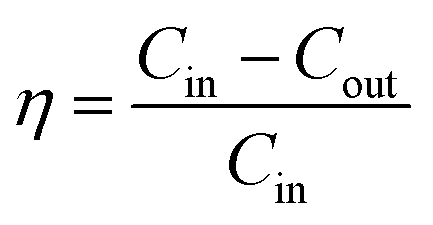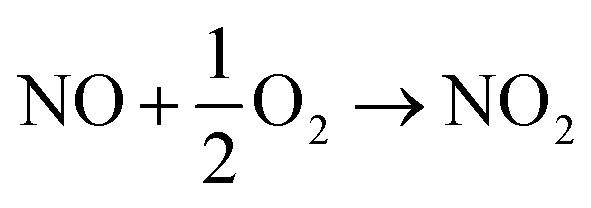DOI:
10.1039/C8RA06376A
(Paper)
RSC Adv., 2018,
8, 32138-32145
Simultaneous absorption of NO and SO2 by combined urea and FeIIEDTA reaction systems
Received
29th July 2018
, Accepted 5th September 2018
First published on 17th September 2018
Abstract
SO2 and NO emitted from coal-fired power plants have caused serious air pollution in China. In this work, a novel mixed absorbent, FeIIEDTA/urea, was employed for simultaneous removal of SO2 and NO in a packed tower, with a corresponding optimal ratio of 0.014 mol L−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5%. The effects of various factors, such as mixed absorbent constitutions, reaction temperature, pH, O2 concentration, as well as concentrations of SO2 and NO, on simultaneous removal were investigated. The desulfurization efficiency was 95–99% in all tests, whereas denitrification was affected significantly by various conditions. NO removal efficiency decreased increasing oxygen concentration as well as increasing NO concentration. With an increase in temperature, pH, or SO2 concentration, NO removal efficiency increased first and then decreased. Under optimal conditions, SO2 removal efficiency was 100% and NO removal efficiency could exceed 91% within 80 min. The reaction mechanism was speculated according to relevant literature.
:5%. The effects of various factors, such as mixed absorbent constitutions, reaction temperature, pH, O2 concentration, as well as concentrations of SO2 and NO, on simultaneous removal were investigated. The desulfurization efficiency was 95–99% in all tests, whereas denitrification was affected significantly by various conditions. NO removal efficiency decreased increasing oxygen concentration as well as increasing NO concentration. With an increase in temperature, pH, or SO2 concentration, NO removal efficiency increased first and then decreased. Under optimal conditions, SO2 removal efficiency was 100% and NO removal efficiency could exceed 91% within 80 min. The reaction mechanism was speculated according to relevant literature.
1. Introduction
Emission of SO2 and NO accounts for 90–95% of NOx in fuel combustion from power plants and results in various environmental problems (e.g., acid rain, photochemical smog, and other pollution) to damage the environment and human health.1 Usually, wet flue gas desulfurization (WFGD) such as the limestone–lime method2 and selective catalytic reduction (SCR)3,4 processes can, respectively, realize SO2 and NO removal for industrial application on a large scale. However, the combination of WFGD and SCR processes for simultaneous desulfurization and denitrification suffer from the need for huge areas of chemical plants, intricate processes, high operating costs, ammonia leakage, and spent catalysts. There is an urgent need for a more economical method for controlling NO and SO2 emission.
The simultaneous absorption technologies of NO and SO2 can be split into “dry” and “wet” processes. The former includes plasma degradation, electron beam radiation photocatalysis, sorbent adsorption, and microwave catalytic decomposition, and necessitates high cost and complex processes to obtain high absorption efficiency of SO2 and NO.5–9 Wet processes applied to industrial furnaces are promising ways to conform to an ultra-low emission standard.
Simultaneous desulfurization and denitrification by wet processes mainly adopt oxidation absorption using KMnO4,10 NaClO2,11 NaClO,12 O3 (ref. 13–15) Fenton reagents,16 UV/H2O2,17 thermal/transition metal ions/persulfate,18 or ultrasound/Fe2+/heat-coactivated persulfate.19 However, oxidation absorption processes are mostly at pilot exploration or laboratory stages due to their significant costs, low oxidation efficiency or secondary pollution. Therefore, trying to exploit new and more promising NOx and SO2 simultaneous removal wet technologies has important theoretical and practical importance.
Reducing absorption with urea ((NH2)2CO) and complexing absorption (e.g., Fe(II)EDTA, hexamminecobalt(II)) are also employed for simultaneous removal of NO and SO2.20,21 However, NO removal through the reduction of (NH2)2CO is not satisfactory. NO removal efficiencies are usually <40% without other additives because of the low solubility of NO in solution. Complex absorption could weaken the impact of the low solubility of NO, but they are easily oxidized, and lose their complexing ability.22 Therefore, simultaneous removal of NO and SO2 by reducing absorption or complexing absorption is barely satisfactory.
Previously, we combined urea and Fe(II)EDTA for NO removal. We noted a synergistic reaction between urea and FeIIEDTA upon NO absorption in which urea could restrain FeIIEDTA oxidation and, importantly, it could react with FeIIEDTA–NO to generate FeIIEDTA, N2, and CO2.23 A mixed absorbent was shown to improve NO removal efficiency significantly, and could maintain >76% for 80 min at optimal conditions in our previous study. However, simultaneous removal of SO2 and NO with a mixed urea ((NH2)2CO) and FeIIEDTA system has not been shown. It is necessary to study the effect of the interrelationship between SO2 and NO on their removal efficiencies by mixed absorbents containing FeIIEDTA and ((NH2)2CO).
We wished to investigate the simultaneous removal of SO2 and NO from flue gas using a mixed urea ((NH2)2CO) and FeIIEDTA system in a “packing tower”. The key influencing factors and mechanism of simultaneous removal of SO2 and NO were investigated. Our results could provide crucial theoretical guidance for follow-up studies and industrial applications of this technology.
2. Experimental section
2.1 Chemicals
FeSO4·7H2O (99.0%), urea (99.0%), C10H14N2O8Na2·7H2O (99.0%), NH4Fe(SO4)2·12H2O (99.0%) and CaCl2 (99.0%) were obtained from Tianjin Kermel Chemical Reagent Co., Ltd. (China). N2 (99.999%), SO2 (99.99%) and NO (99.99%) were provided by Guangzhou Yuejia Gas Co. (China). Other reagents were of analytical grade.
2.2. Experimental apparatus
A schematic diagram of the experimental setup is shown in Fig. 1. The system comprised three main parts: gas-simulation system, packed tower, and a gas sampling-analysis system. Absorption of NO and SO2 was conducted in the packed tower of length 1000 mm and internal diameter 20 mm. The pressure in the packed tower was atmospheric. The concentration of NO and SO2 was measured by a flue gas analyzer (KM950, KANE International) after drying with anhydrous calcium chloride. The resolution and accuracy of this gas analyzer for NO was 1 ppmv and ±5%, respectively.
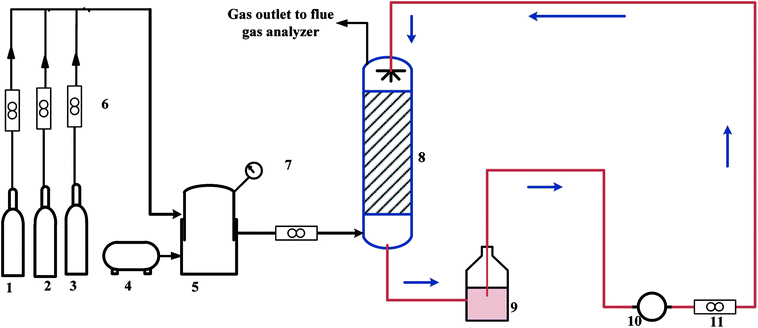 |
| | Fig. 1 Experimental apparatus for NO and SO2 removal (schematic): (1) NO cylinder, (2) N2 cylinder, (3) SO2 cylinder, (4) air compressor, (5) mixed cylinder, (6) gas rotameter, (7) piezometer, (8) packed tower, (9) liquid storage tank, (10) pump, (11) liquid rotameter. | |
2.3. Simultaneous removal of SO2 and NO by urea and FeIIEDTA
At first, the pressure of the mixed gas cylinder was set to 0.2 M Pa. According to the pressure and volume of the mixed gas cylinder, the volume of the desired NO and SO2 concentrations was obtained. NO and SO2 were poured successively into the mixed gas cylinder by adjusting the corresponding rotameter. The desired O2 concentration was obtained by mixing air and N2. The absorbing liquid was prepared by adding urea to FeIIEDTA solution. The pH of the mixed solution was adjusted by NaOH solution and H2SO4. The experiment was started after N2 flowed through the reactor to clean up the remaining air. Absorption was carried out for 80 min, and data were measured every 10 min. The absorbing-liquid volume was ∼300 mL, the gas flow rate was 0.8 L min−1, and the flow rate of the absorption liquid was 45 mL min−1. The effect of mixed absorbent constitutions, temperature, pH, O2 concentration of simulated flue gas, as well as SO2 and NO concentrations on simultaneous desulfurization and denitrification were noted.
2.4. Analytical methods
The concentrations of NO and SO2 were analyzed by a flue gas analyzer (KM950). NO and SO2 removal efficiency were defined as| |
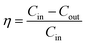 | (1) |
where η is NO or SO2 removal efficiency given as %; Cin is the inlet concentration of NO or SO2 in ppmv; Cout is the outlet concentration of NO or SO2 in ppmv.| |
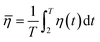 | (2) |
where ![[small eta, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c8.gif) is the average removal efficiency of NO or SO2, T is the total absorption time (min), and η(t) is the removal efficiency of NO or SO2 as a function of time.
is the average removal efficiency of NO or SO2, T is the total absorption time (min), and η(t) is the removal efficiency of NO or SO2 as a function of time.
3. Results and discussion
3.1. Effect of mixed absorbent constitutions on simultaneous removal
Fig. 2 shows the effect of urea concentration on simultaneous removal. As the urea concentration increased from 0 wt% to 10 wt%, the SO2 removal efficiency increased from 94.5% to 99.6%. These experimental results were similar to those in the literature,24 and SO2 removal efficiency using urea solution was almost the same as that of other technologies, all being up to almost 100%. Hence, urea was a good absorbent for SO2. Nevertheless, NO removal efficiency was <40% in all tests, suggesting that NO removal could not proceed well without the help of a complexing agent. This is because the solubility of SO2 in water is 700-times more than that of NO (one volume of water dissolves about 39 volumes of SO2 or ∼0.05 volume of NO at 20 °C).
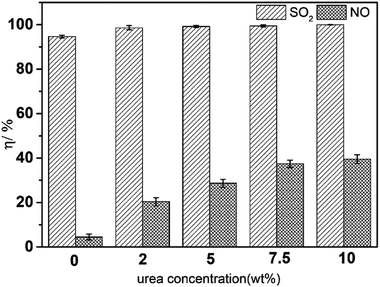 |
| | Fig. 2 Effect of the urea concentration on simultaneous removal. T, 25 °C; pH = 7.0; FeIIEDTA, 0 mol L−1; O2 concentration, 7% (v/v); SO2 concentration, 3000 ppm; NO concentration, 500 ppm. | |
NO could be captured readily by FeIIEDTA in solution to form stable ferrous–nitrosyl complexes FeIIEDTA–NO (eqn (3) and (4)).25
| | |
NO(aq) + FeIIEDTA2− → FeIIEDTA − NO2−
| (4) |
Previously,23 we noted the synergistic effect of (NH2)2CO and FeIIEDTA on NO removal. Urea can react with FeIIEDTA–NO to produce FeIIEDTA for continued complexing of NO, but can also restrain oxidation of FeIIEDTA to FeIIIEDTA. Hence, a combination of FeIIEDTA and urea is suitable and helpful for the removal of NO and SO2.
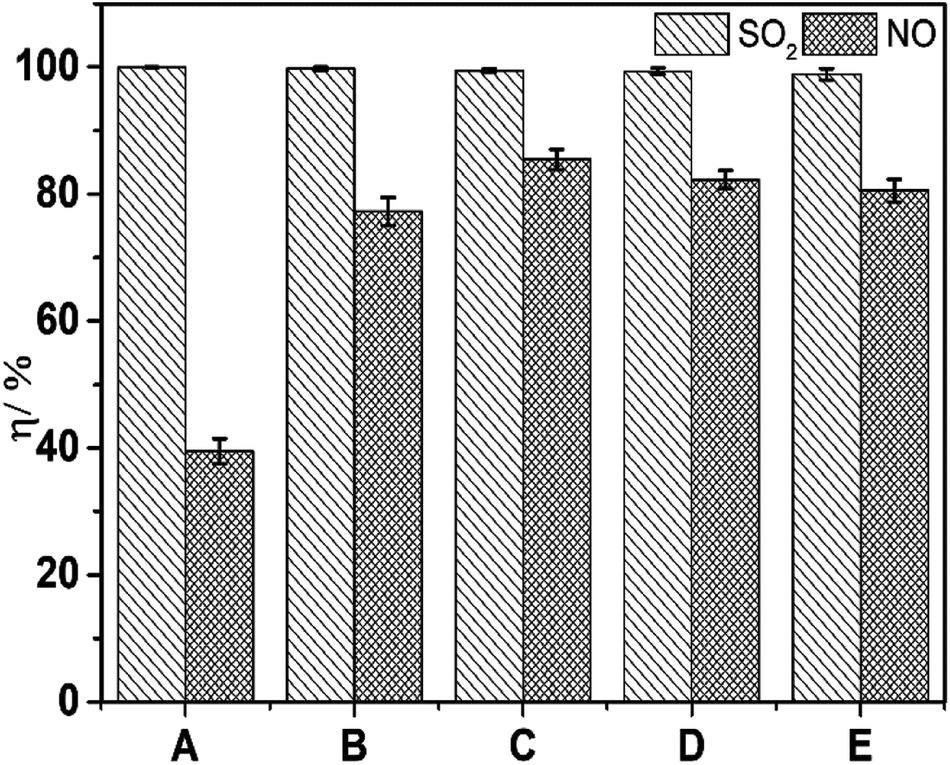
Before seeking an optimal concentration ratio of FeIIEDTA to urea, reagent prices were considered to ensure that industrial scale up is prohibitively expensive. According to the data from the Alibaba Internet website, the prices of industrial FeIIEDTA was ∼$1891/ton ($658.5/kmol), which is approximately five-times higher than that of industrial urea ($366/ton). Then, experiments to determine the optimal concentrations of FeIIEDTA to urea were carried out (Fig. 3).
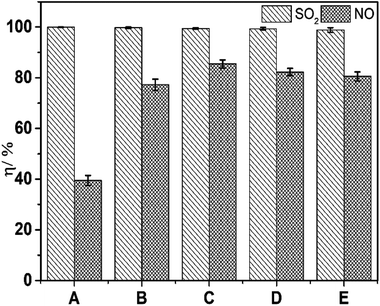 |
| | Fig. 3 Effect of the constitutions of complex absorbents on simultaneous removal. T, 25 °C; pH = 7.0; O2 concentration, 7% (v/v); SO2 concentration, 3000 ppm; NO concentration, 500 ppm; (A), 10 wt% urea; (B), 0.007 mol L−1 FeIIEDTA + 7.5 wt% urea; (C), 0.014 mol L−1 FeIIEDTA + 5 wt% urea; (D), 0.022 mol L−1 FeIIEDTA + 2 wt% urea; (E), 0.028 mol L−1 FeIIEDTA. | |
The prime costs of the five groups from Fig. 3 were identical, and the optimal concentration ratio of FeIIEDTA to urea could be reflected clearly from simultaneous removal efficiencies. As shown in Fig. 3, with the change in the mass ratio, desulfurization efficiency was almost constant at 100%, whereas denitrification efficiency increased significantly. When the concentration ratio of FeIIEDTA to urea increased from 0 mol L−1:10 wt% to 0.014 mol L−1:5 wt%, the average denitrification efficiency increased from 39.4% to 85.5%, because the FeIIEDTA concentration increased. In addition, the viscosity of the absorption liquid increased because the urea concentration was too high, so the liquid diffusivity of NO decreased.26 As the ratio increased further, NO removal efficiency decreased because a too-low urea concentration can result in with high concentrations of FeIIEDTA–NO and FeIIIEDTA accumulating in the complex absorbent. Thus, the optimal ratio of FeIIEDTA to urea was 0.014 mol L−1:5 wt%.
3.2 Effect of temperature on simultaneous removal
The temperature of the absorption liquid plays a significant part in the transfer of NO or SO2 from the gas phase to the liquid phase, and is governed by the diffusion, dissolution and reaction characteristics of the species in solution. The dependence of simultaneous removal on temperature was evaluated. As shown in Fig. 4a, the absorption temperature had a slight effect on desulfurization but a significant effect on NO removal. The denitrification efficiency increased slightly when the reaction temperature was 25–30 °C but, when the reaction temperature increased from 30 °C to 50 °C, there was a decrease in average NO removal efficiency from 86.2% to 58.1%. The effect of temperature on NO removal is highly complicated. On the one hand, a high temperature can accelerate the movement of molecules and increase the chemical rate. As the temperature increases, these factors result in an increase in NO removal efficiency. On the other hand, a lower temperatures is favorable for the formation of iron(II) nitroxyl species.27 Simultaneously, as the temperature increases, the solubility of NO and O2 in the aqueous solution decreased and the consequent decomposition of FeIIEDTA begins to dominate the process. In addition, a higher temperature accelerates the change of FeIIEDTA to FeIIIEDTA. As the temperature increases, these factors can cause a decrease in NO removal efficiency. Hence, the slight decrease and then rapid decrease in NO removal efficiency with an increase in temperature was a result of the interaction of many positive and negative aspects. In summary, 30 °C was the best absorption temperature. However, the temperature of flue gases emitted from industrial boilers is typically ∼100 °C. Hence, before simultaneous desulfurization and denitrification by mixed absorbent FeIIEDTA/urea in industrial applications, flue gases at high temperatures can be treated by different cooling technologies such as water cooling, vaporization cooling, and spray cooling.
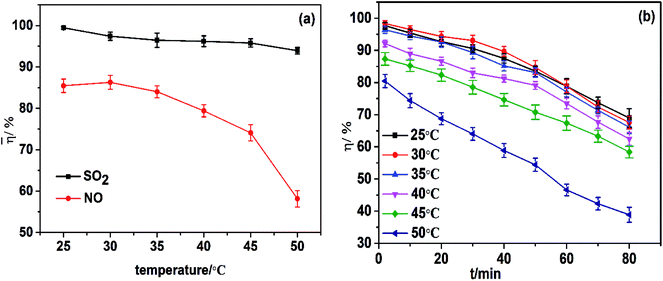 |
| | Fig. 4 (a) Effect of temperature on simultaneous removal. (b) Effect of temperature on NO removal efficiency. pH = 7.0; FeIIEDTA and urea concentration composition, 0.014 mol L−1 FeIIEDTA + 5 wt% urea; O2 concentration, 7% (v/v); SO2 concentration, 3000 ppm; NO concentration, 500 ppm. | |
3.3 Effect of the pH on simultaneous removal
pH is a vital parameter on SO2 and NO removal using a FeIIEDTA and urea mixed solution. To investigate the effect of the initial pH of the mixed solution on simultaneous absorption, experiments were carried out by changing the pH of the absorption liquid from 5.0 to 11.0. Hence, the effect of the pH of mixed absorbents on simultaneous removal was investigated. As shown in Fig. 5a, an alkaline atmosphere was favorable for desulfurization and the average SO2 removal efficiency increased from 95.5% to 100%. Similarly, NO removal efficiency increased from 66.3% to 88.0% with increasing pH from 5.0 to 9.0, which could be because a low pH can promote further oxidation of FeIIEDTA:28| | |
4FeIIEDTA2− + O2 + 4H+ → 4FeIIIEDTA− + 2H2O
| (5) |
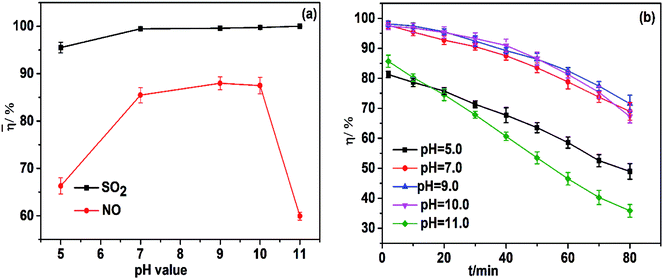 |
| | Fig. 5 (a) Effect of pH on simultaneous removal; (b) effect of pH on NO removal efficiency. T, 25 °C; FeIIEDTA and urea concentration composition, 0.014 mol L−1 FeIIEDTA + 5 wt% urea; O2 concentration, 7% (v/v); SO2 concentration, 3000 ppm; NO concentration, 500 ppm. | |
A sharp decrease in average NO removal efficiency from 87.4% to 60.0% was observed when the pH was between 10.0 and 11.0 because Fe2+ hydrolyzes readily and forms a precipitate in strong alkaline solution,29 which greatly affects NO absorption. Thus, the optimal pH of the mixed absorbent was determined to be 9.0–10.0.
3.4 Effect of O2 concentration on simultaneous removal
Industrial flue gases in China, in general, contain 3–11% (v/v) oxygen, which has a considerable effect on the oxidation of FeIIEDTA.30 The O2 concentration in flue gas was varied from 3% to 11% (v/v) and its effect on simultaneous absorption illustrated in Fig. 6a. SO2 removal efficiency was almost unchanged at ∼99%, and the average NO removal efficiency decreased from 91.9% to 75.6% with an increase in O2 concentration from 3% to 11% (v/v). Low NO removal efficiency under high O2 concentration was due to the oxidation of FeIIEDTA to FeIIIEDTA.
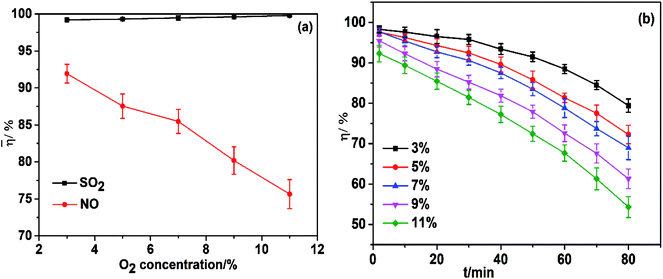 |
| | Fig. 6 (a) Effect of O2 concentration on simultaneous removal; (b) Effect of O2 concentration on NO removal efficiency. T, 25 °C; FeIIEDTA and urea concentration composition, 0.014 mol L−1 FeIIEDTA + 5 wt% urea; pH = 7.0; SO2 concentration, 3000 ppm; NO concentration, 500 ppm. | |
3.5 Effect of SO2 and NO concentrations on simultaneous removal
In practice, the components as well as the concentrations of flue gas are varied with boiler loads and operational conditions. Thus, simultaneous-removal experiments with various concentrations of SO2 and NO were carried out. As shown in Fig. 7a, the effect of SO2 on desulfurization was dependent on its concentration. When the SO2 concentration increased from 500 ppm to 3000 ppm, the average desulfurization efficiency increased from 94.3% to 99.5% and, then, as the SO2 concentration increased further to 4000 ppm, desulfurization efficiency began to decrease. The reason for the increase in SO2 removal efficiency was that the increase in SO2 concentration enhanced mass transfer and then promoted the gas–liquid mass transfer rate, so efficiency was increased. When the SO2 concentration was excessive, despite mass transfer being promoted further, the reaction rate became the controlling step, so SO2 removal resulting from SO2 accumulation decreased.
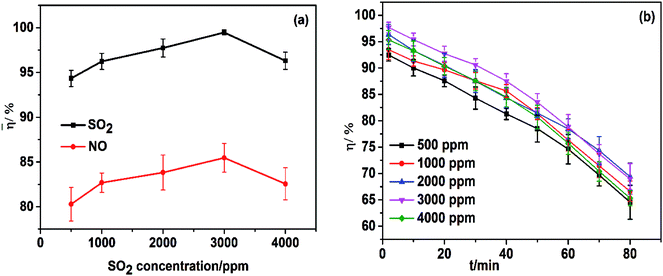 |
| | Fig. 7 (a) Effect of SO2 concentration on simultaneous removal; (b) effect of SO2 concentration on NO removal efficiency. T, 25 °C; FeIIEDTA and urea concentration composition, 0.014 mol L−1 FeIIEDTA + 5 wt% urea; pH = 7.0; O2 concentration, 7% (v/v); NO concentration, 500 ppm. | |
Fig. 7b reveals that NO removal efficiencies decreased with operation time, and when the SO2 concentration increased from 500 ppm to 3000 ppm, the average efficiency of NO removal increased from 80.3% to 85.5%. Then, as the SO2 concentration increased further to 4000 ppm, the average denitrification efficiency decreased to 82.6%. The reasons for the increase in NO removal were as follows:
| | |
2Fe(II)EDTA − NO2− + SO32− → 2Fe(II)EDTA2− + SO3(NO)22− → 2Fe(III)EDTA− + N2O22− + SO32−
| (6) |
| | |
2Fe(III)EDTA− + 2SO32− → 2Fe(II)EDTA2− + S2O62−
| (7) |
| | |
4Fe(II)EDTA − NO2− + 4HSO3− → 4Fe(II)EDTA2− + 2HON(SO3)22− + H2N2O2
| (8) |
| | |
2Fe(III)EDTA− + 2HSO3− → 2Fe(II)EDTA2− + S2O62− + 2H+
| (9) |
Sulfite and hydrosulfite ions can restore the active component FeIIEDTA from FeIIEDTA–NO, and FeIIIEDTA accumulates in the mixed absorption liquid.31 Thus, denitrification efficiency was increased with an appropriate increase in SO2 concentration. However, if the SO2 concentration was >4000 ppm, a small amount of SO2 would compete with NO for the limited complexant (FeIIEDTA) in the gas phase. Moreover, absorption competition between SO2 and NO also occurred in urea solution, resulting in an increase in mass transfer resistance between NO and aqueous phases, from which the denitrification efficiency began to decrease.
The effect of the NO concentration on simultaneous removal is shown in Fig. 8a. An increase in the NO concentration from 300 ppm to 500 ppm could increase SO2 removal efficiencies because NO2 (the oxidation product of NO) could accelerate the oxidation of sulfite to sulfate (eqn (10) and (11)).32
| | |
2NO + O2 + HSO3− + H2O → 3H+ + 2NO2− + SO42−
| (10) |
| | |
2NO + O2 + SO32− + H2O → 2H+ + 2NO2− + SO42−
| (11) |
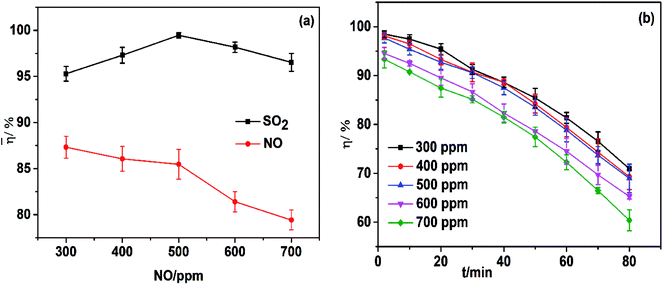 |
| | Fig. 8 (a) Effect of NO concentration on simultaneous removal; (b) effect of NO concentration on NO removal efficiency. T, 25 °C; FeIIEDTA and urea concentration composition, 0.014 mol L−1 FeIIEDTA + 5 wt% urea; pH = 7.0; O2 concentration, 7% (v/v); SO2 concentration, 500 ppm. | |
A further increase in the NO concentration causes severe competition between SO2 and NO for the absorbent, resulting in a decrease in SO2 removal. The increase of the NO concentration would cause a linear decrease in denitrification efficiency, which could be interpreted as a decrease in the molar ratio of oxidant to NO.
3.6 Validation experiments
Comprehensive consideration of the process parameters and experimental data led to optimal experimental conditions for simultaneous removal using a mixed absorbent were determined: 0.014 mol L−1 FeIIEDTA + 5 wt% urea, SO2 and NO initial concentration of ∼3000 and ∼300 ppm, respectively, a temperature of 30 °C, pH of 9.0, and O2 concentration of 3%. In this case, SO2 removal efficiency was 100% and NO removal efficiency could exceed 91% within 80 min under optimal conditions with 300 mL of absorbing liquid volume, gas flow rate of 0.8 L min−1, and flow rate of the absorption liquid of 45 mL min−1.
3.7 Reaction mechanism
The mechanism involved in simultaneous desulfurization and denitrification using FeIIEDTA and urea was speculated according to our previous work and related literature.23,24,31 The desulfurization mechanism was relatively simple because SO2 is highly soluble in water. If the reactions take place in the liquid phase, SO2 can react with the ammonium carbamate generated by urea hydrolysis. The possible forms of sulfur species are SO3, SO2·H2O, HSO3−, SO32−, and SO42−, of which HSO3− and SO42− are the main forms of S4+ species and S6+ species, respectively. In the present study, the reaction between SO32− and urea-hydrolysis products could promote SO2 absorption. Hence, the gas–liquid reaction between SO2 and (NH2)2CO solution was completely gas-film controlled. The related reactions were:| | |
SO2 + H2O → SO2·H2O → HSO3− + H+
| (12) |
| |
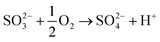 | (14) |
| | |
2(NH2)2CO + H2O → NH2COONH4
| (15) |
| | |
SO32− + 2H+ + NH2COONH4 + O2 → (NH4)2SO4 + CO2
| (16) |
The denitrification mechanism was much more complicated. NO having empty anti-bonding π orbitals can form π bonds by overlapping the d orbit of the central ion Fe2+.33 Thus, NO could be combined readily with FeIIEDTA (eqn (3) and (4)). Meanwhile, in industrial applications, because of O2 in the flue gas, FeIIEDTA is oxidized readily to FeIIIEDTA, and activity is lost (eqn (5)). Hence, during absorption, FeIIEDTA–NO and FeIIIEDTA are the major byproducts. Urea can solve these problems to some degree (eqn (17)). HSO3− and SO32− from the absorption liquid after SO2 removal can also restore FeIIEDTA from FeIIEDTA–NO and FeIIIEDTA (eqn (6)–(9)).
| | |
6FeIIEDTA − NO2− + 2(NH2)2CO → 6FeIIEDTA2− + 5N2 + 2CO2 + 4H2O
| (17) |
Besides being absorbed by FeIIEDTA, some NO is oxidized to NO2 and N2O4, which are dissolved readily in solution, producing nitrous acid and nitric acid, respectively. Subsequently, HNO2 and HNO3 can react with urea to form N2, which can also react with ammonium carbamate to form N2.
| |
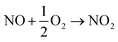 | (18) |
| | |
2NO2 + H2O → HNO2 + HNO3
| (20) |
| | |
N2O4 + H2O → HNO2 + HNO3
| (21) |
| | |
2HNO2 + (NH2)2CO → 2N2 + CO2 + 3H2O
| (22) |
| | |
2HNO2 + NH2COONH4 → 2N2 + CO2 + 4H2O
| (23) |
| | |
6HNO3 + 5(NH2)2CO → 8N2 + 5CO2 + 13H2O
| (24) |
| | |
6HNO3 + 5NH2COONH4 → 8N2 + 5CO2 + 18H2O
| (25) |
The mechanism of simultaneous desulfurization and denitrification using a FeIIEDTA and urea mixed solution is represented in Fig. 9.
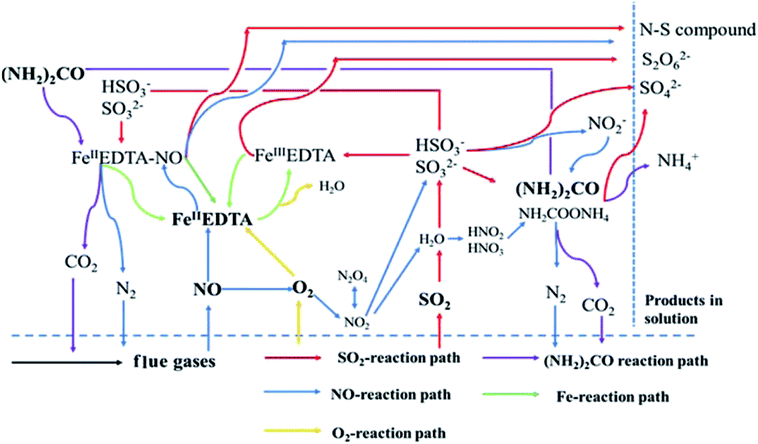 |
| | Fig. 9 Simultaneous desulfurization and denitrification using a FeIIEDTA and urea mixed solution. | |
4. Conclusion
A mixed absorbent of FeIIEDTA/urea was first employed for simultaneous removal of SO2 and NO, and the best mass concentration ratio was 0.014 mol L−1:5 wt%. Desulfurization was affected slightly by different factors, and the SO2 efficiency was 95–99% in all tests, whereas denitrification was affected significantly by various conditions. NO removal efficiency declined with increasing oxygen concentration as well as the increase in NO concentration. However, with increasing temperature, pH, or SO2 concentration, NO removal efficiency initially increased up to the maximum value and then decreased. Under optimal conditions, SO2 removal efficiency was 100% and NO removal efficiency could exceed 91% within 80 min. These research findings suggest that the FeIIEDTA and (NH2)2CO solution could be used as an efficient and low-cost absorbent for simultaneous desulfurization and denitrification.
Conflicts of interest
There are no conflicts to declare.
References
- L. Zheng, G. Liu and C.-L. Chou, Sci. Total Environ., 2007, 384, 374–383 CrossRef PubMed.
- A. Poullikkas, Energy. Technol. Policy., 2015, vol. 2, pp. 92–103 Search PubMed.
- Y. Jin, M. C. Veiga and C. Kennes, J. Chem. Technol. Biotechnol., 2005, 80, 483–494 CrossRef.
- S. Andreoli, F. A. Deorsola, C. Galletti and R. Pirone, Chem. Eng. J., 2015, 278, 174–182 CrossRef.
- L. C. Lau, K. T. Lee and A. R. Mohamed, Fuel, 2011, 90, 1811–1817 CrossRef.
- Z.-s. Wei, H.-j. Niu and Y.-f. Ji, Fuel Process. Technol., 2009, 90, 324–329 CrossRef.
- Y. Han, J. Zhang and Y. Zhao, Energy, 2016, 103, 725–734 CrossRef.
- C.-j. Li, R. Zhao, M.-q. Peng, L. I. U. Hao, Y. U. Gang and X. I. A. Dong-sheng, J. Fuel Chem. Technol., 2015, 43, 1516–1522 CrossRef.
- L. Huang and Y. Dang, Chin. J. Chem. Eng., 2011, 19, 518–522 CrossRef.
- H. Chu, T.-W. Chien and S. Li, Sci. Total Environ., 2001, 275, 127–135 CrossRef PubMed.
- Y. Zhao, T.-x. Guo, Z.-y. Chen and Y.-r. Du, Chem. Eng. J., 2010, 160, 42–47 CrossRef.
- C. V. Raghunath and M. K. Mondal, Chem. Eng. J., 2017, 314, 537–547 CrossRef.
- J. Zhang, R. Zhang, X. Chen, M. Tong, W. Kang, S. Guo, Y. Zhou and J. Lu, Ind. Eng. Chem. Res., 2014, 53, 6450–6456 CrossRef.
- C. Sun, N. Zhao, H. Wang and Z. Wu, Energy Fuels, 2015, 29, 3276–3283 CrossRef.
- Q. Wu, C. Sun, H. Wang, T. Wang, Y. Wang and Z. Wu, Chem. Eng. J., 2018, 341, 157–163 CrossRef.
- Y. Zhao, R.-L. Hao, Q. Guo and Y.-N. Feng, Fuel Process. Technol., 2015, 137, 8–15 CrossRef.
- Y. Liu, J. Zhang, C. Sheng, Y. Zhang and L. Zhao, Chem. Eng. J., 2010, 162, 1006–1011 CrossRef.
- Y. G. Adewuyi, N. Y. Sakyi and M. Arif Khan, Chemosphere, 2018, 193, 1216–1225 CrossRef PubMed.
- Y. Liu, Z. Liu, Y. Wang, Y. Yin, J. Pan, J. Zhang and Q. Wang, J. Hazard. Mater., 2018, 342, 326–334 CrossRef PubMed.
- P. Fang, C. Cen, Z. Tang, P. Zhong, D. Chen and Z. Chen, Chem. Eng. J., 2011, 168, 52–59 CrossRef.
- Y.-P. Mao, W. Bi, X.-L. Long, W.-D. Xiao, W. Li and W.-k. Yuan, Sep. Purif. Technol., 2008, 62, 183–191 CrossRef.
- E. Sada, H. Kumazawa and H. Machida, Ind. Eng. Chem. Res., 1987, 26, 1468–1472 CrossRef.
- F. He, X. Deng and M. Chen, Chemosphere, 2017, 168, 623–629 CrossRef PubMed.
- P. Fang, C. Cen, Z. Tang, P. Zhong, D. Chen and Z. Chen, Chem. Eng. J., 2011, 168, 52–59 CrossRef.
- X. Wang, Y. Zhang, X. Dong, M. Chen, Z. Shi and J. Zhou, Energy Fuels, 2013, 27, 6024–6030 CrossRef.
- J. Wei, Y. Luo, P. Yu, B. Cai and H. Tan, J. Ind. Eng. Chem., 2009, 15, 16–22 CrossRef.
- P. Gans, J. Chem. Soc. A, 1967, 943–946 RSC.
- L. Wang, W. Zhao and Z. Wu, Chem. Eng. J., 2007, 132, 227–232 CrossRef.
- B. Yan, J. Yang, M. Guo, G. Chen, Z. Li and S. Ma, J. Ind. Eng. Chem., 2014, 20, 2528–2534 CrossRef.
- W. Li, L. Zhang, N. Liu, Y. Shi, Y. Xia, J. Zhao and M. Li, Energy Fuels, 2014, 28, 4725–4730 CrossRef.
- E. Sada, H. Kumazawa and Y. Takada, Ind. Eng. Chem. Fundam., 1984, 23, 60–64 CrossRef.
- R. Hao, S. Yang, B. Yuan and Y. Zhao, Fuel Process. Technol., 2017, 159, 145–152 CrossRef.
- J. Enemark and R. Feltham, Coord. Chem. Rev., 1974, 13, 339–406 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Xianhe Dengb and
Jianhua Ding*a
*a,
Xianhe Dengb and
Jianhua Ding*a