DOI:
10.1039/C8RA06365F
(Paper)
RSC Adv., 2018,
8, 31860-31867
Fabrication and catalytic properties of ordered cyclopalladated diimine monolayer : investigation on catalytic mechanism†
Received
28th July 2018
, Accepted 27th August 2018
First published on 12th September 2018
Abstract
“Channel-like” self-assembled monolayers having aliphatic and aromatic diimines (denoted as Si@1DIS, Si@2DIS and Si@3DIS) immobilized on substrates and their palladacycle monolayers (Si@1DIS-Pd, Si@2DIS-Pd and Si@3DIS-Pd) were prepared and characterized. Their catalytic performances were investigated using the Suzuki coupling reaction as a model. Si@3DIS-Pd showed the highest catalytic activity in water without ligands, and better recyclability than that of Si@2DIS-Pd and Si@1DIS-Pd. The reason was the carbon in the aliphatic diimine of Si@2DIS-Pd and Si@1DIS-Pd was easily hydrolyzed because of the active hydrogen of α-C, resulting in poor recyclability. Control of the amount of catalyst could be achieved by modulating the diameter of the channel-like structure, which also affected the catalytic activity. The catalytic process and mechanism were investigated systematically and proposed based on the experimental results obtained by the water contact angle, ultraviolet spectroscopy, X-ray photoelectron spectroscopy, cyclic voltammetry and atomic force spectroscopy. Changes in the morphology of monolayer surfaces during the catalytic process with or without stirring presented a clear process from order to disorder, and indicated that the reaction was a heterogeneous catalytic process occurring on the surface of the catalyst monolayer.
1. Introduction
Palladium (Pd)-catalyzed C–C coupling reactions, such as Suzuki, Heck, Sonogashira as well as other reactions, have important roles in organic chemistry1 and have made major progress,2 including use as homogeneous catalysts. Despite their remarkable usefulness and excellent activity, homogeneous catalysts cannot be recycled, especially in the pharmaceutical industry.3 Therefore, heterogeneous catalysts seem to be preferable because they can be removed by simple filtration leaving few metal residues.4
Studies on Pd catalysts immobilized on different substrates have been described5–9 and applied to catalyze coupling reactions10 due to their excellent stability and versatility towards functional groups. However, some problems associated with heterogeneous catalyst must be solved, such as high loading, heavily reaction condition, aggregation on the substrate, and repeatability.11 Meanwhile, finding the true mechanism is one of the most striking weaknesses of current studies on heterogeneous catalysis.12
Different mechanisms of coupling reactions catalyzed by heterogeneous Pd catalysts have been investigated intensively, including the homogeneous process by leaching Pd species,13 the direct role of Pd nanoparticles on surface sites in the catalytic cycle,14 and surface-driven coupling.15 Despite these efforts, there is uncertain evidence of surface-catalyzed cross-coupling chemistry and some results have been contradictory. Therefore, research should be focused to elucidate the dynamics and true species type of Pd catalyst. Such research may enable development of a rational design for catalysts. Hence, suitable analytical methods must be employed for research.
The surface properties of Langmuir–Blodgett (LB) films16 and self-assembled films17 immobilized on solid substrates can be characterized, identified and evaluated by various measurement techniques. These techniques provide deep insights into catalytic behaviour at the molecular level, such as in the design of ligands containing certain functional groups (e.g., Schiff-bases), which exhibit excellent performance in heterogeneous catalysis.18,19 Our research team focuses on the fabrication and heterogeneous catalytic properties of Schiff-based cyclopalladated LB films20 and self-assembly films, which show superior catalytic activity.21 LB films can be destroyed in organic solvents, heating or distilling, which affects experimental results. Therefore, their self-assembled monolayers linked with substrates were selected to overcome these disadvantages.20b
Herein, a series of aliphatic and aromatic cyclopalladated diimine catalysts in the form of “channel like” self-assembled monolayers were designed and prepared to evaluate their activity and recyclability in the Suzuki–Miyaura coupling reaction. In this way, we aimed to provide an “ideal” template for investigating the heterogeneous catalytic mechanism.
2. Experimental section
2.1 General
Chemical regents were purchased from commercial and experimental sources. The fabrication of cyclopalladated diimines SAM-monolayer (Si@1DIS-Pd, Si@2DIS-Pd and Si@3DIS-Pd), general procedure for the Suzuki cross-coupling reaction as a model, inductively coupled plasma-atomic emission spectroscopy (ICP-AES) analysis and the general procedure for control experiments are presented in ESI.†
3. Results and discussion
3.1 Preparation of self-assembled monolayers of cyclopalladated diimines (Si@1DIS-Pd, Si@2DIS-Pd and Si@3DIS-Pd)
The channel-like cyclopalladated diimines grafted on substrates, such as silicon, were fabricated as depicted in Scheme 1. The channel diameter could be adjusted by designing the ligands. Characterization of cyclopalladated diimine self-assembled monolayers (Si@1DIS-Pd, Si@2DIS-Pd and Si@3DIS-Pd), including the immobilization process, were carried out and interpreted as shown below.
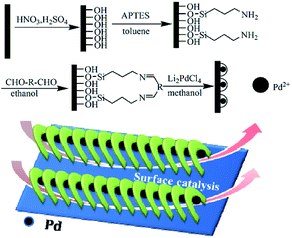 |
| | Scheme 1 Preparation of cyclopalladated diimine self-assembled monolayers (R = –(CH2)n–: n = 0, Si@1DIS-Pd; n = 3, Si@2DIS-Pd; R = –C6H4–, Si@3DIS-Pd). | |
3.1.1 UV-vis absorption spectra of the preparation process of catalytic monolayers. UV spectra of the fabrication of monolayers of cyclopalladated diimines (Si@1DIS-Pd, Si@2DIS-Pd and Si@3DIS-Pd) modified on quartz, along with each step, were measured (Fig. 1). For Si@3DIS-Pd, absorption was not observed for hydrophilic quartz (Si–OH, black line) and the absorption increased gradually due to the amine (Si@APTES, red line). Then, a peak at 273 nm assigned to benzene appeared after modification with 1,4-phthalaldehyde (Si@3DIS, pink line). To ascertain if a diimine had been formed, a contrast experiment was initiated in which Si@3DIS was reacted with aniline. Results showed no change (Si@aniimine, blue line) compared with that of Si@3DIS, indicating that an ordered diimine had been fabricated. The peak at 273 nm shifted to 265 nm (green line) when Si@3DIS coordinated with Pd due to decreasing conjugation of the diimine.20d The absorption peak shifted to shorter wavelengths gradually, suggesting some interaction among molecules or close molecular packing, such as J-aggregates. Similar results were observed for Si@1DIS-Pd and Si@2DIS-Pd (see ESI, Fig. S1†).
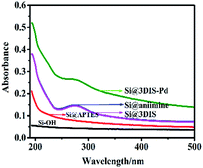 |
| | Fig. 1 UV spectra of the preparation process of Si@3DIS-Pd at different steps. | |
3.1.2 Water contact angle (WCA) analyses. The WCA is a useful way to observe the surface wetting ability related to the structure of the monolayer.22 After pre-treatment with “piranha” solution (Fig. 2), the surface of silicon exhibited higher hydrophilic performance due to hydroxyl groups (5.6°) and the WCA increased to 43.3° due to hydrophobic fatty chains after reaction with APTES. The surfaces linked with 1,4-phthalaldehyde showed a dramatically increased WCA of 78.9° because of introduction of an aryl group. Finally, the WCA of Si@3SID-Pd was 85.7° after coordination with Pd2LiCl4. Changes in the WCA on modified solid surfaces resulted in significant changes in the surface configuration of each step. In the cases of Si@1DIS-Pd and Si@2DIS-Pd, similar results were shown (Fig. S2†).
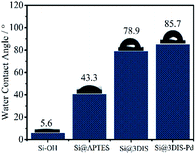 |
| | Fig. 2 Comparison of the WCA in the preparation of cyclopalladated diimine monolayers (Si–OH, Si@APTES, Si@3DIS, and Si@3SID-Pd). | |
3.1.3 Analyses of atomic force microscopy (AFM) images. AFM images of the surface in derivative processes were measured at different steps to provide detailed information on preparation of Si@1DIS-Pd, Si@2DIS-Pd and Si@3DIS-Pd (Fig. 3). AFM images of hydrophilic silicon (Fig. 3a) showed a flat and ordered morphology with a roughness average (Ra) of 0.540 nm and root mean square (Rms) of 0.797. Ordered surfaces with a Ra of 1.151 nm and Rms of 1.570 nm (Fig. 3b) were observed after silanization.23 The AFM images obtained after addition of 1,4-phthalaldehyde (Fig. 3c) also exhibited homogeneous surfaces except for a few clusters, but a higher Ra (2.419 nm) and Rms (4.196 nm) than that observed after silanization (1.151 nm and 1.570 nm, respectively), indicating that a diimine could be formed. AFM of the surface of a cyclopalladated diimine monolayer (Fig. 3d) gave high-density arrays with a Ra of 4.151 nm and Rms of 5.863 nm, suggesting that self-assembly of Pd with Si@3SID had occurred and resulted in changes in morphology. AFM data as well as Ra and Rms values of different processes for Si@1DIS-Pd and Si@2DIS-Pd are provided as shown in Fig. S3a and b† and summarized in Fig. S4.†
 |
| | Fig. 3 AFM images of Si@3DIS-Pd in different preparation steps ((a) Si–OH, (b) Si@APTES, (c) Si@3SID and (d) Si@3SID-Pd). | |
3.1.4 X-ray photoelectron (XPS) spectroscopy. The XPS spectra of Si@3SID-Pd (Fig. 4), Si@1DIS-Pd (Fig. S5†) and Si@2DIS-Pd (Fig. S6†) were obtained, which covered the Si2p, Cl2p, C1s, N1s, and Pd3d energy ranges. The binding energies are summarized in Table S1† for Si@1DIS-Pd and Table S2† for Si@2DIS-Pd. Signals appeared in the spectrum of Cl2p at 197.0 eV BE and Pd3d at 343.2 eV BE and 337.9 eV BE, confirming Pd(II) and chloride on the surface.21h These data provided evidence that Si@1DIS-Pd, Si@2DIS-Pd and Si@3DIS-Pd had been grafted on the wafer surface step by step (Table 1).
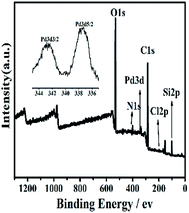 |
| | Fig. 4 XPS spectra of Si@3SID-Pd. Inset: high-resolution XPS of Pd3d. | |
Table 1 Relative element percentage content of Si@3DIS-Pd
| Element |
Pd3d |
Si2p |
Cl2p |
C1s |
N1s |
O1s |
| Atomic% |
0.46 |
8.70 |
0.60 |
56.66 |
5.16 |
26.85 |
3.1.5 Cyclic voltammetry (CV). The electrochemical properties of cyclopalladated diimines (ITO@3DIS-Pd) at different steps were measured (Fig. 5). An oxidation wave at ∼0 V showed that an amine had appeared after grafting with APTES (Fig. 5b). Voltammograms showed a surface wave consisting of oxidation at ∼0.22 V ascribed to a Schiff-base group when ITO@APTES was grafted with 1,4-phthalaldehyde (Fig. 5c). To ascertain if a diimine (ITO@3SID-Pd) had been formed, a contrast experiment was initiated in which ITO@3DIS-Pd was reacted with 2-aminoethyl-ferrocene. No redox waves of ferrocene appeared,20 indicating that a diimine on the surface of the substrate had been prepared (Fig. 5e). An oxidation peak was observed at 0.58 V, which is the characteristic peak of cyclopalladated diimines, suggesting that cyclopalladated diimine was located on the ITO electrode (Fig. 5d). Similar results are presented in Fig. S7 and S8† for ITO@1DIS-Pd and ITO@2DIS-Pd, respectively.
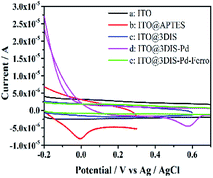 |
| | Fig. 5 Comparison of cyclic voltammograms of ITO@3DIS-Pd during the preparation. (a) ITO; (b) ITO@APTES; (c) ITO@3DIS; (d) ITO@3DIS-Pd and (e) ITO@3DIS-Pd reacted with 2-ethylamino ferrocene. | |
Characterization of the WCA as well as the UV-vis, XPS, AFM and CV data obtained above demonstrated that ordered cyclopalladated diimine self-assembled monolayers were fabricated. Also, the Pd content in cyclopalladated diamine monolayers was 7.7 × 10−10, 45.1 × 10−10 and 188.67 × 10−10 mmol cm−2 for Si@1DIS-Pd, Si@2DIS-Pd and Si@3DIS-Pd, respectively. Therefore, they were used to explore surface chemical reactions using the Suzuki coupling reaction as a model.24
3.2 Catalytic properties and recycling of Si@1DIS-Pd, Si@2DIS-Pd and Si@3DIS-Pd
3.2.1 Investigation of catalytic properties. The Suzuki coupling reaction is usually employed as a template for characterizing catalytic properties.25 The best catalytic conditions for use in subsequent catalytic reactions were: 90 (°C), 2–4 h, K2CO3 as the base, water as the solvent for Si@2DIS-Pd and Si@3DIS-Pd (Table S3†). However, Si@1DIS-Pd showed lower catalytic activity due to the smaller amount of Pd immobilized.Si@3DIS-Pd had higher activity with less catalyst (Table S4†) compared with that of Si@2DIS-Pd (Table S5†), suggesting that catalytic efficiency is dependent upon the orientation, stability and density of catalysts. Higher yields (97–99%) were obtained in the presence of Si@3DIS-Pd in neat water (Table S4,† entries 1–3 and 5–10), even with electron-rich moieties except for o-OCH3–C6H4–Br due to the steric effect (R = o-OMe, entry 4; o-methyl, entry 11). Especially for 4-chlorobenzaldehyde (Table S4,† entry 12), Si@3DIS-Pd was more catalytically active than that of Si@2DIS-Pd (Table S5,† entry 12) due to the higher stability and ordered orientation of Si@3DIS-Pd. The coupling reactions of derivatives of bromoboronic acids with arylbromides were carried out with Si@3DIS-Pd (Table S4,† entries 14–16) to obtain excellent yields. This difference in catalytic activity was attributed to the easy accessibility of substrates with active sites on monolayer surfaces.
3.2.2 Recyclability and stability of Si@3DIS-Pd. Recycling results showed that the yield decreased after eight cycles, but yields of 4-methylbiphenyl maintained at 50% could be obtained over 10 runs. In the case of Si@1DIS-Pd and Si@2DIS-Pd, they could be reused only twice due to easy hydrolysis of fatty Schiff-base groups. UV changes of Si@2DIS-Pd during a catalytic program (Fig. S9†) showed that the peak at 261 nm of a Schiff-base group decreased with increasing time, resulting in the loss of the catalyst. These results showed that Si@3DIS-Pd with an aryl imine was stable. Also, 90–93% yield could be obtained after several cycles in which the reaction time was lengthened to 10 h after eight cycles (red column) (Fig. 6).21h
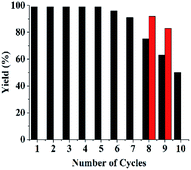 |
| | Fig. 6 Recycling of Si@3DIS-Pd. | |
3.3 Investigation of the catalytic mechanism
Many efforts have been made to illustrate the changes of active centers and interfaces during catalysis. However, most mechanistic studies have only observed the changes of surfaces and structures before and after the heterogeneous catalytic process.14a Detailed research on the whole process needs to be done. It is necessary to carry out leaching studies,4b kinetic studies, hot filtration tests and poisoning tests to solve the question of heterogeneity.26 Hence, the functional surfaces of monolayer can be designed as “bridges” between homogeneous and heterogeneous systems. Morphologic changes of surfaces during catalysis are important factors for elucidating mechanisms.
3.3.1 Hot filtration tests and dynamic studies. When a catalyst was added and removed after 1.5 h, 55% yields were obtained (Fig. 7). Yields did not change until 4 h (Fig. 7, blue line). However, when the catalyst was added to the reaction solution, 95% yield could be obtained after 4 h (Fig. 7, black line). These contrast experiments showed that leaching of active catalysts did not occur from the monolayer of functional catalysts.
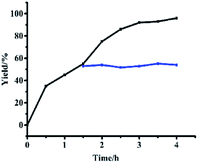 |
| | Fig. 7 Hot filtration test of Si@3DIS-Pd used for catalysis of the Suzuki coupling reaction. | |
3.3.2 Catalyst poisoning. The catalytic activity could be inhibited effectively after mercury addition (Table S5†), in which the yield was 28% (Table S6,† entry 1). Mercury cannot make contact sufficiently with the catalytic active site due to its poor dispersibility in the solvent.21i In the case of the PPh3 ligand, catalytic activity did not decrease (Table S6,† entry 2), which was attributed to ligand exchange with chlorine. However, when thiophene was presented, a significant decrease in activity was observed because the active sites were covered with thiophene (Table S6,† entry 3). These results showed that the catalytic process occurred mainly on the surface of Si@3DIS-Pd monolayers, which acted as a heterogeneous system.
3.3.3 Investigation of the morphology of catalytic surfaces during catalysis with or without stirring. Stirring is an important factor in catalytic processes (particularly in heterogeneous systems). Stirring can improve the reaction rate because of the equilibrium between absorption and desorption on the surface. Therefore, surface changes as imaged with AFM can be described to understand catalytic mechanisms.To elucidate changes on catalytic surfaces related with the catalytic mechanism, the corresponding AFM images as well as Ra and Rms values at different reaction time under stirring (Fig. 8, Table 2) or without stirring (Fig. S10 and S11†) were obtained. AMF (Fig. 8) showed some aggregates of height of 4–5 nm before catalysis (Fig. 8a) but the morphology of the catalyst surface changed dramatically with more aggregates of average height 12 nm in 0.5 h (Fig. 8b), indicating that absorption of substrates occurred on the surface of catalyst monolayers. Round aggregates (30 nm) appeared after 1 h (Fig. 8c), which might have been due to automatic agglomeration of coupling products because the absorption rate was higher than the desorption rate. This phenomenon was the key factor for the catalytic process. It can be observed in Fig. 8d and e that the average height was similar to that at 1 h. Significant morphologic changes on catalyst surfaces were observed during 2.5 h of catalytic processes (Fig. 8f), in which the average height reached ∼120 nm and orientation became irregular. This finding suggested that a violent reaction occurred, accompanied by a sequential adsorption and chemical process (including substrates, intermediates and product), on the surface of the catalyst monolayer. The morphology of the catalyst monolayer turned became more regular during 3–4 h compared with that at 2.5 h, with a height range from 20 nm to 40 nm (Fig. 8g and h). Finally, the ordered catalyst surface could be observed after the used catalyst had been treated by ultrasonic stimulation in water (Fig. 8i). The remarkable changes stated above can be illustrated by active palladacycle on the surface of a catalytic monolayer, including absorption, synergism and adsorption.27 AFM images and Ra/Rms values at different catalytic reaction times without stirring (Fig. S10 and S11†) were also obtained. Results showed that catalytic time must be increased because the covered substrate or product affects the surface catalytic process due to a slower rate of adsorption. This finding was also evidence that stirring could aid surface catalysis (detailed analysis is presented in ESI†).
 |
| | Fig. 8 AFM images of Si@3DIS-Pd monolayer during catalytic process with stirring. (a), (b), (c), (d), (e), (f), (g), (h), (i), respectively, represented before and after catalysis at 0.0 h, 0.5 h, 1.0 h, 1.5 h, 2.0 h, 2.5 h, 3.0 h, 4.0 h and ultrasonic treatment after reaction. | |
Table 2 Ra/Rms data from AFM images of Si@3DIS-Pd used for catalysis at different times
| Time (h) |
0.0 |
0.5 |
1.0 |
1.5 |
2.0 |
2.5 |
3.0 |
4.0 |
Ultrasonic |
| Ra (nm) |
4.15 |
1.65 |
16.18 |
3.14 |
3.99 |
25.28 |
6.69 |
1.82 |
1.70 |
| Rms (nm) |
5.86 |
3.56 |
33.49 |
4.99 |
13.38 |
33.19 |
10.27 |
3.46 |
3.29 |
3.3.4 WCA analyses. The WCA can be used to observe surface wetting ability.22 As catalysis proceeded (Fig. 9), the WCA decreased in 1 h (due to absorption of substrates and salt) and increased before 2.0 h (because of increasing absorption and product). The dramatic decrease in the WAC at 2.5 h was induced by a violent reaction according to AFM (Fig. 8f), and then increased gradually and almost recovered to the original level from 3 h to the end of the reaction, clearly revealing that catalysis proceeded on the surface. A similar phenomenon was presented for catalysis without stirring, in which the catalytic time was prolonged to 24 h (Fig. S12†).
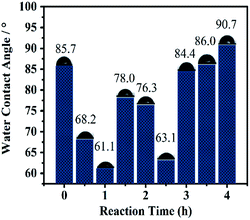 |
| | Fig. 9 Comparison of static water contact angle of Si@3DIS-Pd during the catalytic process under stirring. | |
3.3.5 Analyses of cyclic voltammograms. CV can provide important electrochemical information for catalysts at different reaction times, and help to evaluate surface catalytic process. All cyclic voltammograms displayed a pair of weak and broad redox peaks assigned to Pd(0)/Pd(II) at 0.566 and 0.216 (Fig. 10). The intensity of the peak of Si@3DIS-Pd increased until 2 h, indicating that the catalytic site had lower electroactivity because of redox-active sites covered by some substrates. This finding could be interpreted as the electrode surface being covered gradually by the absorption substrates and intermediates of oxidative addition under catalysis. The peak decreased after 3 h due to adsorption of the coupling product from the catalyst surface. This finding was in agreement with the results of WCA measurement and AFM mentioned above.
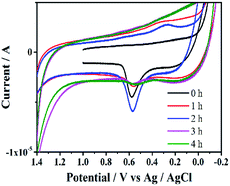 |
| | Fig. 10 Cyclic voltammograms of Si@3DIS-Pd on ITO during the catalytic process with stirring. | |
3.3.6 XPS investigation of the catalytic surface during catalysis with stirring. XPS spectra can give information that can help understanding catalytic mechanisms.15 Changes in active centers were detected through analyses of Si@3DIS-Pd catalysis (Fig. 11). Peaks at 345 eV and 337.8 eV denoted Pd3d 3/2 and Pd3d 5/2 of Pd(II) before the catalytic reaction (Fig. 11a) was observed. Two new peaks at 340.5 eV and 335.3 eV assigned to Pd3d 3/2 and Pd3d 5/2 of Pd(0) appeared because of the reduction of Pd(II) during the catalytic process (Fig. 11b) and shifted obviously to lower energy (341.0 eV and 335.8 eV), which was called the “induction period”, indicating generation of new intermediates.28 These findings were consistent with the results shown in Fig. 8, during which a violent reaction occurred at 2 h. However, the peaks at 341.0 eV and 335.8 eV disappeared after 3 h and only the peak at 337.7 eV remained, implying that Pd(0) was re-oxidized to Pd(II) at the end.29 When the catalytic process occurred, Br 3d peaks at 68.5 eV and 67.4 eV appeared, indicating that the Pd(0) oxidation with Br–Ph occurred on the surface.30 Simultaneously, the B1s peak at 185.5 eV also appeared after 2 h (Fig. 12b), implying that the phenylboronic-acid addition took place. The amount of Pd was 21.7 × 10−10 mmol in solution as measured by ICP-AES, showing almost no leaching of Pd. According to the results presented above, the real active center Pd(0) generated in situ was stabilized by diimine ligands on the surface.31
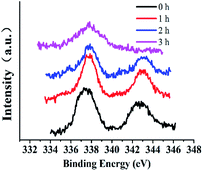 |
| | Fig. 11 The high-resolution XPS of Pd3d during the catalytic process (Si@3DIS-Pd). (a) 0 h, black line; (b) 1 h, red line; (c) 2 h, blue line; (d) 3 h, pink line. | |
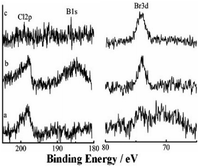 |
| | Fig. 12 High-resolution XPS of B1s, Cl2p and Br3d during the catalytic process at (a) 1 h, (b) 2 h and (c) 3 h. | |
Based on the investigations mentioned above, catalytic processes were proposed (Scheme 2). Ph–Br was absorbed and reacted with Pd(0) species on the surface to form oxidation products, followed by insertion with PhB(OH)2 to give an organometallic. Finally, reductive elimination of intermediates yielded the target molecules, accompanied by the generation of Pd(0) species to complete the catalytic cycle.21
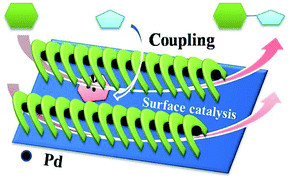 |
| | Scheme 2 Plausible catalytic process. | |
4. Conclusions
Channel-like self-assembled monolayers with aliphatic and aromatic diimines (denoted as Si@1DIS, Si@2DIS and Si@3DIS) immobilized on substrates and their palladacycle monolayers (Si@1DIS-Pd, Si@2DIS-Pd and Si@3DIS-Pd) were fabricated and characterized. Their catalytic properties for the Suzuki coupling reaction were investigated. Si@3DIS-Pd showed higher catalytic activity in neat water and better recyclability (at least eight times) without Pd leaching. These features can meet the specifications of the pharmaceutical industry better than that of Si@2DIS-Pd and Si@1DIS-Pd because the carbon atom in the aliphatic diimine of Si@2DIS-Pd and Si@1DIS-Pd was easily hydrolyzed due to an active hydrogen of α-C, resulting in poor recyclability. Control of the amount of catalyst immobilized could be achieved by adjusting the channel-like structure, which also enhanced the catalytic activity. The catalytic process and mechanism were investigated systematically and proposed based on experimental results. AFM showed stirring to be an important factor in the catalytic process (particularly in a heterogeneous system) which could improve the reaction rate because of the equilibrium between sorption and desorption on the surface. Results indicated that catalysis was heterogeneous on the surface of the monolayer. These results will allow a rational method to enhance the activity, selectivity and application of heterogeneous catalysts.
Conflicts of interest
There are no conflicts.
Acknowledgements
The authors gratefully acknowledge the Chinese Academy of Sciences (XDA09030203), Natural Science Foundation of China (20973157, 21172200) and NSFH (142300410223) for their financial support. The authors thank Professor Luyuan Mao for AFM analyses.
Notes and references
-
(a) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC;
(b) R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114(3), 1783 CrossRef PubMed;
(c) M. Tobisu and N. Chatani, Acc. Chem. Res., 2015, 48(6), 1717–1726 CrossRef PubMed;
(d) T. Iwasaki and N. Kambe, Top. Curr. Chem., 2016, 374(5), 66 CrossRef PubMed;
(e) S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1439 CrossRef PubMed;
(f) N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609 CrossRef.
-
(a) D. Wang, D. Denux, J. Ruiz and D. Astruc, Adv. Synth. Catal., 2013, 355, 129 CrossRef;
(b) X. F. Wu, P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9047 CrossRef PubMed;
(c) C. Torborga and M. Beller, Adv. Synth. Catal., 2009, 351, 3027 CrossRef;
(d) V. Farina, Adv. Synth. Catal., 2004, 364, 1553 CrossRef;
(e) G. Zeni and R. C. Larock, Chem. Rev., 2006, 106, 4644 CrossRef PubMed.
-
(a) J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177–2250 CrossRef PubMed;
(b) M. Butters, D. Catterick, A. Craig, A. Curzons, D. Dale, A. Gillmore, S. P. Green, I. Marziano, J.-P. Sherlock and W. White, Chem. Rev., 2006, 106, 3002 CrossRef PubMed.
-
(a) Á. Molnár, Chem. Rev., 2011, 111, 2251 CrossRef PubMed;
(b) S. Navalona, A. Dhakshinamoorthyb, M. Alvaroa and H. Garciaa, Coord. Chem. Rev., 2016, 312, 99 CrossRef;
(c) S. Paul, M. M. Islam and S. M. Islam, RSC Adv., 2015, 5, 42193–42221 RSC;
(d) S. Santoro, S. I. Kozhushkov, L. Ackermann and L. Vaccaro, Green Chem., 2016, 18, 3471–3493 RSC;
(e) A. Dhakshinamoorthy, M. Opanasenko, J. Čejka and H. Garcia, Adv. Synth. Catal., 2013, 355, 247 Search PubMed.
-
(a) J. J. E. Hardy, S. Hubert, D. J Macquarrie and A. J Wilson, Green Chem., 2004, 6, 53 RSC;
(b) Y. M. Yamada, S. M. Sarkar and Y. Uozumi, J. Am. Chem. Soc., 2012, 134, 3190 CrossRef PubMed;
(c) Y. Li, E. Boone and M. A. El-Sayed, Langmuir, 2002, 18, 4921 CrossRef;
(d) P. Boomi, H. G. Prabu and J. Mathiyarasu, Eur. J. Med. Chem., 2014, 72, 18 CrossRef PubMed.
-
(a) B. Karimi, D. Elhamifar, J. H. Clark and A. J. Hunt, Org. Biomol. Chem., 2011, 9, 7420 RSC;
(b) M. Kim, J. C. Park, A. Kim, K. H. Park and H. Song, Langmuir, 2012, 28, 6441 CrossRef PubMed;
(c) Z. Chen, Z.-M. Cui, F. Niu, L. Jiang and W.-G. Song, Chem. Commun., 2010, 46, 6524 RSC;
(d) J. Zhi, D. Song, Z. Li, X. Lei and A. Hu, Chem. Commun., 2011, 47, 10707 RSC.
-
(a) W. Fang, J. Yang and J. Gong, Adv. Funct. Mater., 2012, 22, 842 CrossRef;
(b) K. Shimizu, R. Maruyama, S. Komai, T. Kodama and Y. Kitayama, J. Catal., 2004, 227, 202 CrossRef;
(c) M. Mora, C. Jiménez-Sanchidrián and J. R. Ruiz, J. Mol. Catal. A: Chem., 2008, 285, 79 CrossRef;
(d) R. M. Mohamed and E. S. Baeissa, Appl. Catal., A, 2013, 464, 218 CrossRef;
(e) P. D. Stevens, G. Li, J. Fan, M. Yen and Y. Gao, Chem. Commun., 2005, 4435 RSC.
-
(a) M. Lv, W. Xie, S. Sun, G. Wu, L. Zheng, S. Chu, C. Gao and J. Bao, Catal. Sci. Technol., 2015, 5, 2925–2934 RSC;
(b) X. Fan, G. Zhanga and F. Zhanga, Chem. Soc. Rev., 2015, 44, 3023–3035 RSC;
(c) H. Sakurai, T. Tsukuda and T. Hirao, J. Org. Chem., 2002, 67, 2721 CrossRef PubMed;
(d) D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782–5816 CrossRef PubMed.
-
(a) M. Opanasenko, P. Štěpnička and J. Čejka, RSC Adv., 2014, 4, 65137–65162 RSC;
(b) S. Navalona, A. Dhakshinamoorthyb, M. Alvaroa and H. Garciaa, Coord. Chem. Rev., 2016, 312, 99–148 CrossRef;
(c) Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC;
(d) A. Dhakshinamoorthy, A. M. Asiric and H. Garcia, Chem. Soc. Rev., 2015, 44, 1922–1947 RSC;
(e) Q. Sun, Z. Dai, X. Meng and F.-S. Xiao, Chem. Soc. Rev., 2015, 44, 6018–6034 RSC;
(f) D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782–5816 CrossRef PubMed;
(g) H. Fan, Z. Qi, D. Sui, F. Mao, R. Chen and J. Huang, Chin. J. Catal., 2017, 38(3), 589–596 CrossRef.
-
(a) R. Franzén and Y. J. Xu, Can. J. Chem., 2005, 83, 266 CrossRef;
(b) M. Lamblin, L. Nassar-Hardy, J.-C. Hierso, E. Fouquet and F.-X. Felpin, Adv. Synth. Catal., 2010, 352, 33 CrossRef;
(c) J. Wei, J. Jiao, J. Feng, J. Lv, X. Zhang, X. Shi and Z. Chen, J. Org. Chem., 2009, 74, 6283 CrossRef PubMed;
(d) S. Ogasawara and S. Kato, J. Am. Chem. Soc., 2010, 132, 4608 CrossRef PubMed;
(e) B. Yuan, Y. Pan, Y. Li, B. Yin and H. Jiang, Angew. Chem., Int. Ed., 2010, 49, 4054 CrossRef PubMed.
- L. Jin and J. Liebscher, Chem. Rev., 2007, 107, 13 Search PubMed.
-
(a) N. T. S. Phan, M. V. D. Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609 CrossRef;
(b) L. Xue and Z. Lin, Chem. Soc. Rev., 2010, 39, 1692 RSC.
-
(a) W. He and Y. Li, Chem.–Eur. J., 2012, 18, 9813 CrossRef PubMed;
(b) J.-S. Chen, A. N. Vasiliev, A. P. Panarello and J. G. Khinast, Appl. Catal., A, 2007, 325, 76 CrossRef;
(c) Z. Niu, Q. Peng, Z. Zhuang and M. Pérez-Lorenzo, J. Phys. Chem. Lett., 2012, 3, 167 CrossRef;
(d) S. MacQuarrie, J. H. Horton, J. Barnes, K. McEleney, H.-P. Loock and C. M. Crudden, Angew. Chem., 2008, 120, 3324 CrossRef.
-
(a) P. J. Ellis, I. J. S. Fairlamb, S. F. J. Hackett, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2010, 49, 1820 CrossRef PubMed;
(b) L. Shao, B. Zhang, W. Zhang, S. Y. Hong, R. Schlçgl and D. S. Su, Angew. Chem., 2013, 125, 2168 (Angew. Chem., Int. Ed., 2013, 52, 2114) CrossRef;
(c) B. M. Choudary, S. Madhi, N. S. Chowdari, M. L. Kantam and B. J. Sreedhar, J. Am. Chem. Soc., 2002, 124, 14127 CrossRef PubMed.
-
(a) J. J. Davis, K. S. Coleman, K. L. Busuttil and C. B. Bagshaw, J. Am. Chem. Soc., 2005, 127, 13082 CrossRef PubMed;
(b) J. J. Davis, C. B. Bagshaw, K. L. Busuttil, Y. Hanyu and K. S. Coleman, J. Am. Chem. Soc., 2006, 128, 14135–14141 CrossRef PubMed.
- A. K. Kakkar, Chem. Rev., 2002, 102, 3579 CrossRef PubMed.
-
(a) A. Ulman, Chem. Rev., 1996, 96, 1533–1554 CrossRef PubMed;
(b) A. K. Kakkar, Chem. Rev., 2002, 102, 3579–3588 CrossRef PubMed;
(c) Y. Yang, S. Wang, Y. Wang, X. Wang, Q. Wang and M. Chen, Biotechnol. Adv., 2014, 32(7), 1301–1316 CrossRef PubMed;
(d) A. Indra, C. S. Gopinath, S. Bhaduri and G. K. Lahiri, Catal. Sci. Technol., 2013, 3, 1625–1633 RSC;
(e) T. Gupta and M. E. van der Boom, J. Am. Chem. Soc., 2006, 128, 8400–8401 CrossRef PubMed;
(f) J. Hu, Y. Wang, M. Han, Y. Zhou, X. Jiang and P. Sun, Catal. Sci. Technol., 2012, 2, 2332–2340 RSC;
(g) C. Ren, J. Zhang, M. Chen and Z. Yang, Chem. Soc. Rev., 2014, 43(21), 7257–7266 RSC;
(h) L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117(23), 6414–6415 CrossRef.
-
(a) H. P. Mungse, S. Verma, N. Kumar, B. Sain and Om P. Khatri, J. Mater. Chem., 2012, 22, 5427–5433 RSC;
(b) D. Domin, D. Benito-Garagorri and K. Mereiter, Organometallics, 2005, 24(16), 3957–3965 CrossRef;
(c) P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410 RSC;
(d) D. V. Jawale, E. Gravel, C. Boudet, N. Shah, V. Geertsen, H. Li, I. N. N. Namboothiri and E. Doris, Catal. Sci. Technol., 2015, 5, 2388–2392 RSC;
(e) E. F. McCord, S. J. McLain and L. T. J. Nelson, Macromolecules, 2001, 34(3), 362–371 CrossRef;
(f) Q. Zhao, B. Chan, W. Zhang, L. Yang, G. Zhang, F. Zhang and X. Fan, Ind. Eng. Chem. Res., 2014, 53, 4232–4238 CrossRef;
(g) H. P. Mungse, S. Verma, N. Kumar, B. Sain and O. P. Khatri, J. Mater. Chem., 2012, 22, 5427 RSC;
(h) H. Su, S. Wu, Z. Li, Q. Huo, J. Guan and Q. Kan, Appl. Organomet. Chem., 2015, 29, 462 CrossRef;
(i) P. K. Khatri, S. Choudhary, R. Singh, S. L. Jain and O. P. Khatri, Dalton Trans., 2014, 43, 8054 RSC;
(j) Q. Zhao, C. Bai, W. Zhang, Y. Li, G. Zhang, F. Zhang and X. Fan, Ind. Eng. Chem. Res., 2014, 53, 4232 CrossRef;
(k) Z. Li, S. Wu, D. Zheng, J. Liu, H. Liu, H. Lu, Q. Huo, J. Guan and Q. Kan, Appl. Organomet. Chem., 2014, 28, 317 CrossRef.
-
(a) M. Rezaeivala and H. Keypour, Coord. Chem. Rev., 2014, 280, 203–245 CrossRef;
(b) P. A. Vigato, V. Peruzzo and S. Tamburini, Coord. Chem. Rev., 2012, 256, 953–1114 CrossRef;
(c) C. J. Whiteoak, G. Salassa and A. W. Kleij, Chem. Soc. Rev., 2012, 41, 622–661 RSC;
(d) G. S. Smith and S. F. Mapolie, J. Mol. Catal. A: Chem., 2004, 213(2), 187–192 CrossRef;
(e) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38(2), 606–631 RSC;
(f) A. El-Faham and F. Albericio, Chem. Rev., 2011, 111(11), 6557–6602 CrossRef PubMed;
(g) M. Semler, J. Čejka and P. Štěpnička, Catal. Today, 2014, 227, 207–214 CrossRef.
-
(a) Y. Zhao, N. Zhao, M. Zou, T. Li, G. Li, J. Li and Y. Wu, Appl. Organomet. Chem., 2016, 30, 540–549 CrossRef;
(b) Z. Fu, T. Li, B. Mu, L. Mao, G. Li, W. Xu and Y. Wu, J. Mol. Catal. A: Chem., 2012, 363, 200 CrossRef;
(c) N. Zhao, F. Wang, M. Zhou, T. LI, H. Liu, W. Xu and Y. Wu, Chin. J. Catal., 2013, 34(8), 1583–1588 CrossRef;
(d) B. Mu, T. Li, C. Li, P. Liu, W. Shang and Y. Wu, Tetrahedron, 2009, 65, 2599 CrossRef.
-
(a) H. Liu, T. Li, X. Xue, W. Xu and Y. Wu, Catal. Sci. Technol., 2016, 6, 1667 RSC;
(b) N. Zhao and T. Li, ChemCatChem, 2013, 5, 1481 CrossRef;
(c) Z. Fu, T. Li, X. He, J. Liu, W. Xu and Y. Wu, J. Mol. Catal. A: Chem., 2014, 395, 293 CrossRef;
(d) Y. Li, Z. Fu, L. Rao, L. Wang, R. Li, T. Li and Y. Wu, J. Chin. Chem. Soc., 2014, 61, 397 CrossRef;
(e) H. Liu, X. Xue, T. Li, J. Wang, W. Xu, M. Liu, P. Chen and Y. Wu, RSC Adv., 2016, 6, 84815 RSC;
(f) K. Zou, H. Liu, T. Li, P. Chen, M. Liu and Y. Wu, RSC Adv., 2015, 5, 16654 RSC;
(g) Z. Fu, J. Liu, X. He, T. Li and Y. Wu, RSC Adv., 2014, 4, 26413 RSC;
(h) Z. Fu, N. Zhang, T. Li, W. Xu and Y. Wu, J. Colloid Interface Sci., 2013, 394, 409 CrossRef PubMed;
(i) Z. Q. Xue, P. P. Huang, T. S. Li, P. L. Chen, M. H. Liu and Y. J. Wu, Nanoscale, 2017, 9, 781–791 RSC.
-
(a) J. S. Sharp, D. J. Farmer and J. Kelly, Langmuir, 2011, 27, 9367–9371 CrossRef PubMed;
(b) A. W. Adamson and A. P. Gast, The Solid-Liquid Interface—Contact Angle, Academic Press, New York, 1997, pp. 347–383 Search PubMed.
- E. S. Gadelmawla, M. M. Koura and T. M. A. Maksoud, Roughness parameters, J. Mater. Process. Technol., 2002, 123, 133–145 CrossRef.
-
(a) B. Yuan, Y. Pan, Y. Li, B. Yin and H. Jiang, Angew. Chem., Int. Ed., 2010, 49, 4054–4058 CrossRef PubMed;
(b) S. Ogasawara and S. Kato, J. Am. Chem. Soc., 2010, 132, 4608–4613 CrossRef PubMed.
-
(a) G. Zeni and R. C. Larock, Chem. Rev., 2006, 106, 4644–4680 CrossRef PubMed;
(b) A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef PubMed;
(c) J. Wei, J. Jiao, J. Feng, J. Lv, X. Zhang, X. Shi and Z. Chen, J. Org. Chem., 2009, 74, 6283–6286 CrossRef PubMed.
- J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–324 CrossRef.
-
(a) K. Köhler, W. Kleist and S. S. Pröckl, Inorg. Chem., 2007, 46, 1876–1883 CrossRef PubMed;
(b) F. Y. Zhao, M. Shirai, Y. Ikushima and M. Arai, J. Mol. Catal. A: Chem., 2002, 180, 211–219 CrossRef.
-
(a) Z. Zhang, T. Sun, C. Chen, F. Xiao, Z. Gong and S. Wang, ACS Appl. Mater. Interfaces, 2014, 6(23), 21035–21040 CrossRef PubMed;
(b) H. Li, G. A. Grasa and T. J. Colacot, Org. Lett., 2010, 12(15), 3332–3335 CrossRef PubMed.
- M. Choi, D. H. Lee, K. Na, B. W. Yu and R. Ryoo, Angew. Chem., Int. Ed., 2009, 48(20), 3673–3676 CrossRef PubMed.
- J. Zou, S. G. Stewart, C. L. Raston and K. S. Iyera, Chem. Commun., 2011, 47, 1803–1805 RSC.
-
(a) C. Röhlich, A. S. Wirth and K. Köhler, Chem.–Eur. J., 2012, 18, 15485–15494 CrossRef PubMed;
(b) F. Rajabi and W. R. Thiel, Adv. Synth. Catal., 2014, 356(8), 1873–1877 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06365f |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Pingping Huang
a,
Pingping Huang












