
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly of an oligo(p-phenylenevinylene)-based molecule on an HOPG surface: insights from multi-scale simulation and STM observation
Yuan Qin a,
Yingying Yanga,
Man Yao*a,
Xiaowan Xuea,
Xudong Wanga,
Hao Huanga,
Ting Chenb,
Dong Wangb and
Lijun Wanb
a,
Yingying Yanga,
Man Yao*a,
Xiaowan Xuea,
Xudong Wanga,
Hao Huanga,
Ting Chenb,
Dong Wangb and
Lijun Wanb
aSchool of Materials Science and Engineering, Dalian University of Technology, Dalian 116024, China. E-mail: yaoman@dlut.edu.cn
bKey Laboratory of Molecular Nanostructure and Nanotechnology, Institute of Chemistry, Chinese Academy of Sciences and Beijing National Laboratory for Molecular Sciences, Beijing 100190, China
First published on 12th September 2018
Abstract
To gain knowledge of the most important weak interactions for supramolecular self-assembly and observe molecular structure for self-assembled architectures, the two-dimensional self-assembly of an oligo(p-phenylenevinylene)-based molecule (AS-OPV) on highly oriented pyrolytic graphite has been investigated. Accurate atomic configuration for the AS-OPV self-assembled pattern has been identified by means of multi-scale simulation combined with scanning tunneling microscopy (STM) experiments. The weak interactions which contribute to the formation of AS-OPV self-assembly are studied by analysis of non-covalent interactions existing in the system and theoretical calculation of their energy values. Investigation of the molecular structure of self-assembly and STM images at a certain temperature range is performed by molecular dynamics and density functional theory simulations. This work paves the way to explore the contribution of weak interactions for the self-assembly system, as well as providing a reference to observe the possible self-assembled structure at temperatures not convenient for direct experimental observation.
1. Introduction
Oligo(p-phenylenevinylene) (OPV) and its derivatives have been widely investigated in recent years and have attracted the increasing attention of supramolecular chemists due to their π-conjugated systems, excellent emission and charge transport properties.1–4 In order to create molecule-based devices and ordered supramolecular nanostructures, molecular self-assembly is proposed as a useful approach.5–8 Self-assembled OPV-based materials are found to have potential application as organic electronic devices in the field of organic field effective transistors,9,10 sensing,11,12 photovoltaic cells,13–15 and light-emitting diodes.16In building and controlling molecular self-assembly, weak non-covalent interactions including van der Waals (vdW), electrostatic, hydrogen bonding and π–π stacking interactions are often found to play crucial roles.17–22 Therefore, research on the non-covalent interactions will be conducive to the regulation of the self-assembly process. A lot of OPV-based molecules are reported to be able to form an ordered two-dimensional (2D) self-assembly on highly oriented pyrolytic graphite (HOPG) surfaces which have been extensively utilized as the substrates for self-assembled systems.23,24 However, it is still unclear what kinds of non-covalent interactions govern the formation of self-assembly. These unsolved issues set obstacles for understanding and regulating the self-assembly process. Apart from the structure of self-assembled molecule, the external environment also has a great influence on self-assembly.25–27 Temperature is one of the important factors in self-assembly process, which has the potential to modify the 2D arrangement and affect its stability.28,29 And then, these changes that may occur in self-assembly will be directly related to the efficiency and stability of molecular devices.30,31 As a consequence, it is necessary to investigate the configuration for OPV-based self-assembly in a wide temperature range.
We employ density functional theory (DFT) and molecular dynamics (MD) methods in conjunction with STM observation to uncover the non-covalent interactions that contribute to the formation of OPV-based supramolecular self-assembly and observe the molecular structure of self-assembled architecture at different temperatures. The feasibility and rationality of this theoretical simulation approach are supported by the good agreement of computations of unit cell parameters and STM image with experimental observations. The atomic geometry of OPV-based self-assembly at room temperature is determined by MD simulation. The weak non-covalent interactions which may contribute to the self-assembly are analyzed by quantitative calculation of their energies. In order to study the structural characters of this self-assembled system at other temperatures, MD simulation at a certain temperature range from 248 K to 373 K is performed. Finally, STM images of OPV-based self-assembly are predicted by DFT simulation.
2. Methods
2.1 STM investigation
AS-OPV molecule (Fig. 1), namely OPV substituted by two aldehydes at both ends, is home-synthesized based on the reported literature.32 Molecular adlayer is prepared and STM measurement is preformed according to the previous work.23 Acetone is bought from Sigma-Aldrich with purity higher than 99.9%. A drop of acetone solution containing the AS-OPV molecule (<10−4 M) was directly deposited onto a freshly cleaved and atomically flat HOPG surface for the preparation of oligomer assembly. Before STM measurement, the as-prepared sample was placed in the air for 5 minutes until the solvent evaporated. STM experiments were carried out by means of a NanoScope IIIa (Digital Instruments) at room temperature. The tunneling tips were prepared by mechanically cutting Pt/Ir wire (90/10). More details about the experimental setup and procedures see the ref. 23 and 32.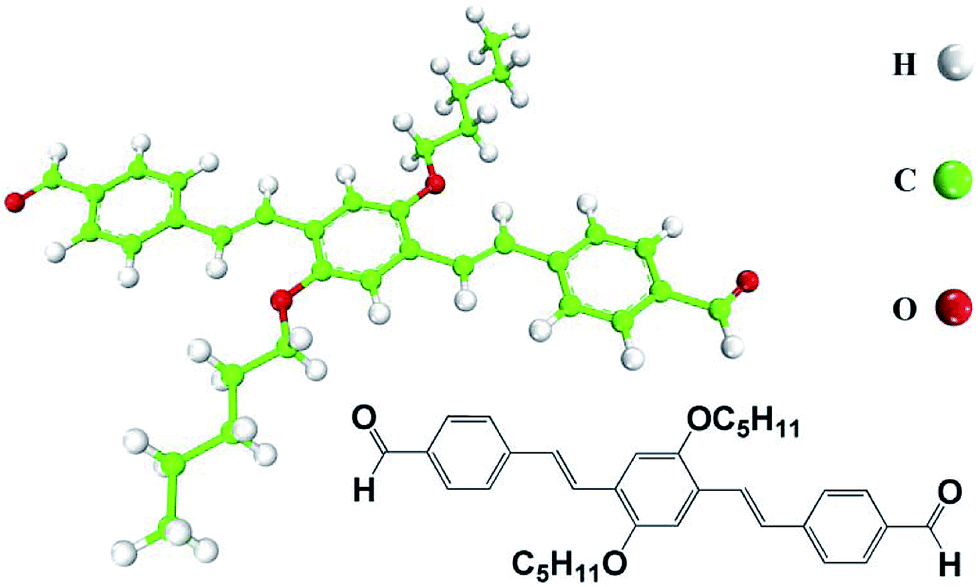 | ||
| Fig. 1 Chemical structure and model of AS-OPV. Hydrogen atoms are shown in white, carbon atoms in green and oxygen atoms in red. | ||
2.2 Computational details
In this research, molecular mechanics (MM) and MD were used to simulate the molecular geometry and energy, DFT was used to calculate the electronic properties. The simulated model used in this work was simplified to be in vacuum as the solvent does not play a decisive role in the formation of this AS-OPV self-assembly and it has evaporated before STM measurement. The simulations were carried out with BIOVIA Materials Studio 2017 R2 software suite.33 The computational details are as follows.Periodic boundary conditions were utilized to describe 2D periodic self-assembly arrangement on HOPG. The Forcite module was used in MM and MD calculations. Several commonly used force fields such as CVFF,34 Universal,35 and PCFF36 were applied to optimize the unit cell. After comparing the calculated and experimental values of the unit cell parameters, CVFF was selected to simulate the AS-OPV system. Smart minimizer approach was performed for the structure optimization. The convergence thresholds adopted to achieve the geometry optimization were energy variation smaller than 2.0 × 10−5 kcal mol−1, maximum force per atom of 0.001 kcal mol−1 Å−1 and maximum displacement of 1.0 × 10−5 Å. Electrostatic and vdW interactions were calculated using the Ewald and atom-based method, respectively. MD simulations were carried out with the NVT ensemble using the Nosé thermostat37 at temperatures ranging from 248 K to 573 K. During the simulation, the molecules were fully relaxed and allowed to desorb. The 1 ns simulations with a time step of 1 fs for the statistical analysis were subsequently performed after MD equilibration at the above temperatures. DFT calculations were carried out with the CASTEP module using Perdew–Burke–Ernzerhof functional of the generalized gradient approximation38 and ultrasoft pseudo-potential.39 The energy cutoff and self-consistent field tolerance were set to 380 eV and 5.0 × 10−7 eV per atom, respectively. STM images were simulated on the basis of Tersoff–Hamann approach40 with a positive bias voltage of 670 mV. In order to predict the STM images at different temperatures, first MD was applied to obtain the self-assembled structure at the simulated temperatures. And then the structures obtained at each simulated temperatures were extracted. Finally, STM simulation was performed with the above structures by DFT calculations. As a result, the STM images at different temperatures were achieved.
3. Results and discussion
3.1 Configuration identification of AS-OPV self-assembled pattern
The STM experiment observed that AS-OPV self-assembly pattern can form at ambient conditions. A typical STM image displaying the adsorption of AS-OPV on HOPG surface is shown in Fig. 2. It can be seen that the AS-OPV molecules form an ordered self-assembled architecture (Fig. 2a) and this is the only ordered phase observed. According to the STM image and chemical structure of AS-OPV molecule (Fig. 1), we have speculated the AS-OPV self-assembly structure and superimpose its model on STM image as shown in Fig. 2b. The white rectangle in Fig. 2b indicates the unit cell of AS-OPV self-assembly. In the AS-OPV molecular model (Fig. 1), the distance between two aldehyde groups is 21.048 Å. Taking into account the unit cell parameters obtained by STM observation of a = 15 ± 1 Å and b = 31 ± 1 Å, it can be conjectured that there is no more than two AS-OPV molecules in the unit cell. Bright features in the STM images are likely to be the backbones of AS-OPV and the dark features are attributed to alkyl chains. The angle between axis a of the unit cell and axis of molecule at the corner of the unit cell is about 63°, and the small cross angle between the axis of molecule in the corner and that in the center is about 54°.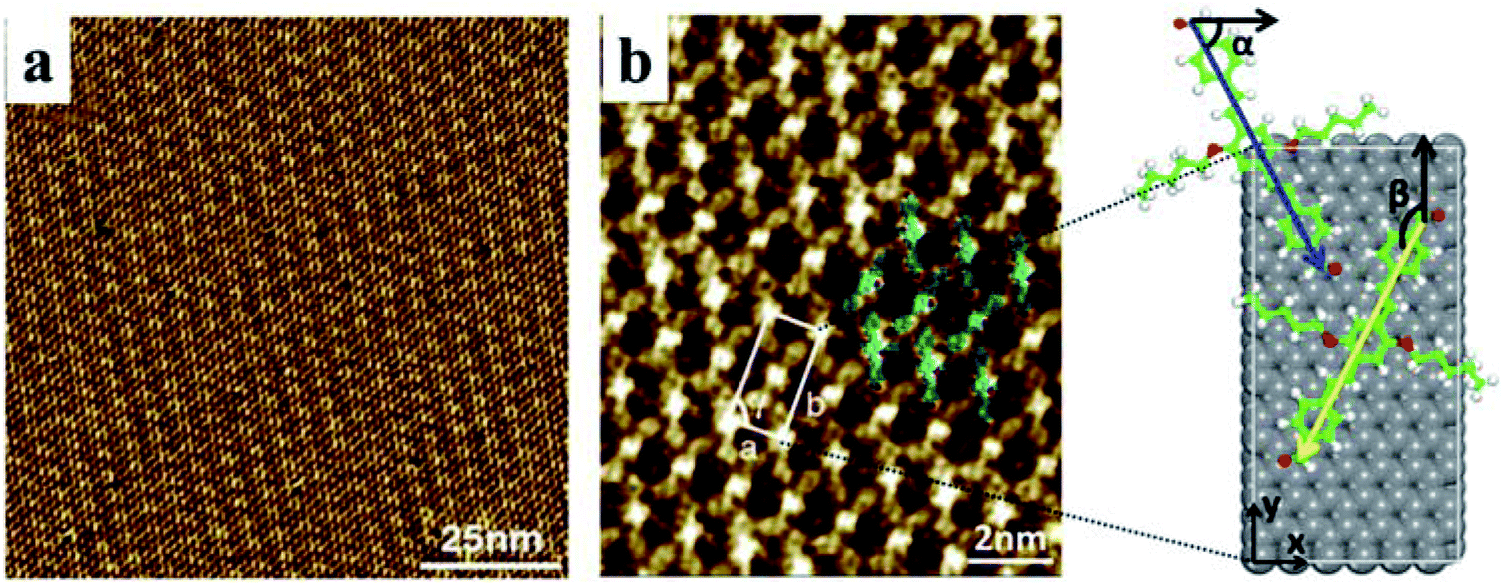 | ||
| Fig. 2 (a) Large-scale STM image (It = 655 pA, Vbias = 670 mV). (b) High-resolution STM image (It = 712 pA, Vbias = 670 mV) and unit cell model of AS-OPV self-assembly. The white rectangle indicates the unit cell and α, β, γ represent the unit cell parameters. Blue and yellow arrows are the backbone axis for the two molecules in the unit cell. Black arrows are the x-axis and y-axis. HOPG substrate is shown in grey. | ||
In order to select the suitable force field to simulate this assembly system, several common force fields listed in Table 1 were used for geometry optimization of the unit cell shown in Fig. 2b. The calculated results of these force fields are not much different from the experimental values. Among these force fields, CVFF presented best. In the case of CVFF, the parameter of optimized unit cell for a is 14.783 Å, in the range of experimental observation of 15 ± 1 Å; b is 29.859 Å, very close to the experimental value of 31 ± 1 Å; and γ is 90.007°, nearly equal to the experimental value. Moreover, the angle α between one molecular axis (blue arrow) of the molecule at the corner and x-axis is 63.146°; the angle β between the other molecular axis (yellow arrow) of the molecule at the center and y-axis is 153.039°. These calculated results are in very good agreement with the experimental data compared with the other force fields. As a result, CVFF was selected as the appropriate force field for simulating AS-OPV self-assembly.
| Calcd value | Exptl value | |||
|---|---|---|---|---|
| Universal | PCFF | CVFF | ||
| a (Å) | 14.663 | 14.531 | 14.783 | 15 ± 1 |
| b (Å) | 29.621 | 29.356 | 29.859 | 31 ± 1 |
| γ (deg) | 90.003 | 89.996 | 90.007 | 90 ± 1 |
| α (deg) | 60.286 | 62.705 | 63.146 | 63 ± 1 |
| β (deg) | 151.763 | 152.711 | 153.039 | 153 ± 2 |
To confirm whether the proposed configuration in Fig. 2b is correct and gain further insight into the atomic geometry of AS-OPV self-assembly, STM simulation was performed with the optimized structure. Fig. 3 shows the simulated and experimental STM images of AS-OPV self-assembly. The unit cell is indicated by white rectangle. It can be seen that the STM images of simulation and experiment are very similar. According to the simulated STM image, the brightest area (bright yellow) is located at the center and four vertices of the white rectangle, followed by the bright area at the rectangular diagonals. These bright features correspond to molecular groups with high electron density. Combined with the unit cell model, it can be identified that the center, vertices and diagonals with bright features are occupied by AS-OPV backbones; the dark areas are attributed to the alkyl chains attached to the backbones. This structure identification confirms the conjecture mentioned above and proves the rationality of the unit cell model in Fig. 2b. Therefore, exact geometry information of AS-OPV self-assembly at the atomic level was verified through the above process.
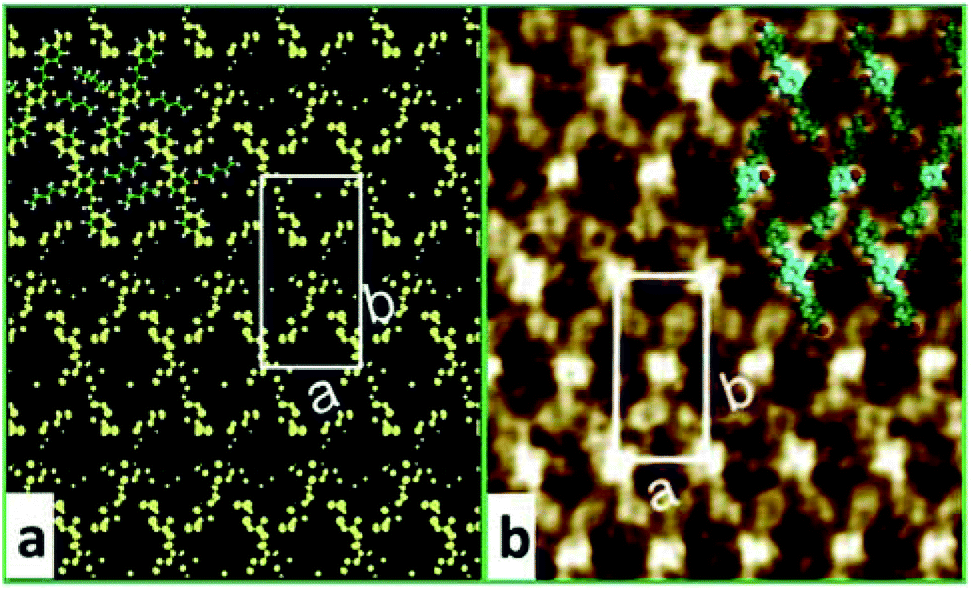 | ||
| Fig. 3 (a) Simulated and (b) experimental STM images of AS-OPV self-assembly at 298 K. The white rectangle indicates the unit cell. | ||
To explore the formation of AS-OPV self-assembly by means of theoretical simulation method, MD simulation was performed at 298 K with a 3 × 5 supercell which obtained by expanding the Y-axis of the unit cell by 3 times and expanding the X-axis by 5 times. The final snapshot for a run time of 1 ns after MD equilibrium is shown in Fig. 4. According to Fig. 4a, AS-OPV molecules form 2D molecular network. The adjacent AS-OPV molecules are staggered and arranged in a close-packed adlayer. As the side view in Fig. 4b shows, the adlayer lies flat on the substrate with the benzene rings parallel to HOPG surface. The partial enlarged view shows the AS-OPV molecules in two adjacent rows are orderly arranged in staggered relation (Fig. 4c). MD simulation reveals that AS-OPV molecules are able to maintain a well-organized arrangement and the self-assembly is stable at room temperature.
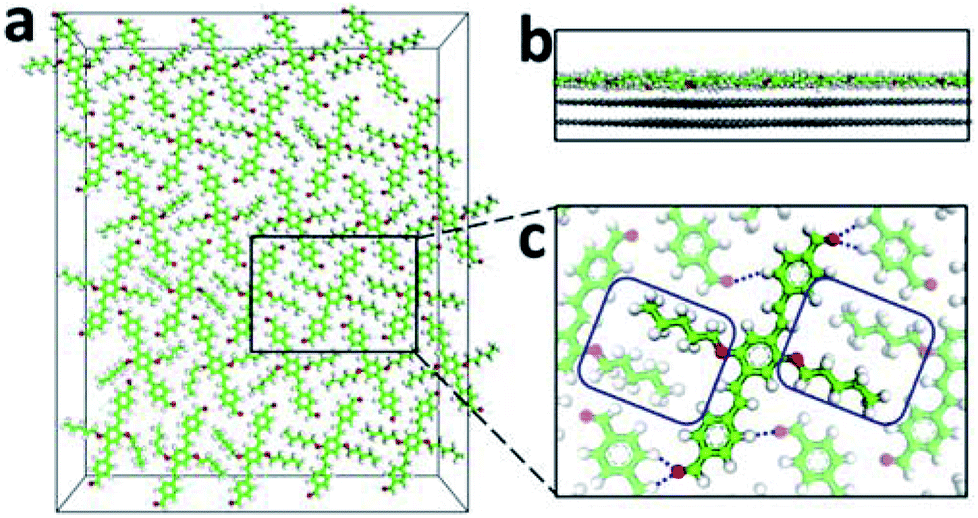 | ||
| Fig. 4 Snapshot of the 3 × 5 supercell for AS-OPV system after 1 ns MD simulation. (a) Top view. (b) Side view. (c) Partial enlarged view. For clarity, the HOPG substrate is not shown in (a) and (c). The blue dotted lines represent the intermolecular hydrogen bondings. | ||
3.2 Analysis of weak interactions in AS-OPV self-assembly
2D self-assembly of supramolecular structures is governed by molecule–molecule and molecule–substrate interactions. For an explicit understanding of self-assembly formation, weak non-covalent interactions in the AS-OPV system are necessary to be analyzed. Fig. 4c shows the alkyl chains marked in blue rectangle interdigitate with each other and form vdW interaction between the adjacent AS-OPV molecules. In addition, the oxygen of the aldehydes interacts with hydrogen of AS-OPV backbones to form hydrogen bonding (blue dotted line). The vdW interaction and hydrogen bonding are helpful for the AS-OPV molecules to organize into a well-defined and close-packed adlayer. According to our statistical calculations, the average bond length of hydrogen bondings in the adlayer is 2.67 ± 0.19 Å and this value is within the common bond length range of 2.2 Å to 3.2 Å for weak hydrogen bonding. Therefore, it can be inferred that the hydrogen bondings existing in the adlayer are mostly weak hydrogen bondings.The reason why molecules can lie flat on the substrate and form 2D assembled structure is also closely related to the interactions between adlayer and substrate. The calculated average distance from centroids of benzene rings in AS-OPV backbone to the top layer of HOPG is 3.52 ± 0.02 Å, suggesting π–π stacking interactions occur. Moreover, the alkyl chains that attached to the benzene rings form vdW force with the HOPG substrate. The vdW force as well as π–π stacking interactions are essential for the adsorption of AS-OPV molecules onto HOPG surface and the structural stability of self-assembly.
To visually illustrate the changes of the electronic structure for adsorbed molecule and substrate material, the electron density difference (EDD) of AS-OPV adsorbing on HOPG is extracted. The EDD is defined as
| Δρ = ρtotal − (ρAS-OPV + ρHOPG) | (1) |
Fig. 5 shows the structure and EDD of AS-OPV adsorbed on HOPG. It is obviously that the electron density of both the AS-OPV molecules and substrate are changed. From the EDD through slice 1 of the adsorption layer (Fig. 5b), the charge accumulation regions are located in AS-OPV backbones; and the charge depletion regions are located in alkyl chains. In contrast to EDD through slice 2 which is very close to the substrate (Fig. 5c), the charge accumulation regions for the top layer of HOPG locate exactly below the area of charge depletion in Fig. 5b, and the case of charge depletion regions is just the opposite. The charge attraction existing between adlayer and substrate contributes to the adsorption assembly.
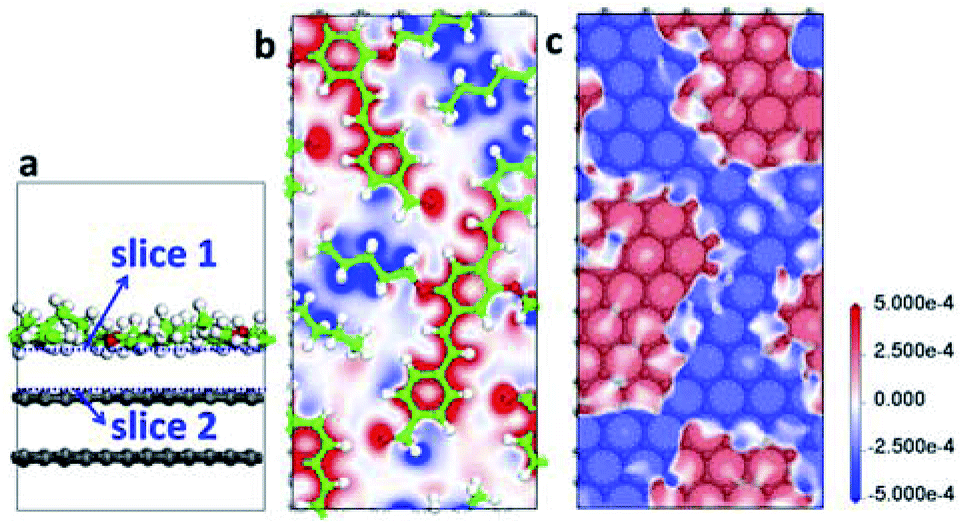 | ||
| Fig. 5 Structure and EDD of AS-OPV molecules adsorbed on HOPG. (a) Side view of unit cell structure. (b) EDD through slice 1. (c) EDD through slice 2. The blue dashed lines in (a) indicate the slices chosen for EDD. The charge accumulation and depletion regions in EDD are represented by red and blue, respectively. | ||
3.3 Exploration of molecular structure and STM image at different temperatures
The above simulation of AS-OPV self-assembly at room temperature is in line with experimental phenomenon, demonstrating the reliability of the theoretical calculations. To explore whether the AS-OPV self-assemblies can be formed in a wider temperature range and investigate the structural stability, MD simulations were performed at different temperatures. From the snapshots after AS-OPV systems have reached MD equilibrium (Fig. 6a–c), the molecules at all simulated temperatures organize into close-packed adlayers by interdigitating with each other. As shown in Fig. 6a and c, the AS-OPV systems exhibit well-ordered assemblies that are very similar to the case of 298 K in Fig. 6b. In addition, the simulations at other temperatures such as 273, 323 and 348 K can also obtain orderly patterns and their molecular structures are similar to the case as Fig. 6a–c show. Given the above, it is reasonable to speculate that AS-OPV self-assemblies are probably to be formed at 248, 273, 323, 348 and 373 K as well as room temperature. We have further extended the simulated temperatures to a higher level until 573 K and found the following results: AS-OPV self-assembled structure is able to main an ordered pattern when the temperature is lower than 400 K; as the simulated temperature continues to rise, part of the ordered configuration of the adlayer is progressively disrupted; when the simulated temperature is higher than 500 K, more than half of the ordered structure is disrupted.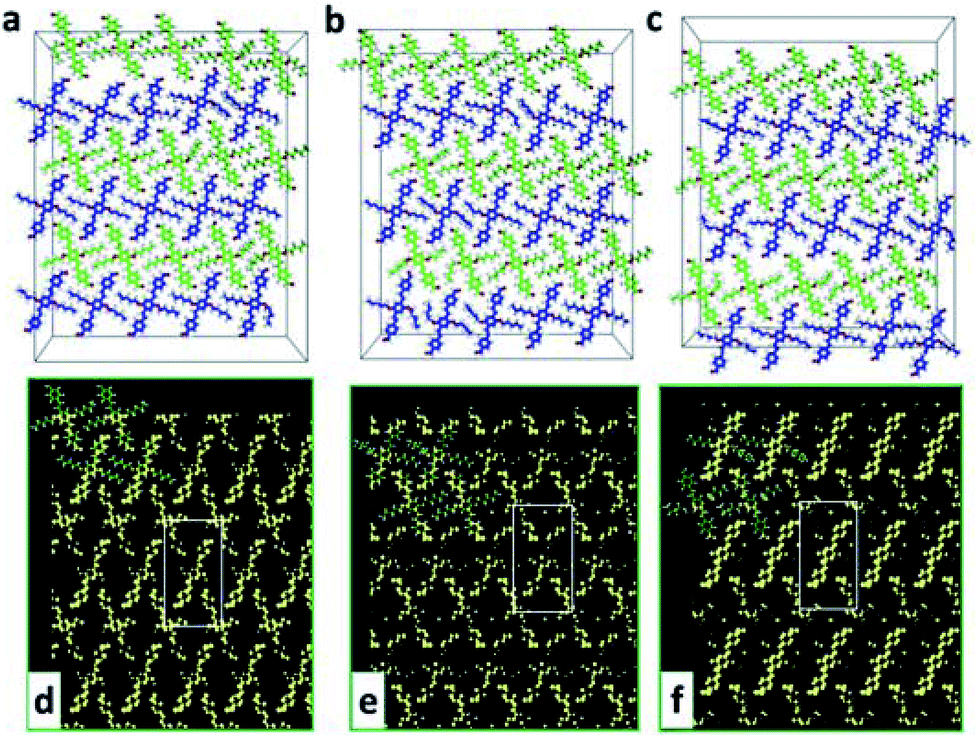 | ||
| Fig. 6 Snapshots of AS-OPV system after 1 ns MD simulations at different temperatures: (a) at 248 K; (b) at 298 K; (c) at 373 K. For clarity, the HOPG substrate is not shown and the carbon atoms of AS-OPV in neighbouring rows are distinguished by green and blue. Simulated STM images of AS-OPV self-assembly at different temperatures: (d) at 248 K; (e) at 298 K; (f) at 373 K. The white rectangles indicate the unit cells. | ||
Adsorption energies (Ead) were also calculated to further explore whether the adsorption of self-assembly was easy to occur at different temperatures. We define the Ead as
| Ead = Etotal − EAS-OPV − EHOPG | (2) |
| Temperature (K) | Ead (kcal mol−1) | EvdW (kcal mol−1) | Eele (kcal mol−1) |
|---|---|---|---|
| 248 | −94.97 | −26.77 | 7.61 |
| 273 | −94.35 | −25.26 | 7.78 |
| 298 | −94.01 | −24.77 | 7.79 |
| 323 | −93.95 | −24.55 | 7.75 |
| 348 | −93.44 | −24.09 | 7.71 |
| 373 | −92.16 | −23.37 | 7.83 |
The possible STM images of AS-OPV self-assembly at different temperatures are simulated to visually display the ordered self-assembled patterns. As Fig. 6d and f show, the simulated STM images are similar to that at room temperature. The adlayer generate linear structure with neighboring molecules interdigitate with each other at the simulated temperatures. Comparing with the AS-OPV geometry, the bright and dark strips in the linear structure are composed of AS-OPV backbones and alkyl chains, respectively. The simulated images could serve as a reference to make preliminary prediction for AS-OPV assembled patterns at different temperatures.
4. Conclusions
We have investigated the 2D self-assembly of AS-OPV on HOPG surface by means of DFT and MD simulations together with STM investigation. For AS-OPV self-assembly, from tentative structural model based on STM experiment to proof for validity of conjecture, our multi-scale simulations effectively determine the self-assembly atomic geometry and quantitatively analyze the weak interactions that contribute to this assembled system. For the adlayer, the presence of hydrogen bonds and van der Waals interaction between adjacent AS-OPV molecules allows it to be closely aligned in a certain direction. Meanwhile, the π–π stacking, electrostatic and van der Waals interactions between adlayer and substrate are advantageous for the adsorption of AS-OPV molecules, which cause these molecules to form the planar self-assembled structures on HOPG surface. In addition, the possible self-assembly structure and STM images at different temperatures has been theoretically simulated with the above methods. According to the simulation results, AS-OPV self-assembled structure is stable only when the temperature is lower than 400 K. As the temperature continues to rise, the self-assembled ordered structure is gradually broken. When the temperature exceeds 500 K, half of the ordered structure has been disrupted. This research provide an effective way to explore the contribution of weak interactions and accurately atomic configuration for the self-assembly system. Besides, for other similar self-assembled system, our simulation approach will serve as a model to explore the weak interaction existing in assembled architecture, and that is anticipated to be helpful in designing and controlling functional self-assembly.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No. 21233010), Fundamental Research Funds for the Central Universities (Grant No. DUT16ZD102), the Key Laboratory of Solidification Control and Digital Preparation Technology (Liaoning Province). Part of this work was performed using computational resources from Supercomputing Center of Dalian University of Technology.Notes and references
- J. Tang, H. Shi, X. He, Y. Lei, Q. Guo, K. Wang and D. He, Chem. Commun., 2016, 52, 1482–1485 RSC.
- A. Sandeep, V. K. Praveen, K. K. Kartha, V. Karunakaran and A. Ajayaghosh, Chem. Sci., 2016, 7, 4460–4467 RSC.
- S. K. Nisha and S. Asha, J. Mater. Chem. C, 2014, 2, 2051–2060 RSC.
- Y. Hirai, S. S. Babu, V. K. Praveen, T. Yasuda, A. Ajayaghosh and T. Kato, Adv. Mater., 2009, 21, 4029–4033 CrossRef.
- J. Li, S. Wieghold, M. A. ner, P. Simon, M. V. Hauf, E. Margapoti, J. A. Garrido, F. Esch, C.-A. Palma and J. V. Barth, Nano Lett., 2014, 14, 4486–4492 CrossRef PubMed.
- T. Yokoyama, S. Yokoyama, T. Kamikado, Y. Okuno and S. Mashiko, Nature, 2001, 413, 619–621 CrossRef PubMed.
- F. della Sala, S. Neri, S. Maiti, J. L. Chen and L. J. Prins, Curr. Opin. Biotechnol., 2017, 46, 27–33 CrossRef PubMed.
- M. B. Duriska, S. M. Neville, B. Moubaraki, J. D. Cashion, G. J. Halder, K. W. Chapman, C. Balde, J. F. Létard, K. S. Murray and C. J. Kepert, Angew. Chem., 2009, 121, 2587–2590 CrossRef.
- J. H. Dou, Y. Q. Zheng, Z. F. Yao, Z. A. Yu, T. Lei, X. Shen, X. Y. Luo, J. Sun, S. D. Zhang and Y. F. Ding, J. Am. Chem. Soc., 2015, 137, 15947–15956 CrossRef PubMed.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2011, 112, 2208–2267 CrossRef PubMed.
- Y. Sagara, K. Kubo, T. Nakamura, N. Tamaoki and C. Weder, Chem. Mater., 2017, 29, 1273–1278 CrossRef.
- K. K. Kartha, S. S. Babu, S. Srinivasan and A. Ajayaghosh, J. Am. Chem. Soc., 2012, 134, 4834–4841 CrossRef PubMed.
- S. Izawa, K. Hashimoto and K. Tajima, Phys. Chem. Chem. Phys., 2012, 14, 16138–16142 RSC.
- C. Stangel, A. Bagaki, P. A. Angaridis, G. Charalambidis, G. D. Sharma and A. G. Coutsolelos, Inorg. Chem., 2014, 53, 11871–11881 CrossRef PubMed.
- T. Nishizawa, H. K. Lim, K. Tajima and K. Hashimoto, Chem. Commun., 2009, 2469–2471 RSC.
- C. Vijayakumar, V. K. Praveen and A. Ajayaghosh, Adv. Mater., 2009, 21, 2059–2063 CrossRef.
- J. Wang, K. Liu, R. Xing and X. Yan, Chem. Soc. Rev., 2016, 45, 5589–5604 RSC.
- S. Y. L. Leung, S. Evariste, C. Lescop, M. Hissler and V. W. W. Yam, Chem. Sci., 2017, 8, 4264–4273 RSC.
- G. Yang, H. M. Ding, Z. Kochovski, R. Hu, Y. Lu, Y. Q. Ma, G. Chen and M. Jiang, Angew. Chem., Int. Ed., 2017, 56, 1–6 CrossRef.
- J. D. C. González, M. Iyoda and J. P. Rabe, Nat. Commun., 2017, 8, 14717 CrossRef PubMed.
- W. Bai, Z. Jiang, A. E. Ribbe and S. Thayumanavan, Angew. Chem., 2016, 128, 10865–10869 CrossRef.
- L. Zhang, X. Wang, T. Wang and M. Liu, Small, 2015, 11, 1025–1038 CrossRef PubMed.
- Q. Chen, T. Chen, D. Wang, H.-B. Liu, Y.-L. Li and L.-J. Wan, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2769–2774 CrossRef PubMed.
- Z. Guo, I. De Cat, B. Van Averbeke, J. Lin, G. Wang, H. Xu, R. Lazzaroni, D. Beljonne, E. Meijer and A. P. Schenning, J. Am. Chem. Soc., 2011, 133, 17764–17771 CrossRef PubMed.
- Y. Zhao, Q. Zhou, Q. Li, X. Yao and J. Wang, Adv. Mater., 2017, 29, 1603990 CrossRef PubMed.
- L. Cheng, Y. Li, C. Y. Zhang, Z. L. Gong, Q. Fang, Y. W. Zhong, B. Tu, Q. Zeng and C. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 32004–32010 CrossRef PubMed.
- T. Chen, W. H. Yang, D. Wang and L. J. Wan, Nat. Commun., 2013, 4, 1389 CrossRef PubMed.
- U. Mazur and K. Hipps, Chem. Commun., 2015, 51, 4737–4749 RSC.
- D. C. Y. Nguyen, L. Smykalla, T. N. H. Nguyen, M. Mehring and M. Hietschold, Phys. Chem. Chem. Phys., 2016, 18, 24219–24227 RSC.
- A. Bhattarai, K. Marchbanks-Owens, U. Mazur and K. Hipps, J. Phys. Chem. C, 2016, 120, 18140–18150 CrossRef.
- M. O. Blunt, J. Adisoejoso, K. Tahara, K. Katayama, M. Van der Auweraer, Y. Tobe and S. De Feyter, J. Am. Chem. Soc., 2013, 135, 12068–12075 CrossRef PubMed.
- T. Jiu, Y. Li, X. Liu, H. Liu, C. Li, J. Ye and D. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 911–924 CrossRef.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. Probert, K. Refson and M. C. Payne, Z. Kristallogr. - Cryst. Mater., 2005, 220, 567–570 Search PubMed.
- P. Dauber Osguthorpe, V. A. Roberts, D. J. Osguthorpe, J. Wolff, M. Genest and A. T. Hagler, Proteins: Struct., Funct., Bioinf., 1988, 4, 31–47 CrossRef PubMed.
- A. K. Rappé, C. J. Casewit, K. Colwell, W. Goddard III and W. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef.
- H. Sun, S. J. Mumby, J. R. Maple and A. T. Hagler, J. Am. Chem. Soc., 1994, 116, 2978–2987 CrossRef.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef PubMed.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892 CrossRef.
- J. Tersoff and D. Hamann, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 805 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |