, 38, 751–757 CrossRef.
- B. Ozcelik, A. Blencowe, J. Palmer, K. Ladewig, G. W. Stevens, K. M. Abberton, W. A. Morrison and G. G. Qiao, Acta Biomater., 2014, 10, 2769–2780 CrossRef CAS PubMed.
- R. Parboosing, G. Mzobe, L. Chonco and I. Moodley, Med. Chem., 2017, 13, 13–21 CrossRef CAS PubMed.
- H. Weng, J. Zhou, L. Tang and Z. Hu, J. Biomater. Sci., Polym. Ed., 2004, 15, 1167–1180 CrossRef CAS PubMed.
- C.-Y. Ko, L. Wu, A. M. Nair, Y.-T. Tsai, V. K. Lin and L. Tang, Biomaterials, 2012, 33, 876–885 CrossRef CAS PubMed.
- A. Nair, J. Shen, P. Lotfi, C.-Y. Ko, C. C. Zhang and L. Tang, Acta Biomater., 2011, 7, 3887–3895 CrossRef CAS PubMed.
- K. Kratz, T. Hellweg and W. Eimer, Colloids Surf., A, 2000, 170, 137–149 CrossRef CAS.
- A. Burmistrova, M. Richter, M. Eisele, C. Üzüm and R. von Klitzing, Polymers, 2011, 3, 1575–1590 CrossRef CAS.
- S. Lanzalaco and E. Armelin, Gels, 2017, 3, 36 CrossRef.
- M. A. Ramin, L. Latxague, K. R. Sindhu, O. Chassande and P. Barthélémy, Biomaterials, 2017, 145, 72–80 CrossRef CAS PubMed.
- Y. J. Che, D. P. Li, Y. L. Liu, Z. Yue, J. L. Zhao, Q. L. Ma, Q. Zhang, Y. B. Tan, Q. Y. Yue and F. J. Meng, J. Polym. Res., 2018, 25, 169 CrossRef.
- R. M. Staruch, G. E. Glass, R. Rickard, S. P. Hettiaratchy and P. E. Butler, Tissue Eng., Part B, 2017, 23, 183–198 CrossRef CAS PubMed.
- X. Tong, S. Lee, L. Bararpour and F. Yang, Macromol. Biosci., 2015, 15, 1679–1686 CrossRef CAS PubMed.
- M. C. Branco, D. J. Pochan, N. J. Wagner and J. P. Schneider, Biomaterials, 2010, 31, 9527–9534 CrossRef CAS PubMed.
- A. Mirmohammadsadegh, A. Marini, A. Gustrau, D. Delia, S. Nambiar, M. Hassan and U. R. Hengge, J. Invest. Dermatol., 2010, 130, 201–210 CrossRef CAS PubMed.
- M. Henke, R. Laszig, C. Rübe, U. Schäfer, K.-D. Haase, B. Schilcher, S. Mose, K. T. Beer, U. Burger and C. Dougherty, Lancet, 2003, 362, 1255–1260 CrossRef CAS.
- M. O. Arcasoy, X. Jiang and Z. A. Haroon, Biochem. Biophys. Res. Commun., 2003, 307, 999–1007 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Jun Zhoub,
Yihui Huangb,
Amirhossein Hakamivalab and
Liping Tang
ab,
Jun Zhoub,
Yihui Huangb,
Amirhossein Hakamivalab and
Liping Tang