DOI:
10.1039/C8RA06503A
(Paper)
RSC Adv., 2018,
8, 32574-32587
Integration of transcriptome and proteome analyses reveal molecular mechanisms for formation of replant disease in Nelumbo nucifera†
Received
2nd August 2018
, Accepted 2nd September 2018
First published on 20th September 2018
Abstract
The normal growth of Nelumbo nucifera, a widely planted aquatic crop in Asia, was severely ruined by replant disease. The mechanism of replant disease was still unknown in aquatic crops. Complementary transcriptomic and proteomic analyses were performed by comparing seedings of first-year planting (FP) and consecutive planting (CP). 9810 differentially expressed genes (DEGs) were identified between FP and CP. Additionally, 975 differentially expressed proteins (DEPs) were obtained. The correlation of proteome and transcriptome illustrated phenylpropanoid biosynthesis, flavonoid biosynthesis, metabolic pathways, and MAPK signaling pathways were significantly activated. Peroxidase, determined as one of the key proteins in replant disease of N. nucifera, was phylogenetically analyzed. A new depiction of the molecular mechanism causing replant disease in N. nucifera was illustrated. A consecutive monoculture stimulated the generation of reactive oxygen species (ROS) and ethylene, altered the metabolic balance of lignin and flavonoid, and attenuated the activity of antioxidant enzymes through DNA methylation. Therefore, the accumulation of autotoxic allelochemicals and the deficiency of antioxidant enzymes unavoidably suppressed the normal growth and development of replanted N. nucifera.
Introduction
Nelumbo nucifera belongs to Nelumbonaceae, Nymphaeales, a perennial aquatic plant. This genus consists of two species, N. nucifera, located in Asia, Australia and Russia, and Nelumbo lutea, distributed throughout North America.1 N. nucifera is rich in flower color and the number of petals, and is considered an ornamental plant and economic crop. N. nucifera has been cultivated in Asia for more than 7000 years, for its colorful flowers, edible seeds and rhizomes.2 Moreover, N. nucifera is widely used as herbal medicine for curing various health problems such as insomnia, cancer, diabetes, heart and liver diseases.3 Replant disease, or the consecutive monoculture problem, is a common phenomenon in various horticultural, vegetable and medicinal plants.4–6 Serious invasion by microbes and pollen abortion are often detected in replanted N. nucifera, causing a decrease in seed yield and floral buds even under normal cultivation management.7 Further studies also indicate that replant disease resulted in a 14.8% decline in the average seed setting rate, and a 14.3% decrease in the single grain dry weight in replanted N. nucifera.8
Previous studies have mainly tried to explain replant disease based on environmental factors, including the imbalance of soil nutrients, shifts of microbial communities into aggressive pathogens and allelopathic autotoxicity.9–12 A long-time consecutive monoculture of one crop in the same field easily lead to deficiency of imperative nutrients, and accumulation of pathogens.13 Additionally, allelochemicals were continuously released into the rhizosphere of replanted crops, and accumulation of these allelochemicals significantly ruined the normal growth of crops.11,12 Although a consecutive monoculture disrupted the suitable surroundings of replanted crops, how it inhibited their normal growth was still unknown.
Physiological studies have found that a consecutive monoculture resulted in the destruction of the cell membrane, a decline in the dry weight of leaves and roots, and a decrease of root activity in high plants, such as Rehmannia glutinosa, Arachis hypogaea, Pseudostellaria heterophylla, tobacco and cotton.14,15 In addition to the physiological studies, an exploration into the response of genes and proteins to a consecutive monoculture would sincerely facilitate our understanding of the mechanism of replant disease. The genes responding to a consecutive monoculture were already indicated in R. glutinosa by the transcriptome, and the genes involved in ethylene synthesis, such as 1-aminocyclopropane-1-carboxylate oxidase (ACO), and related to disruption of DNA, RNA and protein synthesis, such as DNA-directed RNA polymerase 2B, were significantly increased.13 The change of proteins was also examined in P. heterophylla and R. glutinosa by 2-DE-based proteome,14,15 and 20 proteins associated with photosynthesis and energy metabolism were obviously regulated by replant disease.
To date, the studies on replant disease have focused mainly on terrestrial plants, with a lack of investigation in aquatic botony. Whether these results could be used in aquatic botony still needs to be further researched. Moreover, the integration of the transcriptome and proteome was commonly used to unveil the molecular mechanism of high plants in the face of environmental stresses.16,17 However, this method is rarely used in explaining the formation of replant disease, with no reports on N. nucifera. Following the recent release of the N. nucifera genome,18 the integration of the transcriptome and proteome could be used as imperative methodologies for uncovering the mechanism of replant disease in N. nucifera.
In this study, an integrated analysis of the transcriptome and proteome was first performed to research the potential molecular mechanism of replant disease in N. nucifera. Real-time PCR was operated to validate the expression profile of target genes. Based on the biological function of these target genes, the signal pathway of a consecutive monoculture was illustrated in N. nucifera. These experiments aimed to provide data for unveiling the molecular characteristics of replant disease in N. nucifera, and increase our understanding of replant disease in aquatic botony.
Methods
N. nucifera planting and sample preparation
N. nucifera “Taikonglian 36”, the most distributed seed lotus, was widely planted at Wuhan University, Hubei Province, China. The size of the experimental pools was about 10 m × 5 m, separated by 0.7 m walkways between the pools to avoid the influence of various experimental treatments. Ten pools were separated into two groups, with five pools in each group. In the first group, five pools contained never planted N. nucifera. For the second group, five pools contained consecutively planted N. nucifera, from 2013 to 2016. Twenty rhizomes of the same size were selected randomly, and breeded in ten pools with 2 rhizomes in each pool in April, 2017. The same field management was performed for two groups during the growth and development of N. nucifera. Conveniently, the former was the first-year planting group (FP), and the latter was the consecutively planting group (CP).
RNA sequencing and data analysis
Six rhizomes from various pools were randomly collected from both FP and CP groups, with three samples from each group in July, the most serious stage of replant disease. Three independent biological replicates of rhizomes from FP and CP were harvested for RNA sequencing. A transcriptome library was constructed with the help of Beijing Genomic Institute for transcriptomic analysis (BGI, Wuhan, China).19 The total RNA of rhizomes was isolated by a Trizol reagent, and the RNA quality was checked by an Agilent 2100 Bioanalyzer (Aglient Technologies, Santa Clara, CA, USA). mRNA was enriched by Oligo (dT) magnetic beads, and then interrupted to short fragments with a size of about 200 bp. The short fragments were used as templates to synthesize the double-strand cDNA by random primers. Additionally, sequencing adaptors were ligated to the fragments, and the required fragments were purified and amplified by PCR reaction. The library products were scanned for sequencing analysis via BGISEQ-500 (BGI, Wuhan, China). Raw reads had been uploaded to the NCBI Gene Expression Omnibus (GEO accession: GSE114112; enter token: ybcjymswvjyjrit). The clean reads were obtained by filtering the low-quality reads, and mapped to the genome of N. nucifera (https://lotus-db.wbgcas.cn/) using HISAT.20 Based on genome annotation, the novel transcripts, including coding transcripts and noncoding transcripts, were identified using Cuffcompare,21 and the coding ability of these new transcripts was predicted by CPC.22 All the novel coding transcripts were merged with reference transcripts to obtain a complete reference. Subsequently, the clean reads were mapped to it using Bowtie2.23 The mRNA expression was determined by the RPKM method (reads per kb per million reads).24 FDR (false discovery rate) ≦ 0.001 and the absolute value of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2 ratio ≥ 1 were used as thresholds to define the differentially expressed genes (DEGs). Moreover, all DEGs were mapped to gene ontology terms in the database (GO, https://www.geneontology.org/) for functional annotation, Kyoto Encyclopedia of Genes and Genomes database (KEGG, https://www.genome.jp/kegg/pathway.html) enrichment analysis to determine the main metabolic pathways and signal transduction pathways.
2 ratio ≥ 1 were used as thresholds to define the differentially expressed genes (DEGs). Moreover, all DEGs were mapped to gene ontology terms in the database (GO, https://www.geneontology.org/) for functional annotation, Kyoto Encyclopedia of Genes and Genomes database (KEGG, https://www.genome.jp/kegg/pathway.html) enrichment analysis to determine the main metabolic pathways and signal transduction pathways.
iTRAQ labeling and data processing
The samples used for iTRAQ analysis were the same as those used for RNA sequencing. The quality of proteins was assessed using a protein assay kit (Bio-Rad, CA, USA), and 12% SDS-PAGE (Fig. S1†). iTRAQ analysis was also carried out by Beijing Genomic Institute (BGI, Wuhan, China)25 Proteins were extracted and digested, then reduced, alkylated and digested with trypsin. Then peptides were labeled with iTRAQ tags (Applied Biosystems, Foster City, CA, USA). The peptides were labelled with 114, 116 and 117 iTRAQ reagents for FP, and 118, 119 and 121 for CP. SCX chromatography was used for LC-20AB HPLC (Shimadzu, Kyoto, Japan), and the iTRAQ-labelled peptides were reconstituted with solvent A (5% CAN, pH = 9.8), and loaded onto a 4.6 × 250 mm Ultremex SCX column containing 5 μm particles (Phenomenex). The peptides mixture was separated by a gradient of buffer B (95% CAN, pH = 9.8) at a flow rate of 1 ml min−1. The gradient was set as follows: 5% buffer B for 10 min, 5–35% buffer B for 30 min, and 35–95% buffer B for 1 min. Elution was examined by measuring absorbance at 214 nm, and fractions were collected every 1 min. The peptides were analyzed by nanoelectrospray ionization, followed by tandem mass spectrometry (MS/MS) in a Q-EXACTIVE (Thermo Fisher Scientific, San Jose, CA). The MS/MS results were analyzed with ProteinPilot Software 4.0 (Applied Biosystems) using the Paragon algorithm to determine relative quantification and p-values of the proteins. The criteria with fold-change value ≥1.2 and p-value ≤ 0.05 were used to determine differentially expressed proteins (DEPs). The data was uploaded in iproX (https://www.iprox.org/page/PSV023.html;?url=1525224639418ibTp, R8pv). Correlation analysis was performed in order to understand the relation of the proteome and transcriptome.17
Valuation genes expression by real-time PCR
According to the correlation analysis of the proteome and transcriptome, five categories were created as follows: same trend of DEGs and DEPs, opposite trend of DEGs and DEPs, DEPs and NDEGs (no change of the corresponding DEGs), NDEPs (no change of the corresponding DEPs) and DEGs, NDEPs and NDEGs. Real-time PCR was performed to examine the expression profile of 13 genes with the same trend of DEGs and DEPs, using the materials of roots, leaves and rhizomes in FP and CP.26,27 The total RNA was isolated by an RNAprep Pure lant Kit (TIANGEN, China). The first strand of cDNA was generated by MLV transcriptase (Promega, USA), and diluted 5 fold using a template. Real-time PCR was carried out by SYBR Green Real-time PCR master mix (Vazyme, China), using β-actin as a reference gene (Table S1†).
Isolation and analysis of peroxidase (POD) in N. nucifera
The complete coding sequence of POD gene in N. nucifera (NCBI no. XM_010248162.2) was cloned by reverse transcription, using cDNA from leaves as a template (Table S1†). According to a published DNA sequence of POD gene in N. nucifera (NCBI no. NW_010729341.1), the exons and introns were identified. The characteristics of POD in N. nucifera were studied by PeroxiBase (https://peroxibase.toulouse.inra.fr/). Subsequently, the putative amino acid sequence of POD in N. nucifera was compared with other proteins from NCBI by Clustal W software. Additionally, the subsequent phylogenetic tree was generated by the neighbor-joining method based on the amino acid sequences, using the MEGA software version 4.28
Measurement of the activity of antioxidant enzymes and contents of MDA and H2O2
The various tissues of FP and CP seedings, including roots, rhizomes and leaves, were harvested and washed with distilled water. All the tissues were frozen and homogenated in liquid nitrogen immediately, and stored at −80 °C. For examination of enzyme activity, the materials were ground and diluted in 0.1 M PBS buffer (pH = 7.4), and centrifuged for 30 min at 4 °C. Supernatant was harvested for preparation in the following experiments. Super dismutase (SOD) activity was determined as the amount of enzyme required to result in a 50% inhibition of nitro blue tetrazolium (NBT) photoreduction to formazan at 560 nm.29 Catalase (CAT) activity was calculated by examining the decrease of H2O2 at 240 nm.30 Ascorbate peroxidase (APX) activity was estimated by detecting the substrate of ascorbate at 290 nm.31 POD activity was quantified using guaiacol as substrate, according to the modified method.32 Subsequently, MDA content was examined using the thiobarbituric acid (TBA) method.33 H2O2 content was measured by a modified Ti(IV)-H2O2 method.34
Statistical analysis
Data were expressed as the mean ± SD from three independent biological replicates. Significance was calculated based on a one-way analysis of variance (ANOVA), and the least significant difference (LSD) t-test was performed for differences between groups (p < 0.05).
Results
Characteristics of replant disease in N. nucifera
Replant disease seriously interrupted the normal growth and development of N. nucifera (Fig. 1a and b). Following a consecutive monoculture, the symptoms of replant disease in five-year replanted seedings were more severe than four-year replanted ones, which almost resulted in senescence during a five-year consecutive monoculture (Fig. 1b and c). The morphological changes of rhizomes and leaves were investigated, showing the serious damage in replanted N. nucifera. The rhizomes of replanted N. nucifera were rotten and appeared dark brown in color (Fig. 1d). Moreover, the leaves above water were withered with burned shapes around the edges of the leaves (Fig. 1f).
 |
| | Fig. 1 Replant disease seriously interrupted the normal growth and development of N. nucifera. (a) The growth of FP (control) in normal conditions. (b) The growth of CP (treatment) during a four-year consecutive monoculture. (c) The growth of CP during a five-year consecutive monoculture. (d) The morphological changes of rhizomes between FP and CP. (e) The normal leaves of FP. (f) The leaves of CP. | |
Transcriptome analysis and assembly
Two cDNA libraries were constructed from rhizomes of CP and FP as the control, with three samples for each group. On average 66344252 and 65716271 clean reads were obtained. On average 94.30% of the reads were mapped, and the uniformity of the mapping results suggested that the samples were comparable. Based on genome annotation, 23591 novel transcripts were identified in six rhizomes of FP and CP in total, including 19154 coding transcripts and 4437 noncoding transcripts. Then the coding ability of these new transcripts was predicted. All the novel coding transcripts could be merged with reference transcripts to generate complete references.
DEGs identification and analysis
The Deseq2 and PossionDis algorithms were used to detect the DEGs between CP and FP. A total of 25082 genes were identified in FP and CP, including 9810 DEGs and 15272 no-DEGs. There were 5295 up-regulated DEGs and 4515 down-regulated DEGs (Table S2†). More up-regulated genes than down-regulated genes were obtained in response to the consecutive monoculture. These DEGs were classified with GO classification and functional enrichment according to three ontologies, molecular biological function, cellular component and biological process (Fig. 2a). In the biological process category, the most abundant groups were metabolic process (2180), cellular process (1788), single-organism process (1109) and localization (467). Moreover, genes involved in the response to stimulus (363) and the regulation of biological process (295) accounted for 3.7% and 3.0% of the total DEGs. The most DEGs in the cellular component category were for cells (1367), cell parts (1361) and membranes (1066). In terms of molecular function, the function of most DEGs were catalytic activity (2396), binding (1914) and transporter activity (227), whereas the antioxidant activity function (52) only accounted for 0.53% of the total DEGs.
 |
| | Fig. 2 Functional categories of the DEGs. (a) GO functional annotation of the DEGs. (b) The enriched KEGG pathways of the DEGs. The vertical axis describes the name of the pathway, and the horizontal axis shows the corresponding rich factor of the pathway. Higher rich factors suggest greater degrees of enrichment. | |
DEGs pathways
To screen the imperative pathways, DEGs were mapped to KEGG pathways. DEGs were mainly rich in global and overview maps (1858), carbohydrate metabolism (754), signal transduction (596), translation (481), biosynthesis of other secondary metabolites (368) and metabolism of terpenoids and polyketides (189). The enriched KEGG pathway illustrated that the metabolic pathways and the biosynthesis of secondary metabolites were significant enrichment (Fig. 2b), containing more DEGs than other pathways. Beside these, other pathways including flavonoid biosynthesis, isoflavonoid biosynthesis, plant–pathogen interaction, MAPK signaling pathway, phenylpropanoid biosynthesis and plant hormone signal transduction were also significant.
Proteome analysis and identification of DEPs
In total 398832 spectra were generated in the proteome analysis of FP and CP, and 28070 peptides and 6819 proteins were identified. 975 DEPs were found including 541 up-regulated proteins and 434 down-regulated proteins (Table S3†). Using the Blast2GO program, DEPs were mapped to KEGG data. DEPs mainly related to global and overview maps (164), carbohydrate metabolism (81) and biosynthesis of other secondary metabolites (46), shown in Fig. 3a. Moreover, some DEPs participated in translation (25), signal transduction (18) and environmental adaptation (18). The statistics of the pathway enrichment of DEPs illustrated that metabolic pathways and biosynthesis of secondary metabolites were the main significant pathways. Moreover, phenylpropanoid biosynthesis, flavonoid biosynthesis and starch and sucrose metabolism were also significant (Fig. 3b).
 |
| | Fig. 3 KEGG pathway of DEPs (a) and enrichment analysis (b). (a) The y-axis represents various pathways, and the x-axis indicates the number of proteins. (b) The vertical axis describes the name of the pathway, and the horizontal axis shows the corresponding rich factor of the pathway. Higher rich factors suggest greater degrees of enrichment. | |
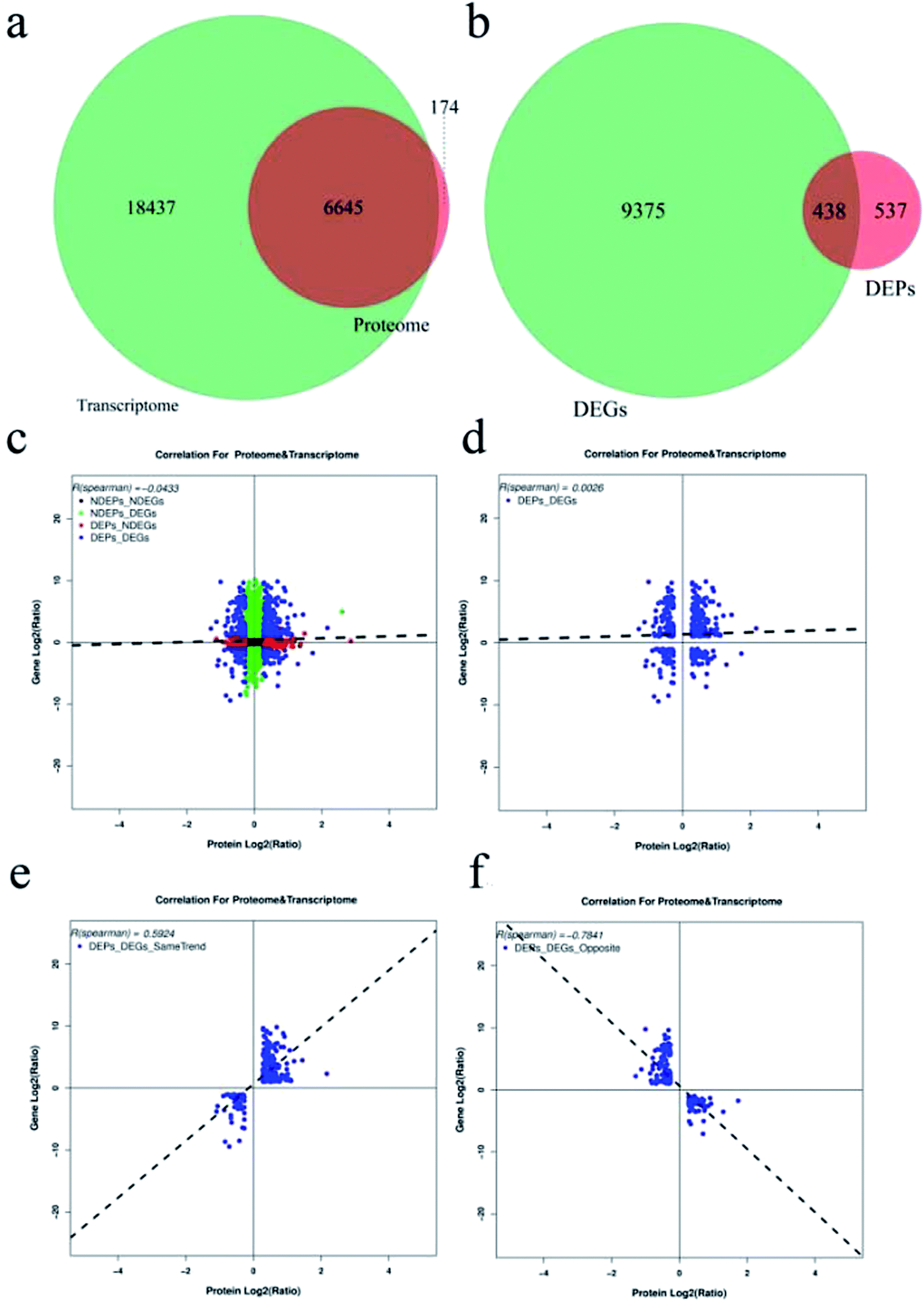
Correlations between the transcriptome and proteome
Integration of the proteome and transcriptome was conducted. 25082 and 6819 genes were identified and quantified in the transcriptome and proteome respectively, whereas only 6645 genes were detected in both analyses (Fig. 4a). 438 DEPs were matched with their DEGs by the analysis of the correlation of 975 DEPs and 9813 DEGs (Fig. 4b). However, a limited correlation was detected between the proteome and transcriptome with the R (spearman) = −0.0433 (Fig. 4c), and a relatively higher correlation of DEPs and DEGs was obtained as R (spearman) = 0.0026 (Fig. 4d). Additionally, the higher positive and negative correlation was indicated in the same trend of DEGs and DEPs (Fig. 4e), and the opposite trend of DEGs and DEPs (Fig. 4f). Among these 438 DEPs, 213 DEPs shared the same tendency of the DEGs, with 225 DEPs showing the opposite tendency of DEGs (Table S4†). The key DEGs with the same trend of DEPs were detected. ACO, DNA (cytosine-5)-methyltransferase 1 (DNMT1), 2-oxoglutarate 3-dioxygenase-like (F3H), polygalacturonase inhibitor-like (PGI), phenylalanine ammonia-lyase-like (PAL), shikimate O-hydroxycinnamoyltransferase (HCT) and cinnamyl alcohol dehydrogenase (CAD) were increased in response to replant disease (Table 1). Whereas asparagine synthetase (ASN) and POD were down-regulated. Moreover, opposite patterns of DEPs and DEGs were also found. For example, pathogenesis-related protein STH-21-like (LOC104588989) was increased in the level of protein during a consecutive monoculture, while it was decreased at the mRNA level. Moreover, chalcone synthase (CHS) and (RS)-norcoclaurine 6-O-methyltransferase-like (6OMT) were significantly decreased in the proteome, and up-regulated in the transcriptome (Table 1).
 |
| | Fig. 4 Correlations between mRNA and protein expression. (a) Venn diagram of genes quantified in the transcriptome (green) and proteome (red). (b) Venn diagram of DEGs (green) and DEPs (red). (c) Scatterplot of the relationship between genes identified in both the transcriptome and proteome. (d) Scatterplot and correlation coefficients between DEGs and DEPs. (e) Scatterplot and correlation coefficients between DEGs and DEPs (the same trend). (f) Scatterplot and correlation coefficients between DEGs and DEPs (the opposite trend). | |
Table 1 DEGS and DEPs in replant diseasea
| Gene ID |
Gene name |
DEGs |
Protein ID |
DEPs |
Molecular function |
| “+” represented fold change was significantly increased (fold-change of DEGs ≥2 or DEPs ≥1.2, p-value ≤ 0.05), “—” represented fold change was significantly decreased (fold-change of DEGs ≥2 or DEPs ≥1.2, p-value ≤ 0.05), “NA” represented no significant fold change was detected. |
| NNU_15912 |
ACO |
+ |
NNU_15912-RA |
+ |
Ethylene biosynthesis |
| NNU_06496 |
F3H |
+ |
NNU_06496-RA |
+ |
Flavonols biosynthesis |
| NNU_02876 |
PGI |
+ |
NNU_02876-RA |
+ |
Plant defense |
| NNU_01774 |
DNMT1 |
+ |
NNU_01774-RA |
+ |
Methylates CpG residues |
| NNU_05129 |
PAL |
+ |
NNU_05129-RA |
+ |
Phenylpropanoid pathways |
| NNU_06315 |
HCT |
+ |
NNU_06315-RA |
+ |
Lignin biosynthesis |
| NNU_24837 |
CAD |
+ |
NNU_24837-RA |
+ |
Lignin biosynthesis |
| NNU_02538 |
PME |
+ |
NNU_02538-RA |
+ |
Modification of cell walls |
| NNU_07527 |
6PGD |
+ |
NNU_07527-RA |
+ |
Pentose phosphate pathway |
| NNU_10554 |
CTPS |
+ |
NNU_10554-RA |
+ |
CTP synthesis |
| NNU_15671 |
SUS |
+ |
NNU_15671-RA |
+ |
Sucrose metabolism |
| NNU_13097 |
ASN |
— |
NNU_13097-RA |
— |
Asp synthesis |
| NNU_04050 |
POD |
— |
NNU_04050-RA |
— |
Antioxidant enzyme |
| NNU_26607 |
CuZnSOD |
— |
NNU_26607-RA |
NA |
Antioxidant enzyme |
| NNU_23981 |
CAT |
— |
NNU_23981-RA |
NA |
Antioxidant enzyme |
| NNU_10113 |
APX |
+ |
NNU_10113-RA |
NA |
Antioxidant enzyme |
| NNU_08709 |
GPX |
— |
NNU_08709-RA |
NA |
Antioxidant enzyme |
| NNU_22334 |
CHS |
+ |
NNU_22334-RA |
— |
Chalcone synthesis |
| NNU_03166 |
6OMT |
+ |
NNU_03166-RA |
— |
(S)-Coclaurine biosynthesis |
| NNU_23196 |
LOC104588989 |
— |
NNU_23196-RA |
+ |
Plant defense |
The correlation of proteome and transcriptome KEGG enrichment
The correlation of proteome and transcriptome KEGG enrichment was examined (Table S5†). The biosynthesis of secondary metabolites, phenylpropanoid biosynthesis, metabolic pathways, stilbenoid, diarylheptanoid and gingerol biosynthesis, flavonoid biosynthesis, plant–pathogen interaction, amino sugar and nucleotide sugar metabolism, MAPK signaling pathway – plant, isoflavonoid biosynthesis and galactose metabolism were all significant pathways in both the proteome and transcriptome (Fig. 5a and b). Starch and sucrose metabolism, nitrogen metabolism, indole alkaloid biosynthesis, pentose and glucuronate interconversions and phenylalanine metabolism were only significant at the proteome level. Moreover, plant hormone signal transduction and cutin, suberine and wax biosynthesis were only significant at the transcriptome level (Fig. 5a and b).
 |
| | Fig. 5 KEGG enrichment correlation of transcriptome and proteome. (a) Number of KEGG enrichment correlation transcriptome and proteome. (b) Scatterplot of KEGG enrichment correlation between the mRNA and protein levels of genes. The pathways in red represent pathways that were significant in both the transcriptome and proteome, and the green pathways are those that were significant in either the transcriptome or proteome. | |
Valuation of the expression of genes during replant disease
In order to examine the data of the proteome and transcriptome, various materials including roots, rhizomes and leaves were harvested from FP and CP. The genes, including DNMT1, F3H, ACO, PGI, ASN, POD, PAL, HCT, CAD, probable pectinesterase (PME), 6-phosphogluconate dehydrogenase (6PGD), CTP synthase (CTPS) and sucrose synthase (SUS), were investigated by real-time PCR (Fig. 6). In response to a consecutive monoculture, DNMT1 mRNA of CP was significantly increased in the roots and rhizomes of N. nucifera, whereas it was down-regulated in the leaves. F3H mRNA was only increased in the rhizome of CP, and CAD mRNA was augmented in the leaves and rhizomes of CP (Fig. 6). Besides these, the mRNA expression of ACO, PGI, PAL, HCT, PME, 6PGD, CTPS and SUS in CP was obviously up-regulated in the roots, rhizomes and leaves, with a decline of ASN and POD mRNA observed in all the materials examined.
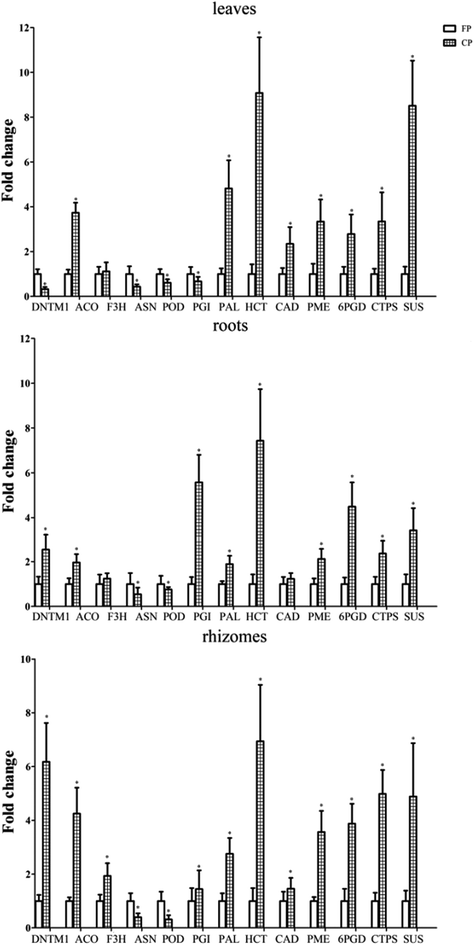 |
| | Fig. 6 Expression pattern of genes with the same trend of DEGs and DEPs. Real-time PCR was performed to examine the change of genes in various tissues of CP, using FP as control. Three independent experiments were conducted, * represents significant differences between FP and CP at p < 0.05, based on LSD test. | |
Examination of the activity of antioxidant enzymes, H2O2 and MDA contents
The activity of antioxidant enzymes and H2O2 content were investigated in CP and FP (Fig. 7). Consistent with the decline in POD transcript and protein, POD activity was significantly decreased in the roots, rhizomes and leaves of CP. As the substrate of POD, H2O2 production in plants was considered as a critical response to biotic and abiotic stresses. However, H2O2 in these tissues of CP was significantly increased, with the higher MDA content. Moreover, enzyme activity of other antioxidant enzymes including SOD, CAT and APX was also attenuated in CP by replant disease (Fig. 7).
 |
| | Fig. 7 Differences in enzyme activity of antioxidant enzymes, H2O2 and MDA content between FP and CP. Three independent experiments were conducted, and p values were generated based on LSD test (*p < 0.05). | |
Isolation and characteristics of the POD gene in N. nucifera
The complete coding cDNA sequence of POD was obtained in N. nucifera with a size of 1456 bp. The open reading frame contained 960 bp, with the start codon ATG and stop codon TGA. The exon-intron architecture was investigated, including three exons and two introns (Fig. 8a). The boundaries between exons and introns followed the GT–AG rule. The putative MW of POD was about 34.2534 KD with pI 9.01. Moreover, the conserved residues involved in the catalytic mechanism, forming disulphide bonds, binding calcium and haem were investigated in POD of N. nucifera by comparing amino acid sequences (Fig. 8b). PeroxiBase predicted that POD of N. nucifera belonged to class III peroxidase by analysis of amino acid sequences, having a farther relationship with class I and class II peroxidase (Fig. 8c).
 |
| | Fig. 8 Isolation and phylogenetic analysis of POD in N. nucifera. (a)The exon–intron architecture of POD gene in N. nucifera was investigated. (b) Amino acid alignment of POD was performed by Clustal W, including POD from N. nucifera (XP_010246464.1), Nicotiana tomentosiformis (XP_009593782.1), Solanum lycopersicum (XP_004234419.1), Herrania umbratica (XP_021275379.1), Durio zibethinus (XP_022739881.1), Gossypium arboreum (XP_017605006.1), Vigna angularis (XP_017433445.1), and Glycine max (XP_003527340.1). The conserved residues involved in the catalytic mechanism are labeled in green, the calcium binding sites are represented by grey, eight cysteines forming disulphide bonds are shaded in yellow, and structural determinants of syringyl peroxidases are shown in red. (c) Phylogenetic analysis of POD was performed by neighbour-joining method using MEGA4 software. The amino acid sequences were obtained from NCBI, including Gossypium raimondii (XP_012441150.1), Solanum pennellii (XP_015069531.1), Populus euphratica (XP_011033755.1), Burkholderia pseudomallei (Q939D2), Escherichia coli (AAC76924.1), Haloarcula marismortui (O59651), Synechococcus elongatus (Q31MN3), Magnaporthe oryzae (A4QUT2), Pisum sativum (P48534), Saccharomyces cerevisiae (P00431), Leishmania major (Q4Q3K2), Arthromyces ramosus (P28313), Coprinopsis cinerea (A8NK72), Phanerochaete chrysosporium (P49012), and Pleurotus eryngii (O94753). POD of N. nucifera is represented with an asterisk. | |
Discussion
A low correlation between the transcriptome and proteome of replant disease
The formation of replant disease was the comprehensive result of the interaction of plants, microbial communities and autotoxic allelochemicals.35 Despite abundant studies on microbial invasion and autotoxic allelochemicals, understanding how plants responded to a consecutive monoculture was key to understanding the molecular mechanism of replant disease. Some genes related to replant disease were already reported in R. glutinosa,13,36 but it was not sufficient to explain the mechanism of replant disease at the single transcript or protein level. In order to accurately investigate the formation of replant disease, this study aimed to screen the target genes based on the correction analysis of the proteome and transcriptome. The correlation between the 975 DEPs and 9810 DEGs generated 438 DEPs, including the same tendency of 213 DEPs and DEGs, and the opposite tendency of 225 DEPs and DEGs. A low correlation was detected between the proteome and transcriptome, due to intermediate and low abundance mRNAs and proteins, the efficiency of translation, codon preference, various status of the tissues and post-translational modification.37
Consecutive monoculture altered the metabolic balance of N. nucifera
Firstly, two elevated proteins in replanted N. nucifera, ATP-dependent 6-phosphofructokinase 3-like and 6PGD, were involved in glycolysis and the pentose phosphate pathway for the production of NADPH, respectively. Secondly, the enzymes catalyzed the synthesis of secondary metabolites were also increased. For example, UMP-CMP kinase 3 (UMK3), CTPS and SUS played important roles in de novo pyrimidine nucleotide biosynthesis, CTP synthesis, sucrose metabolism and the degradation of cellulose or lignocellulose, respectively. Thirdly, two types of enzymes including arogenate dehydratase (ADT) catalyzed conversion of phenylalanine from the shikimate-chorismate pathway, and glucose-6-phosphate 1-dehydrogenase (G6PD) involved in the oxidation of glucose 6-phosphate to 6-phosphogluconolactone were also up-regulated in the replanted N. nucifera. Finally, five proteins consisting of PGI, DNA-damage-repair/toleration protein DRT100-like (LOC104590924), leucine-rich repeat extensin-like protein 4 (LRX4), basic endochitinase-like (LOC104587093) and MLP-like protein 423 (MLP423) participated in MAPK signaling pathway of plant, increasing in replanted N. nucifera. Among these proteins, PGI belonged to the superfamily of leucine reach repeat (LRR) proteins, which played a role as plant defense proteins by reducing the hydrolytic activity of polygalacturonases and favored the accumulation of long-chain oligogalacturonides. However, ASN related to amino acid metabolism was decreased.
Consecutive monoculture stimulated DNA methylation and altered the dynamic balance of antioxidant systems in replanted N. nucifera
ROS was usually released as the normal response to biotic and abiotic stresses,26 acting as a signal molecule to trigger tolerance behavior. However, redundant ROS would make unavoidable oxidative damage to the cell membrane, DNA or proteins. The antioxidant enzymes including SOD, CAT, APX and POD were evoluted for scavenging these abundant ROS. Therefore, the dynamic balance was maintained between ROS-releasing and ROS-scavenging systems in normal conditions. DNA methylation was an epigenetic mechanism regulated by environmental and genomic stresses. Whereas antioxidant enzymes could be suppressed at the RNA and protein levels through DNA methylation. For example, a single CpG dinucleotide within the SOD2 gene close to the transcription initiation site was associated with reduced SOD2 mRNA.38 In the face of environmental stresses, the expression of antioxidant enzymes including SOD2 and GPX1 were regulated by DNA methylation in the skeletal muscle of severely dyslipidemic mice.39 In addition, DNA methylation analyses indicated CpG hypermethylation was detected in promoters of antioxidant genes including SOD1-3 and GPX catalyzed by DNTM1 in guinea pigs exposed to polymerized cell-free hemoglobin.40
As the principal enzyme responsible for maintenance of CpG methylation, the expression of DNMT1 was significantly elevated in replanted N. nucifera. Recently, DNMTs were reported as the downstream targets of the MAPK pathway, and DNA methylation was regulated by the MAPK signal.41,42 In this study, antioxidant systems were disrupted by a consecutive monoculture, with the suppression of POD expression and inhibition of enzyme activity including POD, SOD, CAT and APX. Interestingly, the expression of SOD, CAT and APX was only changed at the transcript level in replanted N. nucifera (Table 1), suggesting post-translational modification occurred in the regulation of antioxidant enzymes. Therefore, the abundant ROS was not eliminated immediately. It seemed that the loss of the ROS-scavenging function and accumulation of ROS in N. nucifera resulted in replant disease. Transcriptome analyses in R. glutinosa also indicated that both attenuation of antioxidant enzymes and up-regulation of ethylene synthesis were involved in replant disease.13,36 However, the calcium signalling pathway promoted in R. glutinosa was not detected in replanted N. nucifera. Additionally, only 20 DEPs associated with photosynthesis and energy metabolism were found in R. glutinosa by the 2-DE-based proteome.15 The different results were probably caused by the various materials used for sequencing. The materials of the transcriptome analyses in R. glutinosa were roots, and leaves were used in the proteome analyses. In our study, the rhizome was used for the transcriptome and proteome analyses, showing more serious damage in replanted seedings. Additionally, the iTRAQ-based proteome was more sensitive than the 2-DE-based proteome, which also resulted in more putative DEPs found in replanted N. nucifera.
The accumulation of lignin was strengthened by a consecutive monoculture with an increase of flavonoid biosynthesis
The phenylpropanoid pathway was considered as an important secondary metabolism pathway in high plants. In this pathway, secondary metabolites such as lignin and flavonoid played key roles in plant growth and disease resistance. As the first and key enzyme between primary and secondary metabolism, PAL controlled the biosynthesis of flavonoid, flavonol, anthocyanins and lignin.43 Our study uncovered that the key proteins associated with lignin synthesis were significantly increased in replanted N. nucifera, including PAL, HCT and CAD. The change of the cell wall was associated with environmental stresses or pathogen invasion, with the modification and subsequent breakdown of lignin.44 Although the accumulation of lignin was a favourable response to defend against pathogens or environmental stresses, growth of the rhizome was coincidentally inhibited.45 Moreover, F3H was considered as one of the main enzymes in the biosynthesis of flavonoid, increasing in the rhizome of replanted N. nucifera. Phenolic acids were the autotoxic allelochemicals, mainly producing through phenylpropanoid and flavonoid pathways. Therefore, up-regulation of proteins related in phenylpropanoid and flavonoid pathways would elevate the production of autotoxic allelochemicals in replanted N. nucifera.
Conclusions
A consecutive monoculture induced the production of allelochemicals, and the allelochemicals released in the surroundings were able to activate ethylene signalling pathways, induce ROS accumulation and disrupt metabolic balance in high plants.46,47 In addition, the role of ethylene and POD in the regulation of lignin was also indicated in transgenic tobacco.48 Based on these results, we hypothesized that ROS and ethylene were released as responsive signals induced by a consecutive monoculture, which would stimulate the following processes, including the accumulation of lignin and flavonoid, and the inhibition of antioxidant enzymes through DNA methylation. The interaction of these negative effects would further result in the production of allelochemicals and serious oxidative damage, considered as the main factors causing replant disease. Although the details of the pathways still need to be confirmed, a new depiction of the harmful mechanism of replant disease in N. nucifera was illustrated based on the analysis of the key pathways (Fig. 9).
 |
| | Fig. 9 Hypothetical model of the mechanism of replant disease in N. nucifera. Ethylene is represented with ET. | |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is financially supported by National Natural Science Foundation of China (no. 31601366), the National Science and Technology Supporting Program (no. 2012BAD27B01) and Fundamental Research Funds for the Henan Provincial Colleges and Universities in Henan University of Technology (no. 2015QNJH07).
References
- J. H. Xue, W. P. Dong, T. Cheng and S. L. Zhou, Nelumbonaceae: systematic position and species diversification revealed by the complete chloroplast genome, J. Syst. Evol., 2012, 50, 477–487 CrossRef.
- J. Shen-Miller, Sacred lotus, the long-living fruits of China Antique, Seed Sci. Res., 2002, 12, 131–143 Search PubMed.
- P. Poornima, C. F. Weng and V. V. Padma, Neferine, an alkaloid from lotus seed embryo, inhibits human lung cancer cell growth by MAPK activation and cell cycle arrest, BioFactors, 2013, 40(1), 121–131 CrossRef PubMed.
- R. S. Utkhede, Soil sickness, replant problem or replant disease and its integrated control, Allelopathy J., 2016, 18, 23–38 Search PubMed.
- X. G. Li, Y. N. Zhang, C. F. Ding, Z. J. Jia, Z. L. He and T. L. Zhang, Declined soil suppressiveness to Fusarium oxysporum by rhizosphere microflora of cotton in soil sickness, Biol. Fertil. Soils, 2015, 51(8), 935–946 CrossRef.
- Z. Y. Zhang, W. X. Lin, Y. H. Yang, H. Chen and X. J. Chen, Effects of Consecutively Monocultured Rehmannia glutinosa L. on Diversity of Fungal Community in Rhizospheric Soil, Agric. Sci. China, 2011, 10(9), 1374–1384 CrossRef.
- Y. J. Lu, J. D. Wu and S. C. Yang, The Obstacles and solution of high yield in seed lotus (Nelumbo nucifera), Journal of Changjiang Vegetables, 2014, 1, 56–58 Search PubMed.
- Z. J. Zhang, Y. C. Guo and S. Z. Guo, Morphological research of flower opening and pollen abortion in replanting and first year planting Nelumbo nucifera, Special Wild Economic Animal and Plant Research, 2002, 4, 6–9 Search PubMed.
- R. Santhanam, V. T. Luu, A. Weinhold, J. Goldberg, Y. Oh and I. T. Baldwin, Native root-associated bacteria rescue a plant from a sudden-wilt disease that emerged during continuous cropping, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(36), 5013–5020 CrossRef PubMed.
- M. Mazzola and L. M. Manici, Apple replant disease: role of microbial ecology in cause and control, Annu. Rev. Phytopathol., 2012, l50, 45–65 CrossRef PubMed.
- L. K. Wu, H. B. Wang, Z. X. Zhang, R. Lin, Z. Y. Zhang and W. X. Lin, Comparative metaproteomic analysis on consecutively Rehmannia glutinosa-monocultured rhizosphere Soil, PLoS One, 2011, 6, e20611, DOI:10.1371/journal.pone.0020611.
- W. X. Lin, L. K. Wu, S. Lin, A. J. Zhang, M. M. Zhou and R. Lin, Metaproteomic analysis of ratoon sugarcane rhizospheric soil, BMC Microbiol., 2013, 13, 135 CrossRef PubMed.
- Y. H. Yang, M. J. Li, P. F. Wang, X. J. Chen, F. Q. Wang and W. X. Lin, Transcriptome-wide identification of the genes responding to replanting disease in Rehmannia glutinosa L. roots, J. Mol. Biol., 2015, 42(5), 881–892 Search PubMed.
- M. Z. Lin, Z. X. Zhang, Z. C. Lin, C. H. You, L. J. Zeng and W. X. Lin, Analysis of differential expression of proteins in replanting disease of Pseudostellaria heterophylla, Acta Prataculturae Sinica, 2010, 6, 197–207 Search PubMed.
- W. X. Lin, C. X. Fang, L. K. Wu, G. L. Li and Z. L. Zhang, Proteomic approach for molecular physiological mechanism on consecutive monoculture problems of Rehmannia glutinosa, OMICS, 2011, 2, 287–296 Search PubMed.
- R. Ghosh, R. C. Mishra, B. Choi, Y. S. Kwon, D. W. Bae and S. C. Park, Exposure to Sound Vibrations Lead to Transcriptomic, Proteomic and Hormonal Changes in Arabidopsis, Sci. Rep., 2016, 6, 33370 CrossRef PubMed.
- M. H. Fan, X. Sun, N. J. Xu, Z. Liao, Y. H. Li and J. X. Wang, Integration of deep transcriptome and proteome analyses of salicylic acid regulation high temperature stress in Ulva prolifera, Sci. Rep., 2017, 7, 11052, DOI:10.1038/s41598-017-11449-w.
- R. Ming, R. VanBuren, Y. Liu, M. Yang, Y. Han and L. T. Li, Genome of the long-living sacred lotus (Nelumbo nucifera Gaertn.), Genome Biol., 2013, 4(5), R41, DOI:10.1186/gb-2013-14-5-r41.
- S. Wang, Z. You, L. Ye, J. Che, Q. Qian and Y. Nanjo, Quantitative proteomic and transcriptomic analyses of molecular mechanisms associated with low silk production in silkworm Bombyx mori, J. Proteome Res., 2014, 13, 735–751 CrossRef PubMed.
- D. Kim, B. Langmead and S. L. Salzberg, HISAT: a fast spliced aligner with low memory requirements, Nat. Methods, 2015, 12(4), 357–360 CrossRef PubMed.
- C. Trapnell, A. Roberts, L. Goff, G. Pertea, D. R. Kelley and H. Pimentel, Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks, Nat. Protoc., 2012, 7(3), 562–578 CrossRef PubMed.
- L. Kong, Y. Zhang, Z. Q. Ye, X. Q. Liu, S. Q. Zhao and L. P. Wei, CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine, Nucleic Acids Res., 2007, 35, W345–W349 CrossRef PubMed.
- B. Langmead and S. L. Salzberg, Fast gapped-read alignment with Bowtie 2, Nat. Methods, 2012, 9, 357–359 CrossRef PubMed.
- A. Mortazavi, B. A. Williams, K. McCue, L. Schaeffer and B. Wold, Mapping and quantifying mammalian transcriptomes by RNA-Seq, Nat. Methods, 2008, 5(7), 621–628 CrossRef PubMed.
- Y. Q. Yin, F. Qi, L. Gao, S. Q. Rao, Z. Q. Yang and W. M. Fang, iTRAQ-based quantitative proteomic analysis of dark-germinated soybeans in response to salt stress, RSC Adv., 2018, 8, 17905–17913 RSC.
- C. Dong, X. F. Zheng, Y. Diao, Y. W. Wang, M. Q. Zhou and Z. L. Hu, Molecular cloning and expression analysis of a catalase gene (NnCAT) from Nelumbo nucifera, Appl. Biochem. Biotechnol., 2015, 177(6), 1216–1228 CrossRef PubMed.
- Y. Y. Zhao, Y. L. Xu, Z. Wang, J. F. Zhang, X. Chen, Z. F. Li, Z. F. Li, L. F. Jin, P. Wei, L. Zhang, X. Q. Zhang, R. Wang and F. Wei, Genome-wide identification and characterization of an amino acid permease gene family in Nicotiana tabacum, RSC Adv., 2017, 7, 38081 RSC.
- K. Tamura, J. Dudley, M. Nei and S. Kumar, MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0, Mol. Biol. Evol., 2007, 24(8), 1596–1599 CrossRef PubMed.
- C. Beauchamp and I. Fridovich, Superoxide dismutase: improved assay and an assay applicable to arylamide gels, Anal. Biochem., 1971, 44(1), 276–287 CrossRef PubMed.
- K. Mizuno, D. Fukuda, M. Kakihara, M. Kohno, T. L. Ha and K. Sonomoto, Purification and gene cloning of catalase from Staphylococcus warneri ISK-1, Food Sci. Technol. Res., 2000, 6(4), 324–329 CrossRef.
- Z. Q. Lu, T. Takano and S. K. Liu, Purification and characterization of two ascorbate peroxidases of rice (Oryza sativa L.) expressed in Escherichia coli, Biotechnol. Lett., 2005, 27(1), 63–67 CrossRef PubMed.
- M. Y. Lee and S. S. Kim, Characteristics of six isoperoxidases from Korean radish root, Phytochemistry, 1994, 35(2), 287–290 CrossRef PubMed.
- Z. Zhang, The Guidance of Plant Physiology Experiments, Chinese Agricultural Science and Technology Press, Beijing, 2004, pp. 120–135 Search PubMed.
- I. B. Ferguson and J. E. Harman, Inhibition by calcium of senescence of detached cucumber cotyledons: effect on ethylene and hydroperoxide production, Plant Physiol., 1983, 71(1), 182–186 CrossRef PubMed.
- Z. F. Li, Y. Q. Yang, D. F. Xie, L. F. Zhu, Z. G. Zhang and W. X. Lin, Identification of autotoxic compounds in fibrous roots of Rehmannia (Rehmannia glutinosa Libosch.), PLoS One, 2012, 7(1), e28806, DOI:10.1371/journal.pone.0028806.
- Y. H. Yang, M. J. Li, X. J. Chen, P. F. Wang, F. Q. Wang and W. X. Lin, De novo characterization of the Rehmannia glutinosa leaf transcriptome and analysis of gene expression associated with replanting disease, Mol. Breed., 2014, 34, 905–915 CrossRef.
- S. P. Gygi, Y. Rochon, B. R. Franza and R. Aebersold, Correlation between protein and mRNA abundance in yeast, Mol. Cell. Biol., 1999, 19(3), 1720–1730 CrossRef PubMed.
- J. Nanduri, V. Makarenko, V. D. Reddy, G. X. Yuan, A. Pawar and N. Wang, Epigenetic regulation of hypoxic sensing disrupts cardiorespiratory homeostasis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(7), 2515–2520 CrossRef PubMed.
- A. Nguyen, N. Duquette, M. Mamarbachi and E. Thorin, Epigenetic Regulatory Effect of Exercise on Glutathione Peroxidase 1 Expression in the Skeletal Muscle of Severely Dyslipidemic Mice, PLoS One, 2016, 11(3), e0151526, DOI:10.1371/journal.pone.0151526.
- O. Rentsendorj, X. Zhang, M. C. Williams, P. W. Buehler and F. D'Agnillo, Transcriptional Suppression of Renal Antioxidant Enzyme Systems in Guinea Pigs exposed to Polymerized Cell-Free Hemoglobin, Toxics, 2016, 4(1), 6, DOI:10.3390/toxics4010006.
- O. Kwon, S. J. Jeong, S. O. Kim, L. He, H. G. Lee and K. L. Jang, Modulation of E-cadherin expression by K-Ras; involvement of DNA methyltransferase-3b, Carcinogenesis, 2010, 31(7), 1194–1201 CrossRef PubMed.
- P. H. Su, Y. W. Lin, R. L. Huang, Y. P. Liao, H. Y. Lee and H. C. Wang, Epigenetic silencing of PTPRR activates MAPK signaling, promotes metastasis and serves as a biomarker of invasive cervical cancer, Oncogene, 2013, 32(1), 15–26 CrossRef PubMed.
- M. A. Naoumkina, Q. Zhao, L. Gallego-Giraldo, X. Dai, P. X. Zhao and R. A. Dixon, Genome-wide analysis of phenylpropanoid defence pathways, Mol. Plant Pathol., 2010, 11(6), 829–846 Search PubMed.
- W. K. Huang, H. L. Ji, G. Gheysen and T. Kyndt, Thiamine-induced priming against root-knot nematode infection in rice involves lignification and hydrogen peroxide generation, Mol. Plant Pathol., 2015, 17, 614–624 CrossRef PubMed.
- R. B. Lima, V. H. Salvador, W. D. Dos Santos, G. A. Bubna, A. Finger-Teixeira and A. R. Soares, Enhanced lignin monomer production caused by cinnamic Acid and its hydroxylated derivatives inhibits soybean root growth, PLoS One, 2013, 8(12), e80542 CrossRef PubMed.
- W. C. Chi, S. F. Fu, T. L. Huang, Y. A. Chen, C. C. Chen and H. J. Huang, Identification of transcriptome profiles and signaling pathways for the allelochemical juglone in rice roots, Plant Mol. Biol., 2011, 77(6), 591–607 CrossRef PubMed.
- F. Cheng, Z. H. Cheng and H. W. Meng, Transcriptomic insights into the allelopathic effects of the garlic allelochemical diallyl disulfide on tomato roots, Sci. Rep., 2017, 6, 38902, DOI:10.1038/srep38902.
- S. Folke, H. Stéphane, L. C. H. Anthony and S. Björn, Enhanced ethylene production and peroxidase activity in IAA-overproducing transgenic tobacco plants is associated with increased lignin content and altered lignin composition, Plant Sci., 2002, 141(2), 165–173 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06503a |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Ran Wang
*a,
Ran Wang