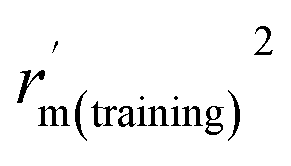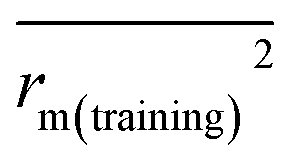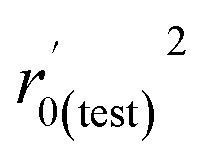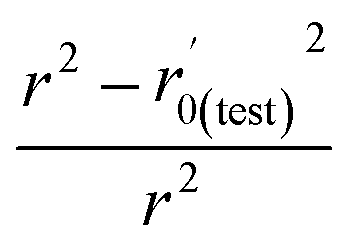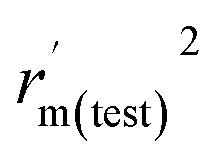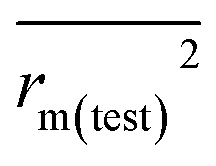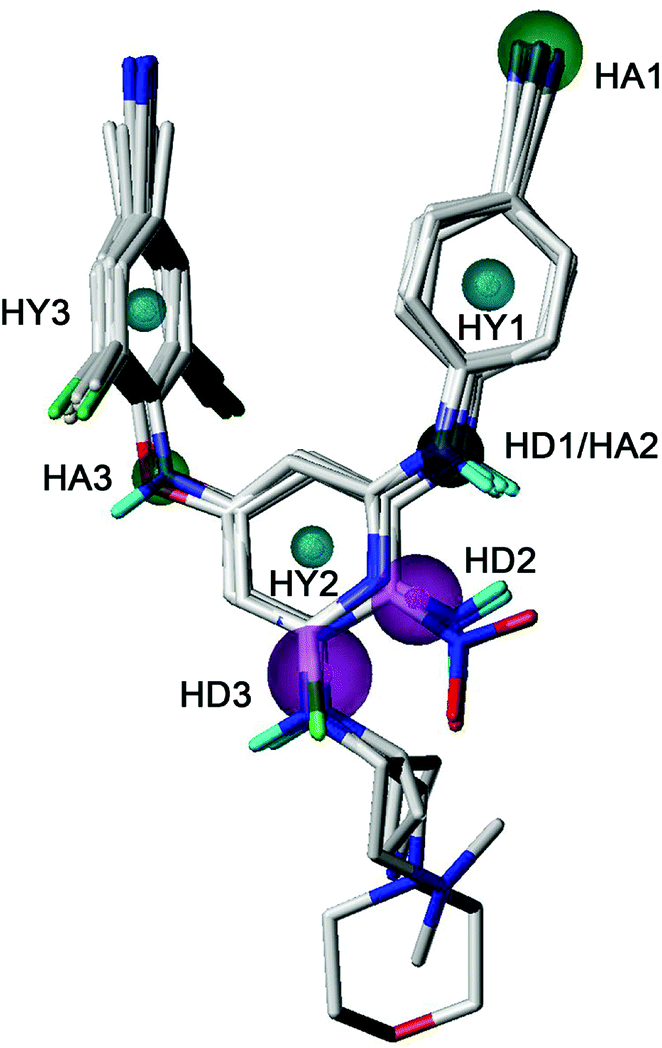DOI:
10.1039/C8RA06475J
(Paper)
RSC Adv., 2018,
8, 40529-40543
In silico studies of diarylpyridine derivatives as novel HIV-1 NNRTIs using docking-based 3D-QSAR, molecular dynamics, and pharmacophore modeling approaches
Received
1st August 2018
, Accepted 26th November 2018
First published on 5th December 2018
Abstract
A series of novel diarylpyridine derivatives has recently been identified as HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs), and most of them exhibited potent activities against HIV-1 strains, with EC50 values in the low nanomolar range. However, the three-dimensional quantitative structure–activity relationships (3D-QSARs) and pharmacophore characteristics of these compounds remain to be studied. In the present study, forty-two diarylpyridine derivatives were firstly docked into HIV-1 reverse transcriptase, and molecular dynamics (10 ns) simulations were further performed to validate the reliability of the docking results, which indicated that residues Lys101, Tyr181, Tyr188, Phe227, and Trp229 might play important roles in binding with these diarylpyridines. The “U”-shaped docking conformations of all compounds were then used to construct 3D-QSAR and pharmacophore models. The satisfactory statistical parameters of CoMFA (qloo2 = 0.665, rncv2 = 0.989, rpred2 = 0.962, etc.) and CoMSIA (qloo2 = 0.727, rncv2 = 0.988, rpred2 = 0.912, etc.) models demonstrated that both constructed models had excellent predictability, and their contour maps gave insights into the structural requirements of the diarylpyridines for the anti-HIV-1 activity. A docking-conformation-based pharmacophore model, containing three hydrophobic centers, three hydrogen-bond acceptors, and three hydrogen-bond donors, was also established. The observations in this study might provide important information for the rational design and development of novel HIV-1 NNRTIs.
Introduction
Human immunodeficiency virus (HIV) reverse transcriptase (RT) is an asymmetric heterodimer which plays a pivotal role in the viral replication stage for catalyzing the replication of single-stranded viral RNA to produce double-stranded DNA before the viral genome is integrated into the DNA of the host.1–4 This characteristic makes the HIV RT an attractive target for the development of anti-HIV drugs.5 To date, many HIV RT inhibitors have been developed, which could be mainly classified into nucleoside RT inhibitors (NRTIs) and non-nucleoside RT inhibitors (NNRTIs).6,7 NRTIs are analogues of the normal dNTP substrates.8 They can compete with endogenous nucleotides at the polymerase active site which could availably terminate the viral DNA synthesis.9 NNRTIs bind to an allosteric site that is an approximately 10 Å distance from the polymerase active site.10 In fact, the structure studies of HIV RT indicate that the NNRTI-binding pocket (NNIBP) does not exist in the absence of NNRTIs.11–14
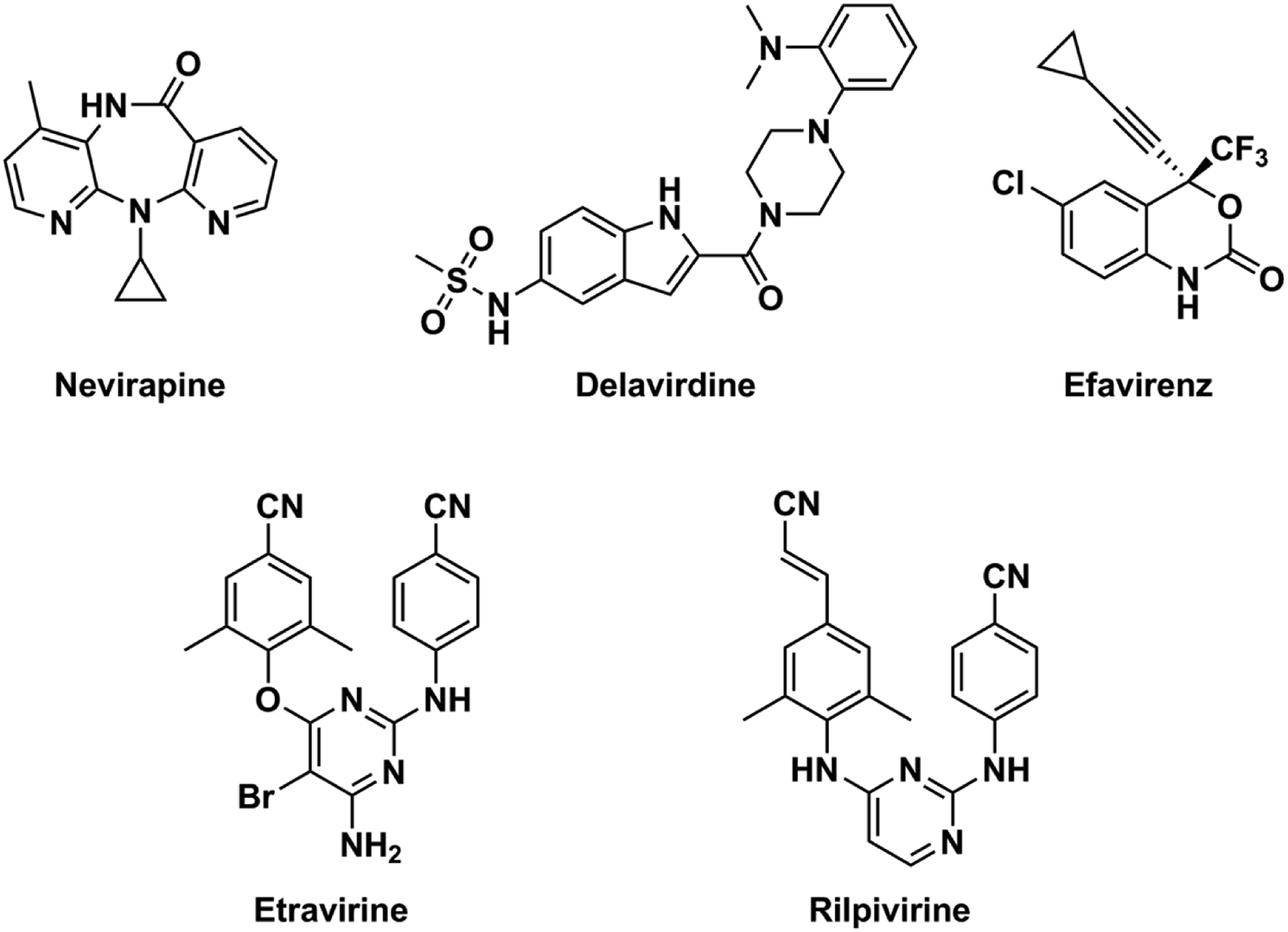
NNRTIs is an important component of highly active antiretroviral therapy, which is a standard treatment for acquired immune deficiency syndrome (AIDS) patients at present.15 Chemical structures of NNRTIs are diverse.16,17 Different from the first-generation NNRTIs that have almost rigid butterfly-like conformation, the second-generation NNRTIs are more flexible and always adopt “U” (or “horseshoe”) conformation to interact with the HIV type 1 (HIV-1) RT (Fig. 1).14,18 The obvious attribution of the second-generation NNRTIs is that they can bind to the RT through torsional flexibility (“wiggling”) and local repositioning (“jiggling”).19 Thus, the characteristic of conformational flexibility makes the second-generation NNRTIs easily adapt to the steric interference caused by different mutations at the allosteric binding site.20 Diarylpyrimidines (DAPYs) is one class of the efficient and promising second-generation NNRTIs with strong resilience to a number of HIV-1 mutants. Etravirine and rilpivirine, as the second-generation NNRTIs of DAPY derivatives, have been approved by U.S. Food and Drug Administration in 2008 and 2011, respectively.21 They have been proved to have highly potent against both WT HIV-1 and many clinically prevalent mutants with low nanomolar IC50 values.22 Although it has been reported that second-generation NNRTIs have remarkable potencies against HIV-1 RT mutants, the drug resistances and severe side effects of NNRTIs were appeared to HIV-infected patients for their long-term usage and remained the major obstacles for prolonged antiretroviral therapy.23 Thus, seeking for the novel and safe NNRTIs with broad spectrum anti-HIV-1 activity is necessary.24 Computer-aided drug design methods, such as molecular docking and pharmacophore models, are often used as effective tools for anticipating the discovery of novel hits by reducing time and costs.
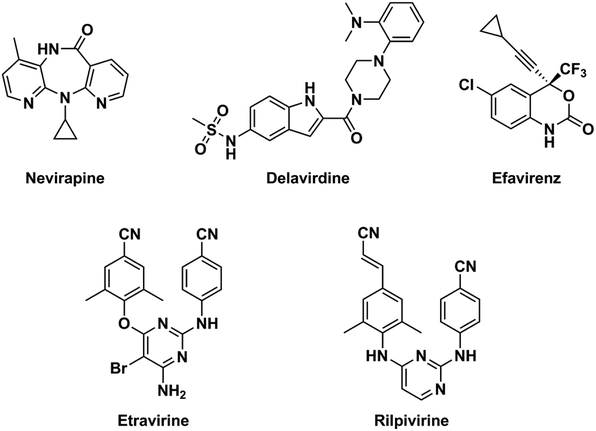 |
| | Fig. 1 Chemical structures of the first-generation (nevirapine, delavirdine, and efavirenz) and the second-generation (etravirine and rilpivirine) NNRTIs. | |
Recently, some novel diarylpyridine derivatives as NNRTIs were designed by replacing the pyrimidine ring of DAPYs with a pyridine ring (Table 1).25,26 Most of them exhibited good antiviral activity against wild-type (WT) HIV-1 strains (EC50 = 0.035–4.41 μM). To understand the three-dimensional quantitative structure–activity relationships (3D-QSARs) and pharmacophore characteristics of these novel diarylpyridines, in the present study, the docking-based 3D-QSAR and pharmacophore modeling studies were performed on the novel series of diarylpyridines. Molecular docking was first performed to investigate the interactions between these diarylpyridines and the HIV-1 RT and to obtain the possible drug-like conformations of them. To verify the rationality of the docking results and analyze the dynamics behavior of the diarylpyridines, molecular dynamics (MD) simulations were carried out on HIV-1-RT-diarylpyridine complexes. The docking conformations of the diarylpyridines were then selected to construct 3D-QSAR models, of which the contour maps could figuratively elucidate the relationships between diarylpyridine structures and their inhibitory activities. Moreover, a pharmacophore model based on the docking poses was constructed and also provided an insight into the key structural features of the diarylpyridines for the activity. This combined ligand- and acceptor-based modeling study might be helpful for designing and developing novel diarylpyridine candidates as HIV-1 NNRTIs.
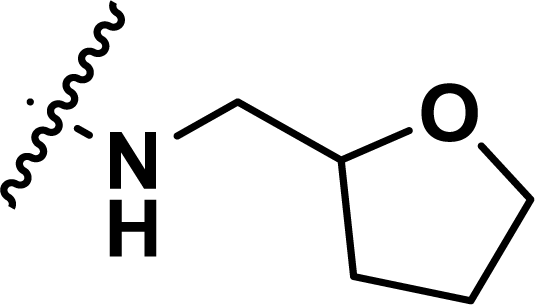
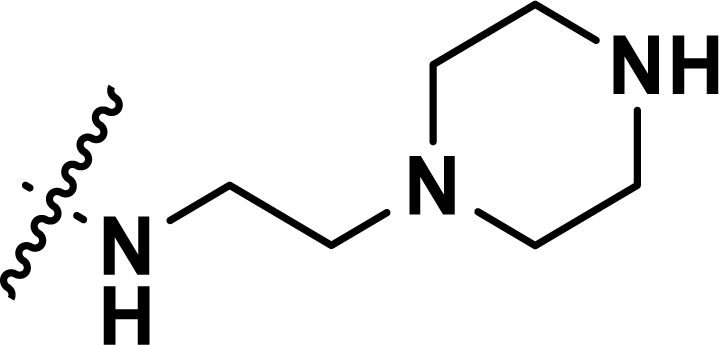
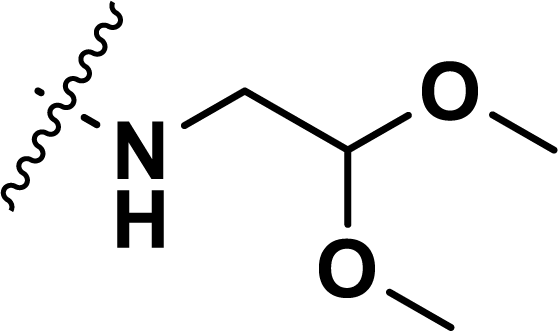
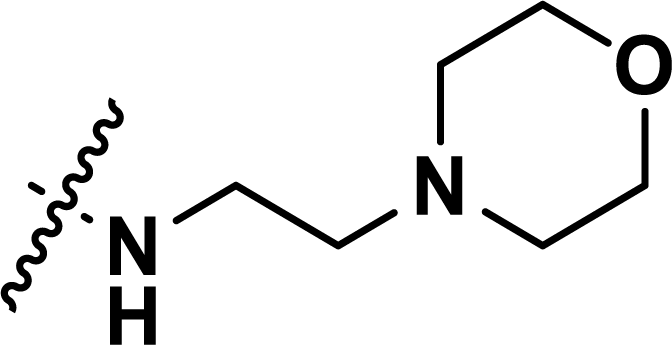
Table 1 Chemical structures, experimental and predicted pEC50 values of diarylpyridine derivatives acting as HIV-1 NNRTIs
Materials and methods
Molecular construction and structure optimization
Forty-two novel diarylpyridine derivatives were selected from the published literatures25,26 for the molecular simulation. Their structures and anti-HIV-1 activities (pEC50 = −log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) EC50) were listed in Table 1. These molecules were then constructed by SYBYL-X 2.1 running on a windows 7 workstation. All the molecules were minimized with Gasteiger-Hückel charges by the default Powell method which used conjugate-gradient minimization.27 The termination and max iterations were set to 0.005 kcal (mol−1 Å−1) and 1000, respectively. Other parameters were set as default values.
EC50) were listed in Table 1. These molecules were then constructed by SYBYL-X 2.1 running on a windows 7 workstation. All the molecules were minimized with Gasteiger-Hückel charges by the default Powell method which used conjugate-gradient minimization.27 The termination and max iterations were set to 0.005 kcal (mol−1 Å−1) and 1000, respectively. Other parameters were set as default values.
Molecular docking
The crystal structure of the HIV-1 RT for molecular docking was available in the Protein Data Bank (PDB ID: 3MEC). The PDB database includes 72 crystal structures of the WT HIV-1 RT.28 3MEC was chosen as a docking receptor because of its high resolution (2.3 Å) and its co-crystallized ligand etravirine (TMC125), which adopt a “U”-shaped conformation29 at the allosteric site.
All co-crystallized water molecules and sulfate ions were removed before docking. The binding pocket was generated by the automatic mode using the extracted ligand TMC125.30 In order to verify the reliability of molecular docking, the cognate ligand TMC125 was extracted and then re-docked into the prepared protein using Surflex-Dock Geom with default parameters. After that, 42 diarylpyridine derivatives were docked into the binding site with the same pattern. All molecules were flexible in the docking process and 20 different conformations were generated for each molecule. The conformation with the highest docking score was selected for the further studies. The visualization of the docking results was performed using PyMOL software.
Molecular dynamics simulation
According to the docking results, the MD simulation was performed on complexes of 3MEC with compounds 11 and 28 using the GROMACS2016.5 software to characterize the stability of protein–ligand interactions. The molecular topology file was generated by acpype, a tool based on Python to use Antechamber to generate topologies for chemical compounds. Both complexes were solvated in a 20 × 20 × 20 Å3 cubic box filling with TIP3P water model, respectively. Seven chloride ions were added to neutralize the charge of the system. The system energy was minimized using AMBER 99SB force field without constraints using the steepest descent integrator for 50000 steps, until a tolerance of 10 kJ mol−1. After equilibrated at 300 K using V-rescale for 100 ps as NVT ensemble, the system was then equilibrated at 1 atm pressure using Parrinello-Rahman algorithm NPT ensemble for 100 ps. Finally, the MD simulation was carried out for 10 ns. The linear constraint solver algorithm, which is three to four times faster than the shake algorithm, was used to constrain the length of covalent bonds.31 The particle-mesh Ewald summation technique was used to compute long-ranged electrostatic interactions.32 The coulomb and van der Waal's cut-offs were set to 1.0 and 1.4 nm, respectively. During the MD simulation, the time step was defined as 2 fs. The coordinate trajectories were written at intervals of 10 ps. Potential energy (PE), hydrogen bond number, root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), radius of gyration (Rg), and solvent accessible surface area (SASA) were calculated by GROMACS inbuilt commands, g_energy, g_hbond, g_rmsd, g_rmsf, g_gyration, and g_sasa, respectively. The free binding energies were calculated using a GROMACS tool for high-throughput MM-PBSA calculations (g_mmpbsa).
3D-QSAR
QSAR is a method to establish computational or mathematical models, which attempt to find a statistically significant correlation between structure and function by a chemometric technique.33 Nowadays, multiple methodologies are maturing for 3D-QSAR studies, such as molecular shape analysis (MSA), hypothetical active site lattice (HASL), comparative molecular field analysis (CoMFA), comparative molecular similarity indices analysis (CoMSIA), and genetically evolved receptor models (GERM). In this study, CoMFA and CoMSIA were performed on 42 diarylpyridine derivatives. The statistical technique for building the models was partial least square (PLS) analysis.
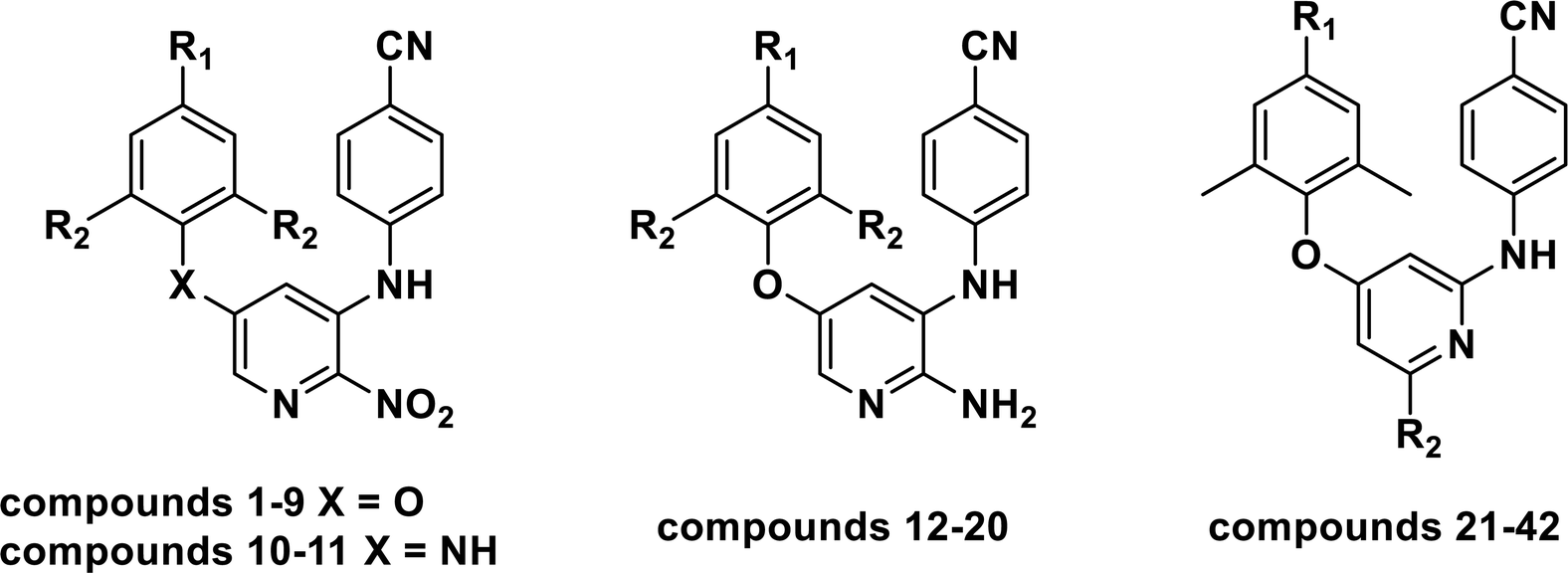
Various statistical parameters were taken into consideration to validate the quality of the constructed CoMFA and CoMSIA models. Cross-validated correlation coefficient with leave-one-out method (qloo2), non-cross-validated correlation coefficient (rncv2), F-statistic values (F), and standard error of estimate (SEE) were calculated to measure the internal prediction of the models. However, these statistical parameters could only judge how well a model may recur the response for the training set molecules34 and could not prove the prediction ability of a model for external molecules. Thus, other metrics, for example, r(test)2, r02, 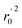 , rm2,
, rm2, 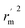 , k, k′, rpred2, root mean square error (RMSE), and mean absolute error (MAE), were considered for external validation of the models.35 r(test)2 is the regression line coefficient of correlation for the test set. r02 (predicted vs. observed activities) and
, k, k′, rpred2, root mean square error (RMSE), and mean absolute error (MAE), were considered for external validation of the models.35 r(test)2 is the regression line coefficient of correlation for the test set. r02 (predicted vs. observed activities) and 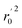 (observed vs. predicted activities) are the regression lines coefficients of correlation with the intercept set to zero. k (predicted vs. observed activities) and k′ (observed vs. predicted activities) are the slops of regression lines. As for a good model, it should satisfy the following conditions: r02 or
(observed vs. predicted activities) are the regression lines coefficients of correlation with the intercept set to zero. k (predicted vs. observed activities) and k′ (observed vs. predicted activities) are the slops of regression lines. As for a good model, it should satisfy the following conditions: r02 or 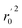 is close to r2; [(r2 − r02)/r2] < 0.1 or
is close to r2; [(r2 − r02)/r2] < 0.1 or 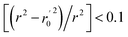 ; 0.85 ≤ k ≤ 1.15 or 0.85 ≤ k′ ≤ 1.15.36 A novel validation parameter rm2 is calculated as the following equation:
; 0.85 ≤ k ≤ 1.15 or 0.85 ≤ k′ ≤ 1.15.36 A novel validation parameter rm2 is calculated as the following equation:
| |
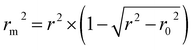 | (1) |
Δ
rm2 and
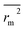
are also important to prove the external prediction of a model. Δ
rm2 is the difference between
rm2 (predicted
vs. observed activities) and
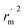
(observed
vs. predicted activities).
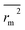
is the average value of
rm2 and
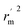
. The values of
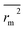
for the training set

and the test set
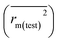
should be more than 0.5. In the case that the previous condition is met, the Δ
rm(training)2 and Δ
rm(test)2 should be less than 0.2.
37 The
rpred2, RMSE, and MAE were calculated by the following equations,
37,38 respectively.
| |
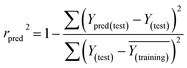 | (2) |
| |
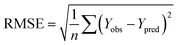 | (3) |
| |
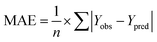 | (4) |
The rpred2 should be more than 0.5 and the MAE value should satisfy the following requirement: MAE ≤ 0.1× training set range.
Furthermore, sensitivity, specificity, accuracy, positive prediction value (PPV), negative prediction value (NPV) and Matthew's correlation coefficient (MCC) were calculated to assess the quality of 3D-QSAR models. Based on a comparison between the predicted and observed values, the compounds can be classified into four categories: true positives (TP), false negatives (FN), false positives (FP), true negatives (TN).39 The above statistical parameters were defined respectively in the following equations:
| |
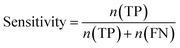 | (5) |
| |
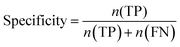 | (6) |
| |
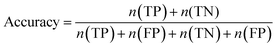 | (7) |
| |
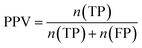 | (8) |
| |
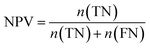 | (9) |
| |
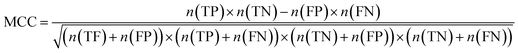 | (10) |
Pharmacophore model
Genetic Algorithm with Linear Assignment of Hypermolecular Alignment of Datasets (GALAHAD) is a novel approach for pharmacophore screening module. The GALAHAD alignment is from a set of ligand molecules that bind at a common target site by Tripos' proprietary technology to generate pharmacophore hypotheses.40 GALAHAD runs in two main stages, including that the ligands are flexibly aligned to each other in internal coordinate space, and then the produced conformations are aligned in Cartesian space. However, these molecules are considered as rigid bodies in the second step. In this study, ten compounds (3, 11, 12, 14, 21, 22, 28, 33, 34, and 40) with high activities and diverse structures were selected to build pharmacophore models. A virtual suggestion according to the experimental activity data is automatically performed to set the parameters of population size, max generations, and mols required to hit for GALAHAD. All conformations of selected molecules were frozen in the setting of advanced parameters.
Results and discussion
Molecular docking
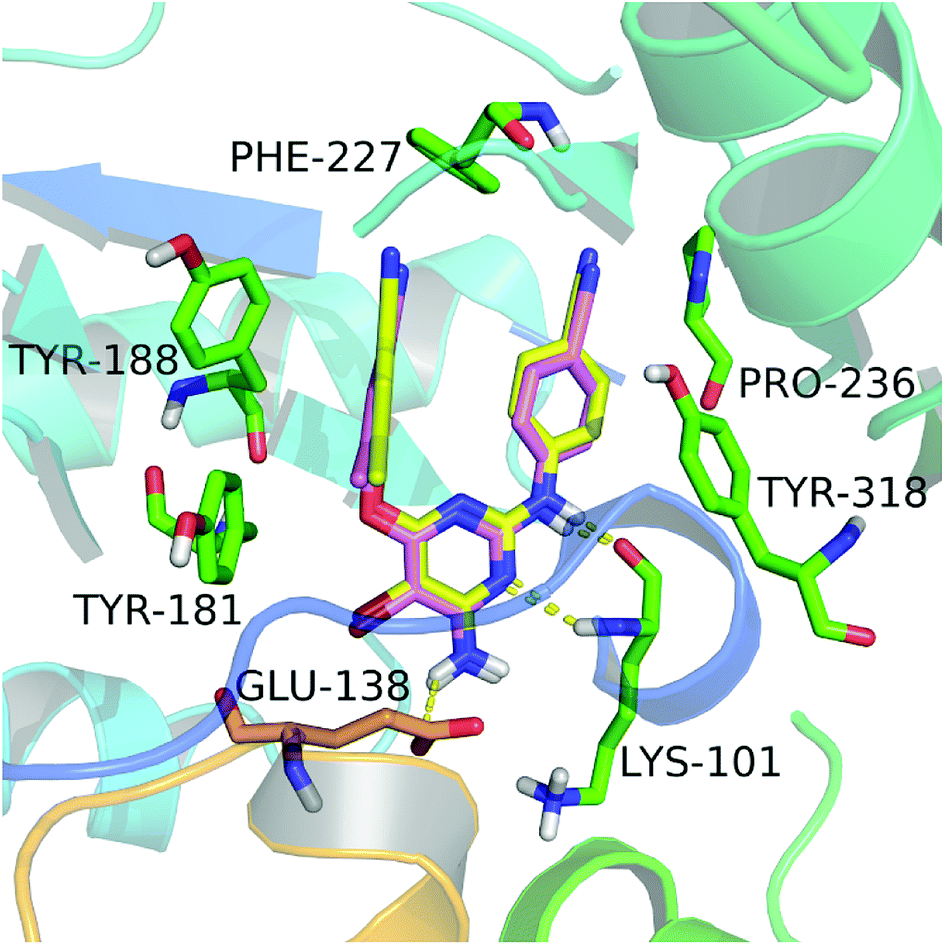
In this study, we firstly applied molecular dockings to investigate the binding mode of the diarylpyridine derivatives in the HIV-1 RT (PDB ID: 3MEC), and the docked conformations of these diarylpyridines were then used to construct the subsequent 3D-QSAR and pharmacophore models. The cognate ligand TMC125 was extracted and then re-docked into the binding site of 3MEC to verify whether the used docking method and parameters were appropriate and reliable. The re-docked conformation was compared with the original conformation of the co-crystal TMC125 (Fig. 2), and the difference between two conformations was very slight with a RMSD of 0.24 Å (<2.0 Å)41 for all atoms of TMC125. The re-docked results showed that TMC125 formed hydrogen bonds with Lys101 (Lys101–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–, 1.78 Å; Lys101–N–H⋯N–, 2.57 Å) and Glu138 (Glu138–C–O⋯H–, 2.39 Å). The bromine substituent of the pyrimidine ring pointed to an opening cavity between Lys101 and Glu138. The left phenyl substituent located at a hydrophobic pocket that formed by Tyr181, Tyr188, Phe227, and Trp229, and interacted with these aromatic residues by π–π stacking. The twist tendency of the re-docked TMC125 and its interaction with the enzyme were similar with the results of a previous study reported by Lansdon et al.29 suggesting that the method of docking was reliable and feasible.
O⋯H–, 1.78 Å; Lys101–N–H⋯N–, 2.57 Å) and Glu138 (Glu138–C–O⋯H–, 2.39 Å). The bromine substituent of the pyrimidine ring pointed to an opening cavity between Lys101 and Glu138. The left phenyl substituent located at a hydrophobic pocket that formed by Tyr181, Tyr188, Phe227, and Trp229, and interacted with these aromatic residues by π–π stacking. The twist tendency of the re-docked TMC125 and its interaction with the enzyme were similar with the results of a previous study reported by Lansdon et al.29 suggesting that the method of docking was reliable and feasible.
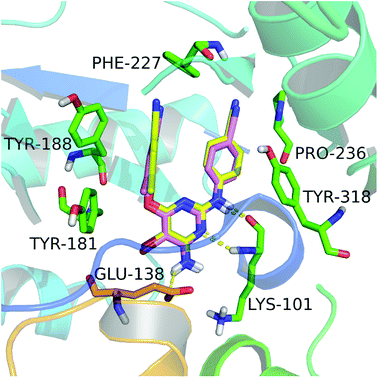 |
| | Fig. 2 The re-docked results of etravirine in the binding pocket of the HIV-1 RT (PDB ID: 3MEC). The re-docked etravirine (pink) almost completely superimposes with the co-crystallized etravirine (yellow). The residues from chains A and B are displayed in green and orange, respectively. Hydrogen bonds are shown as yellow dash lines. | |
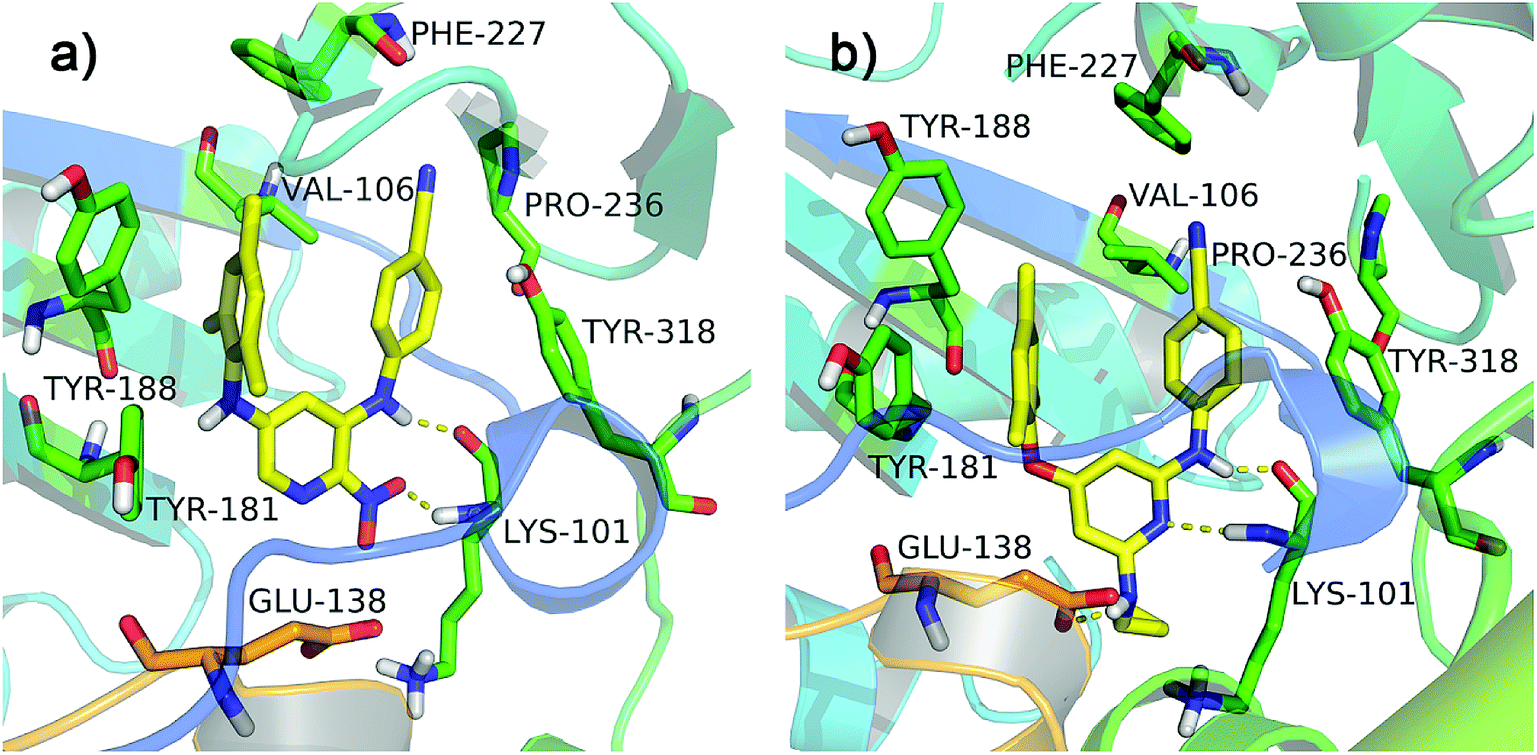
All diarylpyridines were then docked into the NNIBP of the HIV-1 RT using the same docking pattern. All compounds adopted a similar “U” -shape with the original ligand TMC125 in the NNIBP. The similar hydrogen bonds, π–π stacking and hydrophobic interactions were observed in the docking results. The left benzene rings of all compounds interacted with Tyr181, Tyr188, Phe227, and Trp229 by π–π stacking and hydrophobic interactions. Hydrogen bonds were found between Lys101 and the imino group of the right linker, the amino/nitro substituent neighbor to the right linker, or the nitrogen atom of the pyridine ring. The binding patterns of two most active compounds 11 and 28 were shown in Fig. 3a and b, respectively. As for compound 11, the nitro group out of the pyridine ring and the imino group of the right linker hydrogen-bonded with Lys101 (Lys101–N–H⋯O = , 2.02 Å; Lys101–CO⋯H–, 1.89 Å). The nitrogen atoms of the pyridine rings located at the entrance of the opening between Lys101 and Glu138. As shown in Fig. 3b, compound 28 formed two hydrogen bonds with Lys101 (Lys101–N–H⋯N–, 2.27 Å; Lys101–CO⋯H–, 1.65 Å). The imino group of the R2 substituents interacted with Glu138 via a hydrogen bond (Glu138 = O⋯H–, 2.01 Å). In this study, Glu138 was found to be the only amino acid residue of the chain B, which could interact with these diarylpyridine derivatives. This result is in agreement with a previous report by Das et al.42 All found interactions are also consistent with the results from the reported literatures.21,22 It seems that Lys101 is a key residue which could form at least one hydrogen bond with diarylpyridine derivatives.
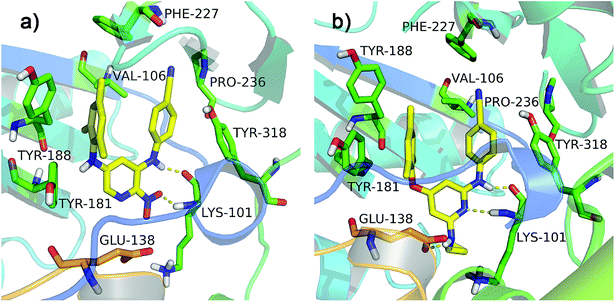 |
| | Fig. 3 The docking results of compounds 11 (a) and 28 (b) in the binding pocket of the HIV-1 RT (PDB ID: 3MEC). The residues from chains A and B are displayed in green and orange, respectively. Hydrogen bonds are shown as yellow dash lines. | |
MD simulations
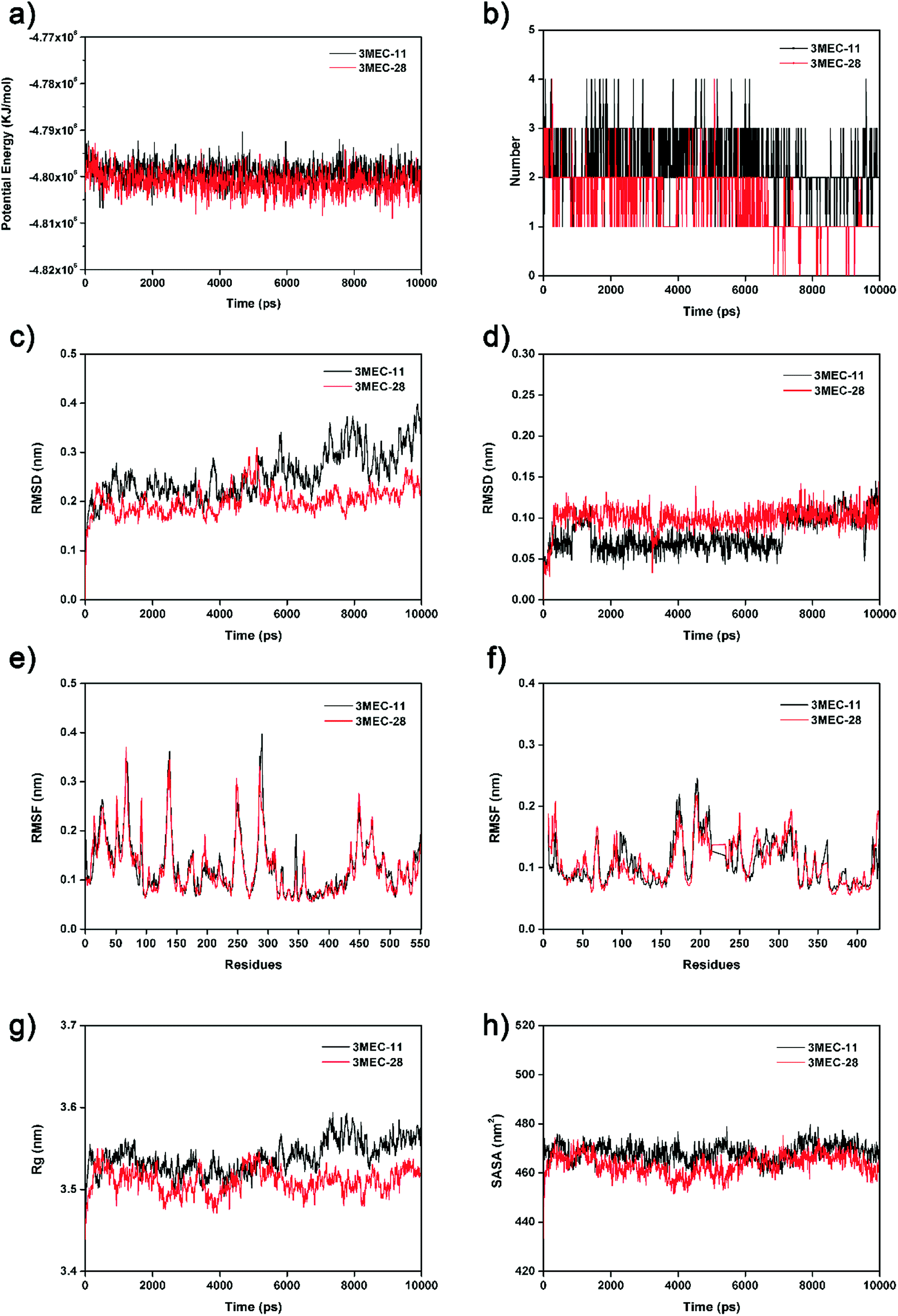
To further validate the docking results and understand the dynamic interactions between ligands and the HIV-1 RT, 10 ns MD simulations were carried on two complexes, 3MEC-11 and 3MEC-28. Fig. 4a showed the average PE values of 3MEC-11 and 3MEC-28 during the 10 ns MD simulations, ranging from −4.812 × 106 to −4.793 × 106 kJ mol−1, which confirmed the stability of two complexes. The total numbers of hydrogen bond interactions of two complexes during the 10 ns MD simulations were shown in Fig. 4b. As shown in Fig. 4c and d, the RMSD values of both protein backbones had a sharply rise during the first 0.3 ns and then remained steady (Fig. 4c); the RMSD values of the total atoms of ligands 11 and 28 maintained at 0.10 and 0.12 nm after 4.0 and 7.5 ns, respectively. These data demonstrated that two systems were stable during the 10 ns MD simulations.
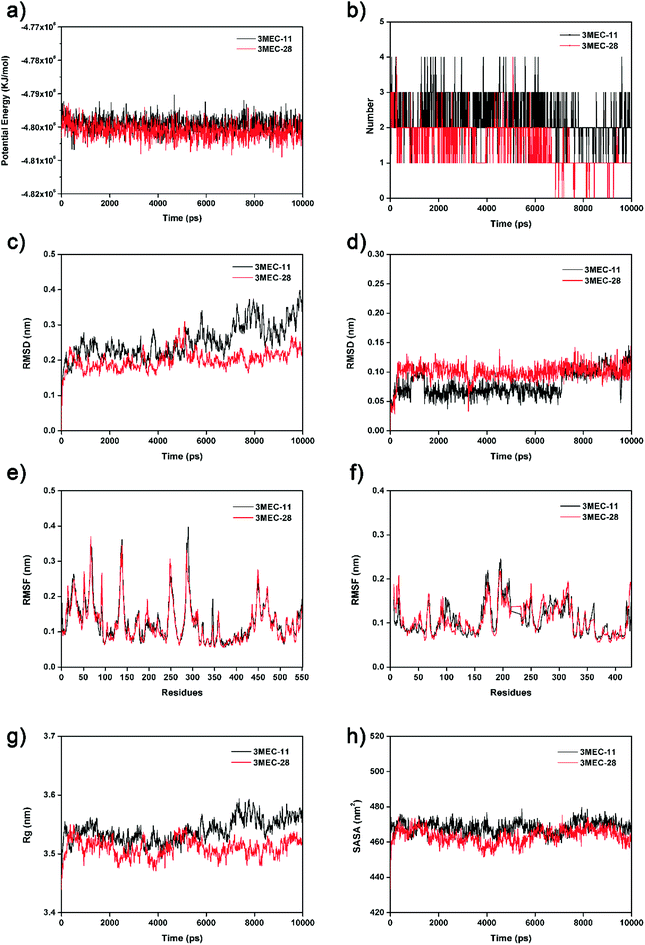 |
| | Fig. 4 The 10 ns MD results of two protein–ligand complexes (3MEC-11 and 3MEC-28). (a) Potential energies. (b) Total number of hydrogen bonds between the ligands and the HIV-1 RT. (c) RMSD values of backbone atoms. (d) RMSD values of the ligands. (e) RMSF values of chain A. (f) RMSF values of chain B. (g) Rg of backbone atoms. (h) SASA of the ligands. | |
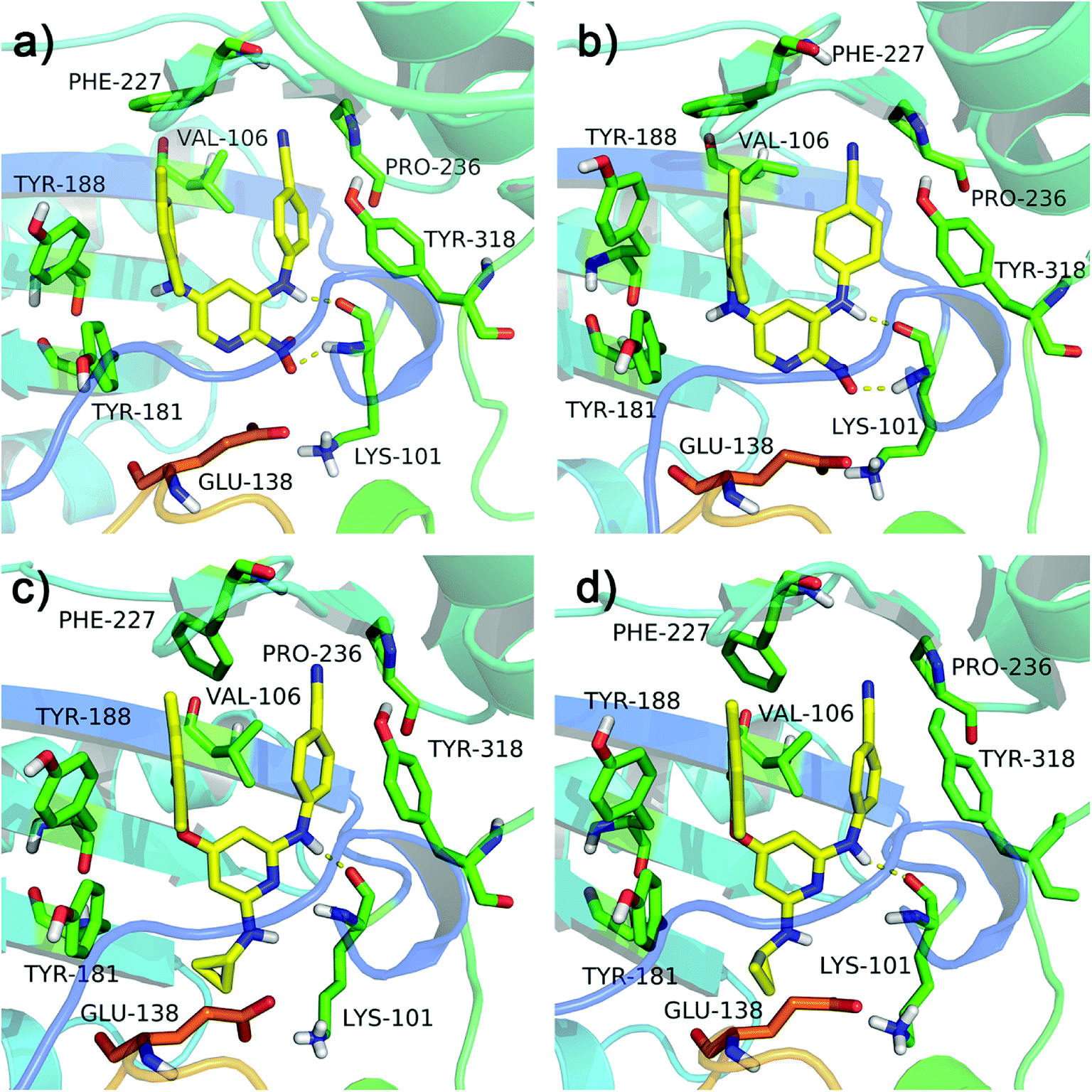
The RMSF values of all residues of chains A and B were lower than 0.40 nm (Fig. 4e and f). Low RMSF values of some key residues in the NNIBP illustrated these residues had good stability in a dynamic environment. The Rg and SASA values of two complexes during the 10 ns MD simulations were shown in Fig. 4g and h, respectively. It could be observed that the Rg and SASA values were equilibrated after a short-time increase at the beginning of the simulations. The binding energies of complexes 3MEC-11 and 3MEC-28 were −37.77 and −36.58 kcal mol−1, respectively. Moreover, the binding modes of complexes 3MEC-11 and 3MEC-28 at 5 ns and 10 ns were shown in Fig. 5. Compounds 11 and 28 maintained a “U”-shape and had the same interactions with the residues of the NNIBP during the MD processes. It should be noted that the hydrogen bonds between compound 28 and Lys101 (chain A) were observed, but the hydrogen bond with Glu138 (chain B) was disappeared. This might be because the distance between the compound 28 and Glu138 was too far at the selected moment for the MD simulation, and this observation also indicated that the Lys101 was a key residue on the other hand. In general, all MD results indicated that complexes 3MEC-11 and 3MEC-28 were structurally stable and the docking results were reliable.
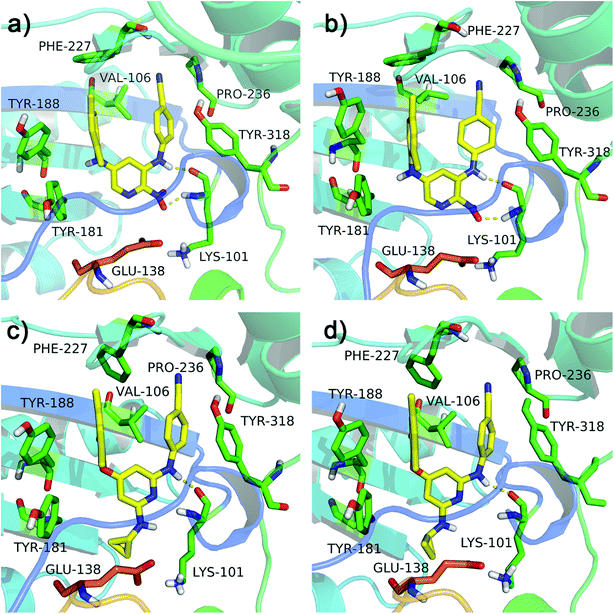 |
| | Fig. 5 Binding modes of the protein–ligand complexes 3MEC-11 at the moment of (a) 5 ns and (b) 10 ns, and 3MEC-28 at the moment of (c) 5 ns and (d) 10 ns during the MD simulations. | |
Statistical results of CoMFA and CoMSIA
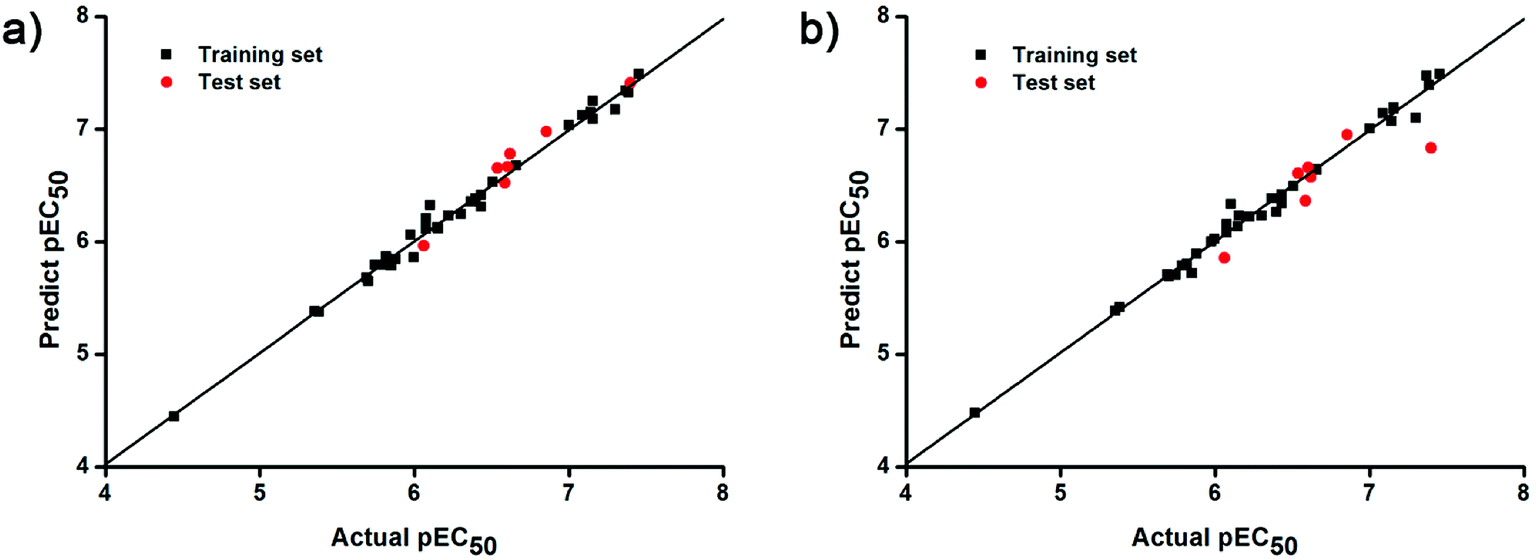
The docking conformations of 42 diarylpyridines were then used for building 3D-QSAR models (Fig. 6). The compounds were divided into a training set (35 compounds) and a test set (7 compounds) randomly. The PLS statistical results were listed in Table 2. All the permutation and combination of steric (S), electrostatic (E), hydrophobic (H), hydrogen-bond donor (D), and hydrogen-bond acceptor (A) fields of CoMSIA were calculated by PLS, and CoMSIA models with low values of qloo2 (<0.5) and rncv2 (<0.9) were not listed in Table 2. The S, E, H, D, and A contributions of CoMSIA (S + E + H + D + A) were 7.5%, 25.4%, 21.1%, 19.5%, and 26.6%, respectively. It could be observed that E and A fields played more significant roles. The CoMFA model (S + E) (qloo2 = 0.665, SEE = 0.084, rncv2 = 0.989) and the CoMSIA model (S + E + H + D + A) (qloo2 = 0.727, SEE = 0.093, rncv2 = 0.988) were used for further analysis. As shown in Table 3, the MAEtraining and MAEtest of CoMFA and CoMSIA were both less than 0.1 × training set range. The values of the slop k and k′ of test set were close to 1. Regarding the values of rm2 metrics, the Δrm(training)2 and Δrm(test)2 of CoMFA and CoMSIA were lower than 0.2; the  and
and 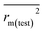 of CoMFA and CoMSIA were higher than 0.5. The correlation between the actual and predicted activities of 35 compounds in the training set and 7 compounds in the test set were shown in Fig. 7. The sensitivity, specificity, accuracy, PPV, NPV, and MCC of CoMFA and CoMSIA were listed in Table 4. The sensitivity values of both models were 96.7%, indicating that 96.7% active compounds were correctly predicted as actives. The specificity of CoMFA and CoMSIA were 91.7% and 83.3%, which manifested the percentages of correctly predicted inactive compounds out of total inactives. The MCC is a correlation coefficient between the observed and predicted binary classifications, and is ranging from −1 to 1, in which the value of 1 signifies a perfect prediction of model. The MCC values of both CoMFA (0.8833) and CoMSIA (0.8220) models were close to 1. All these statistical results of CoMFA and CoMSIA were similar and well within the acceptable limit, which manifested that the developed CoMFA and CoMSIA models with good predictive ability were reliable and reasonable.
of CoMFA and CoMSIA were higher than 0.5. The correlation between the actual and predicted activities of 35 compounds in the training set and 7 compounds in the test set were shown in Fig. 7. The sensitivity, specificity, accuracy, PPV, NPV, and MCC of CoMFA and CoMSIA were listed in Table 4. The sensitivity values of both models were 96.7%, indicating that 96.7% active compounds were correctly predicted as actives. The specificity of CoMFA and CoMSIA were 91.7% and 83.3%, which manifested the percentages of correctly predicted inactive compounds out of total inactives. The MCC is a correlation coefficient between the observed and predicted binary classifications, and is ranging from −1 to 1, in which the value of 1 signifies a perfect prediction of model. The MCC values of both CoMFA (0.8833) and CoMSIA (0.8220) models were close to 1. All these statistical results of CoMFA and CoMSIA were similar and well within the acceptable limit, which manifested that the developed CoMFA and CoMSIA models with good predictive ability were reliable and reasonable.
 |
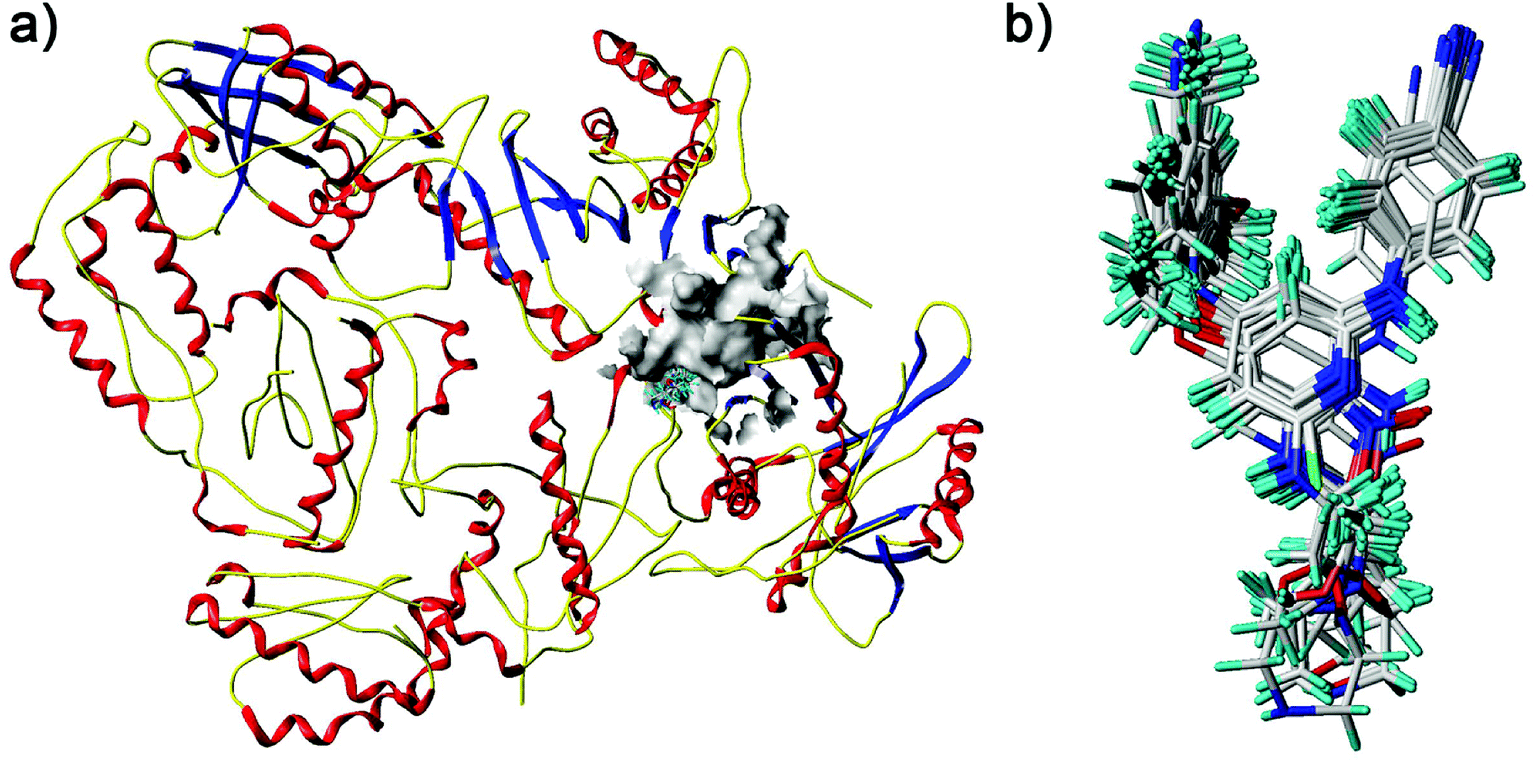
| | Fig. 6 Docked diarylpyridine derivatives in the nonnucleoside binding site of the HIV-1 RT (PDB ID: 3MEC). (a) The surface of the residues which are 5 Å from ligands are shown in gray color. (b) The docking conformations of all the compounds. | |
Table 2 PLS statistical results of the CoMFA and CoMSIA models
| |
Modela |
qloo2 |
N |
SEE |
rncv2 |
F |
Field contribution (%) |
| S |
E |
H |
D |
A |
| S, E, H, D, and A mean steric, electrostatic, hydrophobic, hydrogen-bond donor, and hydrogen-bond acceptor fields, respectively. |
| CoMFA |
S + E |
0.665 |
10 |
0.084 |
0.989 |
221.653 |
0.396 |
0.604 |
|
|
|
| S + E + H + D + A |
0.727 |
12 |
0.093 |
0.988 |
150.555 |
0.075 |
0.254 |
0.211 |
0.195 |
0.266 |
| S + E |
0.613 |
15 |
0.110 |
0.985 |
85.333 |
0.229 |
0.771 |
|
|
|
| E + D |
0.525 |
14 |
0.168 |
0.964 |
38.423 |
|
0.700 |
|
0.300 |
|
| S + E + D |
0.679 |
12 |
0.119 |
0.980 |
90.300 |
0.142 |
0.547 |
|
0.311 |
|
| S + E + A |
0.602 |
11 |
0.146 |
0.969 |
64.793 |
0.128 |
0.473 |
|
|
0.399 |
| S + H + D |
0.543 |
8 |
0.194 |
0.938 |
48.987 |
0.192 |
|
0.383 |
0.424 |
|
| S + H + A |
0.651 |
8 |
0.163 |
0.956 |
70.442 |
0.154 |
|
0.376 |
|
0.471 |
| S + D + A |
0.607 |
15 |
0.112 |
0.985 |
81.432 |
0.205 |
|
|
0.355 |
0.440 |
| CoMSIA |
E + H + D |
0.622 |
14 |
0.081 |
0.992 |
169.485 |
|
0.407 |
0.309 |
0.284 |
|
| E + H + A |
0.601 |
8 |
0.184 |
0.944 |
54.568 |
|
0.359 |
0.264 |
|
0.377 |
| E + D + A |
0.526 |
12 |
0.152 |
0.968 |
54.940 |
|
0.482 |
|
0.188 |
0.330 |
| H + D + A |
0.698 |
11 |
0.113 |
0.981 |
109.043 |
|
|
0.361 |
0.274 |
0.365 |
| S + E + H + D |
0.686 |
13 |
0.074 |
0.993 |
221.377 |
0.105 |
0.379 |
0.264 |
0.253 |
|
| S + E + H + A |
0.640 |
8 |
0.169 |
0.953 |
65.648 |
0.099 |
0.319 |
0.236 |
|
0.346 |
| S + E + D + A |
0.696 |
13 |
0.115 |
0.982 |
90.245 |
0.110 |
0.387 |
|
0.205 |
0.299 |
| S + H + D + A |
0.748 |
10 |
0.116 |
0.979 |
114.403 |
0.091 |
|
0.310 |
0.249 |
0.349 |
| S + E + D + A |
0.670 |
12 |
0.106 |
0.984 |
113.716 |
|
0.279 |
0.232 |
0.211 |
0.278 |
Table 3 Statistical validation parameters of the CoMFA and CoMSIA (S + E + H + D + A) models using training and test set
| Validation parameters |
CoMFA |
CoMSIA |
| Root mean square error of calibration. Root-mean-square error of prediction. |
| rpred2 |
0.962 |
0.912 |
| RMSECa |
0.069 |
0.073 |
| MAEtraining |
0.051 |
0.049 |
| r0(training)2 |
0.989 |
0.988 |
 |
0.989 |
0.988 |
| rm(training)2 |
0.989 |
0.988 |
 |
0.979 |
0.974 |
| Δrm(training)2 |
0.010 |
0.014 |
 |
0.984 |
0.981 |
| RMSEPb |
0.101 |
0.249 |
| MAEtest |
0.110 |
0.181 |
| r2 |
0.955 |
0.659 |
| r0(test)2 |
0.942 |
0.649 |
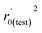 |
0.951 |
0.577 |
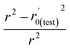 |
0.004 |
0.015 |
| k |
0.993 |
1.018 |
| k′ |
1.007 |
0.982 |
| rm(test)2 |
0.948 |
0.594 |
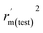 |
0.896 |
0.470 |
| Δrm(test)2 |
0.052 |
0.124 |
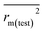 |
0.922 |
0.532 |
| 0.1× training set range |
0.301 |
0.301 |
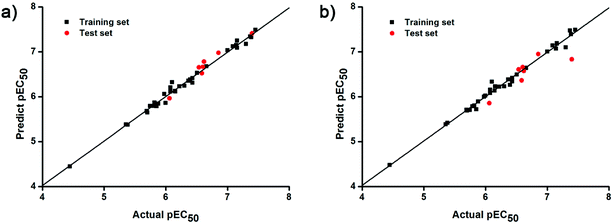 |
| | Fig. 7 Plots of actual versus predicted pEC50 values for training (black squares) and test (red dots) set compounds on the basis of (a) the CoMFA and (b) the best CoMSIA models. | |
Table 4 Sensitivity, specificity, accuracy, PPV, NPV, and MCC values of the CoMFA and CoMSIA (S + E + H + D + A) models
| Model |
Sensitivity |
Specificity |
Accuracy |
PPV |
NPV |
MCC |
| CoMFA |
0.9667 |
0.9167 |
0.9524 |
0.9667 |
0.9167 |
0.8833 |
| CoMSIA |
0.9667 |
0.8333 |
0.9286 |
0.9355 |
0.9091 |
0.8220 |
CoMFA and CoMSIA contour maps
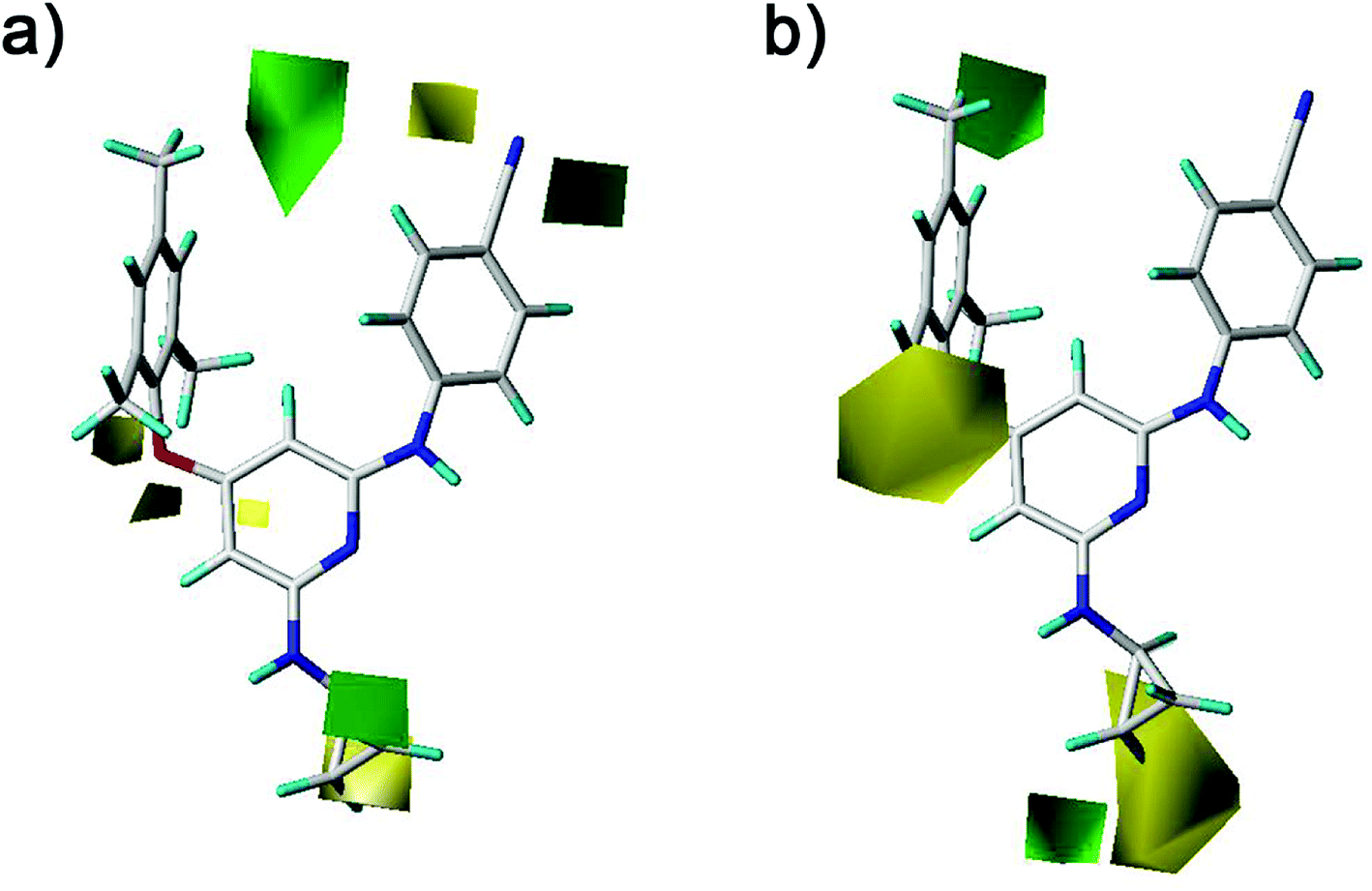
The CoMFA and CoMSIA contour maps were further analyzed to investigate the QSARs of the diarylpyridine derivatives. Compound 28 as a reference molecule was in combination with the contour maps. Fig. 8a and b showed the steric contour maps of CoMFA and CoMSIA models, respectively. The green contours represent that bulky groups are favored at this position and the yellow contours mean that bulky groups are disfavored here. The green contour near the para-position of the left benzene ring indicated that the compound with bulky substituent at this position might be beneficial for the activity. This was consistent with the experimental results that the activities of compounds with a methyl group at the para-position were higher than those of compounds with a hydrogen or a bromine at the same position, for example, 11 (para-CH3) > 10 (para-H), 3 (para-CH3) > 8 (para-Br), and 14 (para-CH3) > 20 (para-Br). It could be observed that three small yellow contours located at the ortho-position of the left benzene ring in Fig. 8a and a middle yellow contour situated at the same place in Fig. 8b, indicating that bulky substituents at the ortho-position were not beneficial for the activity. This visualization might explain why the compound 16 with a methoxy group at the ortho-position has low pEC50 value. It was also in agreement with the order of activity data: 3 (ortho-Br) > 2 (ortho-CH3), 7 (ortho-Cl) > 5 (ortho-CH3), 14 (ortho-Br) > 13 (ortho-CH3), 19 (ortho-Cl) > 17 (ortho-CH3). A yellow contour covered a part of the R2 substituents (compounds 23–42) and was close to the pyridine ring, indicating that the reducing the R2 length might increase the antiviral activity, for example, 28 > 25 and 38 > 35. This result is consistent with a previous conclusion reported by Yang et al.25 Moreover, a green contour neighbor to the terminus of the R2 suggested that the existing of a bulky group at the R2 terminal might enhance the activity.
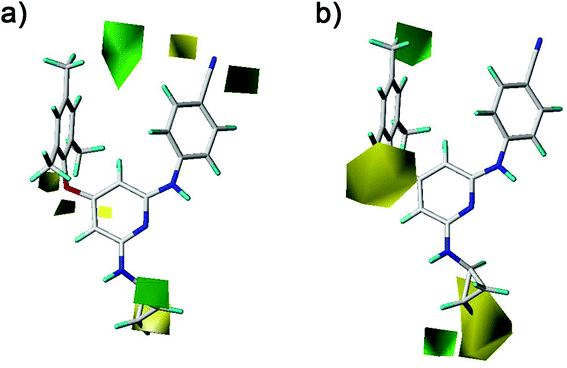 |
| | Fig. 8 Contour maps of steric fields derived from (a) the CoMFA and (b) the best CoMSIA models using compound 28 as a reference molecule. | |
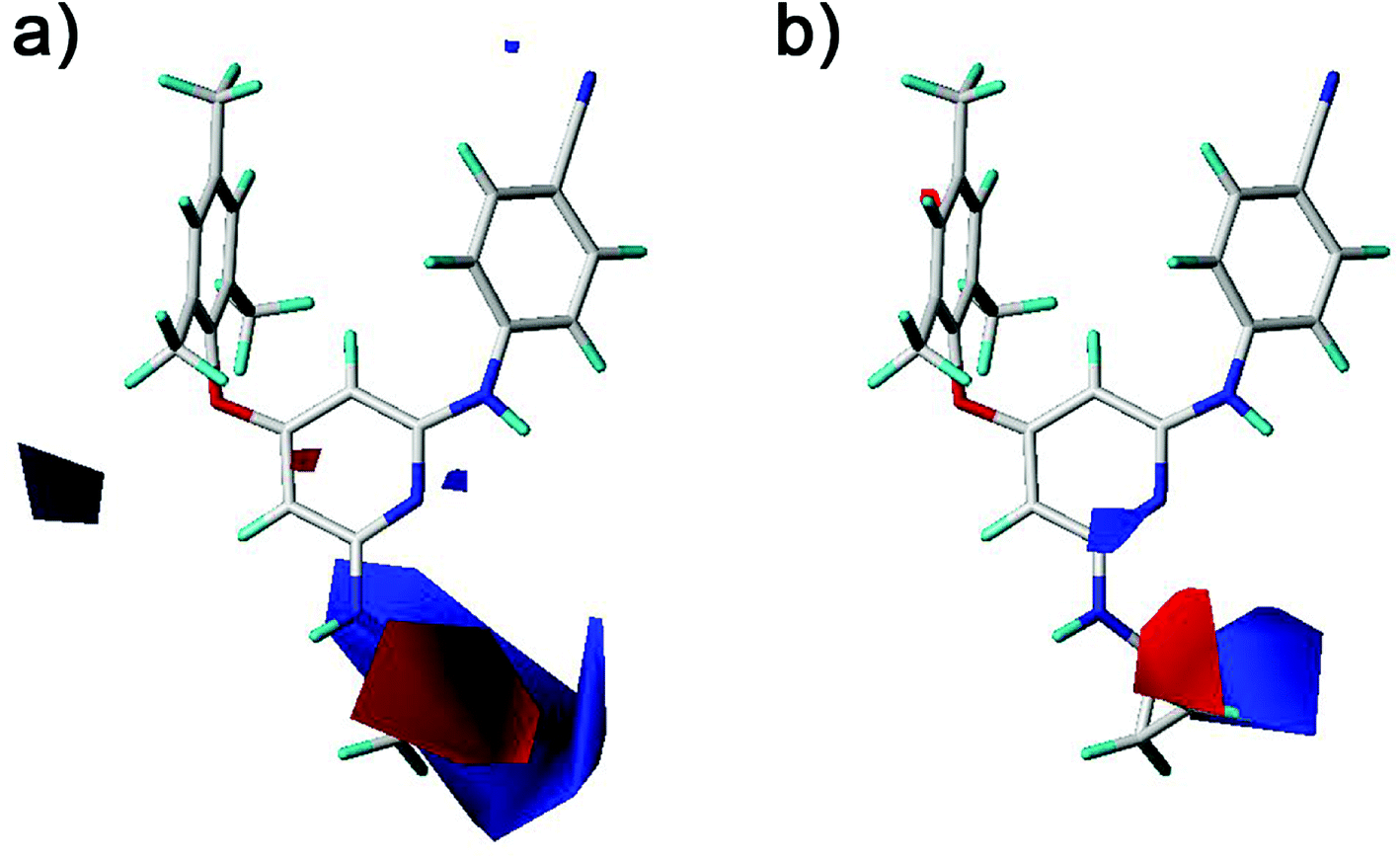
The electrostatic fields are presented as blue contours where electropositive substituents are favorable for the activity and as red contours where electronegative substituents are beneficial (Fig. 9). One blue contour was near the nitro and amino groups which is out of the pyridine ring of compounds 1–20. Although both groups could form hydrogen bonds with the HIV-1 RT, the electrostatic field forces might make their activity different. These results suggested that the amino group was better than the nitro group for the antiviral activity in compounds 1–20. This was supported by the experimental activities as observed in the following compounds, 12 > 1, 13 > 2, 15 > 4, 14 > 5, 18 > 6, 19 > 7, 20 > 9. A red contour located close to the chlorine substituent and the R2 substituents of compounds 21 and 22, revealing the importance of electronegative groups at this position. A blue contour near the linker between the left benzene ring and the pyridine ring was observed in the CoMFA contour maps (Fig. 9a). This indicated that using an electropositive atom as the linker was beneficial for the activity. Compounds 10 (X = NH, pEC50 = 5.975) and 11 (X = NH, pEC50 = 7.398) with a nitrogen atom as the linker exhibited better activities than the corresponding compounds 4 (X = O, pEC50 = 5.693) and 2 (X = O, pEC50 = 6.538) with an oxygen atom as the linker, respectively.
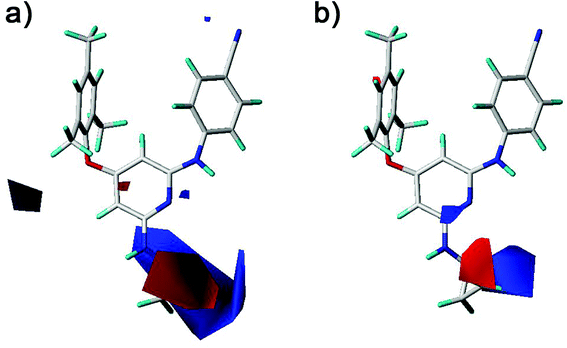 |
| | Fig. 9 Contour maps of electrostatic fields derived from (a) the CoMFA and (b) the best CoMSIA models using compound 28 as a reference molecule. | |
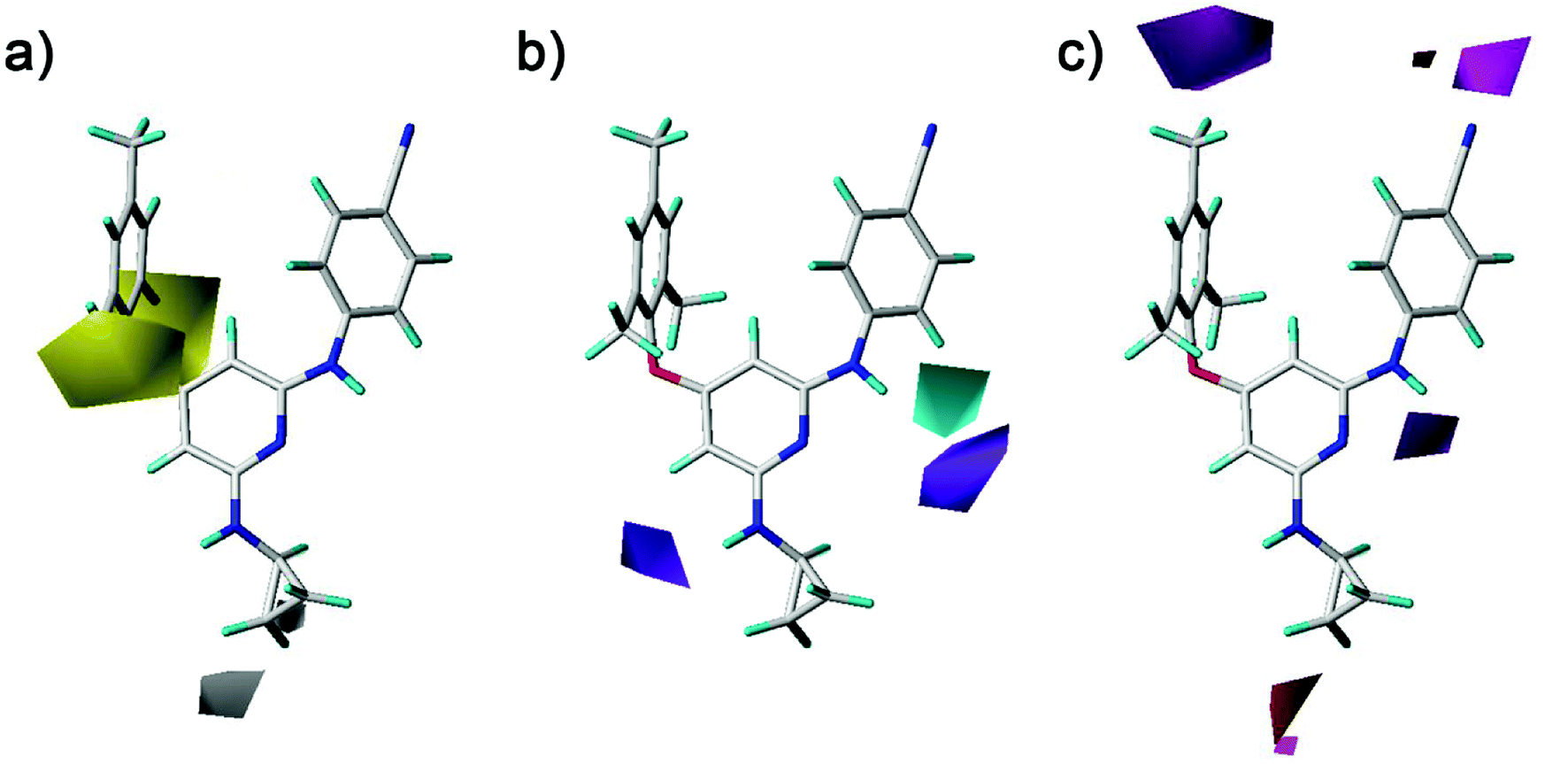
The hydrophobic, hydrogen-bond donor, and hydrogen-bond acceptor contours of the CoMSIA model were shown in Fig. 10. As shown in Fig. 10a, the yellow and white contours mean the hydrophobic substituents are favorable and unfavorable for the activity, respectively. There were two yellow contours that located at the ortho-position of the left benzene ring, indicating that the introduction of hydrophobic groups in this region might enhance the activity. The compounds with the relative hydrophobic substituents (–CH3, –F, –Cl and –Br) at the ortho-position of the left benzene ring exhibited good antiviral activity. A white contour situated at the end of the R2 substituents (compounds 23–42), which pointed to the entrance channel of the binding pocket. Combined with the steric contour maps, this indicated that hydrophilic bulky groups at the R2 terminus were favorable for the activity. As shown in Fig. 10b, a cyan contour near the imino group that connected the pyridine ring with the right benzene ring indicated that a hydrogen-bond donor at this position was advantage of the activity. The previously docked results were also demonstrated this structure was important for the diarylpyridines to form hydrogen bonds with the residue Lys101 of the HIV-1 RT. A purple contour was near the imino group of the R2 substituents, indicating a hydrogen-bond donor was not improper here. As shown in Fig. 10c, the magenta contour indicated hydrogen-bond acceptors were favored and the red contour represented hydrogen-bond acceptors were disfavored. Two magenta contours were near the para-positions of the right and left benzene rings, respectively. However, in the previous docking study, hydrogen bonds were not observed at these positions.
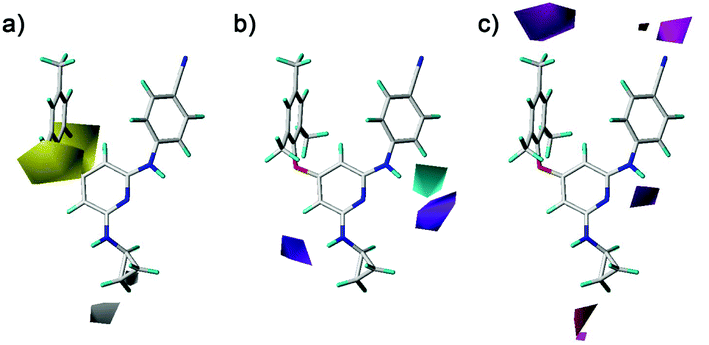 |
| | Fig. 10 Contour maps of (a) hydrophobic, (b) hydrogen-bond donor, and (c) hydrogen-bond acceptor fields of the best CoMSIA model using compound 28 as a reference molecule. | |
Pharmacophore elucidation
GALAHAD was applied to study the pharmacophore characteristics of the diarylpyridine derivatives. A possible pharmacophore model was constructed using ten diarylpyridines with high activities and diverse structures (Fig. 11). The different colors represent different pharmacophore features. As shown in Fig. 11, the built model contained three hydrophobe (HY, cyan) features, three hydrogen-bond acceptors (HAs, green), and three hydrogen-bond donors (HDs, magenta). Three HY features located at the center of the three aromatic rings, respectively. This indicated that maintaining hydrophobic structures in common scaffolds of NNRTI diarylpyridines were necessary to interact with hydrophobic residues in the NNIBP of the HIV-1 RT. The HA3 feature suggested that the atom of the left linker should be a hydrogen-bond acceptor. A HA feature at the cyano group of the right benzene ring indicated that the nitrogen atom of the cyano group might be a hydrogen-bond acceptor candidate. It could be observed that there was a HD/HA feature at the same nitrogen atom that linked the pyridine ring and the right benzene ring. The HD3 feature was a big sphere which covered the nitrogen atom of the R2 substituent of compounds 28, 33, 34, and 40. This indicated that a hydrogen-bond donor at this space had relatively flexible distance to the pyridine ring. In addition, the nitrogen atoms of the nitro groups were out of the sphere HD2 whereas the nitrogen atoms of the amino groups were included in the sphere. This result was consistent with the 3D-QSAR contour maps and experimental activities, indicating an amino group (hydrogen-bond donor) was better than a nitro group (hydrogen-bond acceptor) for improving the activity.
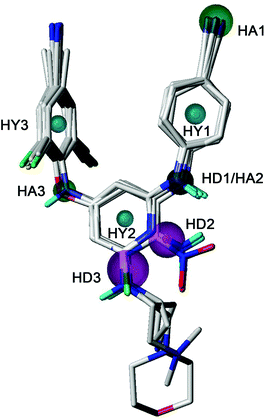 |
| | Fig. 11 The best pharmacophore model aligned with ten diarylpyridine derivatives of HIV-1 RT NNRTIs. The hydrophobic, hydrogen-bond acceptor, and hydrogen-bond donor features are colored in cyan, green, and magenta, respectively. | |
Conclusions
In the present study, a novel series of diarylpyridine derivatives were docked into the HIV-1 RT and the 10 ns MD simulations were performed on complexes of the HIV-1 RT with two diarylpyridine derivatives (compounds 11 and 28). The CoMFA, CoMSIA, and GALAHAD were performed on these docked conformations of the diarylpyridine derivatives to understand their 3D-QSAR relationships and pharmacophore characteristics. The docking and pharmacophore results suggested that diarylpyridine derivatives as NNRTIs should contain hydrophobic parts because the NNIBP of the HIV-1 RT was consisted of hydrophobic residues. The maintenance of the left aromatic ring is necessary to interact with Tyr181, Tyr188, Phe227, and Trp229 by π–π stacking and hydrophobic interactions in the NNIBP. The results also manifested that the group of the right linker should be a hydrogen-bond acceptor/donor to interact with the key residue Lys101 by hydrogen bonds. The statistical results of the 3D-QSAR models demonstrated that the constructed CoMFA and CoMSIA models had preferable predictive ability for novel diarylpyridines or DAPYs. Based on the comprehensive consideration of the docking, 3D-QSAR, and pharmacophore results, the following substituents that are introduced into corresponding positions might be beneficial for improving the anti-HIV-1 activity of diarylpyridines: (1) small hydrophobic substituents at the ortho-positions of the left aromatic ring; (2) bulky groups at the para-position of the left aromatic ring; (3) a short chain as R2 with a bulky hydrophilic terminus; (4) a electropositive substituent as the left linker and a hydrogen-bond donor as the right linker. In conclusion, the molecular modeling results in this paper might provide significant information for the design and development of novel diarylpyridines targeting to the HIV-1 RT.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 21807082, 21877087), Hubei Provincial Natural Science Foundation of China (No. 2017CFB121), Hubei Provincial Department of Education of China (No. Q20171503), and Wuhan International Scientific and Technological Cooperation Project (No. 2017030209020257).
References
- Y. Hsiou, J. Ding, K. Das, J. A. D. Clark, S. H. Hughes and E. Arnold, Structure, 1996, 4, 853–860 CrossRef CAS PubMed.
- T. Ilina, K. LaBarge, S. G. Sarafianos, R. Ishima and M. A. Parniak, Biology, 2012, 1, 521–541 CrossRef CAS PubMed.
- E. D. Clercq, J. Med. Chem., 1995, 38, 2491–2517 CrossRef PubMed.
- H. Mitsuya, R. Yarchoan and S. Broder, Science, 1990, 249, 1533–1544 CrossRef CAS PubMed.
- H. Jonckbeere, J. Anne and E. D. Clercq, Med. Res. Rev., 2000, 20, 129–154 CrossRef.
- D. D. Richman, Nature, 2001, 410, 995–1001 CrossRef CAS PubMed.
- L. R. Boone, Curr. Opin. Invest. Drugs, 2006, 7, 128–135 CAS.
- N. Sluis-Cremer, D. Arion and M. A. Parniak, Cell. Mol. Life Sci., 2000, 57, 1408–1422 CrossRef CAS PubMed.
- R. S. Goody, B. Müller and T. Restle, FEBS Lett., 1991, 291, 1–5 CrossRef CAS.
- E. D. Clercq, Antiviral Res., 1998, 38, 153–179 CrossRef PubMed.
- C. Tantillo, J. Ding, A. Jacobo-molina, R. G. Nanni, P. L. Boyer, S. H. Hughes, R. Pauwels, K. Andries, P. A. Janssen and E. Arnold, J. Mol. Biol., 1994, 243, 369–387 CrossRef CAS PubMed.
- J. Ding, K. Das, C. Tantillo, W. Zhang, A. D. Clark, S. Jessen, X. Lu, Y. Hsiou, A. Jacobo-Molina and K. Andries, Structure, 1995, 3, 365–379 CrossRef CAS.
- J. S. Ren, E. Robert, G. Elspeth, S. Donald, R. Carl, K. Ian, K. James, D. Graham, J. Yvonne, S. David and S. David, Nat. Struct. Biol., 1995, 2, 293–302 CrossRef CAS PubMed.
- J. Ding, K. Das, H. Moereels, L. Koymans, K. Andries, P. A. Janssen, S. H. Hughes and E. Arnold, Nat. Struct. Biol., 1995, 2, 407–415 CrossRef CAS.
- E. L. Asahchop, M. A. Wainberg, R. D. Sloan and C. L. Tremblay, Antimicrob. Agents Chemother., 2012, 56, 5000–5008 CrossRef CAS PubMed.
- P. Zhan, X. Chen, D. Li, Z. Fang, E. D. Clercq and X. Liu, Med. Res. Rev., 2013, 33, E1–E72 CrossRef CAS PubMed.
- E. D. Clercq, Chem. Biodiversity, 2004, 1, 44–64 CrossRef PubMed.
- P. Zhan, X. Liu, Z. Li, C. Pannecouque and E. D. Clercq, Curr. Med. Chem., 2009, 16, 3903–3917 CrossRef CAS.
- G. Liu, W. Wang, Y. Wan, X. Ju and S. Gu, Int. J. Mol. Sci., 2018, 19, 1436 CrossRef PubMed.
- G. Liu, Y. Wan, F. Sai, S. Gu and X. Ju, Mol. Diversity, 2018 DOI:10.1007/s11030-018-9860-1.
- R. Schrijvers, Expert Opin. Pharmacother., 2013, 14, 1087–1096 CrossRef CAS PubMed.
- D. Li, P. Zhan, E. D. Clercq and X. Liu, J. Med. Chem., 2012, 55, 3595–3613 CrossRef CAS PubMed.
- W. Wang, Y. Tian, Y. Wan, S. Gu, X. Ju, X. Luo and G. Liu, Struct. Chem., 2018 DOI:10.1007/s11224-018-1204-3.
- Y. Song, Z. Fang, P. Zhan and X. Liu, Curr. Med. Chem., 2014, 21, 329–355 CrossRef CAS PubMed.
- J. Yang, W. Chen, D. Kang, X. Lu, X. Li, Z. Liu, B. Huang, D. Daelemans, C. Pannecouque and E. D. Clercq, Eur. J. Med. Chem., 2016, 109, 294–304 CrossRef CAS PubMed.
- Z. Liu, Y. Tian, J. Liu, B. Huang, D. Kang, E. D. Clercq, D. Daelemans, C. Pannecouque, P. Zhan and X. Liu, Eur. J. Med. Chem., 2017, 140, 383–391 CrossRef CAS PubMed.
- M. J. D. Powell, Math. Program., 1977, 12, 241–254 CrossRef.
- A. Paneth, W. Płonka and P. Paneth, J. Mol. Model., 2017, 23, 317–325 CrossRef PubMed.
- E. B. Lansdon, K. M. Brendza, M. Hung, R. Wang, S. Mukund, D. Jin, G. Birkus, N. Kutty and X. H. Liu, J. Med. Chem., 2010, 53, 4295–4299 CrossRef CAS PubMed.
- J. Ruppert, W. Welch and A. N. Jain, Protein Sci., 1997, 6, 524–533 CrossRef CAS PubMed.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Chem. Theory Comput., 2008, 4, 1463–1472 Search PubMed.
- D. M. York, T. A. Darden and L. G. Pedersen, J. Chem. Phys., 1993, 99, 8345–8348 CrossRef CAS.
- J. Verma, V. M. Khedkar and E. C. Coutinho, Curr. Top. Med. Chem., 2010, 10, 95–115 CrossRef CAS PubMed.
- A. Tropsha, P. Gramatica and V. K. Gombar, QSAR Comb. Sci., 2003, 22, 69–77 CrossRef CAS.
- P. P. Roy and K. Roy, QSAR Comb. Sci., 2010, 27, 302–313 CrossRef.
- A. Golbraikh and A. Tropsha, J. Mol. Graphics Modell., 2002, 20, 269–276 CrossRef CAS PubMed.
- P. K. Ojha, I. Mitra, R. N. Das and K. Roy, Chemom. Intell. Lab. Syst., 2011, 107, 194–205 CrossRef CAS.
- K. Roy, R. N. Das, P. Ambure and R. B. Aher, Chemom. Intell. Lab. Syst., 2016, 152, 18–33 CrossRef CAS.
- V. P. Zambre, R. Giridhar and M. R. Yadav, Med. Chem. Res., 2013, 22, 4685–4699 CrossRef CAS.
- N. J. Richmond, C. A. Abrams, P. R. Wolohan, E. Abrahamian, P. Willett and R. D. Clark, J. Comput.-Aided Mol. Des., 2006, 20, 567–587 CrossRef CAS PubMed.
- D. Yusuf, A. M. Davis, G. J. Kleywegt and S. Schmitt, J. Chem. Inf. Model., 2008, 48, 1411–1422 CrossRef CAS PubMed.
- K. Das, P. J. Lewi, S. H. Hughes and E. Arnold, Prog. Biophys. Mol. Biol., 2005, 88, 209–231 CrossRef CAS PubMed.
Footnote |
| † Youlan Wan and Yafeng Tian contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*
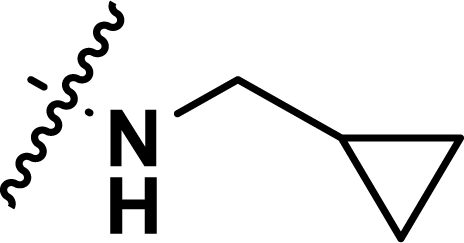
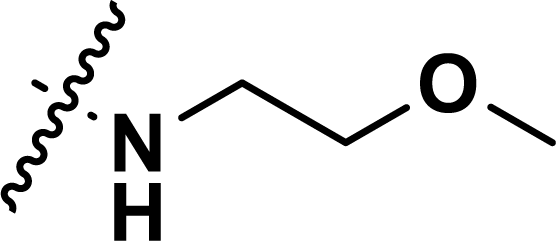
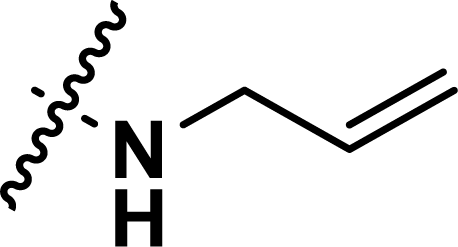
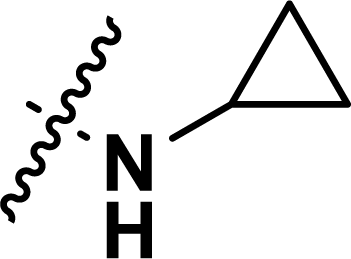


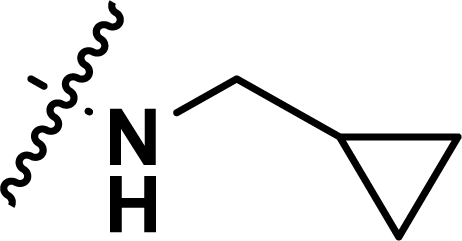
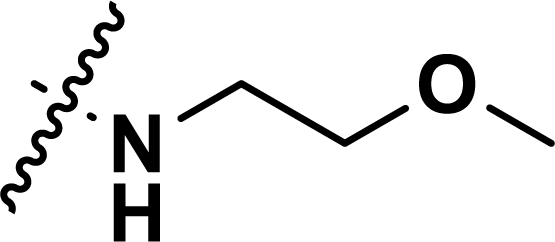
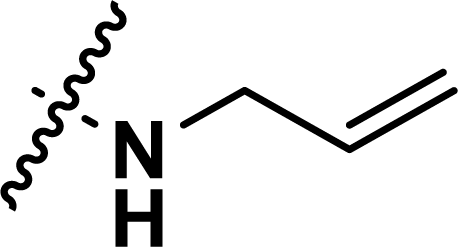
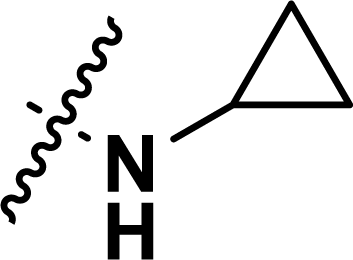
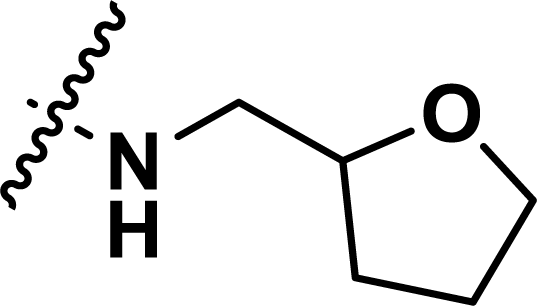
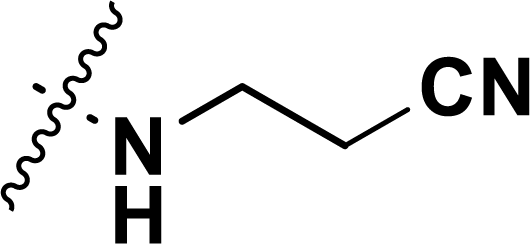
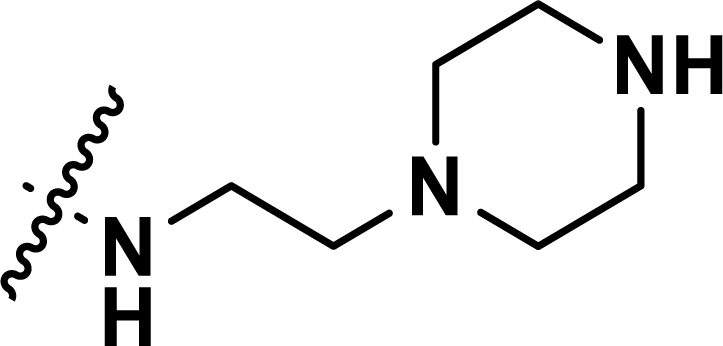
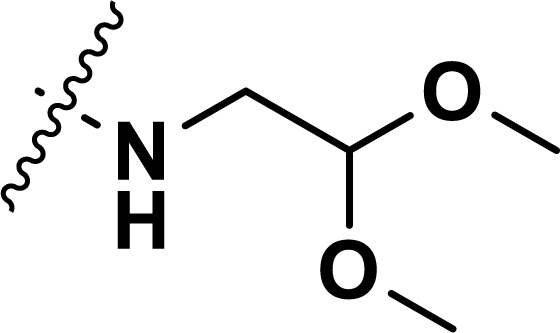
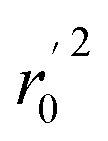 , rm2,
, rm2, 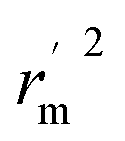 , k, k′, rpred2, root mean square error (RMSE), and mean absolute error (MAE), were considered for external validation of the models.35 r(test)2 is the regression line coefficient of correlation for the test set. r02 (predicted vs. observed activities) and
, k, k′, rpred2, root mean square error (RMSE), and mean absolute error (MAE), were considered for external validation of the models.35 r(test)2 is the regression line coefficient of correlation for the test set. r02 (predicted vs. observed activities) and 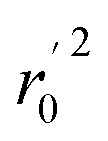 (observed vs. predicted activities) are the regression lines coefficients of correlation with the intercept set to zero. k (predicted vs. observed activities) and k′ (observed vs. predicted activities) are the slops of regression lines. As for a good model, it should satisfy the following conditions: r02 or
(observed vs. predicted activities) are the regression lines coefficients of correlation with the intercept set to zero. k (predicted vs. observed activities) and k′ (observed vs. predicted activities) are the slops of regression lines. As for a good model, it should satisfy the following conditions: r02 or 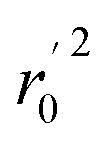 is close to r2; [(r2 − r02)/r2] < 0.1 or
is close to r2; [(r2 − r02)/r2] < 0.1 or 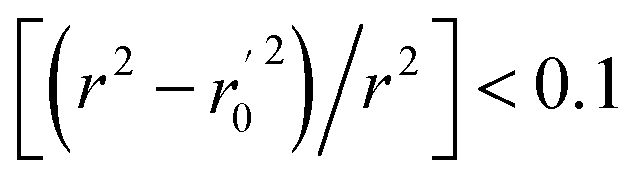 ; 0.85 ≤ k ≤ 1.15 or 0.85 ≤ k′ ≤ 1.15.36 A novel validation parameter rm2 is calculated as the following equation:
; 0.85 ≤ k ≤ 1.15 or 0.85 ≤ k′ ≤ 1.15.36 A novel validation parameter rm2 is calculated as the following equation: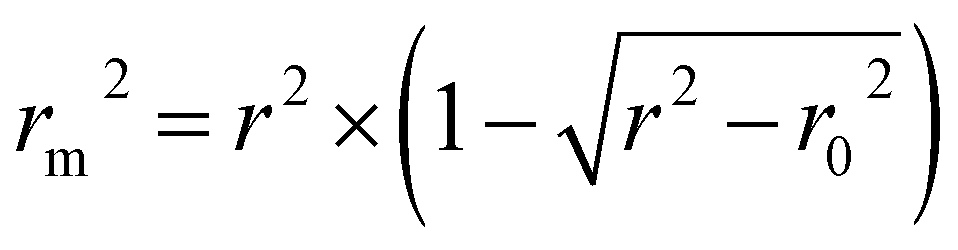
 are also important to prove the external prediction of a model. Δrm2 is the difference between rm2 (predicted vs. observed activities) and
are also important to prove the external prediction of a model. Δrm2 is the difference between rm2 (predicted vs. observed activities) and 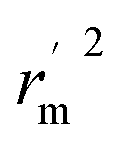 (observed vs. predicted activities).
(observed vs. predicted activities). 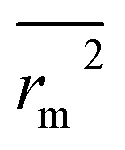 is the average value of rm2 and
is the average value of rm2 and 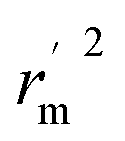 . The values of
. The values of 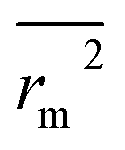 for the training set
for the training set 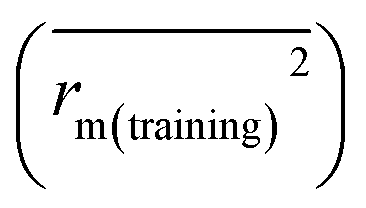 and the test set
and the test set 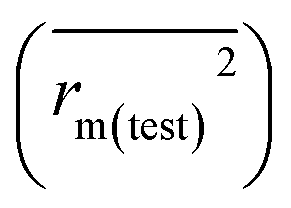 should be more than 0.5. In the case that the previous condition is met, the Δrm(training)2 and Δrm(test)2 should be less than 0.2.37 The rpred2, RMSE, and MAE were calculated by the following equations,37,38 respectively.
should be more than 0.5. In the case that the previous condition is met, the Δrm(training)2 and Δrm(test)2 should be less than 0.2.37 The rpred2, RMSE, and MAE were calculated by the following equations,37,38 respectively.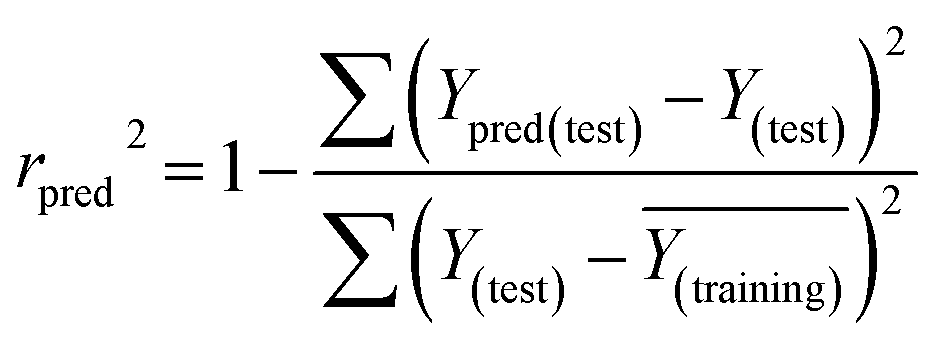

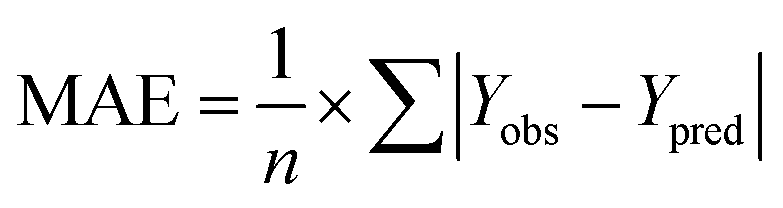
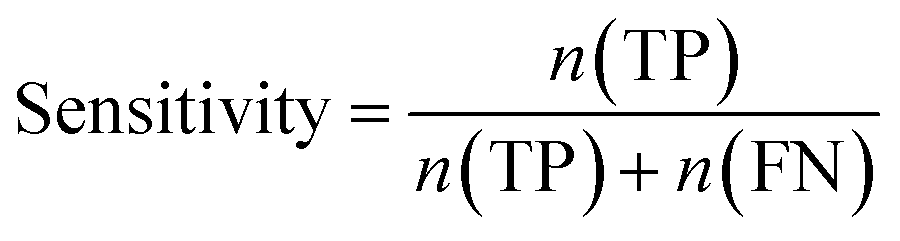

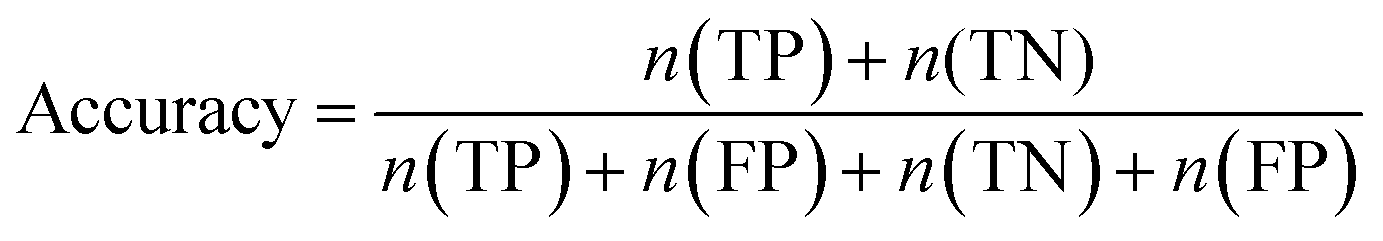
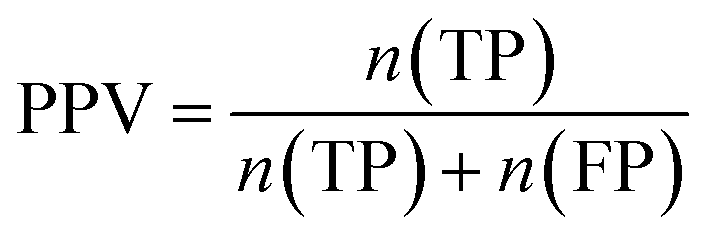
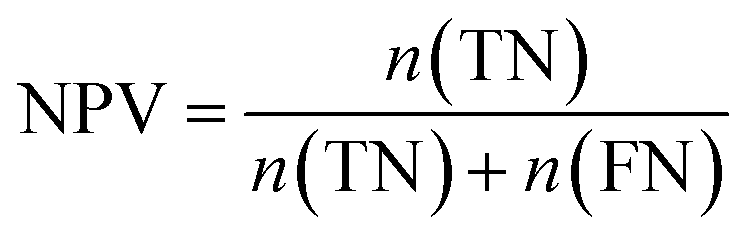
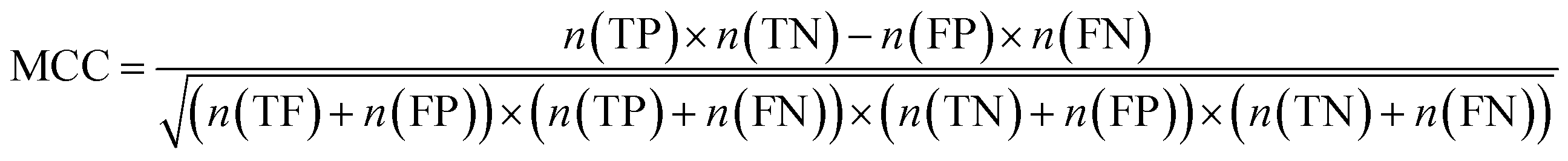
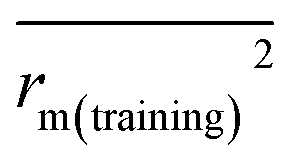 and
and 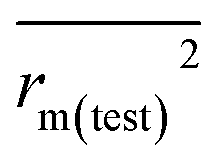 of CoMFA and CoMSIA were higher than 0.5. The correlation between the actual and predicted activities of 35 compounds in the training set and 7 compounds in the test set were shown in Fig. 7. The sensitivity, specificity, accuracy, PPV, NPV, and MCC of CoMFA and CoMSIA were listed in Table 4. The sensitivity values of both models were 96.7%, indicating that 96.7% active compounds were correctly predicted as actives. The specificity of CoMFA and CoMSIA were 91.7% and 83.3%, which manifested the percentages of correctly predicted inactive compounds out of total inactives. The MCC is a correlation coefficient between the observed and predicted binary classifications, and is ranging from −1 to 1, in which the value of 1 signifies a perfect prediction of model. The MCC values of both CoMFA (0.8833) and CoMSIA (0.8220) models were close to 1. All these statistical results of CoMFA and CoMSIA were similar and well within the acceptable limit, which manifested that the developed CoMFA and CoMSIA models with good predictive ability were reliable and reasonable.
of CoMFA and CoMSIA were higher than 0.5. The correlation between the actual and predicted activities of 35 compounds in the training set and 7 compounds in the test set were shown in Fig. 7. The sensitivity, specificity, accuracy, PPV, NPV, and MCC of CoMFA and CoMSIA were listed in Table 4. The sensitivity values of both models were 96.7%, indicating that 96.7% active compounds were correctly predicted as actives. The specificity of CoMFA and CoMSIA were 91.7% and 83.3%, which manifested the percentages of correctly predicted inactive compounds out of total inactives. The MCC is a correlation coefficient between the observed and predicted binary classifications, and is ranging from −1 to 1, in which the value of 1 signifies a perfect prediction of model. The MCC values of both CoMFA (0.8833) and CoMSIA (0.8220) models were close to 1. All these statistical results of CoMFA and CoMSIA were similar and well within the acceptable limit, which manifested that the developed CoMFA and CoMSIA models with good predictive ability were reliable and reasonable.